

Visual Abstract

Key Words: diastolic dysfunction, fibrosis, heart failure, myocardial stunning, stretch
Abbreviations and Acronyms: BP, blood pressure; cTnI, cardiac troponin I; EDPVR, end-diastolic pressure−volume relationship; ΔEDP/ΔEDV, changes in end-diastolic pressure/end-diastolic volume; HFpEF, heart failure with preserved ejection fraction; LV, left ventricular; LVEDP, left ventricular end-diastolic pressure; LVEDV, left ventricular end-diastolic volume; RPO, repetitive pressure overload; PE, phenylephrine; PV, pressure−volume; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
Highlights
-
•
A transient elevation in preload produces mechanical stretch-induced myocyte injury and measurable cardiac troponin I release that is associated with reversible contractile dysfunction and myocyte apoptosis.
-
•
Using a porcine model of intermittent pressure overload, this study demonstrates that repetitive exposure to cyclical elevations in preload elicits significant myocyte loss, yet left ventricular systolic function is preserved and chamber dilatation is absent.
-
•
Instead, myocardial remodeling characterized by myocyte hypertrophy and interstitial fibrosis produces a reduction in left ventricular diastolic compliance that protects the heart from subsequent stretch-induced myocyte injury.
-
•
These results support a novel paradigm that links cardiac adaptations to repetitive stretch-induced injury with the pathogenesis of myocardial stiffening and may explain how reductions in left ventricular diastolic compliance can occur in the absence of sustained hypertension or anatomic hypertrophy.
Summary
Swine subjected to 2 weeks of repetitive pressure overload (RPO) exhibited significant myocyte loss, but left ventricular (LV) systolic function was preserved, and chamber dilatation did not occur. Instead, myocardial remodeling characterized by myocyte hypertrophy and interstitial fibrosis led to a marked reduction in LV diastolic compliance, which protected the heart from stretch-induced myocyte injury and preserved LV ejection fraction without anatomic LV hypertrophy. These results support a novel paradigm that links cardiac adaptations to RPO with the pathogenesis of reduced LV diastolic compliance and may explain how LV stiffening can occur in the absence of sustained hypertension or anatomic hypertrophy.
Cardiomyocyte loss is a central component of the adverse left ventricular (LV) remodeling that often underlies deterioration of cardiac performance and the development of chronic heart failure 1, 2. Although myocyte loss is typically believed to arise as the result of myocardial ischemia, it is increasingly clear that other types of pathophysiological stress can contribute to irreversible myocyte injury (3). Excessive mechanical stretch caused by hemodynamic overload has been implicated as an important mechanism from among the potential nonischemic causes of myocyte death based on clinical data that demonstrated elevated serum cardiac troponin (cTn) concentrations in patients with high filling pressures in the setting of heart failure and other fluid overload states (4). In support of this notion, we recently demonstrated that acutely raising arterial blood pressure to transiently elevate LV preload without ischemia in swine elicited reversible systolic dysfunction (“stretch-induced stunning”) and measurable transcoronary release of cTnI, which indicates myocyte injury (5). Although this response occurred without pathological evidence of necrosis or infarction, post-mortem histopathological analysis revealed an increased number of cardiomyocytes undergoing apoptosis. This indicated that even in the absence of ischemia, transient pressure overload elicits stretch-induced myocyte injury.
It is therefore possible that long-term repetitive exposure to brief episodes of pressure overload could lead to cumulative myocyte loss that results in LV systolic dysfunction in the absence of infarction. This notion was supported by our laboratory’s previous work in swine that involved single and repetitive episodes of “reversible” regional ischemia. In these studies, a single 10-min transient coronary occlusion resulted in stunned myocardium and regionally increased myocyte apoptosis without necrosis in the area at risk of ischemia (6). Although the rate of myocyte apoptosis was low, repeated bouts of demand-induced ischemia distal to a chronic coronary stenosis were shown to lead to chronic myocardial stunning that progressed to hibernating myocardium (7). This progression was accompanied by chronic regional systolic dysfunction, myocyte apoptosis, and substantial regional myocyte loss (8). Thus, it is plausible that repetitive brief episodes of preload elevation, each eliciting stretch-induced injury throughout the entire LV, could lead to cumulative myocyte loss that results in global systolic dysfunction. Clinically, a similar process may occur in older adults who experience repetitive episodes of increased afterload due to aortic stenosis, labile hypertension (9), or age-related aortic stiffening (10) that intermittently impose high levels of mechanical stress on the heart, potentially contributing to adverse LV remodeling and heart failure.
Accordingly, we developed a large animal model of repetitive pressure overload (RPO) to test the hypothesis that repeated short-term elevations in hemodynamic load that stretch the myocardium would elicit chronic stretch-induced stunning with significant cardiomyocyte loss and the development of heart failure with reduced ejection fraction. Our results showed that swine subjected to 2 weeks of RPO exhibited rapid and substantial myocyte loss, yet, in contrast to our hypothesis, LV systolic function was preserved and LV dilatation was absent. Instead, myocardial remodeling characterized by myocyte cellular hypertrophy and interstitial fibrosis produced a marked reduction in LV diastolic compliance. This functions as an adaptive mechanism to protect the heart from subsequent stretch-induced stunning and preserves LV ejection fraction in the absence of overt anatomic LV hypertrophy.
Methods
All procedures and protocols conformed to institutional guidelines for the care and use of animals in research and were approved by the University at Buffalo Institutional Animal Care and Use Committee. Long-term RPO studies were conducted in Yorkshire cross-bred pigs (n = 9; 34 ± 2 kg; 7 males/2 females) that were 3 to 4 months of age (WBB Farm, Alden, New York). Seven additional Yorkshire cross-bred pigs were used as control animals for post-mortem histological measurements (n = 4) and/or admittance catheter-derived LV pressure−volume (PV) relations (n = 5). A flowchart summarizing the sample sizes used for each comparison is included in the Supplemental Figure.
Porcine model of RPO
Animals were initially instrumented with an indwelling jugular vein catheter for blood sampling and phenylephrine (PE) infusion. Following sedation with a telazol/xylazine mixture (0.037 ml/kg intramuscularly), anesthesia was maintained by an infusion of propofol (5 to 10 mg/kg/h). After infiltrating the skin and subcutaneous tissues with marcaine (0.5%), a small incision was made, followed by blunt dissection to isolate the jugular vein. A catheter (18-gauge) was advanced into the vessel over a guidewire and secured to the vessel by fixation ligatures with the proximal end connected to polyethylene tubing. This was tunneled subcutaneously and led out through a small incision between the shoulder blades. Animals subsequently underwent multidetector computed tomography (MDCT) and physiological studies as described in the following sections.
Physiological study protocol
Hemodynamics and echocardiographic indexes of LV function were assessed before, during, and 1 h after PE-induced pressure overload in the closed-chest anesthetized state. On return to the laboratory following MDCT imaging, a micromanometer (5-F, SPR-450, AD Instruments, Inc., Colorado Springs, Colorado) was advanced through the introducer and into the LV under fluoroscopic guidance for assessment of LV pressure, whereas arterial blood pressure (BP) was continuously monitored via the side port. LV volumes and wall thickening were assessed with transthoracic echocardiography (GE Vivid 7, Milwaukee, Wisconsin) 5, 6. Briefly, the LV was imaged in the short- and long-axes projections from a right parasternal approach. Short-axis images were used to assess regional wall thickening, end-systolic and end-diastolic volumes, and LV ejection fraction using American Society of Echocardiography criteria (11). We subsequently infused PE (2 mg/ml infused at 9 ml/h) for 1 h to transiently raise LV systolic and LV end-diastolic pressure (LVEDP). Hemodynamic parameters were continuously recorded, and echocardiography was performed every 15 min throughout the infusion period and for 1 h after PE was stopped. Hemodynamic measurements at selected time points reported in the results section were obtained from 5-s digital averages before and after echocardiographic data collection that were averaged to obtain each value. During the time period of each reported measurement, there was little change in hemodynamic variables due to the stable nature of the experimental preparation. Following data collection at the 1-h post-PE timepoint, catheters were removed, anesthesia was discontinued, and the animal was returned to the animal housing facility. Blood samples for serum troponin I (5) were collected at baseline, as well as 1 and 24 h after PE infusion.
After completing the initial study, the jugular vein catheter was connected to an external programmable infusion pump (OrchesTA Model 500, Instech Laboratories, Plymouth Meeting, Pennsylvania) housed in a standard laboratory animal jacket. The infusion pump was manually programmed to infuse PE to produce RPO for 2 h daily (2 mg/ml infused at 9 ml/h for 2 h/day) for 2 weeks. Thus, the duration of each daily PE infusion was 2 h in conscious animals during the RPO period, whereas an infusion period of 1 h was used in anesthetized animals during physiological studies before and after RPO. On day 14, animals were re-anesthetized for repeat MDCT imaging and a final physiological study. Animals were recovered and a repeat blood sample was obtained 24 h later (day 15). A subset of animals were anaesthetized to obtain LV PV relations as previously described (12) and detailed in the following section. After all measurements and blood sampling was completed, animals were deeply anesthetized with isoflurane (5%) and killed via administration of potassium chloride directly into the LV chamber. The heart was rapidly removed and sectioned for post-mortem tissue sampling.
LV Diastolic Compliance via Admittance Catheter
LV PV loops were obtained from a subset of animals (n = 5) for assessment of diastolic compliance via placement of an admittance catheter (ADV500, Transonic Scisense, Inc., London, Ontario, Canada) following collection of the 24-h post-PE blood sample after 2 weeks of RPO as previously described (12). These were compared to values in normal swine (n = 4). Recent data from our laboratory that compared admittance catheter-derived ventricular volumes to simultaneously acquired contrast-enhanced, MDCT-derived ventricular volumes demonstrated the accuracy of this approach (13). The LV end-diastolic PV relationship (EDPVR) and end-systolic PV relationship (ESPVR) were measured by collecting PV loops during transient preload reductions induced by rapid inferior vena cava occlusion using a 12-F, 20-mm balloon catheter (Z-Med, B. Braun Interventional Systems, Inc., Bethlehem, Pennsylvania) placed via the femoral vein. In each case, LV pressure and volume at end-systole (for the ESPVR) and end-diastole (for the EDPVR) were identified during each cardiac cycle throughout the transient preload reduction period. Nonlinear regression was applied to characterize the slope (i.e., end-systolic elastance) and volume-axis intercept (V0) of the ESPVR using a quadratic fit, in which Pes = a • Ves2 + b • Ves + c, with Pes representing end-systolic LV pressure, Ves representing end-systolic volume, and constants a, b, and c determined by nonlinear least-squares regression (14). For characterization of the EDVPR, an exponential curve fit was applied to calculate the LV diastolic stiffness constant (β) using the equation , where Pes is end-systolic LV pressure, Ves is end-systolic volume, and A and β are empirically determined constants (15). Additional PV loops were obtained during a brief (15 min) infusion of PE to corroborate measurements of changes in end-diastolic pressure/end-diastolic volume (ΔEDP/ΔEDV) during PE made via echocardiographic assessment of LV EDV (n = 3/group). Acquisition and analysis of all LV PV parameters were performed with an IX-228S data acquisition system and PV module data analysis LabScribe2 software (iWorx Systems, Inc., Dover, New Hampshire).
Cardiac MDCT imaging
MDCT imaging was performed before initiating PE infusion at the initial study and 2 weeks after RPO to assess LV remodeling. Under anesthesia (as described previously), the femoral artery and vein were catheterized with introducers (6-F) and the animals were transported to the scanner. Animals were placed in the supine position and scanned on a 64-slice GE Discovery 690 positron-emission tomographic/CT scanner during suspended respiration to minimize movement-related artifact. After scout acquisition to localize the heart, iohexol (Omnipaque, GE Healthcare; 350 mg iodine/ml; 2 ml/kg) was injected via the femoral vein, followed by a 30-ml saline chaser (both infusions at 4 ml/s). First-pass imaging with retrospective electrocardiographic gating was performed to assess LV mass and LV volumes in 8-mm-thick short-axis slices reconstructed at end-diastole (i.e., 95% of the RR interval) and analyzed using ImageJ software (National Institutes of Health, Bethesda, Maryland). The following parameters were used for each scan: gantry rotation time 400 ms, temporal resolution 175 ms, slice thickness 0.625 mm, spatial resolution 0.97 × 0.97 mm (voxel size 0.97 × 0.97 × 0.625 mm), helical pitch variable depending on heart rate (range: 0.20 to 0.26), tube voltage 120 kV, and tube current 600 mA. Upon completion of MDCT imaging, the animals were transported back to the laboratory for physiological studies.
Post-mortem myocardial histopathology
Histopathological data derived from animals subjected to 2 weeks of RPO (n = 8) were compared with values obtained from normal control swine (n = 4; 3 males/1 female) matched for age, sex, and body mass.
Myocyte nuclear density and morphometry
Paraffin-embedded myocardial tissue samples were fixed for morphometry as previously described (16). PAS-stained sections were used to quantify myocyte diameter by counting at least 100 cells from the inner and outer halves of each tissue section. Myocytes were included regardless of size as long as myofilaments could be identified surrounding the nucleus. Myocyte nuclear density was also assessed in periodic acid-Schiff−stained sections as previously described (16).
Interstitial fibrosis
Cardiac fibrosis was assessed via light microscopy of picrosirius red−stained myocardial tissue sections and quantification of collagen volume fraction, as previously described (17).
Myocyte apoptosis
Cardiomyocyte apoptosis was assessed using the In-Situ Cell Death Detection Kit (Roche Diagnostics, Mannheim, Germany) according to manufacturer’s guidelines 5, 6. Briefly, apoptotic cells were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) and epiflourescence with a fluorescein isothiocyanate filter. Samples were co-stained with an F-Actin antibody conjugated to fluorescent Alexa Fluor 555 dye (Alexa Fluor 555 Phalloidin, Life Technologies, Burlington, Ontario, Canada), and co-localization with TUNEL was used to quantify apoptotic cardiomyocytes. At least 100 microscopic fields (132 ± 5 fields; 200×) were examined per sample, and the number of apoptotic myocytes was expressed as TUNEL+ myocytes per square centimeters. Only TUNEL+ nuclei that could be confirmed to be from myocytes via F-Actin co-staining were included.
Capillary density
Myocardial tissue sections were incubated with anti−von Willebrand factor antibodies with appropriate fluorescent-labeled secondary antibodies to identify coronary capillaries, as previously described (16). Vessels were counted in at least 20 random fields from each tissue sample and expressed per tissue area (millimeters squared).
Assessment of serum cTnI
Blood samples were allowed to clot at room temperature for 40 min, centrifuged at 1,500g for 15 min, aliquoted, and frozen for storage at −80°C. Serum was thawed once, and cTnI quantified in duplicate with a porcine specific cTnI enzyme-linked immunosorbent assay kit for serum (Life Diagnostics) according to manufacturer’s instructions. We previously demonstrated that the 99th percentile for normal values of cTnI in swine was 38 ng/l with this assay 5, 6.
Statistical analysis
Data are expressed as mean ± SE. Histopathological, MDCT, and LV PV loop-derived data analysis was performed simultaneously in RPO and control animals in a blinded fashion to ensure scientific rigor. To control for animal growth during the 2-week study period (8 ± 1 kg), LV volumes were expressed relative to body surface area, which was calculated by multiplying body mass (in kilograms) by 0.0734 (18). Differences between time points in physiological studies (e.g., baseline vs. post-PE) were assessed by repeated measures analysis of variance and the post hoc Holm-Sidak test. Although surgical placement of an indwelling jugular vein catheter was successful in all animals, the catheter was displaced in 1 animal 2 days after the initial study. As a result, a total of 8 animals completed the 2-week RPO protocol, and temporal physiological changes between the initial study (n = 9) and after 2 weeks RPO (n = 8) were assessed by unpaired t-tests. Unpaired t-tests were also used to assess differences in PV loop endpoints and post-mortem histopathology between animals subjected to RPO and normal control animals. Statistical analysis was performed with SPSS Statistics 23 (IBM, Armonk, New York), and the acceptable type 1 error rate was prospectively set at 5%.
Results
Hemodynamics and echocardiography after RPO
Hemodynamics and echocardiography before, during, and 1 h after intravenous PE at the initial study and after 2 weeks of RPO are summarized in Tables 1 and 2. On day 1, PE increased peak LV systolic pressure from 102 ± 3 mm Hg to 192 ± 4 mm Hg (p < 0.0001) with a corresponding rise in LVEDP from 11 ± 1 mm Hg to 31 ± 2 mm Hg (p = 0.0002). These returned to baseline within 30 min after PE infusion (Figure 1). Despite normalization of hemodynamics after LV pressure overload, LVEDV remained elevated (from 20 ± 1 ml/m2 to 29 ± 5 ml/m2; p = 0.0565) (Figure 1). As a result, LV ejection fraction declined from 69 ± 3% to 51 ± 7% (p = 0.0217) (Figure 2).
Table 1.
Hemodynamic Response to Intravenous Phenylephrine Before and After 2 Weeks of RPO
| Baseline | 60-Min | 60-Min Post-PE | |
|---|---|---|---|
| Heart rate, beats/min | |||
| Initial | 92 ± 7 | 84 ± 5 | 90 ± 7 |
| 2-week RPO | 96 ± 4 | 87 ± 7 | 97 ± 8 |
| Mean arterial pressure, mm Hg | |||
| Initial | 86 ± 5 | 167 ± 2∗ | 80 ± 5† |
| 2-week RPO | 84 ± 6 | 176 ± 8∗ | 91 ± 9† |
| LV peak systolic pressure, mm Hg | |||
| Initial | 102 ± 3 | 192 ± 4∗ | 99 ± 4† |
| 2-week RPO | 104 ± 5 | 210 ± 10∗ | 112 ± 9† |
| LV end-diastolic pressure, mm Hg | |||
| Initial | 11 ± 1 | 31 ± 2∗ | 12 ± 1† |
| 2-week RPO | 12 ± 1 | 26 ± 1∗ | 12 ± 2† |
| dP/dtmax, mm Hg/s | |||
| Initial | 2,243 ± 88 | 3,290 ± 579 | 1,605 ± 170∗† |
| 2-week RPO | 2,465 ± 201 | 3,632 ± 436∗ | 2,195 ± 278† |
| dP/dtmin, mm Hg/s | |||
| Initial | −2,217 ± 162 | −3,802 ± 675 | −2,108 ± 233 |
| 2-week RPO | −2,016 ± 156 | −4,413 ± 282∗ | −3,023 ± 300∗†‡ |
Values are mean ± SEM.
LV = left ventricular; PE = phenylephrine; RPO = repetitive pressure overload.
p < 0.05 vs. baseline.
p < 0.05 vs. 60-min PE.
p < 0.05 vs. initial.
Table 2.
Selected Echocardiographic Parameters Before, During, and After Intravenous Phenylephrine at the Initial Study and After 2-Weeks of RPO
| Baseline | 60-Min PE | 60-Min Post-PE | |
|---|---|---|---|
| LV end-diastolic volume, ml/m2 | |||
| Initial | 20 ± 1 | 34 ± 4∗ | 29 ± 5∗ |
| 2-week RPO | 20 ± 1 | 22 ± 2† | 17 ± 1†‡ |
| LV end-systolic volume, ml/m2 | |||
| Initial | 6 ± 1 | 21 ± 6∗ | 16 ± 5∗ |
| 2-week RPO | 6 ± 1 | 7 ± 1† | 5 ± 1†‡ |
| Stroke volume, ml/m2 | |||
| Initial | 14 ± 1 | 13 ± 2 | 13 ± 1 |
| 2-week RPO | 14 ± 1 | 14 ± 1 | 12 ± 1 |
| LV ejection fraction, % | |||
| Initial | 69 ± 3 | 46 ± 9∗ | 51 ± 7∗ |
| 2-week RPO | 71 ± 2 | 66 ± 2† | 70 ± 2† |
| Septal wall thickening, mm | |||
| Initial | 5.8 ± 0.4 | 3.8 ± 0.6∗ | 3.6 ± 0.5∗ |
| 2-week RPO | 6.0 ± 0.3 | 5.2 ± 0.4∗ | 5.7 ± 0.3† |
| Posterior wall thickening | |||
| Initial | 5.9 ± 0.4 | 4.1 ± 0.8∗ | 3.8 ± 0.4∗ |
| 2-week RPO | 6.7 ± 0.4 | 6.5 ± 0.4† | 5.8 ± 0.2† |
Values are mean ± SEM.
Abbreviations as in Table 1.
p < 0.05 vs. baseline.
p < 0.05 vs. initial.
p < 0.05 vs. 60-min PE.
Figure 1.

RPO Elicits a Reduction in LV Compliance That Prevents Chamber Distension During an Acute Elevation in Afterload
A 60-min intravenous infusion of phenylephrine (PE) elicited a similar increase in arterial systolic blood pressure (SBP) and left ventricular end-diastolic pressure (LVEDP) at the initial study (blue) and after 2 weeks of repetitive pressure overload (RPO) (purple). At the initial study, this was associated with an increase in left ventricular end-diastolic volume (LVEDV) that persisted after cessation of PE despite normalization of SBP and LVEDP. However, transient PE infusion no longer produced a significant increase in LVEDV when the study was repeated after 2 weeks of RPO, which was indicative of reduced LV compliance after RPO. Values are mean ± SEM. *p < 0.05 versus baseline; †p < 0.05 versus initial.
Figure 2.
Cardiac Adaptations to RPO Prevent Acute Stretch-Induced Stunning and Myocyte Injury During a Transient Elevation in Afterload
The attenuation of LV distension during PE infusion after RPO was associated with a preserved LV ejection fraction and the absence of a significant elevation in serum cardiac troponin I (cTnI) concentrations, indicating that RPO led to an adaptive reduction in LV compliance that protected the heart from stretch-induced stunning and myocyte injury during PE-mediated pressure overload. Values are mean ± SEM. *p < 0.05 versus baseline. Representative videos of short-axis echocardiograms that illustrate the LV response to PE at the initial study (Supplemental Videos 1A, 1B, 1C) and after 2-weeks of RPO (Supplemental Videos 2A, 2B, 2C) are included in the Supplemental Appendix. Abbreviations as in Figure 1.

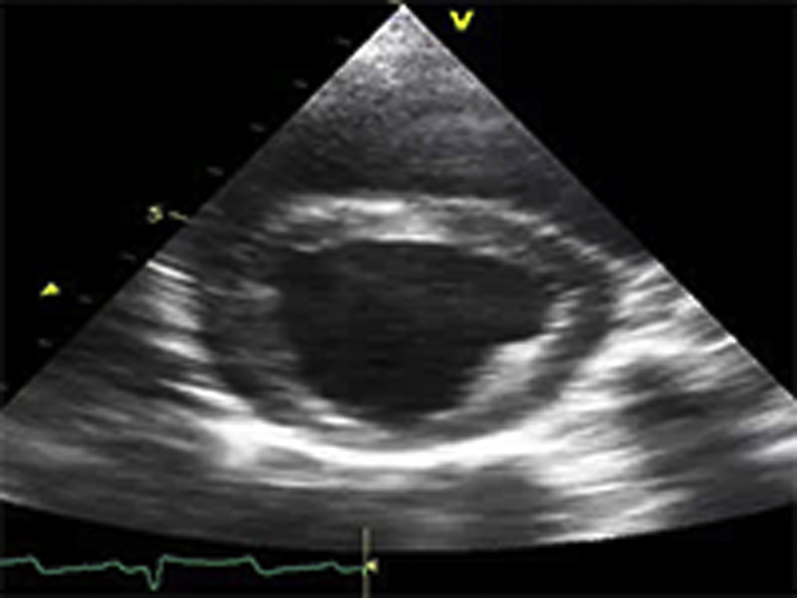
Video Files 1A-1C: Example Left Ventricular Short-Axis Echocardiograms at the Initial Study
Short-axis echocardiograms from the initial study demonstrate normal left ventricular systolic function at rest (arterial blood pressure (BP): 124/78 mmHg; Video 1A), followed by marked dilatation and systolic dysfunction during phenylephrine (PE) infusion (arterial BP: 216/155 mmHg; Video 1B) that persists following cessation of PE despite normalization of arterial BP (arterial BP: 125/86 mmHg; Video 1C).
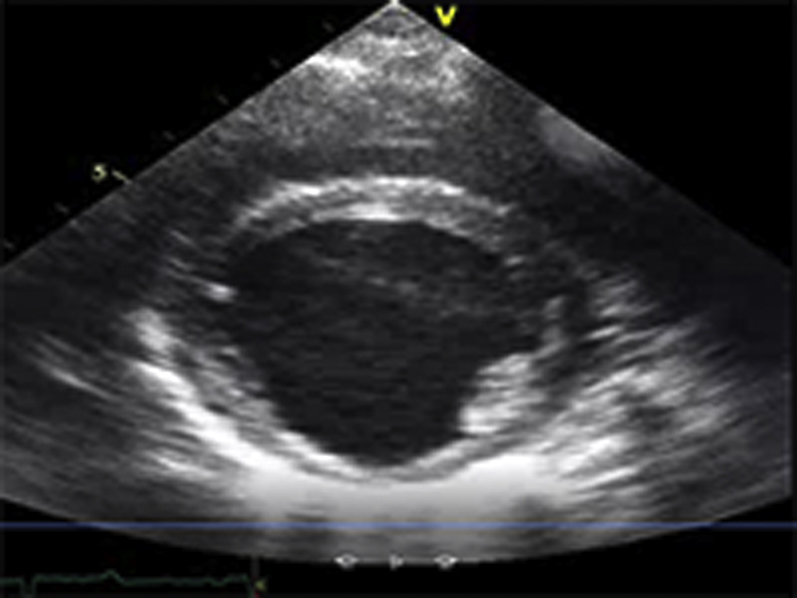
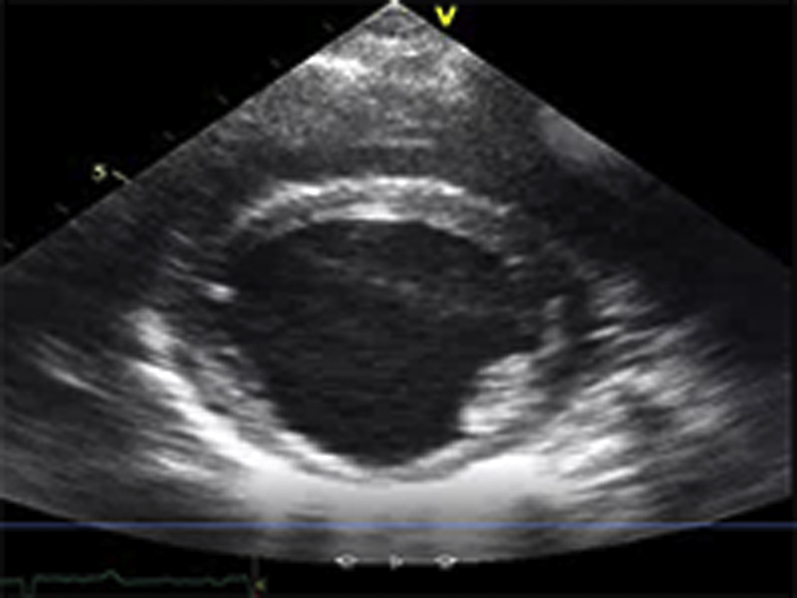
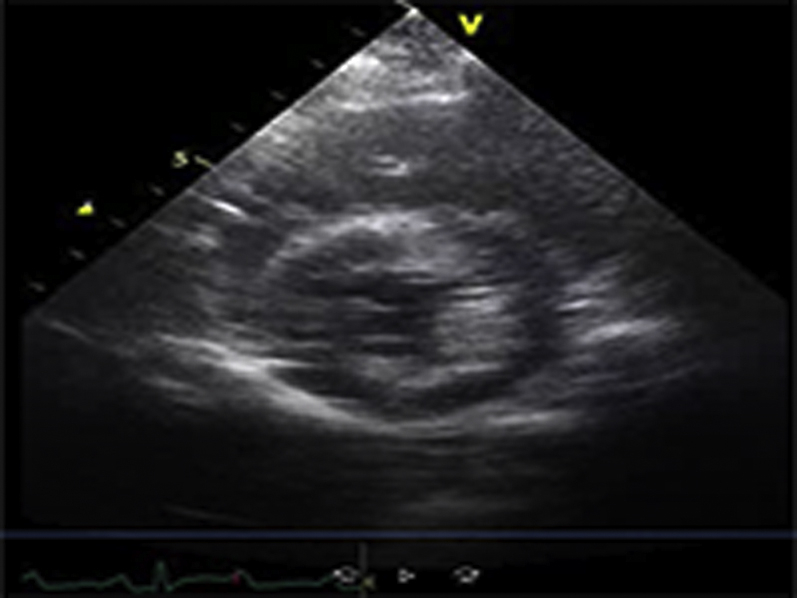
Video Files 2A-2C: Example Left Ventricular Short-Axis Echocardiograms After 2-weeks of Repetitive Pressure Overload
Short-axis echocardiograms from the same animal shown in Video Files 1A-1C obtained after 2-weeks of RPO demonstrate normal left ventricular systolic function at rest (arterial blood pressure (BP): 102/61 mmHg; Video 2A) as well as during (arterial BP: 230/154 mmHg; Video 2B) and after (arterial BP: 132/82 mmHg; Video 2C) phenylephrine (PE) infusion despite a similar arterial BP response to PE at both studies.
*Note: All example echocardiograms were collected from the same animal subject.
After 2 weeks of RPO, hemodynamics and echocardiographic parameters of LV function were normal at rest (Tables 1 and 2). Although PE infusion at this time point produced a comparable increase in LV systolic pressure (from 104 ± 5 mm Hg to 210 ± 10 mm Hg; p < 0.0001) and LVEDP (from 12 ± 1 mm Hg to 26 ± 1 mm Hg; p < 0.0001), echocardiographic assessment of LV volumes revealed a markedly different effect on LV function. In contrast to the initial study, there was only a small, insignificant increase in LVEDV during PE infusion after 2 weeks of RPO (from 20 ± 1 ml/m2 to 22 ± 2 ml/m2; p = 0.4347). This reflected a substantial reduction in myocardial compliance (Figure 1). The absence of LV distension prevented stretch-induced stunning because the LV ejection fraction remained normal during (66 ± 2%) and after (70 ± 2%) PE infusion (Figure 2, left panel). Representative videos of short-axis echocardiograms that illustrated the LV response to PE at the initial study (Supplemental Videos 1A, 1B, and 1C) and after 2 weeks of RPO (Supplemental Videos 2A, 2B, and 2C) are included in the Supplemental Appendix.
Serum troponin I release before and after RPO
Figure 2 (right panel) summarizes the effects of pressure overload on cTnI release. One hour after the initial episode of pressure overload, the LV distension and reduction in systolic function were accompanied by a significant increase in serum cTnI (from 13 ± 9 ng/l to 186 ± 69 ng/l; p = 0.0075) that persisted 24 h later (260 ± 141 ng/l; p = 0.0419 vs. baseline). There was a positive correlation between the degree of LV distension during PE infusion (LVEDV) and serum cTnI concentrations at 1 h (r = 0.75; p = 0.0199) and 24 h (r = 0.77; p = 0.0152) post-PE infusion, which suggested that the extent of LV dilatation and stretch was a determinant of myocyte injury. After 14 days of RPO, transient pressure overload produced minimal LV distention and no increase in serum cTnI (from 5 ± 5 ng/l to 15 ± 10 ng/l; p = 0.2793) (Figure 2). Consistent with this, the number of apoptotic myocytes identified via post-mortem TUNEL staining was not different between animals subjected to 2 weeks of RPO (0.8 ± 0.5 myocytes/cm2) and normal control animals (0.7 ± 0.5 myocytes/cm2; p = 0.9085).
Effect of RPO on MDCT-derived indexes of LV remodeling
Serial cardiac MDCT imaging was performed to assess LV mass and geometry before and after 2 weeks of RPO. RPO did not cause significant anatomic hypertrophy because the LV mass/body weight ratio was similar to that of normal control animals (2.3 ± 0.1 g/kg) at the initial study (2.3 ± 0.1 g/kg; p = 0.9324) and after 2 weeks of RPO (2.4 ± 0.1 g/kg; p = 0.3716). However, it did produce mild concentric remodeling, with an increase in the LV mass/LVEDV ratio from 1.13 ± 0.04 in normal animals to 1.32 ± 0.07 g/ml after RPO (p = 0.0231) (Figure 3).
Figure 3.

RPO Produces Significant Cardiomyocyte Loss and Compensatory Cellular Hypertrophy of Remaining Myocytes
Examples of periodic acid-Schiff−stained myocardial tissue sections from a normal control animal (left) and an animal subjected to RPO (right) illustrate the reduction in myocyte nuclear density and increase in myocyte diameter elicited by RPO. Although RPO did not elicit a change in total LV mass, the LV mass/LVEDV was increased compared with normal control animals (n = 20), which was indicative of concentric LV remodeling in the absence of overt anatomic hypertrophy in swine subjected to RPO. Values are mean ± SEM. Abbreviations as in Figure 1.
Cellular hypertrophy, myocyte loss, and interstitial fibrosis after RPO
Although anatomic hypertrophy was absent, post-mortem histopathological analysis of LV tissue revealed myocyte cellular hypertrophy (myocyte diameter 16.8 ± 0.4 vs. 13.5 ± 0.1 μm; p < 0.0001) in animals subjected to RPO compared with normal control animals (Figure 3). The increase in myocyte size in the face of comparable LV mass reflected substantial global myocyte loss arising from RPO. After only 2 weeks of RPO, myocyte nuclear density declined by nearly 20%, from 1,455 ± 23 nuclei/mm2 in normal control animals to 1,184 ± 29 nuclei/mm2 after RPO (p = 0.0001). This was accompanied by a marked increase in interstitial fibrosis (12.9 ± 1.8% area vs. 6.5 ± 1.5% area; p = 0.0431) (Figure 4). Immunostaining also demonstrated capillary rarefaction with reductions in capillary density in animals subjected to RPO (1,542 ± 26 capillaries/mm2 vs. 1,745 ± 79 capillaries/mm2 in normal control animals; p = 0.0301). Collectively, these data demonstrated that 2 weeks of RPO led to myocardial remodeling characterized by substantial myocyte loss, myocyte cellular hypertrophy, increased interstitial connective tissue, and capillary rarefaction.
Figure 4.

Swine Subjected to RPO Exhibit Increased Interstitial Fibrosis
Picrosirius red−stained tissue sections demonstrate a marked increase in interstitial collagen deposition (red areas) in myocardial samples from animals subjected to RPO compared with normal control swine. Values are mean ± SEM. Abbreviations as in Figure 1.
Reduced myocardial compliance after RPO
The small changes in LVEDV, despite a large increase in LVEDP during PE infusion at the 2-week study, indicated that RPO elicited a significant reduction in LV compliance. This was further evaluated before and after PE infusion using admittance catheter-derived PV loops (Figure 5A) and 2-point diastolic PV curves to quantify the change in diastolic compliance (i.e., ΔEDV/ΔEDP ratio during PE infusion) (Figure 5B). This analysis revealed an approximate 65% reduction in the ΔEDV/ΔEDP ratio (from 1.7 ± 0.2 to 0.6 ± 0.2 ml/mm Hg; p = 0.0025), which demonstrated a marked reduction in LV diastolic compliance after RPO. We also assessed admittance catheter-derived LV PV loops during changes in preload elicited by rapid inferior vena cava occlusion in animals subjected to RPO (n = 5). These also revealed reduced diastolic compliance after RPO as assessed using the EDPVR (Figure 6A). Compared with normal control animals (n = 5), RPO animals exhibited a higher LV stiffness constant (β: 0.053 ± 0.005 vs. 0.035 ± 0.005; p = 0.0258), which reflected an upward and leftward shift of the EDPVR (Figure 6B). This was accompanied by a significant increase in the slope of the ESPVR (from 7.1 ± 0.5 mm Hg/ml to 10.7 ± 1.3 mm Hg/ml; p = 0.0349) without a significant change in the volume axis intercept (from 10.5 ± 3.6 ml to 18.1 ± 4.6 ml; p = 0.2545), which indicated an increase in end-systolic elastance after RPO.
Figure 5.

RPO Reduces LV Diastolic Compliance
(A) Example admittance catheter-derived LV pressure−volume loops at rest and during PE infusion demonstrate that swine subjected to 2 weeks of RPO exhibited a steeper end-diastolic pressure-volume relationship during elevations in afterload. (B) This was visualized via construction of 2-point diastolic pressure−volume curves and quantified via calculation of the ΔLVEDV/ΔLVEDP ratio during acute episodes of PE-induced pressure overload, which was significantly lower after RPO. Values are mean ± SEM. EDPVR = end-diastolic pressure-volume relationship; other abbreviations as in Figure 1.
Figure 6.

RPO Elicits an Upward and Leftward Shift in the LV EDPVR
(A) Example admittance catheter-derived LV pressure−volume loops during transient inferior vena cava occlusion show a steeper EDPVR after 2 weeks of RPO compared with normal control animals. (B) The upward and leftward shift of the EDPVR led to a higher LV diastolic chamber stiffness constant (β) in animals subjected to RPO compared with normal control swine. Values are mean ± SEM; n = 5/group. Abbreviations as in Figures 1 and 5.
Discussion
The results of the present study offered novel insight regarding changes in LV diastolic compliance and the myocardial adaptations to intermittent hemodynamic overload. First, we reproduced our recent finding (5) that an acute elevation in LV preload elicited by a transient increase in arterial BP produced LV distension, reversible stretch-induced myocardial stunning, and a significant rise in circulating cTnI concentrations, which are indicative of myocyte injury in the normal heart. When the heart was exposed to long-term transient elevations in preload via RPO, a reduction in LV compliance developed. This was accompanied by an increase in interstitial connective tissue that ultimately prevented chronic stretch-induced stunning and myocyte injury. Although this adaptive decrease in compliance did not affect LV filling pressures at rest, it was associated with an upward and leftward shift of the EDPVR that resulted in marked elevations in LVEDP, with relatively minor increases in LVEDV when loading conditions were altered. Collectively, these results demonstrated that reductions in LV compliance that were independent of anatomic hypertrophy could occur relatively quickly following repetitive hemodynamic overload and could serve as an adaptive mechanism to protect the heart from subsequent stretch-induced stunning.
Reduced LV diastolic compliance prevents stretch-induced myocyte injury
Among the potential nonischemic causes of myocyte injury, myocyte diastolic stretch from elevations in preload has been implicated as an important mechanism. This is based on preclinical studies that demonstrated apoptosis (19) and troponin I degradation (20), as well as clinical observations that demonstrated elevated cTn concentrations in patients with chronically elevated LV filling pressures in the setting of heart failure (3). Although elevations in circulating cTnI are commonly interpreted as evidence of ischemic myocyte necrosis and is the standard biomarker used to diagnose myocardial infarction, we recently demonstrated that cTnI rapidly rises above normal after brief ischemia (6) and nonischemic myocardial stress. Elevating LVEDP for 1 h elicited prolonged cTnI release with an approximate 6-fold increase in myocyte apoptosis (5). The latter changes occurred without subendocardial ischemia or histopathological evidence of myocyte necrosis. These results complemented earlier studies in isolated myocardium (21) and an intact LV during brief episodes of pressure overload (22), in which time-dependent alterations of the passive length−tension relationship following elevations in systolic and diastolic pressures were ascribed to the viscous properties of creep and stress relaxation (23). Our data demonstrated that creep was not simply a mechanical phenomenon, but was also associated with myocardial damage as assessed by circulating cTnI concentrations and post-mortem measurements of myocyte apoptosis.
The present findings built on this observation by demonstrating that the LV quickly adapts in a fashion that attenuates stretch-induced myocyte injury. Animals subjected to 2 weeks of RPO exhibited cumulative myocyte loss as reflected by a nearly 20% reduction in myocyte nuclear density compared with normal control animals. Nevertheless, after 2 weeks of RPO, they were protected from the development of stretch-induced myocardial injury, and the ejection fraction remained normal before, during, and after preload elevation. This largely reflected reduced diastolic strain arising from increases in interstitial connective tissue. The latter adaptation ultimately prevented further stretch-induced myocyte injury, as indicated by the attenuation of cTnI release following 14 days of RPO. The adaptive reduction in LV compliance also prevented the development of progressive LV dilatation and chronic systolic dysfunction.
The cumulative myocyte loss that developed from RPO in concert with the proliferation of interstitial connective tissue was substantial and of a similar magnitude to that which we previously reported regionally in response to chronic repetitive ischemia (8). In the latter circumstance, the progressive loss of myocytes was accompanied by regional contractile dysfunction and the development of hibernating myocardium. Like chronic hibernating myocardium, the myocyte loss elicited by long-term RPO was accompanied by compensatory cellular hypertrophy of remaining myocytes, which allowed total LV mass to remain normal without the development of anatomic LV hypertrophy. Surprisingly, and in contrast to the deterioration in systolic function from chronic repetitive ischemia, LV ejection fraction was preserved in animals subjected to RPO in the present study. Instead of systolic dysfunction, RPO led to a marked reduction in LV diastolic compliance. This developed in concert with substantial remodeling of the extracellular matrix, because histopathological evaluation of picrosirius red−stained tissue sections revealed that interstitial collagen deposition was nearly doubled in swine subjected to RPO. Thus, the LV response to repetitive myocyte injury appears to be dependent on the nature of the physiological stimulus (i.e., ischemia stretch vs. mechanical stretch). In both circumstances, a constellation of phenotypic changes occurred that appeared to be adaptive in nature. In the case of hibernating myocardium and repetitive ischemia, these included a downregulation in oxidative metabolism that lowered myocardial oxygen demand to protect against further ischemia-induced injury, with only minor increases in interstitial connective tissue (24). In contrast, the myocardial adaptations to RPO led to a reduction in diastolic compliance with preserved systolic function.
The mechanisms responsible for reducing LV compliance might extend beyond the increased interstitial connective tissue demonstrated pathologically. For example, hypertrophied myocytes might also exhibit alterations in cellular proteins responsible for diastolic compliance. In this regard, isoform changes and/or post-translational modifications of the giant sarcomeric protein titin might have contributed to reductions in LV compliance by increasing myocyte-dependent stiffness (25). An adaptive increase in myocyte stiffness was recently shown to occur in remote, noninfarcted myocardium after myocardial infarction via relatively rapid alterations in titin phosphorylation (26). Furthermore, titin was shown to activate signaling pathways that regulate cellular hypertrophy in response to changes in mechanical stress elicited by alterations in hemodynamic load (27), which suggested a potentially important link among titin, myocyte stiffness, and cellular hypertrophy. Further studies will be required to identify myocyte structural alterations and to quantify the extent that these also contribute to myocardial diastolic properties in the present RPO model.
Consequences of myocardial remodeling elicited by RPO
Our findings indicated that the reduction in diastolic LV compliance elicited by RPO might be a double-edged sword. Although these alterations provide protection against stretch-induced myocyte injury, the alterations in passive filling mechanics will elevate LV diastolic pressure at any LV volume and could conceivably limit the extent that cardiac output increases during exercise. This pattern of myocardial remodeling was similar to that observed in many patients with HFpEF. Although the etiology of this clinical syndrome is heterogeneous, and therefore, difficult to fully reproduce in a single experimental model, it is consistently characterized by reductions in LV chamber compliance that compromise the ability of the heart to increase cardiac output because of prominent increases in LV filling pressure during exercise. Several phenotypic characteristics exhibited by the animals subjected to RPO in the present study recapitulated aspects of cardiac remodeling that were reported in a large number of human patients with HFpEF. These included cellular myocyte hypertrophy, interstitial fibrosis, and capillary rarefaction demonstrated histologically, as well as concentric LV remodeling in the absence of anatomic hypertrophy frequently seen on cardiac imaging 28, 29, 30, 31. Thus, it was intriguing to speculate that the myocardial adaptations to intermittent stretch-induced injury from transient pressure overload might lead to reductions in LV diastolic compliance in the absence of chronic hypertension and anatomic LV hypertrophy.
Methodological limitations
Several experimental considerations merit discussion. First, we used intravenous PE to pharmacologically increase arterial BP and elevate LV preload to induce RPO. Although we were unable to completely rule out a direct effect of PE on myocardial remodeling in the present study, several lines of evidence argued against this notion. Although alpha-adrenergic receptors are expressed in the swine heart at similar levels to that observed in humans, this level of expression is markedly lower than that of rodents (particularly rats) from which isolated myocytes are often used to study PE-induced cellular hypertrophy (32). Coronary vascular expression of alpha-adrenergic receptors is minimal or absent in swine (33), and our own data demonstrated that subendocardial perfusion and coronary flow reserve were unaffected by PE infusion at this dose (5), which indicated that PE-mediated coronary vasoconstriction was unlikely to be involved. In addition, a previous study in mice that used a mechanical approach to induce intermittent aortic constriction also observed myocyte cellular hypertrophy in the absence of anatomic hypertrophy (34). Although the LV response to acute pressure overload was not examined in that study, chronic intermittent aortic constriction was associated with a leftward and upward shift of the LV EDPVR and preserved ejection fraction. Although myocyte nuclear density, cTnI, or acute diastolic strain were not reported, the cellular hypertrophy in the absence of anatomic hypertrophy was consistent with stretch-induced myocyte loss. Thus, regardless of whether a mechanical or pharmacological approach is used to elicit RPO, it leads to myocyte loss with a compensatory increase in cardiomyocyte size. Second, it was important to acknowledge that age-, sex-, and body mass−matched swine were used for assessment of histopathology and LV PV analysis in normal control animals, instead of true sham-instrumented animals. Third, conclusions regarding myocyte loss based on changes in myocyte nuclear density assumed that a systematic change in the number of nuclei per myocyte did not occur. However, the observation of myocyte cellular hypertrophy (i.e., increase in myocyte cell diameter) in the absence of a change in LV mass was consistent with the notion that there were fewer myocytes per gram of tissue and net myocyte loss following RPO. Fourth, although microsphere perfusion measurements from our previous study that examined acute pressure overload were consistent with the absence of subendocardial ischemia during PE infusion (5), we did not assess myocardial blood flow in the present study, and therefore, could not completely rule out the presence of subendocardial ischemia during prolonged and/or repeated PE infusions. Finally, although swine subjected to RPO failed to demonstrate overt signs and symptoms of heart failure at rest, several clinical studies of exercise hemodynamics in patients with established HFpEF showed that a large number of patients only exhibited elevated LV filling pressures during exercise and displayed fairly normal resting hemodynamics 35, 36, 37. Data from the present study demonstrated a steeper LV EDPVR and an increased ΔEDP/ΔEDV ratio with increased afterload after RPO and supported the notion that these animals would exhibit impaired diastolic reserve and attenuated increases in cardiac output during exercise. Future studies that use invasive assessment of exercise hemodynamics, which are often neglected in pre-clinical animal studies (38), will be critical to evaluate HFpEF pathophysiology in this model.
Conclusions
In summary, our results demonstrated that repetitive episodes of transient preload elevation elicited by RPO produced profound changes in LV structure and function. These appeared to arise as an adaptive response to prevent the stretch-induced stunning and myocyte injury that initially develop following acute hemodynamic overload in the normal heart. This pattern of remodeling was characterized by reduced LV diastolic compliance, increased interstitial fibrosis, cardiomyocyte cellular hypertrophy, capillary rarefaction, and concentric LV remodeling, thereby recapitulating a phenotype commonly observed in human patients with HFpEF. These changes all developed in the absence of overt anatomic LV hypertrophy and collectively supported a novel paradigm that linked cardiac adaptations to RPO with the pathogenesis of reduced LV diastolic compliance. In contrast to the persistent hemodynamic overload commonly produced in preclinical models, patients might experience intermittent episodes of preload elevation during labile BP elevations associated with mental stress and activities of daily living (9). These transient periods of hemodynamic overload become exacerbated with age-related stiffening of the aorta (10) and could intermittently impose high levels of mechanical stress on the myocardium, leading to chronic myocyte loss via repetitive stretch-induced programmed cell death (19). Further studies will be required to identify the multiple cellular mechanisms involved in the response and to assess the impact of these changes on diastolic and systolic cardiac function during exercise.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: A transient elevation in preload produces mechanical stretch-induced myocyte injury and measurable cTnI release in the absence of ischemia that is associated with reversible contractile dysfunction and myocyte apoptosis. The findings of this study demonstrated that repetitive exposure to cyclical elevations in preload elicited significant myocyte loss, but LV systolic function was preserved and chamber dilatation was absent. Instead, myocardial remodeling characterized by myocyte hypertrophy and interstitial fibrosis produced a reduction in diastolic compliance that protected the heart from subsequent stretch-induced injury.
TRANSLATIONAL OUTLOOK: Long-term repetitive pressure overload−induced interstitial fibrosis and cellular hypertrophy appear to arise as an adaptive response to intermittent episodes of stretch-induced cardiomyocyte apoptosis. This pattern of remodeling produces a reduction in LV diastolic compliance that has the beneficial effect of protecting the heart from stretch-induced injury during subsequent periods of hemodynamic overload. Nevertheless, protection may come at the cost of a compromised ability to increase cardiac output because of prominent elevations in LV filling pressure and inadequate augmentation of stroke volume during exercise, a hallmark characteristic of HFpEF. Because these changes develop without a significant increase in LV mass, they may explain how reductions in myocardial diastolic compliance can occur when persistent hypertension and anatomic LV hypertrophy are absent.
Acknowledgments
These studies could not have been completed without the assistance of Elaine Granica, Rebeccah Young, Beth Palka, and Cheryl Knapp.
Footnotes
This study was supported by the National Heart Lung and Blood Institute (HL-055324, HL-061610, and F32HL-114335), the American Heart Association (17SDG33660200), the National Center for Advancing Translational Sciences (UL1TR001412), the Department of Veterans Affairs (1IO1BX002659), the New York State Department of Health (NYSTEM CO24351), and the Albert and Elizabeth Rekate Fund in Cardiovascular Medicine. Dr. Canty has been a consultant for Lantheus Medical Imaging, Inc. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Whelan R.S., Kaplinskiy V., Kitsis R.N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Ann Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E. Heart failure. J Am Coll Cardiol HF. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Thygesen K., Alpert J.S., Jaffe A.S. Fourth universal definition of myocardial infarction (2018) J Am Coll Cardiol. 2018;72:2231–2264. doi: 10.1016/j.jacc.2018.08.1038. [DOI] [PubMed] [Google Scholar]
- 4.Giannitsis E., Katus H.A. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol. 2013;10:623–634. doi: 10.1038/nrcardio.2013.129. [DOI] [PubMed] [Google Scholar]
- 5.Weil B.R., Suzuki G., Young R.F., Iyer V., Canty J.M., Jr. Troponin release and reversible left ventricular dysfunction after transient pressure overload. J Am Coll Cardiol. 2018;71:2906–2916. doi: 10.1016/j.jacc.2018.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weil B.R., Young R.F., Shen X. Brief myocardial ischemia produces cardiac troponin I release and focal myocyte apoptosis in the absence of pathological infarction in swine. J Am Coll Cardiol Basic Transl Sci. 2017;2:105–114. doi: 10.1016/j.jacbts.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fallavollita J.A., Canty J.M., Jr. Differential 18F-2-deoxyglucose uptake in viable dysfunctional myocardium with normal resting perfusion: evidence for chronic stunning in pigs. Circulation. 1999;99:2798–2805. doi: 10.1161/01.cir.99.21.2798. [DOI] [PubMed] [Google Scholar]
- 8.Lim H., Fallavollita J.A., Hard R., Kerr C.W., Canty J.M., Jr. Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation. 1999;100:2380–2386. doi: 10.1161/01.cir.100.23.2380. [DOI] [PubMed] [Google Scholar]
- 9.Komajda M., Lam C.S. Heart failure with preserved ejection fraction: a clinical dilemma. Eur Heart J. 2014;35:1022–1032. doi: 10.1093/eurheartj/ehu067. [DOI] [PubMed] [Google Scholar]
- 10.Sun Z. Aging, arterial stiffness, and hypertension. Hypertension. 2015;65:252–256. doi: 10.1161/HYPERTENSIONAHA.114.03617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang R.M., Badano L.P., Mor-Avi V. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–270. doi: 10.1093/ehjci/jev014. [DOI] [PubMed] [Google Scholar]
- 12.Weil B.R., Konecny F., Suzuki G., Iyer V., Canty J.M., Jr. Comparative hemodynamic effects of contemporary percutaneous mechanical circulatory support devices in a porcine model of acute myocardial infarction. J Am Coll Cardiol Intv. 2016;9:2292–2303. doi: 10.1016/j.jcin.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konecny F., Smith C., Techiryan G., Canty J.M., Jr., Weil B.R. Dual admittance catheter-derived biventricular pressure-volume analysis in swine: comparison with contrast-enhanced computed tomography during transient pressure overload. FASEB J. 2019;33:531.12. [Google Scholar]
- 14.van der Velde E.T., Burkhoff D., Steendijk P., Karsdon J., Sagawa K., Baan J. Nonlinearity and load sensitivity of end-systolic pressure-volume relation of canine left ventricle in vivo. Circulation. 1991;83:315–327. doi: 10.1161/01.cir.83.1.315. [DOI] [PubMed] [Google Scholar]
- 15.Burkhoff D., Mirsky I., Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. Am J Physiol Heart Circ Physiol. 2005;289:H501–H512. doi: 10.1152/ajpheart.00138.2005. [DOI] [PubMed] [Google Scholar]
- 16.Weil B.R., Suzuki G., Leiker M.M., Fallavollita J.A., Canty J.M., Jr. Comparative efficacy of intracoronary allogeneic mesenchymal stem cells and cardiosphere-derived cells in swine with hibernating myocardium. Circ Res. 2015;117:634–644. doi: 10.1161/CIRCRESAHA.115.306850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schipke J., Brandenberger C., Rajces A. Assessment of cardiac fibrosis - a morphometric method comparison for collagen quantification. J Appl Physiol. 2017;122:1019–1030. doi: 10.1152/japplphysiol.00987.2016. [DOI] [PubMed] [Google Scholar]
- 18.Swindle M.M., Makin A., Herron A.J., Clubb F.J., Jr., Frazier K.S. Swine as models in biomedical research and toxicology testing. Vet Pathol. 2012;49:344–356. doi: 10.1177/0300985811402846. [DOI] [PubMed] [Google Scholar]
- 19.Cheng W., Li B., Kajstura J. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96:2247–2259. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdelmeguid A.A., Feher J.J. Effect of low perfusate [Ca2+] and diltiazem on cardiac sarcoplasmic reticulum in myocardial stunning. Am J Physiol Heart Circ Physiol. 1994;266:H406–H414. doi: 10.1152/ajpheart.1994.266.2.H406. [DOI] [PubMed] [Google Scholar]
- 21.Sonnenblick E.H., Ross J., Jr., Covell J.W., Braunwald E. Alterations in resting length-tension relations of cardiac muscle induced by changes in contractile force. Circ Res. 1966;19:980–988. doi: 10.1161/01.res.19.5.980. [DOI] [PubMed] [Google Scholar]
- 22.LeWinter M.M., Engler R., Pavelec R.S. Time-dependent shifts of the left ventricular diastolic filling relationship in conscious dogs. Circ Res. 1979;45:641–653. doi: 10.1161/01.res.45.5.641. [DOI] [PubMed] [Google Scholar]
- 23.Pinto J.G., Patitucci P.J. Creep in cardiac muscle. Am J Physiol. 1977;232:H553–H563. doi: 10.1152/ajpheart.1977.232.6.H553. [DOI] [PubMed] [Google Scholar]
- 24.Page B., Young R., Iyer V. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res. 2008;102:103–112. doi: 10.1161/CIRCRESAHA.107.155895. [DOI] [PubMed] [Google Scholar]
- 25.LeWinter M.M., Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotter S., Kazmierowska M., Andresen C. Titin-based cardiac myocyte stiffening contributes to early adaptive ventricular remodeling after myocardial infarction. Circ Res. 2016;119:1017–1029. doi: 10.1161/CIRCRESAHA.116.309685. [DOI] [PubMed] [Google Scholar]
- 27.Kotter S., Andresen C., Kruger M. Titin: central player of hypertrophic signaling and sarcomeric protein quality control. Biol Chem. 2014;395:1341–1352. doi: 10.1515/hsz-2014-0178. [DOI] [PubMed] [Google Scholar]
- 28.van Heerebeek L., Borbely A., Niessen H.W. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 29.Kasner M., Westermann D., Lopez B. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57:977–985. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 30.Zile M.R., Baicu C.F., J SI Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohammed S.F., Hussain S., Mirzoyev S.A., Edwards W.D., Maleszewski J.J., Redfield M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinfath M., Chen Y.Y., Lavicky J. Cardiac alpha 1-adrenoceptor densities in different mammalian species. Br J Pharmacol. 1992;107:185–188. doi: 10.1111/j.1476-5381.1992.tb14484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz R., Oudiz R.J., Guth B.D., Heusch G. Minimal alpha 1- and alpha 2-adrenoceptor-mediated coronary vasoconstriction in the anaesthetized swine. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:422–428. doi: 10.1007/BF00169459. [DOI] [PubMed] [Google Scholar]
- 34.Perrino C., Naga Prasad S.V., Mao L. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersen M.J., Olson T.P., Melenovsky V., Kane G.C., Borlaug B.A. Differential hemodynamic effects of exercise and volume expansion in people with and without heart failure. Circ Heart Fail. 2015;8:41–48. doi: 10.1161/CIRCHEARTFAILURE.114.001731. [DOI] [PubMed] [Google Scholar]
- 36.Borlaug B.A., Nishimura R.A., Sorajja P., Lam C.S., Redfield M.M. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3:588–595. doi: 10.1161/CIRCHEARTFAILURE.109.930701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obokata M., Kane G.C., Reddy Y.N., Olson T.P., Melenovsky V., Borlaug B.A. Role of diastolic stress testing in the evaluation for heart failure with preserved ejection fraction: a simultaneous invasive-echocardiographic study. Circulation. 2017;135:825–838. doi: 10.1161/CIRCULATIONAHA.116.024822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roh J., Houstis N., Rosenzweig A. Why don't we have proven treatments for HFpEF? Circ Res. 2017;120:1243–1245. doi: 10.1161/CIRCRESAHA.116.310119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.