

Abstract
Several studies have shown that giant cell tumor of bone frequently exhibits telomeric associations, commonly at chromosome 11p, which is also the location of the H-ras oncogene. In addition, rare H-ras alleles are more common among cancer patients than among healthy controls and point mutations of this oncogene have also been reported in several malignancies. These data led us to investigate gene dosage, restriction fragment-length size, and point mutations for H-ras in giant cell tumor of bone. Quantitative Southern blot analysis revealed no amplification of the H-ras oncogene in tumor DNA compared with DNA from peripheral blood in the same patient or from control subjects. In addition, no point mutations were detected in codons 12, 13, or 61 (mutations of these codons have been reported in other neoplasms] of the H-ras gene. No differences were noted in restriction fragment-length polymorphisms between tumor and peripheral blood in the same patient and no loss of heterozygosity was detected. In addition, there was no increased frequency of rare H-ras alleles (8% of alleles) in giant cell tumor patients compared to controls (21% of alleles) in our study. However, large allele sizes (> 8.5 kb) were significantly overrepresented in GCT patients compared with healthy controls. In our study, three of 12 alleles were found to be rare in the healthy controls but were common among GCT patients. Our data suggest that the H-ras oncogene is unlikely to be the site of a biologically significant primary lesion in GCT tumorigenesis.
INTRODUCTION
Giant cell tumor of bone (GCT) is a benign primary skeletal neoplasm with variable aggressiveness. It has a tendency to recur locally and the ability to develop histologically benign pulmonary metastases. The lesions frequently occur juxta-articularly in the ends of long tubular bones in adults, commonly between the ages of 20 and 50 years. Surgical resection is the primary mode of therapy, and the wider the margin of surgical resection, the lower the recurrence rate. However, wide surgical resections often compromise the musculoskeletal performance and function of an individual.
Cytogenetic analysis of GCT tumor cells shows a proclivity for chromosomal instability manifested as telomeric associations (tas) between various chromosomes, most commonly chromosomes 11p and 19q [1–4]. Interestingly, the H-ras oncogene is mapped to the terminal region of 11p. Members of the ras oncogene family, including H-ras, have frequently been implicated (e.g., point mutations as a mode of activation) as contributing factors in the development of human neoplasms such as breast cancer, bladder carcinoma, and colorectal tumors [5–9]. In addition, it has also been suggested that rare alleles at the H-ras oncogene locus may be more common among patients with certain cancers than among healthy controls [5]. The purpose of this study was to examine the H-ras oncogene for alterations and to consider its potential role in the tumorigenesis of GCT.
MATERIALS AND METHODS
Four patients with histologically proven GCT had 1–2 cm3 of tumor intra-operatively harvested and sterilely transported to the laboratory. Relevant clinical data of the patients studied are summarized in Table 1. None of these patients received chemotherapy or radiation therapy prior to the surgical resection. Cytogenetic analysis was performed on each sample, as previously described [10]. DNA was isolated from each tumor sample as well as from peripheral blood on each patient. Five micrograms of DNA cut with BamHI enzyme were placed in each lane and subsequent electrophoresis was carried out using a 1% agarose gel for 16 hours at 33 volts. Southern blotting on Gene Screen Plus nylon membrane was performed and the filter hybridized for 2 days at 42°C with radiolabeled H-ras probe (730 bp SstI/PstI fragment from Harvey rat sarcoma virus localized to 11pter-pl5.5) and BCR (breakpoint cluster region, 4.1-kb human probe with a 1.2-kb insert localized to 22q11) used as a control probe (both probes from Oncor, Inc., Gaithersburg, MD). The filters were then washed and exposed to high-performance autoradio-graphic film. Densitometric scanning of the individual lanes using BCR as an internal control was performed using an LKB laser densitometer and the relative intensities of bands on genomic Southern blots were computed and copy numbers determined as previously described [11–14],
Table 1.
Clinical data for patients with giant cell tumor of bone
| Subject | Sex | Age (yr) |
Location of tumor | Telomeric associations |
|---|---|---|---|---|
| M.S. | F | 25 | Scapula | + |
| J.S. | F | 23 | Proximal tibia | − |
| G.L. | M | 54 | Distal femur | + |
| M.G. | M | 39 | Proximal tibia | − |
In a similar procedure, restriction fragment-length polymorphisms (RFLP) were analyzed from 33 healthy control individuals, matched for racial background (i.e., Caucasian), for determination of normative allele frequency. These data were compared with the four GCT patients described above and an additional eight GCT patients evaluated at Vanderbilt University during the past 4 years. A marker of known kb sizes was used to construct a logarithmic scale and allele fragment size was determined in DNA from GCT tumor, blood, and normal controls. Chi-square test with Yates’ correction was used to determine statistical significance of the RFLP data.
In addition, DNA from each tumor was amplified by polymerase chain reaction (PCR) using oligonucleotide primers and conditions specific for the amplification of codons 12, 13, and 61, as described by Mitsudomi et al. [9]. Digestion of 30–40 (μl of amplified products with restriction endonucleases MspI or HphI was used to detect point mutations in codons 12 or 13, respectively [9]. Digestion products were subjected to electrophoresis following standard protocols on either a 5% (MspI digests) or 10% (HphI) poly-acrylamide gel, stained with ethidium bromide, and visualized with a UV light source. Controls for the PCR reactions included DNA extracted from the peripheral leukocytes of an unaffected individual and a reaction mixture with all components except the DNA template. In addition, DNA extracted from the cell line HS578T (Oncogene Science, Uniondale, NY) containing a G→A point mutation in codon 12 was used as a positive control. Screening for mutations in codon 61 was performed by direct DNA sequencing analysis of amplified products. PCR products 277 bp in length were generated as described by Mitsudomi et al. [9], purified using the Glassmax spin cartridge system (Gibco BRL, Gaithersburg, MD) and sequenced using the ds cycle sequencing system and an internal primer (5’-AGA CGT GCC TGT TGG ACA TC-3’) lying 35 bp 5’ to codon 61.
RESULTS
Dosage Studies
Tumor DNA was matched with DNA from peripheral blood of four patients (two males and two females) with GCT. Two patients showed tas and one patient (G.L.) had one in 30 cells with a chromosome 11p in tas formation while the other patient (M.S.) did not have 11p involvement. The remaining two GCT patients did not show tas after the analysis of 30 cells from short-term cultures. Densitometric scanning of the autoradiographs indicated a normal copy number with no amplification of the H-ras oncogene from tumor DNA compared with DNA from peripheral blood in the same patient. Additionally, autoradiographic analysis revealed no amplification of the copy number for H-ras relative to peripheral blood DNA from healthy control individuals using BCR as the internal control probe. The average copy number from at least two DNA lanes for tumor DNA was 2.1, compared to an average copy number of 2.3 for the DNA obtained from peripheral blood from the same patients (Table 2).
Table 2.
Densitometric results of copy numbers for H-ras between DNA from giant cell tumor and blood in the same patients
| Subject | DNA type | Average copy numbera |
Allele size (kb) |
|---|---|---|---|
| Healthy control | Blood | 2.0 | 7.4/7.7 |
| M.S. | Tumor | 2.1 | 7.5/7.5 |
| Blood | 2.3 | 7.5/7.5 | |
| J.S. | Tumor | 2.1 | 7.3/7.7 |
| Blood | 2.4 | 7.3/7.7 | |
| G.L. | Tumor | 2.3 | 9.2/9.2 |
| Blood | 2.5 | 9.2/9.2 | |
| M.G. | Tumor | 2.1 | 7.5/8.8 |
| Blood | 2.1 | 7.5/8.8 | |
BCR, a probe from chromosome 22, was used as an internal control for determination of the copy number.
PCR Amplification Studies
Amplified products containing codon 12 were digested with MspI and subjected to polyacrylamide gel electrophoresis (Fig. 2). In all cases, the 316-bp amplified fragments had been cleaved to yield fragments of 235 and 81 bp in length. Only the preservation of wild type DNA sequences in this region would preserve the MspI site. Detection of DNA substitutions in codon 13 required the use of a modified antisense primer that creates an HphI site only if the wild type DNA sequence is retained [9], In all cases (data not shown), the amplified products were reduced in size from 107 bp to 86 and 21 bp, thereby indicating the preservation of the wild type sequence. The identification of mutations in codon 61 required direct DNA sequencing of amplified products and in all cases only the wild type DNA sequence was obtained (data not shown).
Figure 2.
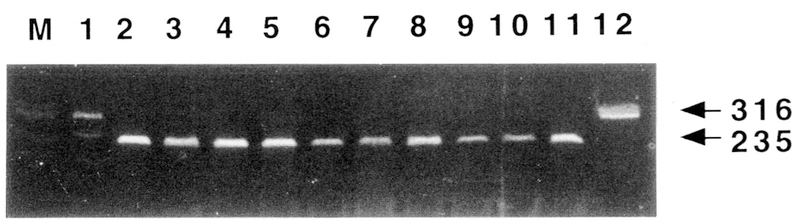
MspI digestion of PCR-amplified DNA from patients with giant cell tumor of bone as compared to positive and negative control DNA. Lane 1, heterozygous positive control showing both the 316-bp (mutant allele) and 235-bp (normal allele) fragments; lanes 2–10, GCT patients showing normal 235-bp fragments after digestion; lane 11, normal control; lane 12, normal control DNA but uncut with MspI showing the 316-bp fragments. Lanes 2 and 4 represent DNA from GCT patients without telomeric associations observed in short-term cultured cells. Lanes 3, 5, and 6–10 represent DNA from GCT patients with telomeric associations observed in short-term cultured cells, with lanes 3, 5, and 6 having 11p terminus involved in telomeric associations in at least one of 30 cells while the remaining GCT patients with telomeric associations did not have 11p involvement.
RFLP Studies
DNA was digested with BamHI and hybridized with the H-ras probe and polymorphic bands ranging in size from 6.4 to 9.5 kb were obtained. We examined RFLP data from patients with GCT (DNA from tumor and blood in the same patient) and compared it to RFLP data from healthy controls (Table 3). The distribution of RFLPs in the two groups is shown in Figure 1. No difference in RFLPs was found between DNA from tumor and that from blood in the same GCT patient. In addition, no loss of heterozygosity for H-ras in tumor DNA was detected in five informative GCT patients.
Table 3.
Distribution of H-ras alleles in control and giant cell tumor (GCT) populations
| Allele size (kb) |
Number of alleles in controls |
Number of alleles in GCT |
χ2 value |
|---|---|---|---|
| Common | |||
| 6.7 | 9 | 0 | 2.28 |
| 6.9 | 7 | 0 | 1.48 |
| 7.0 | 0 | 2 | 2.44 |
| 7.2 | 15 | 3 | 0.60 |
| 7.3 | 1 | 4 | 5.08a |
| 7.4 | 12 | 2 | 0.66 |
| 7.5 | 0 | 4 | 7.92b |
| 7.7 | 9 | 1 | 0.78 |
| 8.8 | 3 | 2 | 0.03 |
| 9.2 | 0 | 3 | 5.10a |
| 9.5 | 0 | 2 | 2.44 |
| Rare | |||
| 6.4 | 2 | 0 | 0.00 |
| 6.6 | 3 | 1 | 0.25 |
| 7.9 | 1 | 0 | 0.28 |
| 8.2 | 1 | 0 | 0.28 |
| 8.3 | 1 | 0 | 0.28 |
| 9.1 | 1 | 0 | 0.28 |
| 9.3 | 1 | 0 | 0.28 |
p < 0.05 for two-tailed test.
p < 0.01 for two-tailed test.
Figure 1.
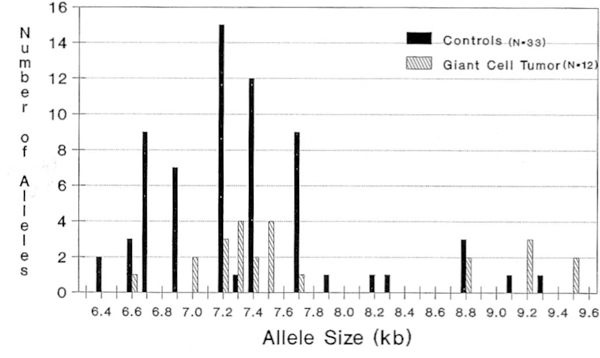
Distribution of H-ras allele size in controls and patients with giant cell tumor of bone.
In our study, common alleles were defined as those occurring at a frequency greater than 5% of the total alleles for each group (e.g., greater than three alleles for controls [33 control subjects representing 66 alleles]). Of the common alleles observed in our healthy controls, three alleles (6.7, 6.9, and 7.7) were rare in GCT patients. Of the common alleles observed in GCT patients, six (7.0, 7.3, 7.5, 8.8, 9.2, and 9.5) were rare in healthy controls (Table 3). Thus, no increased frequencies of rare or uncommon H-ras alleles were noted in patients with GCT when compared to healthy controls. However, five common alleles (7.0, 7.3, 7.5, 9.2, and 9.5) in GCT patients were found to be rare in the healthy controls with a significant difference (p < 0.05, two-tailed χ2 test) χfor three (7.3, 7.5, and 9.2) of the five sites (Table 3). Conversely, three rare alleles (6.7, 6.9, and 7.7) in GCT patients were common in healthy controls. Two alleles (7.2 and 7.4) were common, while seven alleles (6.4, 6.6, 7.9, 8.2, 8.3, 9.1, and 9.3) were rare in both groups. RFLPs for GCT tended to have larger kb sizes (e.g., 9.5), while RFLPs from healthy controls were generally smaller (e.g., 6.4). Large RFLPs (sites > 8.5 kb) were statistically overrepresented (x2 value, 5.36; p < 0.05, two-tailed test) in GCT patients (seven of 24 alleles) compared with healthy controls (five of 66 alleles). Conversely, small RFLPs (sizes < 7.0 kb) were statistically underrepresented (χ2 value, 5.87; p < 0.05, two-tailed test) in GCT patients (one in 24 alleles) compared with healthy controls (21 of 66 alleles). The common RFLPs for GCT were 7.0, 7.2, 7.3, 7.4, 7.5, 8.8, 9.2, and 9.5 kb, representing 22 of 24, or 92%, of the total alleles observed in our sample. The healthy controls had 52 of 66, or 79 %, of their alleles represented by five common RFLPs (6.7, 6.9, 7.2, 7.4, and 7.7). These alleles were similarly seen in healthy controls previously reported [5, 8]. Seven alleles (6.4, 6.6, 7.9, 8.2, 8.3, 9.1, and 9.3) were defined as rare in both populations (controls and GCT patients). Five of these rare RFLPs were observed only in healthy controls and none were observed in GCT patients.
DISCUSSION
Cytogenetic analysis of GCT cells shows a tendency toward tas, with 11p and 19q as the two most commonly involved chromosome regions. Because the H-ras oncogene is located on the terminal region of chromosome 11p and the presence of ras oncogenes has been observed in many tumor types, it has been suggested that H-ras may play a role in the tumorigenesis of GCT [2, 3, 10], Thus, the present study was undertaken to address this hypothesis by examining DNA from GCT patients (tumor and blood samples), with some of these patients showing 11p involvement in tas formation both for amplification of H-ras, for possible RFLP discrepancies compared with healthy controls, and for point mutations of three separate codons reported in other neoplasms.
Southern blot analysis revealed normal copy numbers and therefore no amplification or deletion of the H-ras oncogene using BCR as a control probe between tumor DNA and blood DNA from the same patients. Earlier studies involving TGFB1 located on chromosome 19q also showed normal copy numbers and no amplification of this growth factor gene in GCT [13, 14]. H-ras is polymorphic and shows RFLPs between 6.4 and 9.5 kb in size in our study for both controls and patients with GCT. Certain types of tumors show loss of heterozygosity while others show an increased frequency of rare H-ras alleles in tumor patients when compared to controls [5, 8]. No difference in RFLPs or loss of heterozygosity was detected in our study comparing tumor DNA to DNA from blood in the same GCT patient. In addition, rare alleles were found as frequently in DNA from controls as in GCT tumor DNA, therefore no increased frequency of rare H-ras alleles was observed in GCT tumor DNA. However, we observed that certain rare alleles in controls were found to be common in GCT patients. The significance of the observation is not known.
RFLPs of GCT tumor DNA tended to have larger kb sizes. Significantly more alleles were greater than 8.5 kb in size in GCT patients compared with controls. Alleles for the controls were generally smaller in size. Whether the discrepancy in RFLP size in GCT patients compared with controls represents point mutations affecting the cut site for the BamHI enzyme, thus producing larger allele sizes, or if a true overrepresentation of specific alleles in GCT patients predisposes these individuals to tumor formation is not known at this time. However, no point mutations were recognized in codon 12, 13, or 61 (mutations of these codons have been reported in other neoplasms) with the methodology described in this study.
In summary, our data suggest that deletions, amplifications, or point mutations of the H-ras oncogene are unlikely to be the cause of a biologically significant primary event in GCT tumorigenesis.
Acknowledgments
This research was partially funded by Vanderbilt University Research Council grant number 1-36-425-0401 (H.S.S.) and Biological Research Support Grant #RR-05424 (M.G.B.). We thank Lora Miller, William Wright, and Suzanne Manning for technical assistance and Janie Falkenberg for secretarial assistance.
REFERENCES
- 1.Schwartz HS, Jenkins RB, Dahl RJ, DeWald GW (1989): Cytogenetic analysis of giant cell tumor of bone. Clin Orthop Rel Res 240:250–260. [PubMed] [Google Scholar]
- 2.Bridge JA, Neff JR, Mouron BJ (1992): Giant cell tumor of bone: Chromosomal analysis of 48 specimens and review of the literature. Cancer Genet Cytogenet 58:2–15. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz HS, Butler MG, Jenkins RB, Miller DA, Moses HL (1991): Telomeric associations and consistent growth factor over-expression detected in giant cell tumor of bone. Cancer Genet Cytogenet 56:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardi G, Pandis N, Mandahl N, Heim J, Sfikes K, Willen H, Parragsotopoulos G, Rydholm A, Mitchman F (1991): Chromosomal abnormalities in giant cell tumors of bone. Cancer Genet Cytogenet 57:161–167. [DOI] [PubMed] [Google Scholar]
- 5.Lidereau R, Escot C, Theillet C, Champerme MH, Brumet M, Gept J, Callahan R (1986): High frequency of rare alleles of the human C-Ha-ras-1 proto-oncogene in breast cancer patients. JNCI: 77:697–701. [DOI] [PubMed] [Google Scholar]
- 6.Samtos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M (1982): T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB — and Harvey— MSV transforming genes. Nature 298:343–347. [DOI] [PubMed] [Google Scholar]
- 7.Sidransky D, Tokino T, Hamilton SR, Kinzler KW, Levin B, Frost P, Vogelstein B (1992): Identification of ras oncogene mutations in the stool of patients with variable colorectal tumors. Science 256:102–105. [DOI] [PubMed] [Google Scholar]
- 8.Krontiris TC, DiMartino NA, Colb M, Parkinson DR (1985): Unique allelic restriction fragments of the human Ha-ras locus in leukocyte and tumor DNAs of cancer patients. Nature 313:369–374. [DOI] [PubMed] [Google Scholar]
- 9.Mitsudomi T, Viallet J, Mulshine JL, Linnoila I, Minna JD, Gaz-dar AF (1991): Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene 6:1353–1362. [PubMed] [Google Scholar]
- 10.Schwartz HS, Allen GA, Chudoba I, Butler MG (1992): Cytogenetic abnormalities in a rare case of giant cell osteogenic sarcoma. Cancer Genet Cytogenet 58:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roback EW, Barakat AJ, Dev VG, Mbikay M, Chretien M, Butler MG (1991): An infant with deletion of distal long arm of chromosome 15 (q26.1-qter) and loss of insulin-like growth factor 1 receptor gene. Am J Med Genet 38:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tantravahi U, Kirschner DA, Beauregard L, Page L, Kunkel L, Latt SA (1983): Cytologic and molecular analysis of 46, XXq-cells to identify a DNA segment that might serve as probe for a putative human X chromosome inactivation center. Hum Genet 64:33–38. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz HS, Butler MG (1991): Molecular analysis with TGFB1 probe in giant cell tumor of bone. Transactions of the Orthopaedic Research Society 16:460. [Google Scholar]
- 14.Butler MG, Dahir GA, Schwartz HS (1993): Molecular analysis of transforming growth factor beta in giant cell tumor of bone. Cancer Genet Cytogenet 66:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]