

SUMMARY
Subtypes of nucleus accumbens medium spiny neurons (MSNs) promote dichotomous outcomes in motivated behaviors. However, recent reports indicate enhancing activity of either nucleus accumbens (NAc) core MSN subtype augments reward, suggesting coincident MSN activity may underlie this outcome. Here, we report a collateral excitation mechanism in which high-frequency, NAc core dopamine 1 (D1)-MSN activation causes long-lasting potentiation of excitatory transmission (LLP) on dopamine receptor 2 (D2)-MSNs. Our mechanistic investigation demonstrates that this form of plasticity requires release of the excitatory peptide substance P from D1-MSNs and robust cholinergic interneuron activation through neurokinin receptor stimulation. We also reveal that D2-MSN LLP requires muscarinic 1 receptor activation, intracellular calcium signaling, and GluR2-lacking AMPAR insertion. This study uncovers a mechanism for shaping NAc core activity through the transfer of excitatory information from D1-MSNs to D2-MSNs and may provide a means for altering goal-directed behavior through coordinated MSN activity.
In Brief
Reward outcomes depend on the stimulation protocol used to activate nucleus accumbens medium spiny neuron (MSN) subtypes. In this issue of Neuron, Francis et al. demonstrate rebalanced MSN subtype excitation driven by high-frequency stimulation and substance P release.
Graphical Abstract
INTRODUCTION
The nucleus accumbens (NAc) is a central locus for reward-related behaviors. The major projection neurons that regulate these behaviors, medium spiny neurons (MSNs), are gamma-aminobutyric acid (GABA)ergic neurons that are distinguished by their dopamine receptor expression (expressing either dopamine 1 [D1] or dopamine 2 [D2] receptors), their projections (Albin et al., 1989; Gerfen, 1992; Gerfen et al., 1990; Kupchik et al., 2015; Smith et al., 2013), and their peptide expression (Steiner and Gerfen, 1998; Surmeier et al., 1996). These MSN subtypes differ in their behavioral roles along a ventromedial to dorsolateral gradient (Voorn et al., 2004). More specifically, NAc shell (NAcS) neurons govern motivational value and NAc core (NAcC) neurons underlie goal-directed behavior (West and Carelli, 2016). A wealth of data indicate that the activity of different NAcC MSN subtypes play distinct and often opposing roles in context-dependent reward and aversion (Bariselli et al., 2019; Calipari et al., 2016; Francis and Lobo, 2017; Kravitz et al., 2012; Lobo and Nestler, 2011; Macpherson et al., 2014), where activating D1-MSNs promotes reward-related outcomes and activating D2-MSNs promotes aversive outcomes or blunt reward. However, recent studies have indicated cell-type-specific activation of D2-MSNs, using a stimulation paradigm with prolonged optogenetic depolarization and/or high-frequency stimulation, enhances motivation to obtain food rewards or promotes self-stimulation (Cole et al., 2018; Soares-Cunha et al., 2016, 2018). Additionally, high-frequency (≥50 Hz), but not low-frequency, stimulation of all NAc neurons or NAc D1-MSNs specifically reverses anhedonic outcomes, increasing preference for rewarding stimuli, such as sucrose or social interaction in the absence of stimulation (Francis et al., 2015). Therefore, divergent behavioral outcomes may depend on activation strength.
MSN activity is coordinated on a short timescale by local GABAergic collaterals that allow active MSNs or local GABAergic interneurons that suppress the activity of adjacent MSNs (Dobbs et al., 2016; Taverna et al., 2008; Tepper et al., 2004) or long-range outputs (Edwards et al., 2017; Freeze et al., 2013; Lee et al., 2016), thereby altering the balance between and output from MSN subtypes. Locally, MSNs can modulate activity on a longer timescale by releasing peptides that exert their actions through the control of excitatory input. Local release of dynorphin or enkephalin causes excitatory depression on MSNs (Atwood et al., 2014; Dacher and Nugent, 2011; Tejeda et al., 2017), and substance P can drive potentiation of excitatory transmission (Blomeley and Bracci, 2008; Blomeley et al., 2009). High-frequency stimulation (Bicknell, 1988; Hökfelt, 1991; Qiu et al., 2016; Whim and Lloyd, 1989) or prolonged depolarization (Al-Hasani et al., 2015, 2018) is necessary for peptide release (Hökfelt, 1991; Hökfelt et al., 2000) and is required for fusion of peptide-containing, large dense core vesicles (Cifuentes et al., 2008). Therefore, we predict using high-frequency stimulation paradigms releases peptides and causes lasting functional plasticity changes that work in conjunction with or supersede fast-acting, inhibitory collateral effects, thereby controlling behavior.
In this study, we aimed to assess the consequences of varied D1-MSN stimulation on excitatory transmission on NAc MSN subtypes. We demonstrate strong, high-frequency, but not low-frequency, stimulation of D1-MSNs can indirectly enhance excitatory input on D2-MSNs through the release of substance P from D1-MSNs. This lasting potentiation of excitatory input is post-synaptically expressed, sub-compartment specific, and driven by cholinergic neurons through muscarinic receptor signaling and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) insertion.
RESULTS
NAc Core D1-MSN High-Frequency Stimulation Promotes Excitatory Potentiation on D2-MSNs
AAV-DIO-ChR2 was injected and expressed in the NAcC or NAcS of dynorphin (Dyn)-Cre mice (Figure 1A). Using cell-attached recordings, we found that D1-MSNs displayed high-fidelity action potential firing in response to increasing stimulation frequencies (Figure 1B). To determine the effects of high-frequency stimulation on excitatory transmission, excitatory post-synaptic currents (EPSCs) were evoked with electrical stimulation in the presence of picrotoxin (100 μM) to block GABAA receptor transmission. D1-MSNs were stim ulated with 50-Hz, 473-nm blue light for varying durations. Surprisingly, it was found, 50-Hz trains of NAcC D1-MSN stimulation (10-ms light pulse duration) for either 5 min (5 s light on, 5 s light off) or 15 min promoted a long-lasting excitatory potentiation (LLP) on D2-MSNs (mean ± SE %baseline: 5 min: 141.1% ± 10.55%; 15 min: 158.2% ± 20.19%) but produced excitatory depression on D1-MSNs (5 min:75.10% ± 12.45%; 15 min: 75.86% ± 10.53%; Figures S1A and S1B). These stimulation parameters have been used previously to promote sucrose preference and reverse stress-induced aversion in mice (Francis et al., 2015). To determine the duration of stimulation necessary for the effect, we decreased the stimulation to a single 5-s train of 50-Hz stimulation, which produced the same LLP as 5-min and 15-min stimulation on NAcC D2-MSNs (139.9% ± 6.03%), but had no observable effect on NAcC D1-MSNs (88.58% ±11.08%; Figures 1C, 1E, and 1F). A 50-Hz light train of 1 s was not capable of promoting D2-MSN LLP (92.03% ±13.51%; Figure S1C). Additionally, a 5-s, 50-Hz train with a shortened light pulse duration of 1 ms was not capable of producing D2-MSN LLP (74.81% ± 9.43%; Figure S1D) or high-fidelity D1-MSN firing (Figure S1E). The NAcC D2-MSN LLP was not dependent on potassium channels (Figure S1F) or dependent on GABAA receptor or GABAB receptor transmission (152.8% ± 10.00%; Figure S1G). In contrast to the NAcC, EPSCs in both NAcS MSN subtypes were depressed (Figures 1D and 1E), which was not blocked by norbinaltorphimine (400 nM), a kappa opioid receptor antagonist, in D2-MSNs (61.56% ± 10.78%; Figure S1H). Lower frequency NAcC D1-MSN stimulation at 10 Hz or 20 Hz was not capable of producing LLP on NAcC D2-MSNs (10 Hz: 78.97% ±13.21%; 20 Hz: 77.81% ± 10.60%; Figure 1G), suggesting that high-frequency stimulation is necessary for the local stimulation-induced D2-MSN LLP.
Figure 1. High-Frequency D1-MSN Stimulation Potentiates Excitatory Transmission on D2-MSNs.

(A) Representative expression in the NAc core (NAcC) or shell (NAcS). Red, Dyn-Cre-Tdtomato; green, ChR2-eYFP. AC, anterior commissure. Scale bars: 100 μm.
(B) Cell-attached recordings of D1-MSNs (blue) expressing ChR2 was stimulated with 473-nm blue laser light. Fidelity of D1-MSN firing in cell attached mode is lower at 0.1 mW than 1.0 mW blue light (p < 0.01; n = 5 and 9). Representative traces at 10-, 20-, or 50-Hz stimulation are shown.
(C) NAcC D2-MSNs (gray) were potentiated by a single train of 50-Hz D1-MSN stimulation (p < 0.05; n = 14), and there was no effect on D1-MSNs (blue; p > 0.05; n = 9). Representative paired-pulse excitatory traces pre-stimulation (black) and post-stimulation (red) are shown. Scale bars for synaptic recordings: 100 pA, 25 ms.(D) NAcS D1-MSNs (p < 0.05; n = 8) and D2-MSNs (p < 0.05; n = 9) EPSCs were depressed by D1-MSN 50-Hz stimulation.
(E) Coronal map of EPSC potentiation (green) or depression (red) of MSN subtypes following D1-MSN 50-Hz stimulation.
(F) Potentiation of EPSCs is observed in NAcC D2-MSNs (p < 0.001; n = 9 and 14) and depression of EPSCs in all NAcS MSNs at 20 min from baseline (D1-MSN and D2-MSN; p < 0.01; n = 9 and 8).
(G) 10-Hz (p > 0.05; n = 5) and 20-Hz (p > 0.05; n = 5) stimulation does not alter D2-MSN excitatory transmission.
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S1.
Substance P Release and Neurokinin 1 Receptors Drive the D2-MSN Synaptic Effects of High-Frequency Stimulation
Peptides require high-frequency stimulation for their release (Hökfelt, 1991). The principle excitatory peptide in the striatum, substance P, is exclusively expressed in D1-MSNs and excites about half of the dorsal striatal MSNs (Blomeley and Bracci, 2008; Blomeley et al., 2009). Therefore, we predicted substance P is responsible for the effects of D1-MSN high-frequency stimulation in the NAcC. Indeed, similar to 50-Hz stimulation, substance P (1 μM) produced LLP on NAcC D2-MSNs (131.2% ± 9.15%) but had no effect on NAcC D1-MSNs (106.4% ±7.85%; Figures 2A, 2E, and 2F). Similar to NAcS D1-MSN high-frequency stimulation, substance P (1 μM) depressed excitatory transmission on the NAcS D2-MSNs (82.43% ± 6.67%) and, in contrast to D1-MSN 50 Hz repeated stimulation, had no significant effects on excitatory transmission to NAcS D1-MSNs (109.0% ± 6.20%; Figures 2E, 2F, and S2A). Substance P primarily binds to and activates the neurokinin 1 receptor (NK1R) (Regoli et al., 1987). Therefore, we predicted the NAcC D2-MSN LLP is dependent on the NK1R. Using the NK1R selective antagonist L-733,060 (10 μM), we were able to block the LLP on NAcC D2-MSN caused by substance P bath application (101.1% ±7.07%; Figure 2B). Peptides can act on their receptors at low concentrations (Hökfelt et al., 2000). Additionally, higher concentrations of substance P may have affinity for other NK1Rs at 1 μM. Therefore, we tested the ability of substance P to activate the NK1R at varying concentrations using a bioluminescence resonance energy transfer (BRET) assay. The effective concentration, where the receptor displayed 50% activation (EC50), was 27.9 nM (Figure 2C). Based on these results, we utilized a low concentration of substance P (50 nM) and found this concentration replicated D2-MSN NAcC LLP caused by 1 μM substance P (Figures 2D and 2E). To determine whether high-frequency-driven potentiation on NAcC D2-MSNs is NK1R dependent, we utilized two different selective NK1R antagonists: L-733,060 or the US Food and Drug Administration (FDA)-approved antagonist aprepitant. NK1R antagonism with aprepitant (1 μM) blocked NAcC D2-MSN LLP caused by a single 5-s, 50-Hz train of D1-MSN stimulation (98.02% ± 10.73%; Figure 2G). Additionally, 5 min of NAcC D1-MSN 50-Hz-stimulation-induced potentiation on D2-MSNs was blocked by L-733,060 and produced a transient excitatory depression (79.52% ± 8.22%; Figure S2B). It is possible NK1R could cause excitatory depression by dampening a tonic NK1R tone. However, we found neither L-733,060 nor aprepitant altered synaptic properties in the absence of other stimuli (Figures S2C–S2F). Substance P was able to prevent further potentiation caused by 50-Hz D1-MSN stimulation (2: 140.9% ± 15.02%; 3: 152.5% ± 14.73%; Figure 2H), indicating that substance P and D1-MSN 50-Hz stimulation act through the same mechanism to produce potentiation. Therefore, we conclude high-frequency activation of D1-MSNs drives LLP on NAcC D2-MSNs via a substance P mechanism.
Figure 2. Substance P and 50-Hz D1-MSN Stimulation Drives Excitatory Potentiation through NK1Rs.

(A) EPSCs are enhanced on D2-MSNs after bath application of substance P (p < 0.001; n = 8), but not on D1-MSNs (p > 0.05; n = 5). Representative EPSCs: baseline (black), substance P wash out (red); scale bars for synaptic recordings: 100 pA, 25 ms.
(B) NK1R antagonist L-733,060 blocks substance-P-induced potentiation (p > 0.05; n = 9).
(C) BRET signal in response to different concentrations of substance P.
(D) D2-MSN (p < 0.05; n = 14), but not D1-MSN (p > 0.05; n = 6), EPSCs potentiate in response to substance P bath application.
(E) Coronal map of EPSC potentiation (green) or depression (red) of MSN subtypes following bath application of substance P.
(F) D2-MSN (p < 0.01; n = 24), but not D1-MSN (p > 0.05; n = 8), EPSCs are potentiated at after substance P bath application.
(G) NK1R antagonist aprepitant blocks D1-MSN stimulation-induced potentiation (p > 0.05; n = 8).
(H) Substance P bath application occludes potentiation of D1-MSN 50-Hz stimulation on NAcC D2-MSNs (1 to 2, p < 0.05, n = 7; 2 to 3, p > 0.05, n = 7).
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S2.
LLP on NAcC D2-MSNs Is Post-synaptic
The locus of potentiation (pre- versus post-synaptic) is important in understanding the underlying mechanism of substance-P-induced changes in excitatory transmission in the NAc. We first analyzed paired pulse ratios (PPRs) to examine for a functional change in the probability of release, comparing values before and after 50-Hz stimulation or substance P bath application. PPRs were not different in NAcC MSNs after 50-Hz D1-MSN stimulation or substance P (50 nM; Figures 3A and 3B). Interestingly, using a shorter pulse duration (1 ms), which unmasked a D1-MSN stimulation-induced excitatory depression in NAcC D2-MSNs caused an increase in PPRs (Figure S3A), as was the case with all NAcS MSNs (Figure S3B). These findings indicate NAcC D2-MSNs express potentiation at a post-synaptic locus, which is dependent on stimulation duty cycle, and all NAcS MSNs express pre-synaptic depression. To verify this finding, we examined the amplitude and frequency spontaneous excitatory post-synaptic currents (sEPSCs) to probe for quantal changes that may underlie this specificity. The amplitude of sEPSCs was significantly enhanced on NAcC D2-MSNs (Figures 3C and 3E), but not NAcC D1-MSNs (Figures 3D and 3F), and the frequency remained unchanged in both cell types across conditions, except for a surprising increase in sEPSC frequency in NAcC D1-MSNs after substance P. In agreement with the PPR data, the frequency, but not the amplitude, of sEPSCs was decreased on NAcC D2-MSNs in the short pulse duration condition (Figure S3C) as well as on all NAcS MSNs in the long pulse duration condition (Figures S3D and S3E). Taken together, these results suggest a post-synaptic mechanism drives substance-P-induced LLP on NAcC D2-MSNs, and substance-P-induced excitatory depression on NAcS neurons is pre-synaptic.
Figure 3. D2-MSN Excitatory Potentiation by 50-Hz D1-MSN Stimulation or Substance P Is Post-synaptic.
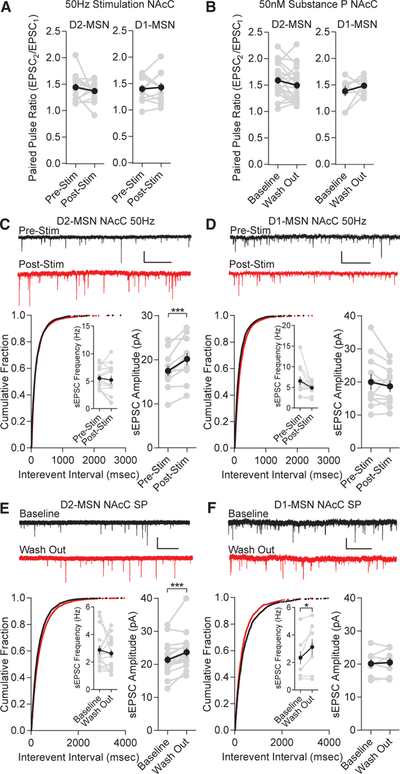
(A) NAcC D1-MSN 50-Hz stimulation does not alter paired pulse ratios (D2-MSNs, p > 0.05, n = 15; D1-MSNs, p > 0.05, n = 11).
(B) NAcC substance P does not alter paired pulse ratios (D2-MSNs, p > 0.05, n = 22; D1-MSNs, p > 0.05, n = 8).
(C) sEPSC frequency is unchanged by D1-MSN 50-Hz stimulation (p > 0.05; n = 12), but amplitude is significantly enhanced (p < 0.001; n = 12). Pre-stim (black) and post-stim (red) are shown; scale bars: 25 pA, 1 s.
(D) Frequency (p > 0.05; n = 11) and amplitude (p > 0.05; n = 11) are unchanged by D1-MSN 50-Hz stimulation.
(E) Frequency is unchanged by substance P (p > 0.05; n = 17), but amplitude is significantly enhanced (p < 0.001; n = 17). Baseline (black) and substance P wash out (red) are shown.
(F) Amplitude is unchanged by substance P (p > 0.05; n = 8), but frequency is significantly increased (p < 0.05; n = 8).
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S3.
Neurokinin Receptor 1 Activation Enhances Activity of NAcC Cholinergic Interneurons
NK1Rs are found on medium and large aspiny neurons within the striatum (Pickel et al., 2000). Using RNAscope in situ hybridization, we found very few NK1R (Tac1r) RNA transcripts coex-pressed with D1 receptor (Drd1) and D2 receptor (Drd2) RNA transcripts in the NAcC (Figure 4A). Because the Tac1r expression was sparse (<2% of cells), we predicted interneurons likely accounted for the expression of the receptor. Choline acetyltransferase (ChAT, Chat)-positive, cholinergic interneurons also express D2 receptors (Le Moine et al., 1990) and may account for a portion of the Drd2/Tac1r RNA coexpression. In accordance with these findings and similar to previous findings (Chen et al., 2001; Pickel et al., 2000), nearly all Chat-expressing neurons in the NAcC and NAcS displayed Chat and Tac1r coexpression across all striatal compartments and bregma locations (Figures 4B and S4A). Somatostatin (Sst) neuron coexpression with Tac1r comprised the residual Tac1r expression not explained by coexpression with Chat (Figure S4B). Additionally, the number of Chat-expressing cells did not differ across all striatal compartments (Figure S4C). These results indicate it is unlikely D1-MSNs cause NK1R-dependent LLP on D2-MSNs via direct collaterals. Rather, D1-MSNs may drive LLP on D2-MSNs through an interneuron-mediated disynaptic mechanism.
Figure 4. ChAT Neuron Activity Is Enhanced by Substance P in a NK1R-Dependent Manner.

(A) Representative RNAscope images of D1 receptor (Drd1, green), D2 receptor (Drd2, red), and neurokinin 1 receptor (Tac1r, gray) RNA transcript merged with DAPI (blue) at 203 and 403 magnification in the NAcC. Few cells display Drd1 and Drd2 coexpression with Tac1r.
(B) Representative RNAscope images at 20× magnification of choline acetyltransferase (Chat, green) and Tac1r (gray) transcript merged with DAPI (blue) in the NAcC, NAcS, and dorsal striatum (DSt). Tac1r expression is coexpressed in nearly all Chat-expressing cells and does not differ across region (p > 0.05; n = 6, 6, and 7).
(C) Substance P enhances firing of ChAT neurons (p < 0.0001; n = 6).
(D) Proportional enhancement in firing rate of ChAT neurons in response to varying substance P concentration.
(E) Proportional change in firing rate of ChAT neurons in response to 100 nM substance P in the presence of varying concentrations of L-733,060.
(F) Substance P causes a lasting increase in ChAT neuron firing rates which is suppressed by L-733,060.
(G) On average, substance P significantly enhances firing rate of ChAT neurons (p < 0.05; n = 7) and the enhancement is blocked by L-733,060 (p > 0.05; n = 7). Representative traces of substance P (black) and substance P+L-733,060 (gray) are shown.
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S4.
ChAT neurons show enhanced firing rates in the dorsal striatum (Aosaki and Kawaguchi, 1996; Govindaiah et al., 2010), and levels of acetylcholine are increased within the striatum by substance P in a NK1R-dependent manner (Anderson et al., 1993; Guzman et al., 1993). Moreover, SST is known to be inhibitory (Grilli et al., 2004; Hou and Yu, 2013; Pittman and Siggins, 1981). Therefore, we decided to explore the effects of substance P on NAcC ChAT neuron firing to assess whether ChAT neurons are involved in driving D2-MSN LLP. We performed cell-attached recordings of NAcC ChAT neurons and, similar to the dorsal striatum, found substance P (1 μM) significantly enhanced firing rates of ChAT neurons in cell-attached (Figure 4C) and whole-cell (Figure S4D) configurations. Although all ChAT cells displayed enhanced firing to 1 μM substance P, a subset exhibited a strong enhancement in firing followed by depolarization block (Figure S4E). To determine the concentration most effective at enhancing ChAT firing rates, without producing depolarization block, we bath-applied substance P at varying concentrations and found the EC50 for substance P was about 43 nM (Figure 4D), which is similar to the EC50 determined by BRET (Figure 2C). Indeed, 50 nM of substance P was able to significantly enhance the firing rate of ChAT neurons, without producing depolarization block (Figure S4F).
We next tested the efficacy of the NK1R antagonist L-733,060 in suppressing enhanced firing at the lowest concentration of substance P (100 nM) capable of producing a near maximal response. L-733,060 displayed an IC50 at about 1.5 μM and a maximal inhibition at 10 μM (Figure 4E): the concentration used to block the substance-P-induced D2-MSN synaptic effect (Figure 2B). Similarly, maximal inhibition of receptor activity in a BRET assay was also observed to be near 10 μM for the NK1R antagonists L-733,060 and aprepitant (Figure S4G). At a low substance P concentration (100 nM), we found the enhanced ChAT firing rate was suppressed by 10 μM L-733,060 (Figures 4F and 4G) or 1 μM aprepitant (Figure S4H), which did not have effects on spontaneous ChAT firing rate in the absence of substance P (Figure S4I). Additionally, baseline firing was not restored until 12 min following the initial application of low concentrations of substance P (11 min: 173.7% ± 20.27%, p < 0.05; 12 min: 147.0% ± 15.22%, p > 0.05; Figure 4F). These results suggest substance P drives a lasting enhancement in spontaneous ChAT firing through NK1Rs. Due to the lack of total suppression by both NK1R antagonists, it is possible another tachykinin receptor could mediate the residual effect. Substance P has lower affinity for the striatal expressing tachykinin receptor neurokinin 3 (NK3R) (Regoli et al., 1987). The enhanced firing rate was not blocked any further by the addition of 1 μM of the NK3R antagonist SB-2220 (Figure S4J), suggesting that NK3Rs do not play a role in the substance-P-induced ChAT excitation. Additionally, inhibition of glutamatergic receptors did not explain the residual effect (Figure S4K). Together, these results demonstrate that low concentrations of substance P cause a long-lasting enhancement in ChAT firing rate via a NK1R mechanism.
D1-MSN High-Frequency Stimulation Drives Neurokinin-Receptor-Dependent Activation of Cholinergic Interneurons
Because substance P can mimic the stimulation-induced LLP on D2-MSNs, we predicted D1-MSNs would enhance NAcC ChAT neuron firing through the same mechanism as substance P. To determine the synaptic connectivity of D1-MSNs to ChAT neurons, we voltage-clamped ChAT neurons (0 mV) in slices expressing ChR2 in D1-MSNs and in the presence of AMPAR and N-methyl-D-aspartate receptor (NMDAR) blockers DNQX and APV, respectively, isolating monosynaptic GABAergic currents from D1-MSNs. D1-MSNs displayed strong synaptic inhibitory post-synaptic currents on ChAT neurons, and these responses were blocked by picrotoxin, demonstrating a robust synaptic D1-MSN to ChAT neuron connection (Figures 5A and S5A). A single 5-s or repeated high-frequency 50 Hz D1-MSN train was sufficient to enhance ChAT firing rate for up to 12 min (11 min: 136.6% ± 10.23%, p < 0.05; 12 min 121.8% ±8.28%, p > 0.05) following optical stimulation, and the lasting effect on firing rate was blocked by aprepitant (1 μM; Figure 5B) or L-733,060 (10 μM; Figure S5C), suggesting the enhanced and lasting spontaneous firing rate is dependent on NK1Rs. Additionally, 5 min of repeated 5-s, 50-Hz trains significantly enhanced firing rate to a similar degree as a single acute train (Figures S5D and S5E), the same as substance P bath application (Figure 4F). Together, these results suggest D1-MSN stimulation drives enhanced ChAT spontaneous firing through activation of NK1Rs.
Figure 5. D1-MSN 50-Hz Stimulation Enhances ChAT Activity by Release of Substance P.

(A) Maximal optogenetically evoked IPSCs from D1-MSNs to ChAT neurons are blocked by picrotoxin.
(B) 50-Hz D1-MSN stimulation enhances ChAT activity (p < 0.0001; n = 6) and is suppressed, but not completely blocked, by aprepitant (p > 0.05; n = 13). Representative traces: vehicle (black) and aprepitant (gray).
(C) 50-Hz D1-MSN stimulation enhanced ChAT firing rate with train durations of 5 s (p < 0.05; n = 10) or 5 s repeated over 5 min (p < 0.05; n = 11).
(D) ChAT firing increased after 50-Hz D1-MSN stimulation (p < 0.01; n = 10), but not 10 Hz (p > 0.05; n = 8) or 20 Hz (p > 0.05; n = 9).
(E) Increased substance P release was measured with an ELISA after 50-Hz stimulation of D1-MSNs in comparison to no stimulation (p < 0.0001; n = 16 and 6), 10 Hz (p < 0.01; n = 7 and 6), and 20 Hz stimulation (p < 0.0001; n = 7 and 6).
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S5.
We next sought to determine parameters necessary for the lasting enhancement in ChAT firing. A 5-s train or repeated 5-s trains were necessary to significantly enhance firing rates of ChAT neurons (Figure 5C). Additionally, 50 Hz, but not 10 Hz or 20 Hz, was necessary for an enhanced firing rate (Figure 5D). To rule out the necessity of GABA disinhibition in causing an enhanced ChAT firing rate, we examined the effect of blocking all GABA transmission on ChAT firing rates. The GABAA receptor antagonist picrotoxin mildly enhanced baseline firing frequency by 1.37 ± 0.66 Hz on average (Figure S5F). However, both GABAA receptor or GABAA and GABAB receptor blockade had no effect on stimulation-induced enhancement in firing rates (Figure S5G), suggesting disinhibition of ChAT neurons is not causing the enhanced firing rate.
Despite the NK1R dependence in ChAT firing, it was unclear whether high-frequency stimulation was sufficient to directly cause release of substance P. We injected the NAcC of Dyn-Cre mice with DIO-ChR2, stimulated acutely prepared NAc slices at varying stimulation frequencies, and collected the artificial cerebrospinal fluid (ACSF) supernatant for ELISA analysis. The concentration of substance P was significantly enhanced by 50-Hz stimulation, but not 10-Hz or 20-Hz stimulation, relative to control slices (Figure 5E). Together, these results demonstrate high-frequency D1-MSN stimulation is sufficient to drive NAcC ChAT excitation through the release of substance P and activation of NK1Rs.
Substance-P-Induced Potentiation on D2-MSNs Requires Muscarinic 1-like Receptor Activation and GluR2-Lacking Receptor Insertion
If ChAT neurons are involved in the potentiation of excitatory transmission on D2-MSNs, we would predict activation of an acetylcholine receptor on MSNs would be responsible for this effect. ChAT neurons make monosynaptic connections with MSNs (Mamaligas and Ford, 2016). Post-synaptically, MSNs express a variety of muscarinic receptors (Goldberg et al., 2012), which are positioned to receive direct acetylcholine input. To determine whether cholinergic interneurons are acting through muscarinic receptor signaling on D2-MSNs, we bath applied substance P (1 μM) in the presence of the broad muscarinic antagonist atropine (1 μM). Atropine blocked excitatory potentiation on D2-MSNs caused by substance P (Figure S6A). Most notably, MSNs express the excitatory muscarinic-1 receptor (M1R) (Bernard et al., 1992; Goldberg et al., 2012; Yan et al., 2001), which is known to be involved in promoting long-term potentiation in the brain (Dennis et al., 2016; Zhao et al., 2018) and is required for long-term potentiation in the striatum (Calabresi et al., 1999). We recorded evoked EPSCs on D2-MSNs before and after bath application of substance P or high-frequency D1-MSN stimulation in the presence of pirenzepine (1 μM), a M1R-like antagonist. Pirenzepine blocked excitatory potentiation on D2-MSNs normally caused by substance P (100 nM or 1 μM) bath application (100 nM: 103.1% ± 12.95%; 1 μM: 89.88% ± 9.14%) or 50-Hz D1-MSN stimulation (107.1% ± 6.541%; Figures 6A, 6B, and S6B), suggesting that activation of these receptors is necessary for excitatory potentiation on D2-MSNs. As a control, we tested whether pirenzepine had a baseline effect on ChAT activity or synaptic properties. Pirenzepine neither affected ChAT firing rate at baseline nor the enhancement in firing frequency due to substance P (Figures S6C and S6D). Additionally, pirenzepine did not affect synaptic properties in D2-MSNs (Figure S6E). Acetylcholinesterase is the primary enzyme responsible for the breakdown of acetylcholine. Because it is predicted that acetylcholine release is enhanced following D1-MSN 50-Hz stimulation and acetylcholinesterase is found in a larger number of cells within the NAcS (Figure S6F), we examined whether we could reverse stimulation-induced depression or cause LLP in the NAcS by elevating acetylcholine levels through blockade of acetylcholinesterase. Blocking acetylcholinesterase with ambenonium (40 nM) did not alleviate the depression on NAcS D2-MSNs caused by D1-MSN 50-Hz stimulation (Figure S6G). The difference in plasticity across the NAcC and NAcS may also be explained by the amount of M1R expression (Chrm1). Using RNAscope, we found no difference in Chrm1 RNA expression in Drd2-positive cells across all striatal compartments (Figure S6H), suggesting differences in M1R expression in D2-MSNs also do not explain the difference between NAcC and NAcS plasticity. These results suggest pirenzepine acts at NAcC D2-MSNs to block potentiation caused by D1-MSN substance P release, and thus, activation of the M1R is required for NAcC D2-MSN LLP.
Figure 6. D2-MSN AMPAR Insertion by Substance P or D1-MSN 50-Hz Stimulation Is M1R Dependent.

(A) D2-MSN excitatory potentiation by substance P is blocked by pirenzepine (p > 0.05; n = 9).
(B) D2-MSN excitatory potentiation by D1-MSN 50-Hz stimulation is blocked by pirenzepine (p > 0.05; n = 9).
(C) AMPAR-evoked current-voltage relationship indicates inward rectification is enhanced at +40 mV by substance P (p < 0.05; n = 7 and 10) and the rectification index is significantly decreased (p < 0.05; n = 7 and 10). Representative traces of substance P (gray) and substance P in the presence of pirenzepine (black) are shown. Scale bars: 100 pA, 25 ms.
(D) Inward rectification of AMPAR currents is enhanced at +40 mV by D1-MSN 50-Hz stimulation (p < 0.05; n = 10 and 12), and the rectification index is significantly decreased (p < 0.05; n = 10 and 12). Representative traces of D1-MSN 50-Hz stimulation (gray) and 50 Hz stimulation in the presence of pirenzepine (black) are shown.
(E) NASPM suppresses substance-P-potentiated EPSCs to control levels. Representative traces: baseline (black); substance P wash out (red); and NASPM (blue; p < 0.05; n = 5 and 4).
(F) NASPM suppresses stimulation-potentiated EPSCs to control levels. Representative traces: pre-stimulation (black); post-stimulation (red); and NASPM (blue; p < 0.05; n = 7 and 7).
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S6.
To determine how D2-MSN LLP is expressed, AMPAR and NMDAR current-voltage relationships were assessed. It was found substance P or 50 Hz stimulation caused significant inward rectification in AMPAR responses at +40 mV, and pirenze-pine treatment prevented this rectification (Figures 6C and 6D). Current-voltage relationships of NMDAR currents were not different between cells across all conditions (Figures S6I and S6J). These results imply that an insertion of GluR2-lacking AMPARs caused the potentiation observed in response to substance P or D1-MSN 50-Hz stimulation conditions. To verify the rectification observed was due to insertion of GluR2-lacking AMPARs, we utilized the inhibitor 1-naphthyl acetyl spermine trihydrochloride (NASPM) (100 μM), which blocks GluR2-lacking AMPARs. D2-MSN EPSCs with substance P treatment (2: 134.0% ± 15.84%; 3: 103.8% ± 9.90%) or D1-MSN 50-Hz stimulation (2: 133.1% ± 11.15%; 3: 87.98% ± 2.67%) displayed a strong sensitivity to NASPM, which caused a de-potentiation of EPSCs to control levels (Figures 6E and 6F). Overall, these results suggest activation of muscarinic receptors, via substance-P-induced ChAT firing, drive GluR2-lacking AMPAR insertion and LLP on NAcC D2-MSNs.
To determine the intracellular signaling mechanism that causes AMPAR insertion, we examined a primary signaling pathway for M1R signaling pathway: G-protein-mediated phospholipase C (PLC) activation and internal calcium release. To broadly block G-protein signaling, we utilized an internal solution containing a non-hydrolyzable guanosine diphosphate (GDP), GDPβS (1 μM), in place of guanosine triphosphate (GTP). Blocking G-protein signaling prevented D1-MSN 50-Hz stimulation-mediated D2-MSN LLP and unmasked a pre-synaptic excitatory depression on D2-MSNs (64.90% ± 8.38%; Figure 7A), as evidenced by an increased PPR (Figure S7A). Additionally, in a separate set of cells, GDPβS prevented inward rectification of AMPAR currents at +40 mV caused by stimulation that remained intact in cell pairs (cells within the stimulation region and ≤150 mm from the first recorded cell; Figure 7B). Inhibition of PLC with U-73122 (3 μM) produced a similar depression to blocking G-protein signaling (66.06% ± 8.70%; Figure 7C) and prevented inward rectification (Figure 7D). Next, we used 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) in the internal solution to chelate calcium and prevent calcium signaling. At a 10-mM concentration, BAPTA blocked the LLP on average (92.94% ± 17.84%) but failed to block LLP in all cells (3/9 cells potentiated; Figure 7E), suggesting a localized synaptic effect may be responsible for the LLP. Relative to paired cells recorded without BAPTA internal solution, BAPTA prevented inward rectification on average (Figure 7F). Depletion of calcium stores with cyclopiazonic acid (CPA) blocked LLP on D2-MSNs (80.18% ± 7.07%; Figure 7G) and prevented inward rectification (Figure 7H). Interestingly, in the presence of GDPβS, U-73122, and CPA, but not BAPTA, PPR was significantly increased (Figures S7A–S7D), suggesting blocking this pathway converts the LLP to a pre-synaptic form of excitatory depression. The lack of ability to detect a change in PPR in the BAPTA condition was likely due to the subset of cells that potentiated. Taken together, these results suggest M1R signaling drives D2-MSN LLP through PLC activation by G-protein signaling and subsequent internal calcium release.
Figure 7. D1-MSN-Stimulation-Induced D2-MSN LLP Requires M1R Signaling.

(A) GDPβS-mediated block of G-protein signaling prevents LLP of NAcC D2-MSNs following D1-MSN 50-Hz stimulation (p < 0.05; n = 8).
(B) Inward rectification of AMPAR currents at +40 mV is blocked by GDPβS compared to D2-MSN pairs (+40 mV: p < 0.01; rectification index p < 0.05; n = 6 and 6).
(C and D) Internal solution with the PLC antagonist U-73122 (C) promotes stimulation-induced depression on D2-MSNs (p < 0.05; n = 9) and (D) prevents inward rectification (p > 0.05; n = 9).
(E and F) Calcium chelation with BAPTA prevents (E) LLP (p > 0.05; n = 9) and (F) inward rectification compared to D2-MSN pairs (+40 mV: p < 0.0001; rectification index p < 0.05; n = 7 and 7).
(G and H) CPA application to deplete internal calcium stores blocks (G) LLP (p > 0.05; n = 8) and (H) inward rectification (p > 0.05; n = 8).
Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics, see Table S1. See also Figure S7.
DISCUSSION
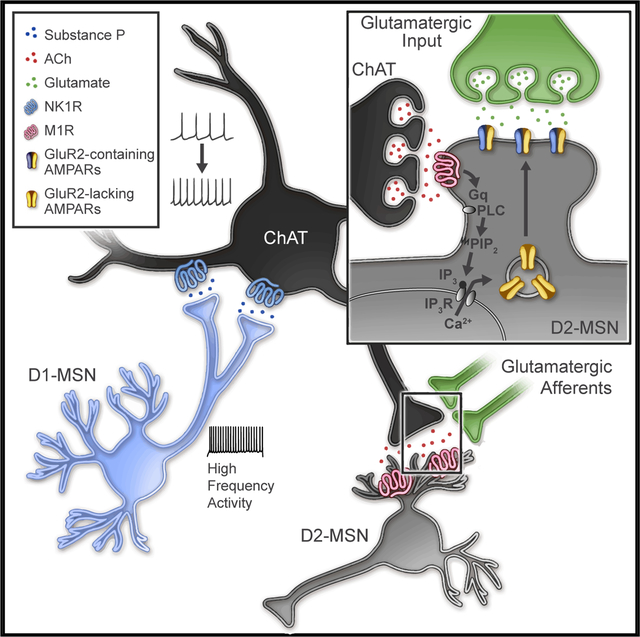
Differential behavioral outcomes to MSN subtype stimulation is thought to drive balanced collateral inhibition, where enhancing activity of one MSN subtype suppresses activity of opposing MSN subtypes. In this study, we report a mechanism in which strong activation of one MSN subtype, D1-MSNs, drives indirect lateral excitation of the opposing subtype, D2-MSNs, in the NAcC (Figure 8). This high-frequency, high-duty cycle D1-MSN stimulation drives release of substance P and activation of the NK1R, which robustly enhances ChAT neuron firing for tens of minutes. This activity putatively enhances acetylcholine release and, via M1Rs, promotes a long-lasting, post-synaptic enhancement of excitatory synaptic input on D2-MSNs. The potentiation is dependent on neither a previously described disinhibition through GABA receptor signaling (Govindaiah et al., 2010; Tejeda et al., 2017) nor a reduced potassium channel function (Day et al., 2008; Shen et al., 2007) but rather due to enhanced AMPAR insertion. This study reveals a mechanism in which local circuits rebalance excitation between MSN subtypes in a long-lasting manner. These findings have compelling implications for the interpretation of frequency-dependent, MSN-specific activation in the context of reward and aversion.
Figure 8. Model of Disynaptic Substance P Action in the NAcC.

High-frequency activation with sustained depolarization of NAcC D1-MSNs causes release of substance P, activation of NK1Rs on ChAT neurons, and increased ChAT activity. Acetylcholine release is enhanced by increased ChAT activity activating M1Rs and a Gq-protein signaling cascade. G-protein activation of PLC putatively activates PIP2-IP3-IP3 receptor signaling, releasing calcium from internal stores promoting calcium-mediated insertion of calcium-permeable, GluR2-lacking AMPARs.
Substance P promotes distinct excitatory effects on various medial-lateral and dorsal-ventral sub-compartments within the striatum. Substance P drives post-synaptic LLP of D2-MSNs in the NAcC, pre-synaptic depression of excitatory transmission in the NAcS, and pre-synaptic potentiation of excitatory input in the dorsal striatum (Blomeley and Bracci, 2008). Despite these differences, ChAT neuron firing rates increase in both dorsal striatal (Aosaki and Kawaguchi, 1996; Govindaiah et al., 2010) and NAcC compartments of the striatum in response to substance P. Moreover, Tac1r and Chat coexpression is equivalent across NAc sub-compartments, indicating differential signaling at D2-MSN synapses may play a vital part in determining the location (i.e., pre-synaptic versus post-synaptic) and direction (i.e., potentiation or depression) of plasticity. Additionally, the stimulation, release, and overall effects of peptides may differ across the NAcC and NAcS. For instance, NAcS D1-MSN stimulation drives excitatory synaptic depression on both MSN sub-types that are pre-synaptic. D1-MSNs also express and release dynorphin, which acts pre-synaptically to depress excitatory transmission (Tejeda et al., 2017). Despite the nature of dynorphin action within the shell, the excitatory depression due to high-frequency NAcS D1-MSN stimulation does not rely on kappa receptor activation, is likely due to substance P, and may act through a distinct multi-synaptic, pre-synaptic mechanism. The NAcS results may point to sub-compartment-specific peptide release mechanisms or differences in responsivity by various excitatory inputs onto MSNs (Tejeda et al., 2017; Voorn et al., 2004). Retrograde tracing studies (Li et al., 2018; Voorn et al., 2004) and terminal expression (Britt et al., 2012; Stuber et al., 2011; Tejeda et al., 2017) demonstrate differential innervation of excitatory inputs to NAcC and NAcS regions, and expression at these terminals may be more or less sensitive to substance P, acetylcholine, or other modulators, like dopamine (see below). For instance, NAcS nicotinic receptor expression may cause depression instead of potentiation, and this pre-synaptic depression could be unmasked following blockade of post-synaptic mechanisms, which contribute to synaptic depression in the NAcC.
ChAT neuron activity and release of acetylcholine plays a critical role in behavior by acting through pre-synaptic and post-synaptic mechanisms (Lim et al., 2014). Pre-synaptically, acetylcholine can act through nicotinic receptors. Direct alteration of nicotinic receptor function is not likely the cause of D2-MSN long-lasting excitatory potentiation, as muscarinic antagonists can completely block substance-P-induced potentiation. However, pre-synaptic expression of nicotinic receptors and ChAT activation can cause dopamine release (Cachope et al., 2012; Threlfell et al., 2012), in addition to the release of other neurotransmitters (MacDermott et al., 1999), and pre-synaptic NK1R alone can enhance dopamine release (Guzman et al., 1993). Although we cannot completely rule out a role for dopamine in modulating the plasticity seen here, it is unlikely that post-synaptic dopamine receptors on D2-MSNs are involved in the long-term plasticity observed. D2 receptors are inhibitory G-protein coupled receptors (GPCRs) that reduce excitability of D2-MSNs (Hernandez-Lopez et al., 2000) and coordinated activation of nicotinic receptors, and release of dopamine promotes long-term depression in the striatum (Partridge et al., 2002; Wang et al., 2006). Additionally, post-synaptic excitatory D1 receptors are not likely to be involved, as LLP is observed in D2-MSNs and low overlap in D1 and D2 receptor expression is observed in the NAc (Kupchik et al., 2015).
However, excitatory muscarinic receptors, which are expressed post-synaptically on MSNs (Yan et al., 2001), are necessary for LLP on NAcC D2-MSNs. Despite the ability of acetylcholine to affect both MSN subtypes, the effects of low concentrations of substance P are specific to D2-MSNs through M1R activation. M1Rs are expressed in both MSN subtypes. Activity of M1Rs has been shown to facilitate long-term potentiation in the hippocampus (Dennis et al., 2016) via the insertion of AMPARs (Zhao et al., 2018) and is required for long-term potentiation in the striatum through canonical long-term potentiation mechanisms (Calabresi et al., 1999). Though the lack of effect on D1-MSNs is curious, we predict that the differential expression of muscarinic receptors in MSNs is governed by the expression of the inhibitory muscarinic receptor M4 in D1-MSNs (Yan et al., 2001), which opposes activation of M1Rs and suppresses glutamatergic excitation (Pancani et al., 2014), thereby negating downstream effects of excitatory M1R-mediated calcium signaling. Although it is hypothesized M1Rs act through mechanisms related to long-term potentiation, additional studies examining specific intracellular mechanisms linking M1R signaling to AMPAR insertion remain to be conducted.
In this study, we have demonstrated that only high-frequency (50-Hz), but not low-frequency (≤20-Hz), stimulation is capable of releasing substance P from D1-MSNs. Peptide release requires higher frequency stimulation (Hökfelt et al., 2000), and peptides are differentially released by varying stimulation protocols (Bick-nell, 1988; Qiu et al., 2016), suggesting that cells tightly regulate mechanisms of peptide release to control neuronal activity. Previous studies have demonstrated NAcS D1-MSNs can release dynorphin with 10-Hz stimulation and longer depolarization times (10 ms; Al-Hasani et al., 2015), and we reveal here substance P can be released from D1-MSNs by 50-Hz stimulation and longer depolarization times (10 ms). Interestingly, lower pulse width light (1 ms) is not capable of producing this effect and biases excitatory changes to pre-synaptic depression. This study may help inform mechanistic underpinnings of NAc deep brain stimulation, which utilizes high-frequency electrical stimulation protocols (using 130-Hz stimulation) to alleviate depression symptoms in humans (Bewernick et al., 2010; Schlaepfer et al., 2013). Optogenetic high-frequency stimulation of all NAc neuronal subtypes or D1-MSNs promotes similar outcomes to human deep brain stimulation in mice (Francis et al., 2015). Moreover, high-frequency (40-Hz) D2-MSN activation or long light pulse stimulation (1 s) is rewarding (Cole et al., 2018; Soares-Cunha et al., 2016, 2018), suggesting strong activity of any MSN subtype within striatum may produce coordinated rewarding effects. Although we observed high-fidelity firing rates of MSNs at higher frequencies, in vivo, MSN firing rates are thought to be substantially lower (Mahon et al., 2003). It is possible these stimulation parameters may induce artificial upstates, which naturalistically are driven by distinct excitatory inputs (Gruber and O’Donnell, 2009; O’Donnell and Grace, 1995) and may allow for coincident activity and synaptic plasticity. In terms of anti-depressant deep brain stimulation, it is conceivable these protocols release peptides or, in the case of repeated, long-lasting stimulation, deplete peptides (Weldon et al., 1990) within various striatal subregions to rebalance excitation, thereby altering reward circuits and behavior. In vivo studies will be necessary to determine how these ex vivo results translate behaviorally.
The transfer of excitation to NAcC D2-MSNs from D1-MSNs may play a strong role in governing learned responses to salient rewarding and aversive signals. Substance P is released in the striatum by a variety of behavioral stimuli, including stressful events and drugs of abuse (Commons, 2010; Sandweiss and Vanderah, 2015), stimuli which are thought to enhance D1-MSN activity (Calipari et al., 2016). Enhancing activity of D1-MSNs would likely increase the probability of strong excitatory stimuli promoting the release of substance P and subsequent D2-MSN excitatory plasticity. The activity of both MSNs is required for goal-directed behavior (Gruber et al., 2009). Although NAc D1-MSN activity is required for driving conditioning of reward (Francis and Lobo, 2017) or aversion-related outcomes (Carlezon and Thomas, 2009) and direct activation of D1-MSN activity can modulate aversive or anhedonic outcomes to repeated stress (Francis et al., 2015), suppression of D2-MSN activity impairs behavioral flexibility (Macpherson et al., 2016), enhances responses to stressful stimuli (Francis and Lobo, 2017; Francis et al., 2015), and can promote aversion (Kravitz et al., 2012). Thus, the lasting excitation on D2-MSN may promote attention to behaviorally salient information. Although, additional studies parsing out the role of substance P release in various goal-directed behaviors are necessary for a more precise interpretation of how these electrophysiological results translate to behavioral outcomes. Overall, we demonstrate differential stimulation parameters of MSN subtypes produce previously unexplored and lasting plasticity effects, which have strong relevance for interpreting stimulus-driven, reward-related behavioral outcomes.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Co-corresponding authors, Antonello Bonci (Antonello.Bonci@nih.gov) or T. Chase Francis (chase.francis@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult male and female (3–6 months) Dyn-Cre-Tdtomato and A2a-Cre-Tdtomato mice on a C57wt/Bl6J were used for all experiments. Mice were bred in house and Dyn-Cre-Tdtomato mice were generated by crossing Ai9-Tdtomato mice with Dyn-Cre mice. Similarly, A2a-Cre-Tdtomato mice were generated by crossing Ai9-Tdtomato with A2a-Cre mice. Mice were group housed, fed ad libidum, and were on a reversed 12hr light/dark cycle. All studies were approved by NIDA ACUC and conducted in accordance with the NIDA ACUC and NIH guidelines.
METHOD DETAILS
Surgical Methods
Mice used in optogenetic stimulation experiments were anesthetized with ketamine/xylazine and scalps were cleaned with alternating ethanol and betadine application. Scalps were locally anesthetized with lidocaine and mice were stereotactically injected bilaterally with 0.5 μL AAV5-DIO-ChR2(H134)-eYFP (UNC Vector Core, Chapel Hill, NC) using Hamilton Neurosyringes 2–4 weeks prior to slice preparation and recording. The NAcC (A/P: +1.6, Lat: +1.5, D/V: −4.4, 10° angle) and the NAcS (A/P: +1.3; Lat: +1.65; D/V: −4.5; 12° angle) were targeted. Mice were injected with warm saline (0.25 mL) to replenish fluids and carprofen (5 mg/kg) for pain relief during recovery.
Electrophysiology
Coronal NAc slices (300 μm) were prepared in ice cold N-Methyl D-Glucamine (NMDG) artificial cerebrospinal fluid (ACSF) containing (in mM): 92 NMDG, 20 HEPES, 25 glucose, 30 sodium bicarbonate, 1.2 sodium phosphate, 2.5 potassium chloride, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 10 magnesium, 0.5 calcium chloride, osmolarity 305–310 mOsm, pH 7.34. Immediately following slicing, slices were transferred to 32°C NMDG solution for 2–3 min. Slices were then transferred to HEPES holding solution for 1hr before recording. HEPES holding solution was the same as NMDG solution but replaced NMDG with 92 mM sodium chloride and contained 1 μM magnesium chloride and 2 mM calcium chloride. Recording was performed at 33°C and slices were perfused with ACSF (2.5 mL/min) containing (in mM): 125 sodium chloride, 26 sodium bicarbonate, and 11 glucose, 2.5 potassium chloride,1.25 sodium phosphate, 2.4 calcium chloride, 1 magnesium chloride, osomolarity 305–310 mOsm, pH 7.34. For synaptic recordings, picrotoxin (100 μM) was added to the ACSF and a cesium methanesulfonate internal solution was used containing (in mM): 117 cesium methanesulfanate, 20 HEPES, 2.8 sodium chloride, 4 magnesium ATP, 0.3 sodium GTP, 0.4 EGTA, pH 7.30, osmolarity 280–287 mOsm. For IV curves, spermine (100 μM) was added to the pipette solution. In some cases, as specified in the text, potassium gluconate internal solution was used containing (in mM): 135 potassium gluconate, 10 HEPES, 4 potassium chloride, 4 magnesium ATP,0.3 sodium GTP, pH 7.30, osmolarity 285–287 mOsm.
Whole-cell voltage clamp of MSNs (−70 mV holding) was performed under differential interference contrast visual guidance on a BX61 Olympus microscope using 1.8–3.0 MU resistance borosilicate glass pipettes. Signals were amplified and digitized using a Mulitclamp 700B amplifier (4 kHz low-pass Bessel filter) and Digidata 1322 digitizer (10 kHz), respectively (Molecular Devices, Sunnyvale, CA USA). For sEPSCs, traces were filtered offline at 1 kHz. For representative traces, signals were filtered offline at 1 kHz and averaged across 7–10 sweeps. MSNs were identified as Tdtomato positive or negative using a mercury arc lamp and RFP filter. Cell-attached recordings were performed with 3–4 MU resistance pipettes and signals were filtered at 1 kHz. Cholinergic interneurons were identified by cell size (40–60 μm soma diameter) and high baseline firing rates (Bennett et al., 2000). For electrical stimulation, a concentric bipolar stimulating electrode (FHC, Bowdoin, ME) was used. Evoked currents were stabilized for 5–10 min prior to data acquisition. For optical stimulation, a fiber attached to a DPSS 473 nm, 100 mW laser (OptoEngine LLC, Midvale, UT) was affixed to the stimulating electrode to stimulate the region at the tip of the electrode. Stimulation was performed at frequencies described in the text at intensities of 0.3–1.0 mW. Stimulation at varying frequencies was performed at a 50% duty cycle. Repeated stimulation was performed with 5 s of stimulation (light on) followed by 5 s of no stimulation (light off) for the time specified. Unique antagonists used are specified in the Key Resources Table. Aprepitant was dissolved in 100% DMSO and DMSO vehicle was used for all experiments used for direct comparison to the antagonists effects. All other drugs were dissolved in water or ACSF.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| AAV5-DIO-ChR2(H134R)-eYFP | UNC Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aprepitant | Selleck Chem | Cat#: S1189 |
| L-733,060 hydrochloride | Tocris | Cat#: 1145 |
| Pirenzepine dihydrochloride | Tocris | Cat#: 1071 |
| SB22220 | Tocris | Cat#: 1393 |
| Substance P | Tocris | Cat#: 1156 |
| Guanosine 5′-O-(2-Thiodiphosphate) trilithium salt | Santa Cruz Biotechnology | Cat #: sc-295029 |
| NASPM trihydrochloride | Tocris | Cat #: 2766 |
| BAPTA-tetracesium salt | Thermofisher Scientific | Cat#: B1212 |
| Spermine tetrahydrochloride | Sigma-Aldrich | Cat #: S2876 |
| Critical Commercial Assays | ||
| Substance P ELISA | R and D Systems | Cat #: SKGE007 |
| RNAscope multiplex fluorescent manual assay | ACD Bio | Cat #: 320850 |
| Experimental Models: Organisms/Strains | ||
| B6;129S-Pdyntm1.1(cre)Mjkr/LowlJ (Dyn-Cre) | Jackson Laboratories | Stock #: 027958 |
| Tg(Adora2a-cre)KG139Gsat/Mmucd (A2a-Cre) | Mutant Mouse Resource and Research Centers (MMRRC) | Stock#: 031168-UCD |
| B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Ai9) | Jackson Laboratories | Stock #: 007909 |
| Oligonucleotides | ||
| Mm-Drd1-C1 (nucleotide target region 444–1358) | ACD Bio | GenBank#: NM_010076.3 |
| Mm-Drd2-C2 (nucleotide target region 85–1153) | ACD Bio | GenBank#: NM_010077.2 |
| Mm-Tacr1-C3 (nucleotide target region 845–1775) | ACD Bio | GenBank#: NM_009313.5 |
| Mm-Sst-C1 (nucleotide target region 18–407) | ACD Bio | GenBank#: NM_009215.1 |
| Mm-Chat-C2 (nucleotide target region 1090–1952) | ACD Bio | GenBank#: NM_009891.2 |
| Mm-Chat-C1 (nucleotide target region 1090–1952) | ACD Bio | GenBank#: NM_009891.2 |
| Mm-Ache-C2 (nucleotide target region 316–1318) | ACD Bio | GenBank#: NM_001290010.1 |
| Mm-Chrm1-C1 (nucleotide target region 851–1994) | ACD Bio | GenBank#: NM_001112697.1 |
| Software and Algorithms | ||
| pClamp | Molecular Devices | Version 10 |
| Graphpad Prism | Graphpad Software | Version 6 and version 8 |
Bioluminescence Resonance Energy Transfer Assay
Gq protein engagement assay uses Renilla luciferase-fused NK1R and mVenus-fused Gαq for the resonance energy transfer (RET) pair. As previously reported (Yano et al., 2017), cells were harvested, washed, and resuspended in phosphate-buffered saline (PBS). Approximately 200,000 cells/well were distributed in 96-well plates, and 5 μM coelenterazine H (substrate for Bioluminescence RET (BRET)) was added to each well. One minute after addition of coelenterazine H, substance P was added to each well. In antagonist mode, L-733,060 or aprepitant was added 15 min before the addition of 100 nM substance P. The BRET signal from the same batch of cells was determined as the ratio of the light emitted by mVenus (510–540 nm) over RLuc (485 nm) in Mithras LB940 (Berthold Technologies, Bad Wildbad, Germany). Results are calculated for the BRET change (BRET ratio for the corresponding drug minus BRET ratio in the absence of the drug). In parallel, the fluorescence of the acceptor was quantified with excitation at 500 nm and emission at 540 nm for 1 s recording to confirm the constant expression levels across experiments.
RNAscope in situ hybridization
Brains were dissected rapidly, flash frozen, and stored at −80°C. Small brain slices (16 mm) of the striatum were prepared on a Leica CM3050S cryostat at −20°C and mounted directly on microscope slides. RNAscope (multiplex fluorescent manual assay) was run according to manufacturer’s specifications (ACD Bio, Newark, CA). Briefly, slices were dehydrated with ethanol and fixed in 4% formalin. Slices were washed and treated with a protease solution. Probes were incubated for 2 hr at 40°C. Commercially available probes from ACD Bio were used (see Key Resources Table). Signals were amplified, washed, and DAPI solution was applied for < 20 s. Slides were coverslipped and imaged on a confocal microscope (Olympus). Counts were made using ImageJ software (NIH, Bethesda, MD). Cells displaying fluorescent signal within two standard deviations of the mean fluorescence of the probe within perinuclear coexpression regions of the other probes were determined to be positive for RNA expression. For quantification of Chrm1 RNA in Drd2+ cells, 25 cells from a 40x image were randomly chosen from 40–100 Drd2+ cells within each image and traces were drawn perisomatically to quantify density of RNA expression. Density for each cell was subtracted from background density. For each animal, 6 slices (2 hemispheres for each Bregma location: anterior +1.70, intermediate +1.42, and posterior +0.98) were used for cell counting or quantification. Counts and quantified RNA expression were averaged across hemispheres then averaged across Bregma regions for each animal. Individual points in figures are representative of one animal.
Substance P ELISA
Coronal NAc slices (150 mm) expressing ChR2 in D1-MSNs were prepared in the same solutions as slice preparation for electrophysiology. After slicing and a 1–2 hr incubation in a HEPES holding solution, slices were transferred to individual wells in a 36-well culture plate and 300 μL of oxygenated normal ACSF was added to each chamber. Each stimulation condition (no stimulation, 10 Hz, 20 Hz, or 50 Hz) was performed in its own plate, in parallel to other conditions, to prevent effects due to light contamination and each slice was placed in a new well. Slice without ChR2-eYFP expression were not used. Stimulation was delivered with an optic fiber placed above slices and 473 nm blue light stimulation (1–3 mW) or no stimulation was performed for 15 min. Supernatant was collected in 100 μL aliquots and frozen at −20°C. For quantification of Substance P release was quantified using a substance P standard curve from a commercial ELISA kit following manufacturer guidelines (R and D Systems, Cat: KGE007). A blank control consisting of 1:1 ELISA diluent:ACSF was run with each assay to account for any potential background absorbance due to ACSF alone.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data and statistical analysis were performed with Prism 6 or 8 (GraphPad Software). Electrophysiological, RNAscope, and ELISA experiments utilized mice, slices, or samples from different litters. The sample size used in each experiment was based on previous electrophysiological and RNAscope experiments (Tejeda et al., 2017), BRET experiments (Yano et al., 2017), or ELISA experiments (Al-Hasani et al., 2015). For time course data, repeated-measure ANOVAs were performed followed by multiple comparison post hoc tests. For most time course experiments, the more rigorous Geisser-Greenhouse’s correction was performed to avoid the assumption of sphericity. Unless otherwise specified, values reported in the text are expressed as a percent of baseline, and are the mean change 15 min after the start of substance P bath application or stimulation (20 min from the start of the experiment). For two sample data, paired or unpaired t tests were performed. For one sample data, one sample t tests were performed and the experimental values and actual means were reported. For bar graph data with more than 2 groups, one-way ANOVAs were used. All n reported for electrophysiological experiments are cells and 3–8 mice were used per experimental condition, with no more than 3 cells per experimental condition per animal. In electrophysiology experiments, cells were only excluded if they were deemed to be a statistical outlier as determined by a Grubbs outlier test. No more than 1 cell was excluded in each group. Recordings that did not survive at least 5 min after stimulation or bath application of a drug were not included in the analysis. In RNAscope data, each point represents a single animal which was derived from an average across three Bregma locations averaged bilaterally, 3–4 animals were used for each experiment, the experimenter was uninformed about the predicted results, and no slices were excluded. For ELISA quantification, the experimenter was blind to all conditions, results were replicated in three independent assays, and samples were only excluded if they fell outside of the range of the standard curve. In all graphs, means are reported as mean ± SEM and p values reported in figures and legends are from post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For exact statistics see Table S1.
Supplementary Material
Highlights.
High-frequency activation of NAc D1-MSNs causes release of substance P
D1-MSN high-frequency activation potentiates excitatory transmission on D2-MSNs
D1-MSN high-frequency activation drives enhanced cholinergic interneuron firing
D2-MSN AMPA receptor insertion by substance P requires M1 receptor signaling
ACKNOWLEDGMENTS
We would like to thank Drs. Hugo Tejeda and Marco Pignatelli for critical guidance on the development of experiments, Dr. Sergi Ferré for sharing instrumentation for the BRET study, and Dr. Mary Kay Lobo for the initial development of high-frequency stimulation protocols used in the study. This work is funded by NIH/NIDA IRP and NIH/NIGMS 1FI2GM128622–01.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2019.05.031.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, et al. (2015). Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, Wong JT, Mabrouk OS, McCall JG, Schmitz GP, Porter-Stransky KA, Aragona BJ, Kennedy RT, and Bruchas MR (2018). In vivo detection of optically-evoked opioid peptide release. eLife 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Chase TN, and Engber TM (1993). Substance P increases release of acetylcholine in the dorsal striatum of freely moving rats. Brain Res. 623, 189–194. [DOI] [PubMed] [Google Scholar]
- Aosaki T, and Kawaguchi Y (1996). Actions of substance P on rat neostriatal neurons in vitro. J. Neurosci 16, 5141–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, and Lovinger DM (2014). Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat. Neurosci 17, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariselli S, Fobbs WC, Creed MC, and Kravitz AV (2019). A competitive model for striatal action selection. Brain Res. 1713, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, and Wilson CJ (2000). Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J. Neurosci 20, 8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Normand E, and Bloch B (1992). Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J. Neurosci 12, 3591–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewernick BH, Hurlemann R, Matusch A, Kayser S, Grubert C, Hadrysiewicz B, Axmacher N, Lemke M, Cooper-Mahkorn D, Cohen MX, et al. (2010). Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol. Psychiatry 67, 110–116. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ (1988). Optimizing release from peptide hormone secretory nerve terminals. J. Exp. Biol 139, 51–65. [DOI] [PubMed] [Google Scholar]
- Blomeley C, and Bracci E (2008). Substance P depolarizes striatal projection neurons and facilitates their glutamatergic inputs. J. Physiol 586, 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeley CP, Kehoe LA, and Bracci E (2009). Substance P mediates excitatory interactions between striatal projection neurons. J. Neurosci 29, 4953–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, and Bonci A (2012). Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, and Cheer JF (2012). Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, and Bernardi G (1999). Activation of M1-like muscarinic receptors is required for the induction of corticostriatal LTP. Neuropharmacology 38, 323–326. [DOI] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, et al. (2016). In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc. Natl. Acad. Sci. USA 113, 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr., and Thomas MJ (2009). Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology 56 (Suppl 1), 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-W, Wei L-C, Liu H-L, Qiu Y, and Chan Y-S (2001). Cholinergic neurons expressing substance P receptor (NK(1)) in the basal forebrain of the rat: a double immunocytochemical study. Brain Res. 904, 161–166. [DOI] [PubMed] [Google Scholar]
- Cifuentes F, Montoya M, and Morales MA (2008). High-frequency stimuli preferentially release large dense-core vesicles located in the proximity of non-specialized zones of the presynaptic membrane in sympathetic ganglia. Dev. Neurobiol 68, 446–456. [DOI] [PubMed] [Google Scholar]
- Cole SL, Robinson MJF, and Berridge KC (2018). Optogenetic self-stimulation in the nucleus accumbens: D1 reward versus D2 ambivalence. PLoS ONE 13, e0207694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons KG (2010). Neuronal pathways linking substance P to drug addiction and stress. Brain Res. 1314, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, and Nugent FS (2011). Opiates and plasticity. Neuropharmacology 61, 1088–1096. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, and Surmeier DJ (2008). Differential excitability and modulation of striatal medium spiny neuron dendrites. J. Neurosci 28, 11603–11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC, Broad LM, Fitzjohn SM, Isaac JTR, and Mellor JR (2016). Activation of muscarinic M1 acetylcholine receptors induces long-term potentiation in the hippocampus. Cereb. Cortex 26, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs LK, Kaplan AR, Lemos JC, Matsui A, Rubinstein M, and Alvarez VA (2016). Dopamine regulation of lateral inhibition between striatal neurons gates the stimulant actions of cocaine. Neuron 90, 1100–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, and Bonci A (2017). Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat. Neurosci 20, 438–448. [DOI] [PubMed] [Google Scholar]
- Francis TC, and Lobo MK (2017). Emerging role for nucleus accumbens medium spiny neuron subtypes in depression. Biol. Psychiatry 81, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, Brooks JM, Iñiguez SD, O’Donnell P, Kravitz A, and Lobo MK (2015). Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol. Psychiatry 77, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, and Kreitzer AC (2013). Control of basal ganglia output by direct and indirect pathway projection neurons. J. Neurosci 33, 18531–18539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR (1992). The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15, 133–139. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr., and Sibley DR (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Ding JB, and Surmeier DJ (2012). Muscarinic modulation of striatal function and circuitry. Handb. Exp. Pharmacol 223–241. [DOI] [PubMed] [Google Scholar]
- Govindaiah G, Wang Y, and Cox CL (2010). Substance P selectively modulates GABA(A) receptor-mediated synaptic transmission in striatal cholinergic interneurons. Neuropharmacology 58, 413–422. [DOI] [PubMed] [Google Scholar]
- Grilli M, Raiteri L, and Pittaluga A (2004). Somatostatin inhibits glutamate release from mouse cerebrocortical nerve endings through presynaptic sst2 receptors linked to the adenylyl cyclase-protein kinase A pathway. Neuropharmacology 46, 388–396. [DOI] [PubMed] [Google Scholar]
- Gruber AJ, and O’Donnell P (2009). Bursting activation of prefrontal cortex drives sustained up states in nucleus accumbens spiny neurons in vivo. Synapse 63, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AJ, Hussain RJ, and O’Donnell P (2009). The nucleus accumbens: a switchboard for goal-directed behaviors. PLoS ONE 4, e5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RG, Kendrick KM, and Emson PC (1993). Effect of substance P on acetylcholine and dopamine release in the rat striatum: a microdialysis study. Brain Res. 622, 147–154. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, and Surmeier DJ (2000). D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J. Neurosci 20, 8987–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hökfelt T (1991). Neuropeptides in perspective: the last ten years. Neuron 7, 867–879. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, and Diez M (2000). Neuropeptides–an overview. Neuropharmacology 39, 1337–1356. [DOI] [PubMed] [Google Scholar]
- Hou ZH, and Yu X (2013). Activity-regulated somatostatin expression reduces dendritic spine density and lowers excitatory synaptic transmission via postsynaptic somatostatin receptor 4. J. Biol. Chem 288, 2501–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, and Kalivas PW (2015). Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci 18, 1230–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moine C, Tison F, and Bloch B (1990). D2 dopamine receptor gene expression by cholinergic neurons in the rat striatum. Neurosci. Lett 117, 248–252. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Weitz AJ, Bernal-Casas D, Duffy BA, Choy M, Kravitz AV, Kreitzer AC, and Lee JH (2016). Activation of direct and indirect pathway medium spiny neurons drives distinct brain-wide responses. Neuron 91, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen Z, Fan G, Li A, Yuan J, and Xu T (2018). Cell-type-specific afferent innervation of the nucleus accumbens core and shell. Front. Neuroanat 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SAO, Kang UJ, and McGehee DS (2014). Striatal cholinergic interneuron regulation and circuit effects. Front. Synaptic Neurosci. 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, and Nestler EJ (2011). The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front. Neuroanat 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, and Siegelbaum SA (1999). Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci 22, 443–485. [DOI] [PubMed] [Google Scholar]
- Macpherson T, Morita M, and Hikida T (2014). Striatal direct and indirect pathways control decision-making behavior. Front. Psychol 5, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson T, Morita M, Wang Y, Sasaoka T, Sawa A, and Hikida T (2016). Nucleus accumbens dopamine D2-receptor expressing neurons control behavioral flexibility in a place discrimination task in the IntelliCage. Learn. Mem 23, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Casassus G, Mulle C, and Charpier S (2003). Spike-dependent intrinsic plasticity increases firing probability in rat striatal neurons in vivo.J. Physiol 550, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaligas AA, and Ford CP (2016). Spontaneous synaptic activation of muscarinic receptors by striatal cholinergic neuron firing. Neuron 91, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, and Grace AA (1995). Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J. Neurosci 15, 3622–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Bolarinwa C, Smith Y, Lindsley CW, Conn PJ, and Xiang Z (2014). M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses. ACS Chem. Neurosci 5, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JG, Apparsundaram S, Gerhardt GA, Ronesi J, and Lovinger DM (2002). Nicotinic acetylcholine receptors interact with dopamine in induction of striatal long-term depression. J. Neurosci 22, 2541–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Douglas J, Chan J, Gamp PD, and Bunnett NW (2000). Neurokinin 1 receptor distribution in cholinergic neurons and targets of substance P terminals in the rat nucleus accumbens. J. Comp. Neurol 423, 500–511. [PubMed] [Google Scholar]
- Pittman QJ, and Siggins GR (1981). Somatostatin hyperpolarizes hippocampal pyramidal cells in vitro. Brain Res. 221, 402–408. [DOI] [PubMed] [Google Scholar]
- Qiu J, Nestor CC, Zhang C, Padilla SL, Palmiter RD, Kelly MJ, and Rønnekleiv OK (2016). High-frequency stimulation-induced peptide release synchronizes arcuate kisspeptin neurons and excites GnRH neurons. eLife 5, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoli D, Drapeau G, Dion S, and D’Orléans-Juste P (1987). Pharmacological receptors for substance P and neurokinins. Life Sci. 40, 109–117. [DOI] [PubMed] [Google Scholar]
- Sandweiss AJ, and Vanderah TW (2015). The pharmacology of neurokinin receptors in addiction: prospects for therapy. Subst. Abuse Rehabil. 6, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer TE, Bewernick BH, Kayser S, Mädler B, and Coenen VA (2013). Rapid effects of deep brain stimulation for treatment-resistant major depression. Biol. Psychiatry 73, 1204–1212. [DOI] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, and Surmeier DJ (2007). Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat. Neurosci 10, 1458–1466. [DOI] [PubMed] [Google Scholar]
- Smith RJ, Lobo MK, Spencer S, and Kalivas PW (2013). Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr. Opin. Neurobiol 23, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Cunha C, Coimbra B, David-Pereira A, Borges S, Pinto L, Costa P, Sousa N, and Rodrigues AJ (2016). Activation of D2 dopamine receptor-expressing neurons in the nucleus accumbens increases motivation. Nat. Commun 7, 11829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Cunha C, Coimbra B, Domingues AV, Vasconcelos N, Sousa N, and Rodrigues AJ (2018). Nucleus accumbens microcircuit underlying D2-MSN-driven increase in motivation. eNeuro 5, ENEURO.0386–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, and Gerfen CR (1998). Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp. Brain Res. 123, 60–76. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, Tye KM, Kempadoo KA, Zhang F, Deisseroth K, and Bonci A (2011). Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature 475, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, and Yan Z (1996). Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci 16, 6579–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Ilijic E, and Surmeier DJ (2008). Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J. Neurosci 28, 5504–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, Lowell BB, Carlezon WA Jr., and Bonci A (2017). Pathway- and cell-specific kappa-opioid receptor modulation of excitation-inhibition balance differentially gates D1 and D2 accumbens neuron activity. Neuron 93, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Koós T, and Wilson CJ (2004). GABAergic microcircuits in the neostriatum. Trends Neurosci. 27, 662–669. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, and Cragg SJ (2012). Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJMJ, Groenewegen HJ, Robbins TW, and Pennartz CMA (2004). Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 27, 468–474. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, and Surmeier DJ (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50, 443–452. [DOI] [PubMed] [Google Scholar]
- Weldon P, Bachoo M, and Polosa C (1990). Depletion by preganglionic stimulation and post-stimulus recovery of large dense core vesicles in synaptic boutons of the cat superior cervical ganglion. Brain Res. 516, 341–344. [DOI] [PubMed] [Google Scholar]
- West EA, and Carelli RM (2016). Nucleus accumbens core and shell differentially encode reward-associated cues after reinforcer devaluation.J. Neurosci 36, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whim MD, and Lloyd PE (1989). Frequency-dependent release of peptide cotransmitters from identified cholinergic motor neurons in Aplysia. Proc. Natl. Acad. Sci. USA 86, 9034–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Flores-Hernandez J, and Surmeier DJ (2001). Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience 103, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Yano H, Provasi D, Cai NS, Filizola M, Ferre S, and Javitch JA (2017). Development of novel biosensors to study receptor-mediated activation of the G-protein alpha subunits Gs and Golf. J. Biol. Chem 292, 19989–19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LX, Ge YH, Xiong CH, Tang L, Yan YH, Law PY, Qiu Y, and Chen HZ (2018). M1 muscarinic receptor facilitates cognitive function by interplay with AMPA receptor GluA1 subunit. FASEB J. 32, 4247–4257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.