

Cellular communication network factor 1 (CCN1) is a dynamically expressed, matricellular protein required for vascular development and tissue repair. The CCN1 gene is a presumed target of Yes-associated protein (YAP), a transcriptional coactivator that regulates cell growth and organ size.
KEYWORDS: CCN1, YAP, angiogenesis, extracellular matrix, ischemic retinopathy
ABSTRACT
Cellular communication network factor 1 (CCN1) is a dynamically expressed, matricellular protein required for vascular development and tissue repair. The CCN1 gene is a presumed target of Yes-associated protein (YAP), a transcriptional coactivator that regulates cell growth and organ size. Herein, we demonstrate that the CCN1 promoter is indeed a direct genomic target of YAP in endothelial cells (ECs) of new blood vessel sprouts and that YAP deficiency in mice downregulates CCN1 and alters cytoskeletal and mitogenic gene expression. Interestingly, CCN1 overexpression in cultured ECs inactivates YAP in a negative feedback and causes its nuclear exclusion. Accordingly, EC-specific deletion of the CCN1 gene in mice mimics a YAP gain-of-function phenotype, characterized by EC hyperproliferation and blood vessel enlargement. CCN1 brings about its effect by providing cells with a soft compliant matrix that creates YAP-repressive cytoskeletal states. Concordantly, pharmacological inhibition of cell stiffness recapitulates the CCN1 deletion vascular phenotype. Furthermore, adeno-associated virus-mediated expression of CCN1 reversed the pathology of YAP hyperactivation and the subsequent aberrant growth of blood vessels in mice with ischemic retinopathy. Our studies unravel a new paradigm of functional interaction between CCN1 and YAP and underscore the significance of their interplay in the pathogenesis of neovascular diseases.
INTRODUCTION
Extracellular matrix (ECM) proteins occupy a significant part of the extracellular environment of blood vessels and play crucial roles in vascular physiology, homeostasis, and responses to ischemic injury. In addition to the common structural basement membrane and constitutively expressed ECM proteins, predominantly made up of collagens, glycoproteins, and proteoglycans, the pericellular matrix includes mesh-like link proteins, such as those of the cellular communication network (CCN) family (1). Typically, these matricellular proteins are highly expressed in development and following injurious stimuli, but their mechanisms of action are incompletely understood. The CCN1 protein, in particular, is a functionally multifaceted 41-kDa polypeptide with regulatory rather than structural roles in the extracellular environment (2). CCN1 plays essential roles in stem cell lineage commitment and regulates inflammation, wound healing, and tumorigenesis in the adult. In vitro studies have shown that CCN1 activities are context and cell type dependent and rely on CCN1 interaction with various receptors, including integrins (e.g., ανβ3, αΜβ2, α6β1, αIIbβ3, αMβ2, and αDβ2), which reprograms gene expression toward adhesion, migration, differentiation, and connective tissue remodeling (3, 4). However, the sheer scope of the pathways activated by CCN1 raises the question of how specificity in the cellular response is achieved in physiologically relevant processes, such as angiogenesis.
Global CCN1 deficiency in mice impaired blood vessel bifurcation in the chorionic plate, resulting in placental hypovascularization and embryonic lethality (5). Our previous studies showed that the EC-specific loss of CCN1 function induced the formation of a dysmorphic dysfunctional vessel network with an altered hierarchical organization into arteries, capillaries, and veins (6). CCN1 signals reprogram the gene expression of Dll4 of the Notch signaling pathway and SHP-1, a protein phosphatase that dephosphorylates specific tyrosine residues of vascular endothelial growth factor (VEGF) receptor 2 (VEGF-R2). Under ischemic conditions, CCN1 gene expression was abnormally depressed, causing angiogenic imbalance and an exuberant neovascularization characteristic of ischemic retinopathy in humans (7). Despite such a profound effect on vascular cell behavior and pathology, the mechanisms underpinning CCN1 gene regulation, molecular and functional interactions, and protein signaling remain largely unaddressed issues.
Published gene set enrichment analyses from knockout animals and primary tumors identified CCN1 to be a bona fide target of the Yes-associated protein (YAP) (8). YAP is the core component of the Hippo pathway, which comprises a key kinase cascade associated with cell growth and organ size regulation. YAP becomes transcriptionally inactive when it is phosphorylated and sequestered in the cytoplasm, which results in growth inhibition (9). Conversely, active nonphosphorylated YAP rapidly translocates into the nucleus and interacts with DNA-binding transcription factors (e.g., RUNX3, p73, AP-1, serum response factor [SRF], and transcriptional enhanced associate domain [TEAD]) to turn on the expression of growth-promoting and apoptosis-inhibiting genes (10). The binding of all TEAD proteins to their DNA-binding elements within a promoter target required transcriptional coactivation by YAP exclusively (11). Accordingly, constitutive activation of YAP induced excessive cell proliferation and the formation of oversized organs. The inputs that regulate YAP activation/inactivation relate to cellular events, including adhesion, cell-cell interactions, and cell polarity, all of which are critical in angiogenesis (12). Whether dedicated extracellular signaling factors and receptors regulate YAP signaling is not fully understood. Our earlier work and that of others have shown that cells under mechanical tension activate CCN1 gene expression and YAP transcriptional activities (13–15). As a transcriptional coactivator, YAP controls gene transcription on a genomewide scale, yet YAP deletion phenocopied the vascular defects associated with CCN1 deficiency in mice (16, 17). Interestingly, constitutive activation of YAP or the loss of its upstream negative regulators led to striking tissue overgrowth, as seen with endothelial cell (EC) hyperproliferation and aberrant enlargement of blood vessels following deletion of CCN1 in mice (6, 18). Here, we present in vitro and in vivo evidence of YAP-dependent CCN1 gene regulation and CCN1 signal-induced inhibition of YAP during the physiological and pathological growth of blood vessels. We uncovered a negative-feedback loop involving CCN1 signals and YAP function through matrix unstiffening that is critical for blood vessel normal growth and regeneration under ischemic conditions.
RESULTS
Expression profile of CCN1 and YAP during retinal vascular development.
In the retina of newborn rodents, blood vessels emerge from the optic nerve and give rise to interconnected vascular plexuses during the first postnatal week. Thus, the retina is a suitable model to study the regulation and functional interactions among genes in the context of the spatiotemporal progression of organ vascularization and relevant pathologies. To understand the complex regulatory roles of CCN1 and YAP during blood vessel development, we first studied their levels of expression and mapped their location in the retinal tissues and cells. Using a CCN1-GFP bacterial artificial chromosome transgenic mouse, which carries the gene for a green fluorescent protein (GFP) reporter downstream of the CCN1 promoter, we observed that the GFP signals were particularly associated with ECs (Fig. 1A). As the primary capillary plexus expands during the first postnatal week, GFP:CCN1 was localized in the leading vascular front, i.e., in the tip ECs that possess numerous filopodia and the trailing stalk ECs that form lumenized structures. The GFP fluorescence was four times stronger in the vascular front toward the peripheral zone than in the central zone (Fig. 1C and D). In the fully developed capillaries around the optic nerve head, the level of CCN1 was only residual, which is in agreement with previous data indicating that the highest CCN1 levels are largely associated with ECs undergoing angiogenesis (i.e., tip and stalk ECs) (6). A strong but uniformly distributed YAP signal was associated with the developing retinal vasculature throughout the capillary plexus (Fig. 1B to D). Transverse sections showed that YAP was located predominantly in the nucleus of the blood vessels in the advancing front (Fig. 1E). In striking contrast, YAP was excluded from the nucleus and largely seen in the cytoplasm of the fully developed vasculature around the optic nerve head. Cytoplasmic YAP was also detected in glial fibrillary acidic protein (GFAP)-positive astrocytes of the retina, even though these cells do not express CCN1 (Fig. 1F and G). Thus, the cellular localization of CCN1 and YAP is tightly programmed both spatially and temporally during sprouting angiogenesis.
FIG 1.

Expression pattern of CCN1 and YAP in developing vessels of the mouse retina. (A) Retinal flat mounts of CCN1:GFP reporter transgenic mice stained with isolectin B4 (IB4). Arrows show tip cells. (B) Retinal flat mounts labeled with YAP antibody alone (a, c, and d) or in combination with IB4 (b). (C and D) Quantification of CCN1:GFP and YAP signals in central and peripheral zones of the P7 retina, delineated by a virtual red line (C). The CCN1:GFP and YAP signals were normalized to those for IB4-stained blood vessel surface. Values are means plus SE. *, P < 0.01 versus central zone (n = 4). (E) Transverse sections of central and peripheral areas of the retinas of CCN1:GFP mice stained with YAP antibody (a, c, e, g). The cells’ nuclei were stained with DAPI (b, c, f, g). The retinal layers indicated are the ganglion cell layer (GCL), the inner nuclear layer (INL), and the outer nuclear layer (ONL). (F) Retinal flat mount labeled with YAP and GFAP antibodies. (G) Transverse sections of P7 mouse retinas stained with YAP and GFAP antibodies.
CCN1 gene transcription in ECs is YAP dependent.
CCN1 is known as an immediate early responsive gene which is transcriptionally activated through Rho GTPase signals, directly sensing changes in the globular actin (G-actin) concentration (13). Two pathways sensitive to actin dynamics are potentially involved in CCN1 gene expression: the first is YAP and its DNA-binding partners of the TEAD family, and the second is myocardin-related transcription factor A (MRTF-A) and its DNA-binding partner serum response factor (SRF). Indeed, the CCN1 promoter contains two TEAD-YAP binding sequences (one proximal and one distal to the transcription start site) and one functional serum response CArG box (Fig. 2A). As most MRTF-A target genes remain silent, despite the constitutive expression of MRTF-A in the nucleus (13, 19), we examined the role of YAP as a determining factor of CCN1 gene expression. CCN1, YAP, and TEAD1 expression was measured in human retinal EC (HREC) cultures exposed to jasplakinolide, a naturally occurring cell-permeant cyclodepsipeptide that rapidly induces actin polymerization and F-actin stabilization (20). As shown in Fig. 2B and C, jasplakinolide treatment rapidly but transiently increased CCN1 protein levels. This increase in CCN1 coincided with a decrease in YAP phosphorylation on S127 and an increase of its translocation into the nucleus (Fig. 2D). YAP and TEAD1 exhibited basal constitutive gene expression in control and treated cells.
FIG 2.

Active YAP induces CCN1 gene transactivation in HRECs. (A) Schematic layout of the CCN1 promoter with the relative location of the CArG and TEAD consensus sequences in the mouse genome and their conservation in human and mouse. dist, distal; prox, proximal. (B and C) Protein levels of CCN1, pYAP, YAP, TEAD1, and GAPDH. (B) Protein levels were determined by immunoblotting after cell treatment with jasplakinolide (Jasp.; 0.1 μM). (C) Densitometric analysis of the protein bands. Values are means plus SE (n = 3). To facilitate comparisons, CCN1 protein levels in control nontreated cells were set to 1 and those of pYAP were set to 5. *, P < 0.004 versus the value at time zero. au, arbitrary units. (D) Immunostaining of control and jasplakinolide-stimulated cells with YAP antibody. Cell nuclei were stained with DAPI (as shown in panel b for panel a and as shown in panel d for panel c). (E) CCN1 promoter reporter activity in jasplakinolide (Jaspl.)-treated HRECs for 1 h (n = 3). (F and G) YAP and MRTF-A enrichment of the endogenous CCN1 promoter determined by a quantitative ChIP assay in control and jasplakinolide-treated cells. Values are means ± half of the range from three experiments using different cell preparations.
Transient-transfection experiments were carried out using two constructs with a luciferase (luc) reporter gene driven by either a 2,398-bp or a 2,200-bp CCN1 promoter fragment. Following jasplakinolide treatment, the 2,398-bp promoter fragment induced a substantial 39-fold increase in CCN1 promoter activity, while the shorter 2,200-bp fragment lacking the MRTF-A-SRF binding site induced a 19-fold increase (Fig. 2E). Mutation of the distal TEAD sequence did not significantly affect the reporter activity, but mutation of the TEAD sequence proximal to the transcription initiation site completely repressed the promoter activity. Thus, the proximal TEAD and CArG box sequences contain the essential stimulatory elements required for full-blown expression of the CCN1 gene. At the same time, the CArG box alone is not sufficient because mutation of the proximal TEAD site completely abolished the promoter activity. We further examined a potential cooperative interaction between the CArG box SRF and TEAD via their transcriptional coactivator partners, MRTF-A and YAP. Quantitative chromatin immunoprecipitation (ChIP) assays using YAP- and MRTF-A-specific antibodies showed that jasplakinolide treatment induced 3.4- and 4.1-fold enrichment of YAP and MRTF-A, respectively, in the CCN1 promoter region encompassing the proximal TEAD sequence (Fig. 2F and G). A 1.7- and 1.5-fold enrichment of YAP and MRTF-A, respectively, was seen at the distal TEAD site as well, even though this site did not significantly influence CCN1 promoter activity. Taken together, our results indicate that YAP-TEAD is a primary driver of CCN1 gene transcription through the proximal site and that MRTF-A–SRF has a synergistic effect in response to changes in actin dynamics.
YAP is required for CCN1 gene transactivation during in vivo angiogenesis.
We measured CCN1 gene transcripts using quantitative PCR (qPCR) during postnatal retinal vascular development. CCN1 transcript levels were the highest at postnatal day 7 (P7), when the primary capillary plexus had formed (Fig. 3A). Using ChIP analysis of the proximal TEAD site, we compared YAP enrichment in the peripheral (with ongoing vessel sprouting) and central (with completed vascular growth) retinal areas of a P7 mouse (Fig. 3B). YAP enrichment was stronger in the peripheral zones (+39%) than in the central regions, which is consistent with YAP-dependent transactivation of the CCN1 gene in the area of active blood vessel formation (Fig. 3C). MRTF-A enrichment was also increased by 23% in the peripheral retina, supporting the observation that it acts together with YAP to augment CCN1 gene expression (Fig. 3D). Since MRTF-A and TEAD are constitutively expressed in the cells and their localization in the nucleus is independent of their activity (21) and because mutation of the proximal TEAD site nearly completely abolished CCN1 activation in ECs, we postulated that YAP activation is the primary determining factor of CCN1 promoter activity in vivo as well. To test this hypothesis, we generated mice with either EC-specific or ubiquitous deletion of YAP (hereafter referred to as EC-ΔYAP and UB-ΔYAP mice, respectively) using the Cdh5-Cre ERT2 and UBC-Cre ERT2 mice with the YAPflox/flox alleles (Fig. 3E). ChIP assay analysis showed that YAP enrichment of the endogenous CCN1 promoter was reduced by 73% and 85% in the developing retina of the EC-ΔYAP and UB-ΔYAP mouse mutants, respectively (Fig. 3F and G), indicating that YAP was efficiently depleted in both mutant mice. It is noteworthy that the EC-specific loss of YAP resulted in no overt vascular defects (Fig. 3H to J). Both wild-type and EC-ΔYAP animals exhibited a similar vessel morphology and hierarchical organization, despite a 21% reduction of cell proliferation (i.e., bromodeoxyuridine [BrdU]-positive [BrdU+] cell counts) in EC-ΔYAP mouse retinas (Fig. 3K). However, the ubiquitous loss of YAP showed a significant reduction of the vascular area, junctional density, and radial expansion of the vascular network compared to the findings in YAP+/+ mice. EC proliferation was reduced by 60% compared to that in YAP+/+ mouse retinas. The number of apoptotic cells was not significantly altered in the UBΔYAP and EC-ΔYAP mouse retinas compared to that in wild-type mice (Fig. 3L). This is consistent with the finding of Watson et al. that apoptosis does not influence the number of vessels generated during angiogenesis (22).
FIG 3.

YAP-dependent transactivation of the CCN1 gene during retinal vascular development. (A) CCN1 mRNA levels determined by qPCR in mouse retinas at P3, P7, and P14. Data shown are means ± SE (n = 4). (B to D) YAP enrichment of the endogenous CCN1 promoter determined by ChIP assay. DNA was immunoprecipitated from retinal homogenates pooled from 10 mouse eyes of each genotype (YAP+/+, EC-ΔYAP, and UB-ΔYAP). YAP- and MRTF-A-immunoprecipitated DNA was quantified by qPCR. (E) Schematic diagram showing conditional targeting of the YAP genomic locus with Cdh5-Cre ERT2 and UBC-Cre ERT2 mice. (F and G) YAP enrichment of the proximal TEAD site of the endogenous CCN1 gene was analyzed by ChIP assay. (H) Representative immunofluorescence images of IB4-stained retinal flat mounts at P6 of YAP+/+, EC-ΔYAP, and UB-ΔYAP mice. Vascular features were analyzed with AngioTool software (as shown in panel b for panel a, as shown in panel d for panel c, and as shown in panel f for panel e). The outline of the vasculature is shown in black, the vasculature skeleton representation is shown in red (top row), and branching points are shown in green (bottom row). (I to K) Quantitative analysis of vascular parameters (i.e., vascular area [I], junction density [J]) of representative retinas from wild-type and mutant mice. **, P < 0.05 versus YAP+/+ mice (n = 4) of the same age. (K) Quantitative analysis of EC proliferation at P6, as determined by BrdU incorporation. Equivalent areas of retinas from YAP+/+, EC-ΔYAP, and UB-ΔYAP mice were compared. Data are means ± SE. *, P < 0.05 versus YAP+/+ mice; **, P < 0.001 versus YAP+/+ mice (n = 4). (L) Quantification of TUNEL-positive cells in retinal sections of wild-type and YAP mutant mice (n = 6 for each group). Data are presented as means ± SE (n = 6).
Vascular sprouts of UB-ΔYAP mutant retinas were characterized by the appearance of short stubs with blunted edges lacking filopodial extensions (Fig. 4A). As a transcriptional coactivator, YAP-TEAD targets a gene program of cell proliferation and EC differentiation that includes a large group of genes involved in cytoskeleton remodeling, cell adhesion/extracellular matrix remodeling, and VEGF–VEGF-R2 signaling (23). We examined the expression of genes representative of the above-mentioned categories in the retinas of EC-ΔYAP and UB-ΔYAP mutant mice. Quantitative PCR analysis confirmed that the ubiquitous deletion of YAP significantly reduced by more than 50% the expression of most cytoskeletal, ECM, VEGF signaling, and cell cycle genes (Fig. 4B to E). Expression of these genes has been associated with the YAP-TEAD interaction with distant enhancers through chromatic looping (24). However, EC-specific deletion of YAP resulted in a less than 50% reduction of the expression of most genes, suggesting that YAP loss in ECs only was not sufficient to induce impactful alterations of vessel growth- and expansion-related genes like global YAP deletion did. Of note, the expression of the YAP paralogue transcriptional coactivator with the PDZ-binding motif (TAZ) was not altered upon the EC-specific or global deletion of YAP, which ruled out a potential compensatory relationship between YAP and TAZ. Similarly, the expression of VEGF-A and VEGF-R2 was not significantly altered upon EC-specific or global deletion of YAP (Fig. 4F).
FIG 4.
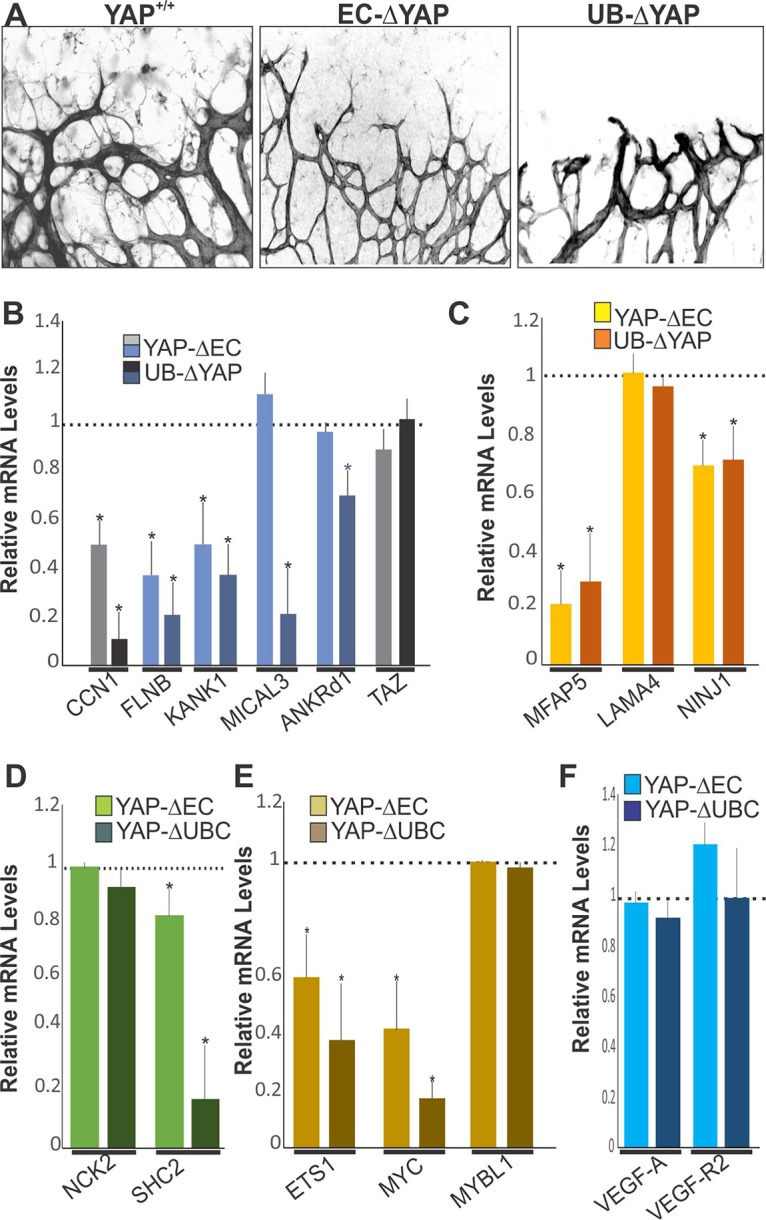
Vascular and molecular alterations associated with YAP deletion. (A) IB4 staining of P5 retinal flat mounts from YAP+/+, EC-ΔYAP, and UB-ΔYAP mice. (B to E) Relative mRNA levels of YAP target genes in retinal lysates from YAP+/+, EC-ΔYAP, and UB-ΔYAP mice. Expression of TAZ was also included. The mRNA levels in YAP+/+ mice were set to 1. Each measurement was performed in triplicate (n = 3). (F) Relative mRNA levels of VEGF-A and VEGF-R2 in retinal lysates from YAP+/+, EC-ΔYAP, and UB-ΔYAP mice (n = 3).
Regulation of YAP activity by CCN1.
When phosphorylated, YAP is sequestered in the cytoplasm in an inactive form. In contrast, dephosphorylated YAP translocates into the nucleus and promotes the transcription of gene targets. Since YAP is regulated by mechanical cues defined by the composition of the ECM, we asked whether the YAP-induced secretion of CCN1 in the extracellular environment contributes to the feed forward of feedback cycles that affect YAP activity. First, we examined whether there is a potential functional cross talk between CCN1 and YAP by determining the effects of adenovirus (Ad)-mediated CCN1 overexpression on YAP activation. HREC cultures were plated at a subconfluent density because the nuclear localization of YAP was sensitive to cell density (i.e., activating YAP signals predominate in a low cell density, whereas inhibitory signals predominate in crowded cells) (25). As shown in Fig. 5A, exposure of HRECs to increasing levels of CCN1 under a low fetal serum concentration proportionally increased the levels of inactive phosphorylated YAP (pYAP). Concordantly, cells transfected with a GFP-tagged YAP construct showed a strong YAP:GFP signal in the nucleus, whereas those overexpressing CCN1 were enriched with a cytoplasmic YAP:GFP signal (Fig. 5B).
FIG 5.

Expression of CCN1 controls YAP cellular localization and activity in HRECs. (A) Expression of YAP and pYAP in HRECs expressing increasing concentrations of Ad-CCN1 (MOI, 1 to 5), as determined by Western blotting and densitometric scanning of protein bands. (B) Cytoplasmic localization of YAP in HRECs overexpressing CCN1. Cells were transduced with either Ad-luc or Ad-CCN1 (3 MOI) and further transfected with GFP-tagged YAP (i.e., pEGFP-C3-hYAP1). (C and D) Expression of CCN1, YAP, and pYAP in HRECs transduced with Ad-CCN1 and exposed to VEGF (50 ng/ml) for 4 h. *, P < 0.0003 (n = 3). (E) Effects of CCN1 on YAP transcriptional activity, as determined upon cell transfection with the 8×GTIIC-lux plasmid. *, P < 0.001 (n = 3). (F) Effect of function-blocking integrin antibodies (Ab) on CCN1-induced YAP phosphorylation. *, P < 0.0004; **, P < 0.0003 (n = 3). (G and H) Effects of small Rho GTPases and pharmacological inhibitors on CCN1-mediated YAP inactivation. *, P < 0.05 versus conditions with VEGF (n = 3); NS, not significant. (I) Phosphorylation status of potential YAP kinases (e.g., Erk1/2, Akt, and Src kinase) upon VEGF stimulation of CCN1-overexpressing cells. Protein lysates were analyzed by Western immunoblotting with the indicated antibodies. Cntl, control; Veh, vehicle; pYAP, pErk1/2, pAkt, and pSrc kinase, phosphorylated YAP, Erk1/2, Akt, and Src kinase, respectively.
VEGF activates YAP via its effects on the actin cytoskeleton (23). Stimulation of HRECs with VEGF reduced the level of YAP phosphorylation, indicating that YAP became active. In the same cells, overexpression of Ad-CCN1 increased the level of YAP phosphorylation (Fig. 5C and D). Thus, CCN1 signals interfere and counter VEGF-induced YAP activation. In VEGF-stimulated cells, CCN1 overexpression reduced the transcriptional activity of YAP-TEAD by 80%, as determined by a luciferase reporter driven by TEAD response elements (i.e., 8×GTIIC-lux) (Fig. 5E).
CCN1 is a bona fide ligand for various integrin receptors (26). To gain a broader understanding of the impact of CCN1 and YAP cross talk in VEGF signaling, we determined whether neutralization of various integrin unit subtypes rescues YAP dephosphorylation and its nuclear localization induced by CCN1. As shown in Fig. 5F, blockade of either β1, β3, or αvβ3 integrin rescued VEGF-induced YAP dephosphorylation in CCN1-expressing cells. Thus, YAP inactivation is, at least in part, dependent on the CCN1 interaction with the β1 and β3 integrin subtypes. As protein kinases downstream of Rho GTPases mediate CCN1-integrin signaling, we determined whether inhibition of Rho GTPases reversed the effect of CCN1 on YAP activation. When cotransfected with the p8×GTIIC-luciferase reporter and a dominant negative form of either RhoA, Cdc42, or Rac1, YAP activation was not affected by CCN1 (Fig. 5G). However, cell treatment (after 3 h of VEGF stimulation) with either latrunculin B, MK-2206, or an Src kinase inhibitor, each of which targets actin polymerization, Akt/PKB, and Src kinase, respectively, reversed, at least in part, the effects of CCN1 on YAP-TEAD activity (Fig. 5H). Cell treatment with the extracellular signal-regulated kinase 1 and 2 (Erk1/2) inhibitor had no effect. Fully confirming these associations, increased YAP phosphorylation in VEGF-treated CCN1-overexpressing cells increased the phosphorylation of potential upstream YAP kinases, including phosphorylated Akt (pAkt) and Src kinase (Fig. 5I). Thus, both Akt and Src kinase might operate in a common cytoskeletal actin-sensitive pathway, leading to YAP phosphorylation and nuclear exclusion in response to CCN1 signals.
EC-specific deletion of CCN1 resulted in a gain of YAP function.
Since the in vitro data presented above establish a link between CCN1 signals and YAP activity, we hypothesized that the EC-specific loss of CCN1 increases active YAP and results in a gain of YAP function. To explore the premise, mice with an EC-specific deletion of CCN1 were produced by crossing of CCN1flox/flox mice with transgenic Cdh5-Cre-ERT2 Cre recombinase-expressing deleter mice (Fig. 6A). CCN1 expression in the retina of mutant mice was significantly reduced (>82%) at both the mRNA and the protein levels (Fig. 6B and C). Concomitantly, the phosphorylation of YAP on conserved serine residue Ser112 (homologous to Ser127 in human YAP) was markedly decreased in CCN1 mutant mice compared to wild-type mice (Fig. 6C and D). Analysis of the vascular phenotype of the CCN1 mutants during rapid growth cycles of ECs at P6 showed dramatic changes in the retinal vasculature: reduced forward progression and increased radial expansion with widely lumenized vessels and less “gappiness” among vascular branches (Fig. 6E to H). Vascular specification into arteries, capillaries, and veins was lost in EC-ΔCCN1 mice, which is consistent with previous reports (6). The retinal vessels of wild-type CCN+/+ mice were slender and well organized into arteries, capillaries, and veins compared with those of CCN1 mutant mice. We further studied the phenotypic plasticity of ECs by their proliferation index and differentiation into tip and stalk cells. As shown in Fig. 6I and J, the number of endothelial tip cells located at the retinal vascular edge was significantly higher in mutant mice than in wild-type control mice. The BrdU+ cell count at the vascular edge showed a 3-fold increase in EC-ΔCCN1 mouse retinas than in CCN1+/+ mouse retinas (Fig. 6K), which is consistent with increased YAP activity and EC proliferation. We also examined the expression of genes previously identified in genomic analyses to be YAP targets (27). As shown in Fig. 6L, the expression of six YAP target genes was significantly increased in the retinas of CCN1 mutant mice. These targets include genes involved in actin cytoskeleton-mediated processes, such as cell motility (ANKrd1), growth (MYC, ETS1), actin dynamics (FLMA), and VEGF signaling (SHC2). Together, these observations demonstrate CCN1’s involvement in YAP inactivation during sprouting of blood vessels and further evidenced the CCN1-dependent regulation of YAP during vascular development.
FIG 6.

EC-specific deletion of CCN1 increased YAP activation and EC proliferation during vessel development. (A) Schematic representation of EC-specific deletion of CCN1 in mice. (B) CCN1 mRNA levels in CCN1+/+ and EC-ΔCCN1 mouse retinas. **, P < 0.001 versus CCN1+/+ mice (n = 4). (C and D) Expression of CCN1, pYAP, and YAP in retinal protein lysates from control CCN1+/+ and EC-ΔCCN1 mice. **, P < 0.001 versus YAP of CCN1+/+ mice (n = 4). (E) IB4-stained retinal flat mounts of CCN1+/+ and EC-ΔCCN1 mice. (F) The outline of blood vessels, the vasculature skeleton, and the branching points are highlighted in yellow, red, and green, respectively. (G and H) Quantitative analysis of vascular areas and junctional density of representative retinas from CCN1+/+ and EC-ΔCCN1 mice. **, P < 0.003 versus CCN1+/+ mice (n = 4) of the same age. (I and J) Vascular fronts of flat-mounted IB4-stained CCN1+/+ and EC-ΔCCN1 mouse retinas. IB4 staining was overexposed to visualize tip cell filopodia (arrows). Tip cells were counted in four equivalent areas of each retina. *, P < 0.05 versus CCN1+/+ retinas (n = 6). (K) Quantitative analysis of EC proliferation, as determined by BrdU incorporation. Equivalent areas of CCN1+/+ and EC-ΔCCN1 mouse retinas were compared. Data are means plus SE. *, P < 0.05 versus CCN1+/+ retinas (n = 4). (L) Relative mRNA levels of YAP target genes in retinal lysates of CCN1+/+ and EC-ΔCCN1 mice. Data are means ± SE (n = 3).
CCN1-YAP interplay involves changes in actin cytoskeleton and matrix compliance.
CCN1 is a small ECM protein that localizes pericellularly and regulates cell matrix tension. Our qualitative analyses showed that the HRECs’ spread area was increased in VEGF-stimulated cells and decreased when cells expressed CCN1 (Fig. 7A). This is because cells attach to a small area and thus experience low mechanical stress when they secrete and adhere to CCN1. Conversely, CCN1-expressing cells spread to a greater extent when plated on a stiff substrate, like type I collagen. CCN1-expressing cells displayed reduced stress fibers and YAP nuclear exclusion, whereas those plated on type I collagen showed extensive stress fibers and a mixed nuclear and cytoplasmic localization of YAP (Fig. 7B). Under these conditions, YAP nuclear import correlated with YAP-TEAD transcriptional activity, as determined by a luciferase reporter activation assay (Fig. 7C). Thus, cellular tension and the organization of stress fibers that generate pulling forces against the ECM are determining factors of CCN1-induced YAP inactivation (28, 29). Since the destabilization of stress fibers inhibits YAP activity, we determined which subset of the F-actin network is modulated by CCN1. For this purpose, HRECs were treated with either SMIFH2 or CK666. SMIFH2 and CK666 inhibit the formation of F-actin bundles (formin dependent) and the F-actin branched networks that sustain lamellipodium formation (Arp dependent), respectively. As shown in Fig. 7D, YAP activity was unaffected by CK666 but was significantly increased in SMIFH2-treated cells, suggesting that CCN1 targets F-actin formin-dependent bundles.
FIG 7.

CCN1 induces YAP phosphorylation through actin capping/severing proteins. (A) Mean spread area of HRECs cultured under the indicated conditions. *, P < 0.05 (n = 20 to 45 cells). (B) Close-up view of YAP (green)- and phalloidin (Phal; red)-stained HRECs following Ad-CCN1 and/or VEGF treatment. The additional effect of substrate stiffness was tested by plating the Ad-CCN1-transduced cells on type I collagen-coated wells. (C) Effects of CCN1 and type I collagen on VEGF-induced YAP activation determined by the 8×GTIIC-lux reporter activity. *, P < 0.003 (n = 4). (D) Effects of formin- and Arp-dependent F-actin inhibitors (SMIFH2 [1, 10, 50 μM] and CK666 [1, 10, 50 μM], respectively) on CCN1-dependent regulation of YAP activity determined by the 8×GTIIC-lux reporter activity (n = 4). (E and F) Depletion of CapZ or gelsolin with siRNAs rescued YAP inhibition by CCN1. (E) Phosphorylation of YAP was analyzed by Western blotting upon transfection of the cells with CapZ or gelsolin siRNAs. (F) Densitometric measurement of the protein bands. *, P < 0.004 (n = 3). (G) IB4-stained retinal flat mounts of P5 mice upon intravitreous injection of the SMIFH2 inhibitor (1 μl, 10 μM) or gelsolin siRNA. (H) A representative image of the phosphorylation pattern of YAP in retinal lysates analyzed by Western blotting upon SMIFH2 inhibitor or gelsolin siRNA injection. Animal treatment was as described in the legend to panel G. siCtrl, siCpz, and siGlsn or siGln, control, Cpz, and gelsolin siRNAs, respectively.
Moreover, it is recognized that as cells adhere to specific (self-made or exogenous) substrate, they adjust the tension and overall organization of their actin cytoskeleton by engaging numerous actin-binding proteins (30). Among these, gelsolin and CapZ are known to be organizers of F-actin distribution and dynamics that increase the turnover of F-actin by severing microfilaments and preventing filament reannealing and polymerization (31). Accordingly, depletion of either gelsolin or CapZ with small interfering RNAs (siRNAs) rescued YAP nuclear localization in CCN1-treated VEGF-stimulated cells Fig. 7E and F. We further examined the effects of depletion of actin-capping proteins on vascular growth and morphogenesis in postnatal mouse retinas. Intravitreous injection into the mouse eye of either gelsolin siRNA or SMIFH2 produced the same set of characteristic abnormalities in the developing retinal vasculature as CCN1 deletion did, which included the formation of a denser and more highly anastomosed superficial capillary plexus (Fig. 7G), and YAP phosphorylation was simultaneously reduced (Fig. 7H). Collectively, these findings indicate that CCN1-induced YAP inactivation is, at least in part, dependent on actin-capping proteins.
AAV-mediated expression of CCN1 in the endothelium inhibits YAP activation and reduced abnormal neovascular growth in the retina.
Since our studies showed that the interplay among CCN1 signaling, cytoskeletal architecture, and YAP transcriptional activation normalizes angiogenesis in the retina, we postulated that they could serve as potential molecular tools to correct vascular abnormalities. Therefore, we investigated the functional relevance of CCN1-YAP cross talk in pathological angiogenesis using mice with oxygen-induced retinopathy (OIR), a model of ischemia-induced VEGF-dependent retinal neovascularization (32, 33). Mice were subjected to hyperoxia from P7 to P12 to induce vaso-obliteration and normoxia until P17 to produce maximal preretinal neovascularization (Fig. 8A and B). Abnormal retinal neovascular tufts invaded the normally avascular vitreous, which leads in humans to retinal detachment and blindness (34). In this model, the steady-state mRNA levels of CCN1 were significantly reduced at P12 and P17 (i.e., >80% and >65%, respectively) compared with those under normoxic conditions (35). Therefore, we examined whether the ectopic expression of either CCN1 or a dominant negative form of YAP unable to interact with TEAD (i.e., dn-YAP) could rescue the abnormal vascular phenotype in OIR. To this end, CCN1 and dn-YAP were ectopically expressed through adeno-associated virus type 6 (AAV6)-mediated gene transfer. Retro-orbital (RO) injection of AAV6-GFP into P4 mouse pups allowed the targeted and highly efficient expression of GFP in the ECs of retinal blood vessels (Fig. 8C). Quantitative analyses showed that preretinal neovascular tufts, which can be seen abundantly in the central and midperipheral retina in AAV6-GFP-injected eyes, were markedly reduced (<80%) in mice injected with AAV6-CCN1 (Fig. 8D and E). Analysis of retinal protein and mRNA contents showed that CCN1 mRNA and CCN1 protein levels increased at P17 in AAV6-CCN1-treated mice compared with AAV6-GFP-treated mice (Fig. 8F). Importantly, the levels of active nonphosphorylated YAP, which were increased in retinal protein lysates of AAV6-GFP-injected OIR mice, were decreased in AAV6-CCN1-injected eyes, indicating that CCN1 affected neovascular tuft formation, at least in part, by preventing YAP activation. In addition, AAV6-mediated expression of CCN1 induced, at least in part, revascularization of the retina by virtue of its own proangiogenic activity. Meanwhile, injection of AAV6–dn-YAP reduced both CCN1 gene expression and neovascular growth in the retina (<60%) (Fig. 8D to G). Interestingly, large areas in the central retina remained avascular, indicating that dn-YAP suppressed both normal and abnormal vascular growth in retinas with OIR. Of note, expression of neither CCN1 nor dn-YAP affected VEGF levels in the retina (Fig. 8H). Thus, CCN1 functions in tandem with YAP to control the pathological growth of blood vessels in the model of OIR. Figure 9 illustrates the regulatory loop model between CCN1 and YAP in action during VEGF-induced blood vessel growth and regeneration.
FIG 8.

AAV-mediated expression of CCN1-induced YAP phosphorylation and reduced neovascular growth in retinas with OIR. (A and B) Representative flat-mount preparations of IB4-stained retinas from control and OIR mice at P12 and P17. (C and D) Flat-mount images of IB4-stained retinas from OIR mice at P17 following retro-orbital injection of AAV6-GFP, AAV6–dn-YAP, or AAV6-CCN1. (E) Percentage of neovascular tufts determined by computer-assisted image analysis. **, P < 0.05 versus conditions with AAV6-GFP. (F) Quantitative analysis of CCN1 mRNA levels in the retinas of OIR mice that were injected with AAV6-GFP, AAV6–dn-YAP, or AAV6-CCN1. Data are means ± SE. *, P < 0.05 versus conditions with AAV6-GFP; **, P < 0.001 versus conditions with AAV6-GFP. (G) Qualitative analysis of CCN1, pYAP, and YAP proteins by Western immunoblotting in retinal lysates from OIR mice following retro-orbital injection at P4 of either AAV6-GFP, AAV6–dn-YAP, or AAV6-CCN1. (H) Relative mRNA levels of VEGF-A in retinal lysates of the AAV6-GFP, AAV6–dn-YAP, or AAV6-CCN1 mouse groups.
FIG 9.
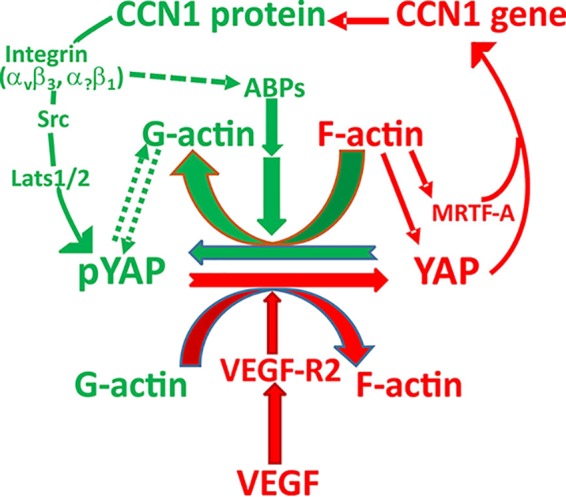
Schematic model of feed-forward and feedback regulation among CCN1, YAP, and VEGF in sprouting angiogenesis. VEGF signaling through VEGF-R2 promotes a YAP-mediated transcriptional program that controls cytoskeleton dynamics and maintains a feed-forward loop that ensures continuous sprouting. CCN1, a component of the YAP transcriptome, provides negative feedback on YAP activity by inducing a cytoskeletally relaxed state conducive to YAP phosphorylation/inactivation. Subsequently, high and high-to-low branching densities occur, providing specific vascular patterns. Lats, large tumor suppressor kinase; ABPs, actin binding proteins.
DISCUSSION
We present herein evidence of a dual requirement and hitherto undescribed regulatory loop between CCN1 and YAP during vascular development and regeneration. First, we show that YAP activates the CCN1 gene promoter via TEAD binding to the consensus cis element most proximal to the initiation of the transcription site. The MRTF-A–SRF complex also influences the CCN1 promoter, and the combined transcriptional coactivation by YAP and MRTF-A was required for a full-blown induction of CCN1 gene transcription. These findings are consistent with those of previous genomic studies that showed that expression of MRTF-SRF genomic targets is dependent on YAP-TEAD activity and, reversibly, that YAP-TEAD target gene expression is also dependent on MRTF-SRF signaling (21). Cross talk between these two pathways may arise as a consequence of two of their properties. (i) YAP and MRTF physically interact with one another in the context of their promoter targets (e.g., the CCN1 promoter). Indeed, the YAP-MRTF interaction was shown to be mediated by the YAP WW domain and a conserved PPXY motif of MRTFs (36). The YAP-MRTF interaction allows their recruitment to DNA independently of their own DNA-binding partners (e.g., TEAD and SRF). (ii) Like the CCN1 gene, YAP and MRTF are highly sensitive to the monomeric actin (G-actin) concentration in cells (13, 37). Cell adhesion and spreading alone directly initiate YAP activation, but additional cytoskeletal events, such as the conversion of G-actin to F-actin, amplify TEAD-YAP activity by further activating YAP and MRTF-A. According to a study by Pawłowski et al., binding to G-actin prevents YAP and MRTF from interacting with the importin family of nuclear import factors (38). Furthermore, our in vivo data corroborated our findings in cultured ECs in vitro. The proximal TEAD-YAP binding site of the endogenous CCN1 promoter was more enriched with both YAP and MRTF-A in retinal areas of active angiogenesis than in those with fully formed vessels, and the loss of YAP function substantially reduced the CCN1 promoter occupancy by transcriptional coactivators.
Interestingly, EC-specific deletion of YAP resulted in a mere 50% reduction of CCN1 gene transcription, despite a high recombination efficiency of YAP deletion in ECs, whereas the ubiquitous deletion of YAP induced a greater reduction of CCN1 transcript levels (92%). There are three possible explanations for this observation. First, both endothelial and nonendothelial YAP-dependent mechanisms regulate CCN1 gene transcription and sculpt the vascular environment. Such inputs become abrogated only following the ubiquitous deletion of YAP. This possibility is supported by the observation that the ubiquitous deletion of YAP produced more pronounced vascular defects and molecular alterations than EC-specific YAP deletion alone. In the retina, YAP is expressed by astrocytes and, potentially, retinal ganglion cells (RGCs); both are major sources of VEGF, a key angiogenic factor that establishes and maintains a functional retinal vascular network. The importance of astrocyte- and RGC-derived VEGF has previously been shown by the complete absence of a retinal vascular plexus in RGC- and astrocyte-deficient mice (39, 40). VEGF recruits YAP as a transcriptional coactivator to control the expression of genes required for EC motility, adhesion, and proliferation (23). The dramatic alterations of vascular ECs at the vascular front in mice with ubiquitous YAP deficiency mimicked the phenotypes associated with the small Rho family GTPase Cdc42, which induces the filopodial formation of tip cells and promotes luminogenesis during vascular development (41). Thus, CCN1 gene expression may be influenced by YAP activity in different cell types in the retina. Second, CCN1 and YAP are also highly expressed in the vascular smooth muscle cells of blood vessels (42). Smooth muscle-specific knockout of YAP was shown to produce major vascular defects, including thinning of the arterial wall and aneurysmal enlargement of the blood vessel lumen. Vascular hypoplasia was largely due to increased expression of cell cycle arrest genes, such as Gadd45α and Trp63. Since the bidirectional signals between ECs and mural cells, such as smooth muscle cells, regulate vessel plasticity, the global loss of YAP function likely affects the cellular cross talk between ECs and smooth muscle cells. Cell-cell communication is critical for the angiogenic process to proceed properly during both normal development and regeneration. Third, a potential compensation for YAP by its paralogue, TAZ, is possible but unlikely. Indeed, global YAP or TAZ deletion in mice produced distinct phenotypes/outcomes, suggesting that YAP and TAZ regulate different sets of genes (17, 43). Studies using the double knockout of YAP and TAZ produced vascular defects that were more severe than those resulting from the single knockout of each of these genes, which may argue for the additive effects of the deletion of both genes (44). Taken together, our results support a model whereby the molecular alterations produced by YAP deletion at the global tissue level determine the subsequent alterations of gene expression and vascular defects more than those produced by YAP deletion at the EC level do.
Another interesting outcome of this study is the uncovering of a negative feedback loop in which increased CCN1 protein levels regulate YAP nuclear localization and activity. CCN1 is an integrin-binding protein involved in EC adhesion and migration. As a matricellular protein, CCN1 has both ECM and growth factor-like properties (45). As a chemotactic molecule, CCN1 promotes the conversion of cells from a stationary to a motile phenotype, a process that is critical for controlling developmental processes and tissue remodeling. It is recognized that as cells adhere to a specific (self-made or exogenous) substrate, they adjust the tension and overall organization of their actin cytoskeleton by engaging many actin-binding proteins (46). Cells gauge their adhesive cues alongside soluble growth factors to make a net cell cycle engagement decision. Concordantly, stimulation with VEGF alone increased cell spreading and YAP nuclear localization, while overexpression of CCN1 prior to VEGF stimulation reduced the cell spread area and favored YAP nuclear exclusion. In contrast, cells plated on a stiff collagen substrate spread to a significantly greater extent, overriding the effects of CCN1 on VEGF-stimulated cells. Conceivably, stiffer substrates, such as type I collagen, allow the homogeneous distribution of growth factor receptors and signal transduction cascades (e.g., the Erk1/2 signal) across the cell and reduce the growth factor threshold, forcing the cells to shift to proliferation. Therefore, the predominance of CCN1 in the extracellular microenvironment provides the cells with a compliant surface incompatible with YAP activation and cell growth. This hypothesis is supported by our finding that the EC-specific loss of CCN1 in mice increased the level of active YAP by depriving the CCN1 signal negative feedback, unleashing the uncontrolled growth of ECs and the formation of enlarged dysmorphic blood vessels in the postnatal retina. Mechanistically, CCN1 binding to integrins β1 and β3 and phosphorylation of phosphatidylinositol 3-kinase and Src were required for the CCN1-induced phosphorylation of YAP and its cytoplasmic retention. Whether CCN1-derived signals control YAP phosphorylation by direct activation of YAP kinases or inhibition of specific phosphatases is unknown. A recent genetic screen identified 16 kinases which, when silenced, led to YAP dephosphorylation and nuclear translocation even under the high-cell-density conditions in which the Hippo signaling kinases are typically activated (47). This suggests that both canonical and noncanonical kinases and/or phosphatases may transduce CCN1 signals to YAP and contribute to the molecular cascade of the CCN1-YAP regulatory loop. Further studies of the CCN1 signaling kinome are warranted.
Our data further showed that the CCN1-induced reorganization of the actin cytoskeleton regulates YAP phosphorylation and activity. Cytoskeletal actin functions as a platform that facilitates the transmission and transduction of CCN1-integrin signals to YAP. The state of F-actin assembly/disassembly is monitored by actin-capping and -severing proteins, like CapZa and gelsolin. The use of siRNAs directed against gelsolin and CapZ reversed the changes induced by CCN1, resulting in YAP activation and nuclear localization. CapZ induces F-actin depolymerization, converting F-actin to G-actin, while gelsolin, a filament-severing and -capping protein, shortens the average length of filaments by binding to the side of F-actin and cutting it into two pieces (30). Depletion of gelsolin or CapZ increased the formation of stress fibers, mimicking the physiological state of actin when the cells were plated on a stiff collagen substrate. This remarkable actin-dependent interplay between CCN1 and YAP appears to hold in vivo as well. Indeed, intravitreal injection of the SMHIF2 inhibitor, which specifically inhibits formin-mediated actin nucleation, produced exuberant YAP activation and EC hyperproliferation, which resulted in the enlargement of blood vessels. Thus, selective disruption of formin-dependent actin cables affects the vascular phenotype like CCN1 deletion does.
Together, our data highlight a major angiomodulatory pathway involving CCN1, YAP, and VEGF. Their distinctive temporal and spatial expression patterns and biological activities appear to be intertwined and to regulate one another. At first, parallel gradients of CCN1 and VEGF form in the vicinity of vascular sprouts. The VEGF gradient forms through the degradation and/or sequestration of VEGF within the matrix, while the CCN1 gradient forms via YAP-CCN1 interregulation. The robustness of the transition between these two functional states is governed by interlocking of positive and negative feedback loops. Via its effects on the actin cytoskeleton, VEGF promotes a YAP-mediated transcriptional program that controls cytoskeleton dynamics and maintains a feed-forward loop that ensures continuous sprouting. Soon after, CCN1, a component of the YAP transcriptome itself, provides negative feedback on YAP activity, allowing correct sprouting patterns across a spectrum of high to low branching densities. In essence, CCN1 provides local control of YAP-dependent migratory gene expression in tip cells while allowing trailing stalk cells (with higher YAP activity and a lower CCN1 level) to proliferate and lumenize. The possibility of feedback signals other than those of CCN1 to YAP also cannot be ruled out. The complexity of this cross talk may be further amplified when both CCN1 and YAP signals converge on those of the Notch, Wnt, transforming growth factor β (TGF-β), and G protein-coupled receptor (GPCR) signals (6, 48, 49). Future studies will map out other components of this CCN1-YAP regulatory circuit.
Finally, the mouse OIR model demonstrates that the dysregulation of CCN1-YAP cross talk is critical in the pathogenesis of neovascular diseases. Hyperoxia-induced vaso-obliteration, which is due to excessive EC apoptosis from oxidative stress (50), decreased YAP activity and CCN1 gene expression. Likewise, the retinopathy response associated with OIR reduced CCN1 gene expression, even though the relative ischemia subsequent to vaso-obliteration is known to stimulate proangiogenic factor synthesis (e.g., VEGF, insulin-like growth factor). Such fluctuations create an angiogenic imbalance considered to be an important risk factor for retinopathy in humans (51). The excessive production of VEGF is the main cause of abnormal neovascular growth, as VEGF-specific antagonists markedly suppress retinal neovascularization in mice and primates with ischemic retinopathy (52). As a direct consequence of high VEGF levels, active nonphosphorylated YAP levels are robustly elevated during the neovascular phase of OIR. Thus, we can reasonably conclude that the excessive and abnormal growth of blood vessels was due to reduced CCN1 expression and the subsequent loss of CCN1 negative feedback on YAP activation. As a proof of concept, the AAV-mediated overexpression of CCN1 increased phosphorylated YAP levels, reduced neovascular tuft formation, and allowed, at least in part, revascularization of the retina. It is noteworthy that the ectopic expression of CCN1 had no effect on VEGF expression. Likewise, dn-YAP blocked all vascular growth, suggesting that YAP mediates the effects of VEGF on neovascular tuft formation. Taken together, our findings demonstrate that the VEGF-YAP-CCN1 regulatory axis is critical for the development and stability of retinal blood vessels and that any dysregulation of this circuit will shift vessel regeneration into a pathological state. Hence, therapeutic approaches designed to intervene in the CCN1-YAP angiomodulatory pathway may help revert the pathological growth of blood vessels characteristic of vascular diseases of the eye.
MATERIALS AND METHODS
Reagents, antibodies, and vectors.
All chemicals used were of reagent grade and included IB4 (2 μg/ml; Vector Laboratories), jasplakinolide (0.5 μM; EMD Millipore), 4-hydroxytamoxifen (4HT; catalog number H7904; Sigma), latrunculin B (0.5 μM; catalog number L5288; Sigma), rat tail type I collagen (100 μg/ml; produced in the B. Chaqour lab using pepsin digestion), PD09059 (20 μM; catalog number 444966; Calbiochem), SMIFH2 (1 to 50 μM; Millipore-Sigma), MK-2206 (1 μM; EMD Millipore), CK666 (1 to 50 μM; Millipore-Sigma), VEGF165 (VEGF-A isoform; 50 ng/ml; R&D), and 4′,6-diamidino-2-phenylindole (DAPI). The antibodies used in this study were as follows: polyclonal anti-CCN1 antibody (1:250; Biovision Inc.), anti-YAP (1:250 for immunofluorescence; Santa Cruz), anti-YAP (1:500 for Western blotting; Cell Signaling), anti-YAP (1:100 for ChIP; Active Motif), pYAP-S127 (which recognizes pYAP-S112; 1:500; Cell Signaling), anti-collagen IV (1:500; Developmental Studies Hybridoma Bank), anti-MLK1/MRTF-A (1:100; a generous gift from R. Prywes), anti-MLK1/MRTF-A (1:150; Santa Cruz Biotech), anti-TEAD1 (Santa Cruz Biotech), anti-pAkt (1:250; Cell Signaling), anti-Src kinase (1:250; Fisher Scientific), anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase; 1:1,000; Santa Cruz Biotech), anti-BrdU–Alexa Fluor 488 (1:500; Molecular Probes), and fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG and tetramethyl rhodamine isocyanate-conjugated goat anti-rabbit IgG antibodies (1:1,000; Vector Laboratories). siRNAs targeting gelsolin and CapZ were obtained from Qiagen. Plasmid vectors, including 8×GTIIC-Lux, YAP-GFP, CCN1, and the dn-YAP/S94A YAP mutant, were from Addgene. The dominant negative forms T19N RhoA, T17N Cdc42, and T17N Rac were a gift from K. M. Hahn (UNC). Adenoviral vectors Ad-CCN1 and Ad-luc were generated and amplified as previously described (13).
Generation of mice with CCN1 and YAP conditional alleles.
All procedures involving experimental animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals (53), and all animal experiments were approved by the Institutional Animal Care and Use Committee of SUNY Downstate Medical Center, New York, NY. All mice used were of the C57BL/6J genetic background. CCN1:GFP mice carrying GFP under the control of the CCN1 promoter and CCN1flox/flox and YAPflox/flox mice carrying loxP-flanked sequences around exons 1 and 2 were previously described (6, 32, 54). Tg(Cdh5-Cre ERT2) and Tg(UBC-Cre ER) transgenic mouse lines expressing the Cre recombinase gene under the control of the Cdh5 and ubiquitin C gene promoters, respectively, have been previously described (55, 56). Mice carrying the CCN1flox/flox allele were bred to Cdh5-Cre ERT2 mice to generate EC-ΔCCN1. Mice carrying the YAPflox/flox allele were bred to Cdh5-Cre ERT2 and UBC-Cre ER mice to generate EC-ΔYAP and UB-ΔYAP mice, respectively. The genotype was determined by PCR to identify mice with floxed alleles and hemi- and homozygous floxed alleles with or without the Cre allele. A solution of 4-hydroxytamoxifen (4HT) was dissolved in ethanol at 10 mg/ml, and then 4 volumes of corn oil were added. 4HT samples were further diluted in corn oil prior to intraperitoneal injection of 100 μl to mouse pups. Recombination levels in mice with the Cre allele were determined by qPCR and Western blotting to determine recombination efficiency, as previously described (35, 57).
Recombinant AAV vector generation and RO injection.
AAV6 vectors expressing cDNA encoding dn-YAP or CCN1 under the control of the cytomegalovirus promoter were used, as this isotype induced a relatively uniform expression of a reporter transgene in the vascular wall following systemic injection of the vector. AAV vectors were produced by triple transfection of 293T cells with the pAV-FH AAV vector, the Ad helper vector, and a vector encoding the Rep and serotype-specific Cap proteins, followed by iodixanol gradient centrifugation and purification by anion-exchange chromatography. Viral titers were determined as the number of genomic copies (GC) per microliter using quantitative real-time PCR (>1 × 1013 to 1014 GC/ml). AAV packaging, purification, and quality control were performed by Vigene Biosciences. The AAV-GFP control was obtained from the Penn Vector Core at the University of Pennsylvania. For RO injection, mouse pups were anesthetized using isoflurane anesthesia prior to retro-orbital injection of the recombinant viral vectors (4 × 1013 vector genomes).
Intravitreal injections.
Intravitreal injection of pharmacological inhibitors behind the ora serrata and directly into the vitreal cavity of one eye was performed using a 33-gauge needled syringe at postnatal day 3 (P3). In the contralateral eye, an equal volume of vehicle (dimethyl sulfoxide) was injected. Similarly, the mice were given a 1-μl intraocular injection of 5 μmol/μl gelsolin siRNA in one eye and 5 μmol/μl control siRNA in the other eye. No untoward effects (e.g., redness or opacity) were noted from injection.
Immunohistochemical staining and analysis of angiogenesis in the mouse retina.
The mouse eyes were fixed in 4% paraformaldehyde for 2 h. The retinas were dissected, laid flat on SuperFrost Plus-coated slides, and permeabilized in 0.1% Triton X-100 at room temperature for 20 min. Blood vessels were visualized by IB4 staining as previously described (6). Images were captured using a Leica DM5000 B fluorescence microscope. The peripheral retina was defined as the distal-most 20% of the vascular plexus (within 400 μm of the leading edge of the growing superficial capillary plexus), wherein active sprouting and vessel specification take place. The central retina was defined as the remaining area around the optic nerve head. Vascular parameters were measured using AngioTool software (58). By assessing the variation in foreground and background pixel mass densities across an image, the software determines morphological and spatial parameters. Four fluorescent images per retina were taken from 4 or 5 mice and analyzed.
OIR and retinal neovascularization image analyses.
Ischemic retinopathy was produced in C57BL/6J mice (29). Both male and female mice (5 or 6 mice per group) were used. Since postnatal weight gain and litter size are independent factors in OIR (59), consistent weights of mice in litters of the same size were used when making comparisons. Neonatal mice and their nursing dams were exposed to 75% oxygen in a Pro-Ox 110 chamber oxygen controller from Biospherix Ltd. (Redfield, NY) between P7 and P12 and then returned to normoxia until P17. Quantification of vascular obliteration and preretinal neovascular tufts in retinal flat mounts was performed at P12 and/or P17. The area of preretinal neovascularization was calculated by selecting tufts, which appear more brightly stained than normal vasculature, based on pixel intensities. The results for selected regions were then summed to generate the total area of neovascularization. The avascular areas and zones of neovascularization were expressed as a percentage of the total retinal surface area.
BrdU incorporation assay.
BrdU was administered at 10 mg/kg of body weight intraperitoneally. The retinas were further digested with proteinase K (10 μg/ml), fixed in 4% paraformaldehyde, treated with DNase I (0.1 unit/ml) for 2 h at 37°C, and incubated with anti-BrdU antibody. The number of BrdU-positive cells in equivalent areas of IB4-stained retinas from control and mutant mice was determined.
TUNEL staining.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was performed with an ApopTag fluorescein in situ apoptosis detection kit (EMD Millipore, Billerica, MA) according to the manufacturer’s protocol. TUNEL-positive nuclei were counted, and the number was normalized to the number of DAPI-positive nuclei.
Immunohistochemistry and Western immunoblotting.
Fixed and permeabilized retinal mounts were stained with IB4 as described above and/or with the antibody indicated in the text. Immunodetection was performed with either rhodamine- or fluorescein-conjugated anti-mouse or anti-rabbit immunoglobulin secondary antibody diluted in blocking solution. Retinal mounts were washed several times in phosphate buffer solution between incubations. Images were acquired using a Leica DM5500B fluorescence microscope. For retinal protein analysis, mouse eyes were enucleated, and the retinas were carefully dissected and homogenized in lysis buffer containing 1% Triton X-100, a 1% volume of phosphatase, and a protease inhibitor mixture. Protein samples (25 μg) were fractionated in a 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane, and Western blot analysis was performed by incubation with primary antibodies and then secondary antibodies. Immunodetection was performed using enhanced chemiluminescence (Pierce). Protein bands were quantified by ImageJ software (National Institutes of Health).
Cell culture, treatment, and adenoviral infection.
In vitro studies were performed with primary human retinal endothelial cells (HRECs; System Biology) maintained in culture according to the manufacturer’s instructions. The HREC phenotype was validated using several criteria: (i) a cobblestone morphology appearance, (ii) positive staining for factor VIII, (iii) uptake of acetylated low-density lipoprotein, and (iv) CD-31 and IB4 positivity. Cells were propagated in 35-mm dishes in predefined endothelial growth medium containing 10% fetal bovine serum (FBS; Atlanta Biological Inc.). Cells at 60% confluence were treated as described in the text and further processed for molecular and immunohistochemical analyses. For cell infection with adenoviral vectors, HRECs were first incubated in serum-free medium for up to 3 h with adenoviral vectors expressing the CCN1 (Ad-CCN1) or luciferase (Ad-luc) transgene. Cells were further incubated in serum-containing medium for 16 h. Viral vectors were used at multiplicities of infection (MOIs) ranging from 1 to 5.
Total RNA isolation and quantitative real-time PCR.
Total RNA was extracted from either cells or tissues using an RNeasy kit (Qiagen), and the quantity and quality of RNA were determined. RNA was converted using SuperScript III reverse transcriptase (Invitrogen), followed by quantitative real-time PCR using the TaqMan technology on a StepOne ABI sequence detection system. Highly specific primers were designed using Web-based primer design programs. The transcript amount (determined by the −2ΔΔCT threshold cycle [CT] method) was obtained by normalization to an endogenous reference (18S rRNA) relative to a calibrator.
Transient transfection, mutagenesis, and reporter assay.
Transfection was then performed using the Lipofectamine 2000 transfection reagent in serum-free medium. Cells were allowed to recover for 16 h in fresh medium containing 10% serum. Cells were transfected in triplicate with a mixture of DNAs of the desired expression vectors driven by the promoters of interest and a Cypridina luciferase vector. At 24 h after transfection, relative luciferase activity was measured in medium and cell lysates using a BioLux assay system (New England Biolabs). All experiments included a negative control that served as a baseline indicator of luciferase activity.
YAP-TEAD activity.
HRECs (50,000/dish) were first transduced with either Ad-GFP or Ad-CCN1 (MOI, 3) and incubated for 24 h. The cells were then transfected with the 8×GTIIC-luciferase plasmid (100 ng) together with the Cypridina luciferase reporter plasmid to adjust for transfection efficiency. After 24 h, the cells were incubated in low fetal serum concentrations (2%) overnight, followed by stimulation with VEGF (100 ng/ml) for the time period indicated in the text. Medium and cell lysates were then subjected to testing with a dual-luciferase assay system as described above.
ChIP assay.
DNA-protein complexes were fixed by directly adding formaldehyde (1%) to cultured cells or freshly isolated retinas. For retinal tissue, DNA was immunoprecipitated from retinal homogenates pooled from 10 mouse eyes of each genotype. The samples were then homogenized in lysis buffer. The nuclei were collected by microcentrifugation and then resuspended in sonication buffer containing 0.1 g of glass beads. The chromatin solution was precleared by centrifugation and incubated at 4°C with 1 μg of affinity-purified polyclonal antibody or no antibody for 16 h. Immunoprecipitation was continued for an additional 24 h, followed by elution of immune complexes. Cross-links were reversed by the addition of NaCl, and RNA was removed by the addition of 10 μg of RNase A per sample, followed by incubation at 65°C for 5 h. The pellets were collected by microcentrifugation. Real-time PCR was performed on 1 ng genomic DNA from ChIP experiments using the following primers: 5′-ACACACAAAGGTGCAATGGA-3′ and 5′-GTGACGTCGGCTCTGTCTG-3′ for human CCN1prox (ChIP), 5′-GAAAGGGCACTTTGGAATGA-3′ and 5′-GGCCCTTAGTGCTAATGCTG-3′ for human CCN1dist (ChIP), 5′-AAAAGGTGCAACGGAGCCAG-3′ and 5′-GTGTTGCAGTGACGTAGCTC-3′ for mouse CCN1prox (ChIP), and 5′-TCTCCGAGATTGCCCAGTTG-3′ and 5′-CGTTCAAACAGACACGCCAT-3′ for mouse CCN1dist (ChIP).
Statistical analyses.
Statistical analyses were performed using Prism software for Windows from GraphPad Inc. (San Diego, CA). To test for differences among several means of significance, a one-way analysis of variance with the Newman-Keuls multiple-comparison test was used. Where appropriate, a post hoc unpaired t test was used to compare two means/groups, and P values of <0.05 were considered significant.
ACKNOWLEDGMENT
We thank Genesis Lopez and Melanie Vinueza for their technical contributions. We thank all past and present lab members for their contributions to the generation and characterization of genetically modified animals and for helpful discussions during the preparation of the manuscript. We are grateful to Fernando Camargo (Harvard Stem Cell Institute) for providing YAPflox/flox mice and K. M. Hahn (UNC) for the generous gift of Rho GTPase dominant negative forms.
This work was supported in part by grants from the National Eye Institute of the National Institutes of Health (grants EY022091-05A1 and EY024998) to B.C.
REFERENCES
- 1.Bishop PN. 2015. The role of extracellular matrix in retinal vascular development and preretinal neovascularization. Exp Eye Res 133:30–36. doi: 10.1016/j.exer.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 2.Krupska I, Bruford EA, Chaqour B. 2015. Eyeing the Cyr61/CTGF/NOV (CCN) group of genes in development and diseases: highlights of their structural likenesses and functional dissimilarities. Hum Genomics 9:24. doi: 10.1186/s40246-015-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jun JI, Lau LF. 2011. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10:945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang R, Amir J, Liu H, Chaqour B. 2008. Mechanical strain activates a program of genes functionally involved in paracrine signaling of angiogenesis. Physiol Genomics 36:1–14. doi: 10.1152/physiolgenomics.90291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. 2002. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol 22:8709–8720. doi: 10.1128/mcb.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chintala H, Krupska I, Yan L, Lau L, Grant M, Chaqour B. 2015. The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development 142:2364–2374. doi: 10.1242/dev.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasan A, Pokeza N, Shaw L, Lee HS, Lazzaro D, Chintala H, Rosenbaum D, Grant MB, Chaqour B. 2011. The matricellular protein cysteine-rich protein 61 (CCN1/Cyr61) enhances physiological adaptation of retinal vessels and reduces pathological neovascularization associated with ischemic retinopathy. J Biol Chem 286:9542–9554. doi: 10.1074/jbc.M110.198689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Pasolli HA, Fuchs E. 2011. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A 108:2270–2275. doi: 10.1073/pnas.1019603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plouffe SW, Hong AW, Guan KL. 2015. Disease implications of the Hippo/YAP pathway. Trends Mol Med 21:212–222. doi: 10.1016/j.molmed.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu FX, Guan KL. 2013. The Hippo pathway: regulators and regulations. Genes Dev 27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. 2001. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 15:1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaqour B. 2013. Molecular control of vascular development by the matricellular proteins CCN1 (Cyr61) and CCN2 (CTGF). Trends Dev Biol 7:59–72. [PMC free article] [PubMed] [Google Scholar]
- 13.Hanna M, Liu H, Amir J, Sun Y, Morris SW, Siddiqui MA, Lau LF, Chaqour B. 2009. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem 284:23125–23136. doi: 10.1074/jbc.M109.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura I, Rosenbloom J, Macarak E, Chaqour B. 2001. Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am J Physiol Cell Physiol 281:C1524–C1532. doi: 10.1152/ajpcell.2001.281.5.C1524. [DOI] [PubMed] [Google Scholar]
- 15.Piccolo S, Dupont S, Cordenonsi M. 2014. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev 94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 16.Mo FE, Lau LF. 2006. The matricellular protein CCN1 is essential for cardiac development. Circ Res 99:961–969. doi: 10.1161/01.RES.0000248426.35019.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG, Magnuson TR, O'Neal W, Milgram SL. 2006. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol 26:77–87. doi: 10.1128/MCB.26.1.77-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, Halder G. 2011. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J 30:2325–2335. doi: 10.1038/emboj.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du KL, Chen M, Li J, Lepore JJ, Mericko P, Parmacek MS. 2004. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J Biol Chem 279:17578–17586. doi: 10.1074/jbc.M400961200. [DOI] [PubMed] [Google Scholar]
- 20.Holzinger A, Meindl U. 1997. Jasplakinolide, a novel actin targeting peptide, inhibits cell growth and induces actin filament polymerization in the green alga Micrasterias. Cell Motil Cytoskeleton 38:365–372. doi: 10.1002/(SICI)1097-0169(1997)38:4<365::AID-CM6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Foster CT, Gualdrini F, Treisman R. 2017. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev 31:2361–2375. doi: 10.1101/gad.304501.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watson EC, Koenig MN, Grant ZL, Whitehead L, Trounson E, Dewson G, Coultas L. 2016. Apoptosis regulates endothelial cell number and capillary vessel diameter but not vessel regression during retinal angiogenesis. Development 143:2973–2982. doi: 10.1242/dev.137513. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L, Jain RK, Mazzone M, Schmidt T, Fischer T, Halder G, Ruiz de Almodóvar C. 2017. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev Cell 42:462–478.e7. doi: 10.1016/j.devcel.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. 2015. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaqour B, Goppelt-Struebe M. 2006. Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J 273:3639–3649. doi: 10.1111/j.1742-4658.2006.05360.x. [DOI] [PubMed] [Google Scholar]
- 27.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. 2013. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 29.Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. 2011. Hippo pathway regulation by cell morphology and stress fibers. Development 138:3907–3914. doi: 10.1242/dev.070987. [DOI] [PubMed] [Google Scholar]
- 30.dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. 2003. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 31.Cooper JA, Schafer DA. 2000. Control of actin assembly and disassembly at filament ends. Curr Opin Cell Biol 12:97–103. doi: 10.1016/S0955-0674(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 32.Lee S, Elaskandrany M, Ahad A, Chaqour B. 2017. Analysis of CCN protein expression and activities in vasoproliferative retinopathies. Methods Mol Biol 1489:543–556. doi: 10.1007/978-1-4939-6430-7_46. [DOI] [PubMed] [Google Scholar]
- 33.Chaqour J, Lee S, Ravichandra A, Chaqour B. 2018. Abscisic acid—anti-angiogenic phytohormone that modulates the phenotypical plasticity of endothelial cells and macrophages. J Cell Sci 131:jcs210492. doi: 10.1242/jcs.210492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartnett ME. 2015. Pathophysiology and mechanisms of severe retinopathy of prematurity. Ophthalmology 122:200–210. doi: 10.1016/j.ophtha.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Elaskandrany M, Lau LF, Lazzaro D, Grant MB, Chaqour B. 2017. Interplay between CCN1 and Wnt5a in endothelial cells and pericytes determines the angiogenic outcome in a model of ischemic retinopathy. Sci Rep 7:1405. doi: 10.1038/s41598-017-01585-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim T, Lim DS. 2016. The SRF-YAP-IL6 axis promotes breast cancer stemness. Cell Cycle 15:1311–1312. doi: 10.1080/15384101.2016.1161994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu OM, Miyamoto S, Brown JH. 2016. Myocardin-related transcription factor A and Yes-associated protein exert dual control in G protein-coupled receptor- and RhoA-mediated transcriptional regulation and cell proliferation. Mol Cell Biol 36:39–49. doi: 10.1128/MCB.00772-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pawłowski R, Rajakylä EK, Vartiainen MK, Treisman R. 2010. An actin-regulated importin alpha/beta-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J 29:3448–3458. doi: 10.1038/emboj.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH, Honore JC, Kermorvant-Duchemin E, Varma DR, Tremblay S, Leduc M, Rihakova L, Hardy P, Klein WH, Mu X, Mamer O, Lachapelle P, Di Polo A, Beausejour C, Andelfinger G, Mitchell G, Sennlaub F, Chemtob S. 2008. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 14:1067–1076. doi: 10.1038/nm.1873. [DOI] [PubMed] [Google Scholar]
- 40.Tao C, Zhang X. 2016. Retinal proteoglycans act as cellular receptors for basement membrane assembly to control astrocyte migration and angiogenesis. Cell Rep 17:1832–1844. doi: 10.1016/j.celrep.2016.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, Stump P, Byerly L, Donaldson M, Hao J, Fruttiger M, Lu QR, Zheng Y, Lang RA, Xin M. 2017. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc Natl Acad Sci U S A 114:10918–10923. doi: 10.1073/pnas.1704030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Hu G, Liu F, Wang X, Wu M, Schwarz JJ, Zhou J. 2014. Deletion of yes-associated protein (YAP) specifically in cardiac and vascular smooth muscle cells reveals a crucial role for YAP in mouse cardiovascular development. Circ Res 114:957–965. doi: 10.1161/CIRCRESAHA.114.303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N, Makita R, Kurihara H, Morin-Kensicki EM, Nojima H, Rossant J, Nakao K, Niwa H, Sasaki H. 2009. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 16:398–410. doi: 10.1016/j.devcel.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Artap S, Manderfield LJ, Smith CL, Poleshko A, Aghajanian H, See K, Li L, Jain R, Epstein JA. 2018. Endocardial Hippo signaling regulates myocardial growth and cardiogenesis. Dev Biol 440:22–30. doi: 10.1016/j.ydbio.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan L, Chaqour B. 2013. Cysteine-rich protein 61 (CCN1) and connective tissue growth factor (CCN2) at the crosshairs of ocular neovascular and fibrovascular disease therapy. J Cell Commun Signal 7:253–263. doi: 10.1007/s12079-013-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berrier AL, Yamada KM. 2007. Cell-matrix adhesion. J Cell Physiol 213:565–573. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 47.Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong K-K, Wong K-K, Kim C, Camargo FD. 2014. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 16:108–117. doi: 10.1038/ncb2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaqour B. 2016. Regulating the regulators of angiogenesis by CCN1 and taking it up a Notch. J Cell Commun Signal 10:259–261. doi: 10.1007/s12079-016-0328-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, Gerhardt H, Engelhardt B. 2015. Wiring the vascular network with neural cues: a CNS perspective. Neuron 87:271–296. doi: 10.1016/j.neuron.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 50.Kowluru RA, Kanwar M. 2009. Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Radic Biol Med 46:1677–1685. doi: 10.1016/j.freeradbiomed.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartnett ME. 2010. The effects of oxygen stresses on the development of features of severe retinopathy of prematurity: knowledge from the 50/10 OIR model. Doc Ophthalmol 120:25–39. doi: 10.1007/s10633-009-9181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimoto K, Kubota T. 2012. Anti-VEGF agents for ocular angiogenesis and vascular permeability. J Ophthalmol 2012:852183. doi: 10.1155/2012/852183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 54.Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. 2011. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144:782–795. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim KH, Chen CC, Alpini G, Lau LF. 2015. CCN1 induces hepatic ductular reaction through integrin alphavbeta(5)-mediated activation of NF-kappaB. J Clin Invest 125:1886–1900. doi: 10.1172/JCI79327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. 2007. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan L, Lee S, Lazzaro DR, Aranda J, Grant MB, Chaqour B. 2015. Single and compound knock-outs of microRNA (miRNA)-155 and its angiogenic gene target CCN1 in mice alter vascular and neovascular growth in the retina via resident microglia. J Biol Chem 290:23264–23281. doi: 10.1074/jbc.M115.646950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zudaire E, Gambardella L, Kurcz C, Vermeren S. 2011. A computational tool for quantitative analysis of vascular networks. PLoS One 6:e27385. doi: 10.1371/journal.pone.0027385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanhaesebrouck S, Daniels H, Moons L, Vanhole C, Carmeliet P, De Zegher F. 2009. Oxygen-induced retinopathy in mice: amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr Res 65:307–310. doi: 10.1203/PDR.0b013e3181973dc8. [DOI] [PubMed] [Google Scholar]