

Abstract
Oncolytic viruses, including the oncolytic rhabdovirus VSV‐GP tested here, selectively infect and kill cancer cells and are a promising new therapeutic modality. Our aim was to study the efficacy of VSV‐GP, a vesicular stomatitis virus carrying the glycoprotein of lymphocytic choriomeningitis virus, against prostate cancer, for which current treatment options still fail to cure metastatic disease. VSV‐GP was found to infect 6 of 7 prostate cancer cell lines with great efficacy. However, susceptibility was reduced in one cell line with low virus receptor expression and in 3 cell lines after interferon alpha treatment. Four cell lines had developed resistance to interferon type I at different levels of the interferon signaling pathway, resulting in a deficient antiviral response. In prostate cancer mouse models, long‐term remission was achieved upon intratumoral and, remarkably, also upon intravenous treatment of subcutaneous tumors and bone metastases. These promising efficacy data demonstrate that treatment of prostate cancer with VSV‐GP is feasible and safe in preclinical models and encourage further preclinical and clinical development of VSV‐GP for systemic treatment of metastatic prostate cancer.
Keywords: immunotherapy, interferon, metastatic prostate cancer, oncolytic virus, VSV‐GP
Short abstract
What's new?
While oncolytic viruses are promising antitumor agents, those currently under development exhibit variability between patients in their effectiveness against tumors. In this study, multiple prostate cancer cell lines were found to differ in their susceptibility to killing by the oncolytic rhabdovirus VSV‐GP. Despite this, VSV‐GP was highly effective in prostate cancer mouse models. When administered intratumorally or intravenously, VSV‐GP produced long‐term remission of subcutaneous tumors and bone metastases. The data suggest that, with further development, VSV‐GP could be a clinically valuable agent for the systemic treatment of metastatic prostate cancer.
Abbreviations
- BLI
bioluminescence imaging
- GFP
green fluorescence protein
- HSV
herpes simplex virus
- IFN‐I
interferon type I
- IFNα
interferon alpha
- IFNβ
interferon beta
- ISG
interferon‐stimulated gene
- JAK1
Janus kinase 1
- Luc
firefly luciferase
- mCRPC
metastatic castration‐resistant prostate cancer
- MOI
multiplicity of infection
- OV
oncolytic virus
- PCa
prostate cancer
- PFU
plaque forming unit
- s.c.
subcutaneous
- STAT1
signal transducer and activator of transcription 1
- VSV
vesicular stomatitis virus
- VSV‐GP
VSV pseudotyped with the glycoprotein of lymphocytic choriomeningitis virus
Prostate cancer (PCa) is the leading cause of cancer in men both in the United States and Europe.1, 2 Despite the high 5‐year survival rate when detected at local stage, metastatic castration‐resistant prostate cancer (mCRPC) still has a dismal prognosis.3
Oncolytic viruses (OVs), which selectively infect and kill tumor cells, have shown promising results in preclinical models and clinical trials in a wide range of tumor types both as monotherapies and in combination with other compounds.4, 5, 6 Moreover, the OV Talimogene Laherparepvec (Imlygic®, T‐VEC) has recently been licensed in the United States and Europe for the treatment of melanoma.7, 8 In the few early‐phase clinical trials that have included PCa patients, safety and efficacy data were also encouraging.9 However, most of the OVs currently under development need to be applied intratumorally, as pre‐existing or rapidly induced neutralizing antiviral antibodies limit the systemic delivery to the tumor tissue and thereby the efficacy in metastatic disease. Among the OVs currently in the development pipeline, vesicular stomatitis virus (VSV) carrying the envelope protein (GP) of the lymphocytic choriomeningitis virus, VSV‐GP, has several outstanding features: (i) There is practically no pre‐existing immunity against VSV‐GP in the general population and VSV‐GP does not readily induce neutralizing antibodies.10 This potentially allows effective repeated systemic delivery of VSV‐GP to the primary tumor and to metastases. (ii) Natural VSV infection generally does not cause severe disease in humans and VSV‐GP has completely lost the residual neurotoxicity of wild‐type VSV.11 (iii) VSV‐GP has a fast replication cycle of only 6–8 hr, resulting in rapid cell lysis and high virus titers.12 (iv) Infection is efficiently controlled by the IFN‐I‐induced antiviral response,13 which has been shown to be disrupted in most cancer types,14 making VSV selective for a broad range of tumors.
We here report successful treatment of PCa with VSV‐GP in a panel of PCa cell lines and patient‐derived primary PCa cultures, and in mouse models. Furthermore, systemic application of VSV‐GP induced complete remission in a bone metastasis mouse model.
Materials and Methods
Ethics statement
Permission for the use of patient specimens has been given by the Ethical Committees of Medical University of Innsbruck (Nr.:UN4837:317/4.7).
Animal experiments were performed in compliance with the national animal experimentation law (“Tierversuchsgesetz”) and animal trial permission was granted by national authorities (Bundesministerium für Wissenschaft, Forschung und Wirtschaft, BMWF‐66.011/0119‐II/3b/2012 and BMWFW‐66.011/0041‐WF/V/3b/2016).
Cell lines and viruses
PC3, PC‐3M‐Luc, Du145, LNCaP, VCaP, LAPC4, 22Rv1 and the murine TRAMP‐C1 cell lines were obtained from ATCC (LGC Standards, Heidelberg, Germany) and maintained as previously described.15 Human cell lines were routinely authenticated by STR profiling as described before.16 Hamster BHK‐21 cells and murine L929 cells were obtained from ATCC and used for virus production and titration as described previously.17 BHK‐21 cells were maintained in Glasgow minimum essential medium (GMEM) (Gibco, Carlsbad, CA) supplemented with 10% heat inactivated FCS (PAA Laboratories, Vienna, Austria), 5% tryptose phosphate broth (Gibco), 100 units/ml penicillin (Gibco) and 0.1 mg/ml streptomycin (Gibco). L929 cells were maintained in high‐glucose DMEM (Lonza, Basel, Switzerland) supplemented with 10% heat inactivated FCS, 4 mM l‐glutamine (Gibco), 100 units/ml penicillin and 0.1 mg/ml streptomycin.
VSV‐GP, VSVncp‐ΔG‐GFP*GP, VSV‐GP‐GFP and VSV‐GP‐Luc have been described before.17 A schematic representation of the VSV‐GP variants used is shown in Supporting Information 1.
Patients and clinical samples
Patient specimens were obtained from six patients undergoing radical prostatectomy at the Department of Urology of the Medical University of Innsbruck. Patient information is detailed in Supporting Information, Table 1. Primary cells were obtained and cultured as previously described.18 For experiments, primary cells were seeded without STO mouse embryonic fibroblast feeder layer.
Tropism assay
Cells were plated in 12‐well plates to obtain monolayer cultures and infected with 10‐fold serial dilutions of VSVncp‐ΔG‐GFP*GP. Sixteen hours postinfection, cells were harvested and analyzed for GFP expression using flow cytometry. Samples showing 2%–20% GFP+ cells were used to calculate virus titers using the following formula: (%GFP+ cells) × (dilution factor)/(well volume) and given relative to the reference cell line BHK‐21.
Virus growth kinetics
Cell monolayers were infected with VSV‐GP with an MOI of 0.1. One hour after infection, medium was removed, cells were washed with PBS (PAA Laboratories) and fresh medium was added. Supernatant of infected cells was collected after 24 and 48 hr for analysis of viral titers by TCID50 as described before.17
In vitro cytotoxicity assay
Cells were plated in 96‐well plates and infected with indicated VSV‐GP concentrations. At indicated time points, WST‐I reagent (Roche, Basel, Switzerland) was added to each well and absorbance at 450 nm was measured after 4 hr. Absorbance values were normalized to a nontreated sample. For IFN‐I resistance bioassay, cells were treated with a recombinant universal type‐α IFN (PBL, NJ) at 500 U/ml or left untreated 16 hr prior to virus treatment.
Flow cytometry
Cells growing in monolayers were harvested using 5 mM EDTA (Sigma‐Aldrich, St. Louis, MO), prepared to a single cell suspension and stained for flow cytometry analysis using the following specific antibodies: anti‐α‐dystroglycan from mouse (clone IIH6C4, Merck Millipore, Darmstadt, Germany) 1:500; APC‐conjugated anti‐mouse‐IgM from goat (Jackson ImmunoReasearch, Suffolk, UK) 1:500.
Binding assay
Cells were harvested using 5 mM EDTA and incubated at 4°C with VSV‐GP at an MOI ranging from 100 to 500 for 30 min. Samples were washed, fixed with 4% formaldehyde and prepared for flow cytometry using a LCMV‐GP specific antibody (KL25) as described above. APC‐conjugated anti‐mouse IgG from goat (Life Technologies, Carlsbad, CA) was used as secondary antibody.
IFN‐beta ELISA
Cells were plated in 12‐well plates to obtain monolayer cultures and infected with VSV‐ΔG‐GP‐GFP at an MOI 3. One hour after infection, cultures were extensively washed with PBS and fresh medium was added. Twenty‐four hours after infection, medium was collected and stored at −80°C until ELISA assay was performed in duplicates using a human‐IFNβ ELISA kit (Invitrogen, Karlsruhe, Germany) and following manufacturer's indications. Plates were analyzed in an ELISA microplate reader (Bio‐rad, Hercules, CA).
Preparation of cell lysates and Western blot
Cells were plated in 6‐well plates and treated with 100 U/ml of recombinant universal IFN‐I and cell lysates were prepared 1, 2, 4, 8, 12 and 24 hr after treatment. Cells were lysed in ice‐cold cell‐lysis buffer (50 mmol/l HEPES, pH 7.5; 150 mmol/l NaCl; 1% Triton X‐100; 2% aprotinin; 2 mmol/l EDTA, pH 8.0; 50 mmol/l sodium fluoride; 10 mmol/l sodium pyrophosphate; 10% glycerol; 1 mmol/l sodium vanadate; and 2 mmol/l Pefabloc SC) for 30 min and then centrifuged (15,000g) for 10 min to remove cell debris. Lysates were stored at −80°C until use.
Protein separation was performed by sodium dodecyl sulfate (SDS)‐polyacrylamide gel electrophoresis (PAGE) under standard reducing conditions on a 10% polyacrylamide gel. Proteins were transferred by electrophoresis to a 0.45 µm nitrocellulose membrane (Whatman, Dessel, Germany). Membranes were blocked with TBST‐BSA (TBS containing 0.1% Tween‐20 and 5% Bovine Serum Albumin, Sigma‐Aldrich) and stained overnight at 4°C with primary antibodies. Detection was done using horseradish peroxidase (HRP) specific secondary antibodies. Blots were developed with ECL.19 The following antibodies were used: anti‐pSTAT1‐Tyr701 from rabbit (clone 58D6, Cell Signaling Technology, Leiden, The Netherlands), anti‐STAT1 from mouse (clone 10C4B40, Biolegend, London, UK), anti‐β‐actin from mouse (clone AC‐74, Sigma‐Aldrich), HRP‐conjugated anti‐rabbit from goat and HRP‐conjugated anti‐mouse from goat (Jackson ImmunoResearch).
Real‐time PCR for IFN‐induced genes
Cells plated in 6‐well plates were treated with VSVncp‐ΔG‐GFP*GP (MOI:10). RNA was isolated 24 hr post‐treatment using the RNeasy kit (Qiagen, Hilden, Germany). Synthesis of cDNA was done with the RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Waltham, MA) using polyT oligomers. Real‐time PCR was performed in 20 µl reaction mix containing 0.05 U/µl HOT‐FIREPol DNA polymerase (Solis BioDyne, Tartu, Estonia), 2 µl 10× buffer, 3 mM MgCl2, 0.2 mM dNTPs, 200 nM forward primer, 200 nM reverse primer and 0.5 µM Evagreen (Biotium, Fremont, CA). Preordered primer sets were used, containing specific primers for human IFNβ, OAS1, MX1, ISG15, IFIT1 and using GAPDH as reference gene (Invivogen, San Diego, CA). Reaction was performed in triplicates in a CFX Connect thermocycler (Bio‐rad). Change in gene expression was calculated using the ΔCt method and the reference gene as calibrator or the ΔΔCt method using the reference gene and the PBS treated samples as calibrators.20
Animal models
Tumors were implanted bilaterally by subcutaneous injection of 2x106 Du145 cells in the flanks of Balb/c Rag2−/−γc−/− male mice (bread in‐house) in a final volume of 100 µl or subcutaneous injection of 5 × 106 VCaP cells in the right flank of athymic nude male mice (Janvier, Le Genest St Isle, France) mixed 1:1 with Matrigel (Corning, Wiesbaden, Germany) in a final volume of 100 µl. Tumors size was measured every two days with a caliper and tumor volumes were calculated as length × width2 × 0.4. When tumors reached a size of 0.07 cm3, mice were treated as indicated in the figure legends. Mice were euthanized when tumor size reached 0.8 cm³.
2 × 106 22Rv1 cells were injected subcutaneously in the right flank of athymic nude male mice (Janvier) in a final volume of 100 µl. Tumors were monitored as indicated before. When tumors reached a size of 0.05 cm3, mice were treated as indicated in the figure legends. Mice treated at a tumor size of 0.07 cm3 with 107 pfus VSV‐GP‐Luc or VSV‐GP‐GFP were used for BLI or tumors were resected and prepared for histological analysis respectively.
For the syngeneic model, 2 × 106 TRAMP‐C1 cells were injected subcutaneously in the right flank of male C57BL/6JRj mice (Janvier) in a final volume of 100 µl. Tumors were monitored as indicated before. When tumors reached a size of 0.07 cm3, mice were treated as indicated in the figure legends. Mice were euthanized when tumors reached 0.8 cm3.
For the bone metastasis model, PC‐3M‐Luc cells were prepared to a 2.5 × 107/ml cell suspension and 10 µl of this cell suspension were injected in the left tibia of athymic nude male mice (Janvier). Tumor engraftment and growth was monitored by bioluminescence imaging (BLI). When bioluminescent signal was higher than 106 p/sec/cm2/sr, mice were treated intravenously with a single tail vein injection of VSV‐GP (5 × 107pfus, 100 µl) or left untreated. Tumors were further monitored every 4 days by BLI.
Bioluminescence imaging (BLI)
BLI was performed using the Lumina In Vivo Imaging System (IVIS Lumina II, Perkin Elmer, Waltham, MA). Mice were injected intraperitoneally with 1.5 mg D‐Luciferin (Promega GmbH, Mannheim, Germany) and imaged 15–30 min after substrate injection. Exposure times were adjusted to bioluminescence intensity and ranged from 0.5 to 60 sec. For quantification of bioluminescent signal, regions of interest (ROI) were manually defined and the average intensity within the ROI was measured as photons/sec/cm2/sr.
Histology of 22Rv1 tumors
Animals were sacrificed at the indicated time‐points. Tumors were isolated and fixed with 4% paraformaldehyde (Carl Roth, Karlsruhe, Germany) for 16–24 hr and subsequently equilibrated in 30% sucrose solution (Sigma‐Aldrich). Tissue was embedded in tissue Tec O.C.T freezing medium (Sakura Finetek Europe, Alphen aan den Rijn, The Netherlands) and stored at −80°C. Tissue was then sectioned into slices of a thickness of 4–7 µm using a cryostat (Microm HM560, ThermoFisher Scientific), thaw‐mounted onto polylysine coated slides (ThermoFisher Scientific) and stored at −20°C. For antibody staining, slices were thawed and dried at room temperature and blocked 3 hr with blocking buffer containing 5% normal goat serum (Sigma‐Aldrich), 1% BSA and 0.2% Triton (Carl Roth) in PBS. Afterward, slices were incubated overnight with anti‐active‐caspase 3 antibody (R + D Systems, Minneapolis, MN) diluted 1:200 in blocking buffer. Detection of primary antibody was done by incubation at room temperature for 30 min with goat‐anti‐rabbit antibody conjugated with Alexa594 (Life Technologies) diluted 1:1000 in blocking buffer containing 0.25 µg/ml DAPI (Sigma‐Aldrich). Finally, slices were embedded in Mowiol‐Dabco mounting medium (Sigma‐Aldrich). Analysis was done using a fluorescence microscope (Eclipse Ti, Nikon CEE GmbH, Vienna, Austria) with a DS‐Fi2 camera (Nikon) and using the NIS‐Elements BR 4.20 software (Nikon)
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (Version 7, GraphPad Software, La Jolla, CA) as indicated in the figure legends. For in vitro experiments and s.c. tumor volumes, statistically significant differences were determined by one‐way ANOVA or two‐way ANOVA with Bonferroni's multiple comparison correction. Kaplan–Meier survival curves were compared using the Log‐rank (Mantel‐Cox) test. Statistically significant differences were encoded as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
VSV‐GP infects and kills PCa cell lines and primary cultures
It has previously been shown that patients differ considerably in their susceptibility to oncolytic virus treatment. We therefore analyzed the susceptibility of different PCa cell lines to VSV‐GP with the aim to elucidate potential determinants of oncolytic virus efficacy. From a panel of 6 human and one murine PCa cell line, VSV‐GP efficiently infected and killed 4: PC3, Du145, 22Rv1 and TRAMP‐C1 cells (Fig. 1 a,c). Infection of these cells yielded high virus titers in PC3 and Du145 and more modest titers in 22Rv1 and TRAMP‐C1 (Fig. 1 b). Interestingly, the bone‐tropic PC‐3M‐Luc cell line showed comparable infection rates to the parental PC3 but slightly reduced virus replication titers and killing rate after VSV‐GP infection (Fig. 1 a–c). Effective killing was also achieved in the LAPC4 cell line, despite less efficient infection and virus production (Fig. 1 a–c). In contrast, inefficient infection, virus production and killing were observed in VCaP and LNCaP cells (Fig. 1 a–c).
Figure 1.
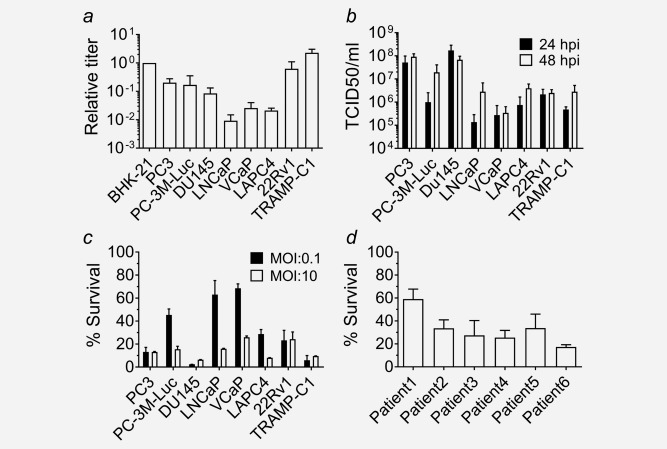
VSV‐GP efficiently infects and kills PCa cell lines and primary cultures. (a) Tropism of VSV‐GP was determined for different human prostate cancer cell lines and a murine cancer cell line (TRAMP‐C1). Cells were infected with 10‐fold serial dilutions of VSVncp‐ΔG‐GFP*GP and sixteen hours post infection, percentage of GFP+ cells was determined by flow cytometry and used to calculate the titer on each cell line. Titers are given as relative to the titer in the control cell line BHK‐21. Experiments were done at least in duplicates. Bars represent mean ± SEM of at least n = 2 independent experiments. (b) Replication kinetics were determined in the same cell line panel. Cells were infected with an MOI of 0.1 with VSV‐GP. Twenty‐four and forty‐eight hours postinfection, supernatant was collected and virus was titrated by TCID50 on BHK‐21 cells. Bars represent mean ± SEM of at least n = 2 independent experiments. (c) For the killing assay, cells were infected with VSV‐GP at an MOI of 0.1 or MOI of 10. Viability of infected cultures was determined 72 hr postinfection using the WST‐1 assay and expressed relative to PBS treated cells. Bars represent mean ± SEM of at least n = 2 independent experiments performed in dodecaplicates. (d) Killing assay was performed in monolayers of primary cultures using an MOI of 0.1 as described in (c).
To study whether infection of VCaP and LNCaP was restricted at the entry level, we analyzed the surface expression of α‐dystroglycan (α‐DG), the receptor mediating VSV‐GP entry,21 and virus binding to the cell surface. The lowest level of α‐DG expression and virus binding was indeed observed in LNCaP cells (Fig. 2). Furthermore, increasing the virus dose by 100‐fold induced killing rates comparable to the sensitive cell lines (Fig. 1 c). Surprisingly, VCaP, showed high levels of α‐DG and the amount of virus bound was comparable to the other more susceptible cell lines (Fig. 2), indicating that virus replication was restricted at a post entry level.
Figure 2.
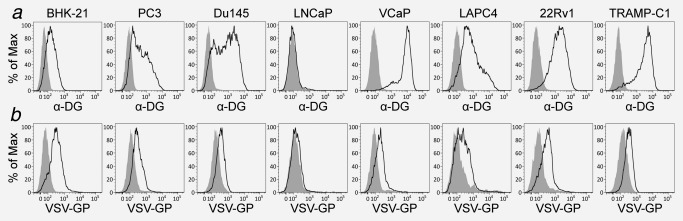
α‐dystroglycan surface expression and virus binding is reduced on LNCaP. (a) Surface expression of α‐dystroglycan was analyzed by flow cytometry using specific antibodies or only secondary antibody as control. (b) Binding capacity of VSV‐GP to prostate cancer cells was analyzed. Cells were prepared to a single‐cell suspension using nonenzymatic methods and infected with VSV‐GP at 4°C or treated with PBS for control. After 30 min, cells were washed several times, fixed, stained with antibodies specific for GP and analyzed by flow cytometry.
To further broaden our in vitro cell panel, we studied the sensitivity of primary cultures of prostate epithelial cells from PCa patient samples. All primary cultures tested were sensitive to VSV‐GP killing, yet killing rates were heterogeneous, ranging from 60% to 20% survival (Fig. 1 d).
Taken together, VSV‐GP could successfully infect and kill PCa cell lines and patient‐derived primary cultures.
Interferon type I induced antiviral responses in PCa cell lines
We have previously shown in mouse models that the efficacy of oncolytic virus treatment can be limited by the interferon response in cancer cells17 and interferon competence of a tumor can determine its susceptibility to oncolytic virus therapy. Therefore, we tested the responsiveness of the PCa cell lines to IFN‐I using recombinant IFN‐α. Treatment with IFN‐α activated the Jak/STAT1 pathway in PC3 and VCaP cells (Fig. 3 c). However, while IFN‐I treatment induced complete protection to VSV‐GP‐induced killing in VCaP cells, killing rates in PC3 and PC‐3M‐Luc cells were only attenuated (Fig. 3 a), indicating that these two cell lines had a reduced sensitivity for IFN‐I signaling. The murine TRAMP‐C1 cell line was also protected from VSV‐GP killing upon IFN‐α treatment (Fig. 3 a). Increasing the virus dose to an MOI of 10 completely overcame the IFN‐α protective effect in TRAMP‐C1 cells and partially in PC3 and VCaP cells (Fig. 3 b). In contrast, no STAT1 protein was detected in LAPC4 and 22Rv1 cells (Fig. 3 d). Consequently, VSV‐GP‐induced killing rates were not influenced by IFN‐I treatment (Fig. 3 a,b). In addition, 22Rv1 cells might be resistant to IFN‐I because of known heterozygous frameshift mutations in the JAK1 gene (p.L431fs*22 and p.K142fs*26).22 Finally, despite activation of STAT1 in the Du145 cell line, no IFN‐α induced protection was observed (Fig. 3 a–c), indicating that the IFN response was blocked downstream of STAT1. Another important aspect of the innate immune response is the capacity of the infected cell to produce IFN‐I upon viral infection, which can act in an auto‐ and paracrine fashion to induce an antiviral response. PC3 was the only cell line able to produce IFN‐I upon virus infection (Fig. 3 e). However, expression of some IFN‐stimulated genes (ISGs) upon VSV‐GP treatment was observed in both IFN‐I sensitive and IFN‐I resistant cell lines (Fig. 3 f), although to a considerably lesser extent in the IFN‐I resistant cells lines.
Figure 3.
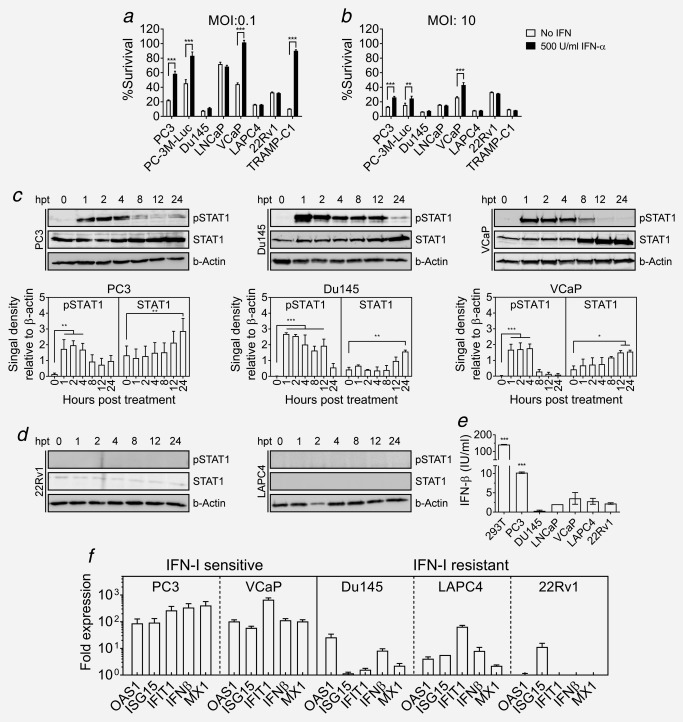
IFN‐I induced antiviral response in PCa cell lines. (a,b) Capacity of PCa cell lines to mount an IFN‐I‐induced antiviral response was studied. Cell monolayers were treated with 500 U/ml of IFN‐I or PBS for 16 hr prior to infection with VSV‐GP at an MOI of 0.1 (a) or MOI of 10 (b). Cell viability of the infected cultures was determined 72 hr postinfection using WST‐I reagent as indicated before. (c) Monolayers of PC3, Du145 and VCaP cell lines were treated with 100 U/ml IFN‐I. At indicated time‐points, lysates were prepared and analyzed for STAT1 expression and phosphorylation, using β‐actin as loading control. Representative blots and quantification of signal density relative to β‐actin are shown. (d) Representative blots of 22Rv1 and LAPC4 cells treated as in (d). (e) Production of IFN‐I by infected cultures was determined. Cell monolayers were infected with VSVncp‐ΔGGFP* GP at MOI of 3 or mock treated with PBS. Supernatant was collected 24 hr postinfection and concentration of IFN‐β was measured using a specific ELISA. Bars represent mean IFN‐β concentration ± SEM in VSVncp‐ΔG‐GFP*GP treated supernatants (***p < 0.001 compared to noninfected). (f) Monolayers of prostate cancer cell lines were treated with VSVncp‐ΔG‐GFP*GP at an MOI of 3 or mock treated with PBS. mRNA was extracted 24 hr post‐treatment, reverse‐transcribed to cDNA and expression of indicated genes was analyzed by real‐time PCR. Fold expression of the selected genes was calculated relative to GAPDH and the mock treated sample.
In summary, the capacity of PCa cell lines to mount an antiviral response was heterogeneous, as were the molecular mechanisms responsible for the lack of such response.
VSV‐GP is effective in PCa subcutaneous tumor models
We then tested whether our in vitro observations correlated with susceptibility to VSV‐GP treatment in mouse models. Initially, we investigated the xenograft model with subcutaneous (s.c.) Du145 tumors. Two intratumoral injections of 107 plaque forming units (PFUs) of VSV‐GP were sufficient to induce full tumor remission in all treated mice, with no tumor relapses detected during the 100‐day follow‐up (Fig. 4 a,b). In addition, in a dose–response experiment in s.c. 22Rv1 tumors, a single intratumoral dose of 2.3 × 108 PFUs of VSV‐GP induced response in 6/7 treated mice, 5/7 showing tumor remission and 1/7 showing stable disease (Fig. 4 c; Supporting Information, 2). All responding mice survived until the end of the study (86 days after treatment) (Fig. 4 d). Reducing the dose to 3.3 × 105 resulted in 3/7 treated mice showing tumor growth delay, 1/7 with stable disease and 2/7 with tumor remission and an overall survival rate of 57.1% (Fig. 4 c,d; Supporting Information, 2).
Figure 4.
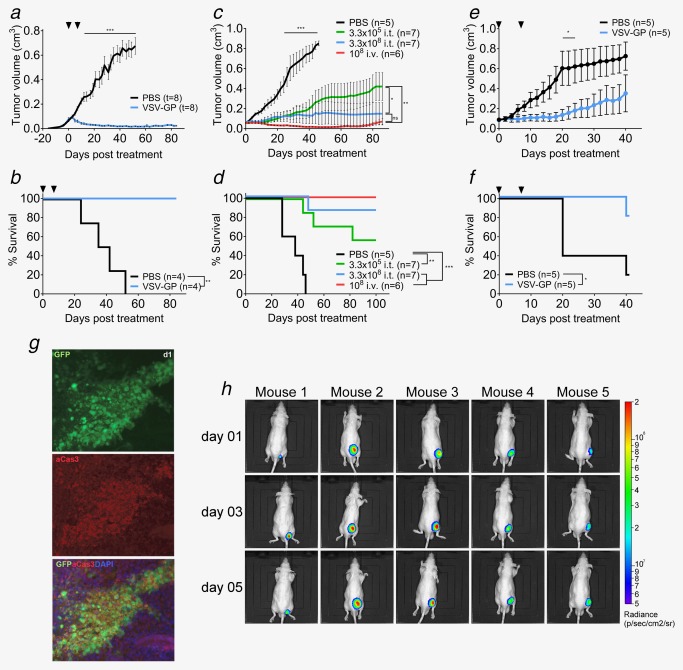
VSV‐GP shows efficacy in PCa xenograft subcutaneous models. (a,b) 2 × 106 Du145 cells were injected subcutaneously into both flanks of Balb/c‐rag−/−γ−/− mice. Tumors were treated at a size of 0.07 cm3 with intratumoral injection of either 107 pfus VSV‐GP or PBS. Treatment was repeated 7 days later. Mean of tumor volume (a) and Kaplan–Meier survival curve (b) are shown. (c,d) 2 × 106 22Rv1 cells were injected subcutaneously into the right flanks of nude mice. Tumors were treated at a size of 0.05 cm³ with intratumoral injection of either 3.3 × 105 pfus VSV‐GP, 2.3 × 108 pfus VSV‐GP or PBS or with intravenous injection of 108 pfus VSV‐GP. Mean of tumor volume (c) and Kaplan–Meier survival curve (d) are shown. (e,f) 5 × 106 VCaP cells mixed 1:1 with matrigel were injected subcutaneously into the right flank of nude mice. Tumors were treated at a size of 0.07 cm³ with an intratumoral injection of either 107 pfus VSV‐GP or PBS. Treatment was repeated 7 days later. Mean of tumor volume (e) and Kaplan–Meier survival curve (f) are shown. (g) Subcutaneous 22Rv1 tumors were treated at a size of 0.07 cm³ with an intratumoral injection of 108 pfus VSV‐GP‐GFP. Tumors were isolated at indicated time points and fixed. Cryo‐slices were prepared and probed with fluorescently labelled antibodies recognizing activated caspase 3. Antibody signal and viral GFP signal were analyzed under the fluorescence microscope. (h) Subcutaneous 22Rv1 tumors treated at a size of 0.07 cm³ with an intratumoral injection of 108 pfus VSV‐GP‐Luc. Representative BLI pictures of treated mice are shown.
We next used two VSV‐GP variants expressing firefly luciferase (VSV‐GP‐Luc) or GFP (VSV‐GP‐GFP) to correlate the therapeutic effect observed with viral replication in the tumor. We were able to detect viral protein expression selectively in the tumor as early as one day after treatment and up to 9 days (Fig. 4 h; Supporting Information, 3) in nude mice bearing s.c. 22Rv1 tumors treated with a single injection of VSV‐GP‐Luc. When 22Rv1 s.c. tumors were treated with VSV‐GP‐GFP, we could detect virus spread within the tumor and co‐localization of GFP signal with activated caspase 3 (Fig. 4 g).
We then tested whether VSV‐GP was effective when administered systemically. Intravenous treatment with 108 PFU of VSV‐GP of s.c. 22Rv1 tumors induced complete responses in 6/6 animals with an overall survival rate of 100% during the 86 days of follow‐up (Fig. 4 c,d; Supporting Information, 2).
Having seen that susceptible tumor in vitro were also susceptible in vivo, we next used a xenograft model with VCaP s.c. tumors to investigate potential in vivo effects in those tumors which were resistant in vitro. Treatment with two intratumoral injections of 107 pfus resulted in stable disease in 3/5 mice and tumor growth delay in 2/5 mice (Fig. 4 e,f). It is important to note that 2/5 tumors treated with PBS also showed growth delay.
Finally, we studied the therapeutic outcome of VSV‐GP treatment in an immune‐competent C57BL/6 model bearing syngeneic TRAMP‐C1 s.c. tumors. Treatment with three intratumoral doses of 108 VSV‐GP pfus resulted in tumor growth delay and a significant increase of the median survival compared to control mice (Fig. 5 a,b). Using the VSV‐GP‐Luc variant, we detected replication of VSV‐GP in the TRAMP‐C1 tumors 24 hr after virus administration (Fig. 5 c). However, VSV‐GP presence in the tumor was shorter than in the xenograft model, indicated by a significant drop of the reporter signal three days after treatment (Fig. 5 c).
Figure 5.
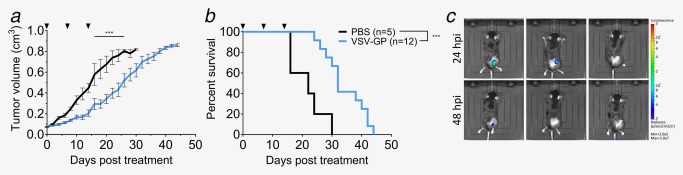
VSV‐GP increases mean survival of TRAMP‐C1 tumor‐bearing mice. (a,b) 2 × 106 TRAMP‐C1 cells were injected subcutaneously into the right flank of C57BL/6JRj mice. Tumors were treated at a size of 0.07 cm³ with intratumoral injection of either 108 PFUs VSV‐GP or PBS. Treatment was repeated 7 and 14 days later. Mean of tumor volume (a) and Kaplan–Meier survival curve (b) are shown. (c) Subcutaneous TRAMP‐C1 tumors treated at a size of 0.07 cm³ with an intratumoral injection of 108 pfus VSV‐GP‐Luc. Representative BLI pictures of treated mice are shown.
In conclusion, VSV‐GP was efficient in the in vivo PCa models tested, independently of the susceptibility observed in vitro. In addition, no signs of viral pathogenicity were observed, despite the immunodeficiency status of some of the models used.
Systemic treatment with VSV‐GP is effective in a bone metastasis model
These results encouraged us to test the feasibility of VSV‐GP treatment in a metastasis model. One of the most frequent metastatic presentations of PCa is bone metastasis,23 which requires systemic treatment. Therefore, we chose a well‐established bone metastasis model of PCa16 to test the feasibility of VSV‐GP treatment in the metastatic setting. Mice carrying PC‐3M‐Luc bone tumors in the left tibia were treated with a single intravenous injection of 5 × 107 PFUs VSV‐GP. Luciferase signal from the tumor was used to measure tumor burden. Tumor remission was observed in 4/5 treated mice (Fig. 6). The one mouse not responding to the first VSV‐GP treatment was treated 24 days after the first treatment with a second intravenous injection of the same dose of VSV‐GP. This led to a decrease of the luciferase signal and reduction of the tumor burden (Fig. 6; Supporting Information, 4). These results indicate that bone metastasis could be treated effectively and safely by systemic application of VSV‐GP.
Figure 6.
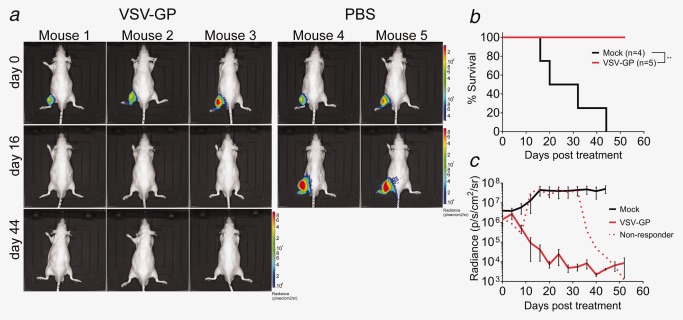
VSV‐GP causes tumor remission in a PCa bone metastasis model. Bone tumors were established by injection of 2.5 × 105 PC‐3M‐Luc cells in the left tibia of nude mice. Tumor growth was monitored by BLI of the luciferase signal. Mice were treated when luciferase signal reached a threshold level of 106 p/sec/cm2/sr with a single intravenous injection of 5 × 107 pfus VSV‐GP or left untreated. (a) Representative BLI pictures, (b) Kaplan–Meier survival curve and (c) kinetics of luciferase signal are shown.
Discussion
Here, we found that VSV‐GP is highly effective in lysing prostate cancer cell lines and in the treatment of prostate cancer xenografts in mice. VSV‐GP systemic treatment induced long‐term complete remission in an s.c. and bone metastasis mouse model of PCa. Oncolytic activity was found to be inhibited by type I interferon in interferon‐competent PCa cell lines and by low level virus receptor expression in one PCa cell line.
In cell cultures, mouse models and in clinical trials, the efficacy of oncolytic viruses has varied considerably with some patients responding dramatically while in many patients no effect has been seen.5 Clinical data for PCa is limited, but Yu et al. have shown in a PTEN−/− mouse PCa model that sensitivity of prostate cancer to the oncolytic virus VSV changes with tumor progression, varying from sensitive in early stages to resistant in established tumors to sensitive again in CRPC.24 The reason for this general variability of oncolytic virus efficacy is not fully understood and biomarkers that predict responsiveness of an individual cancer patient are urgently needed. These markers could not only help stratify patients for treatment, but also would point to mechanisms that limit the therapeutic activity of oncolytic viruses. Understanding these mechanisms is crucial to further improve virotherapy regimen.
Naturally, one major factor determining the efficacy of oncolytic virus therapy is the susceptibility of the specific cancer to virus infection and killing, which, as shown here, can differ considerably between prostate cancer cell lines (Fig. 1). This heterogeneity was further studied using a complementary in vitro panel consisting in primary cultures of prostate epithelial cells from PCa patients. Unfortunately, the epithelial cell population obtained using the current culture protocols shows features of intermediate and basal epithelial cells, while a fully differentiated luminal culture has not yet been achieved.25, 26 Although pathologists diagnose prostate cancer based on the absence of basal cell markers, there are many studies demonstrating the role of basal cells in PCa initiation and progression.27, 28 Therefore, we think that the primary cells used in this study, despite not completely resembling the situation in the patient, are a suitable model to complement our in vitro studies in cell lines supporting the finding of heterogenic responses to VSV‐GP (Fig. 1).
One mechanism of resistance to VSV‐GP infection was found to be the lack of its receptor, as shown for LNCAP cells (Fig. 2). Virus receptor expression in cancer tissue could indeed be a practical biomarker. In addition, a major factor that can limit virus spread in cancers is the innate antiviral immune response induced by type I interferons. Although Stojdl et al. showed that around 80% of the tested tumor cell lines had lost the capacity to mount an antiviral response upon IFN‐I exposure,12, 14 several tumor cell lines are still responsive to IFN‐I.17, 29, 30 Also in PCa, Dunn et al. showed loss of IFN‐I response in some cell lines,31 while other studies have shown that IFN‐I induced genes are active and play an important role in protecting cells from VSV infection.32 Using a larger panel of PCa cell lines, we have now observed considerable heterogeneity in the IFN‐I induced antiviral response. Thus IFN‐I responsiveness is expected to be a predictor for the susceptibility to virotherapy in PCa patients. However, our in‐depth studies showed that IFN‐I responsiveness in PCa is caused by various molecular alterations in different cell lines, which makes biomarker development for interferon‐competence challenging. Nevertheless, resistance to VSV‐GP in interferon competent cancers could potentially be overcome by combining oncolytic virus therapy with inhibitors of the interferon response. Accordingly, we have previously shown that treatment efficacy for an IFN‐I‐competent ovarian carcinoma xenotransplant in mice was enhanced by co‐application of a JAK 1/2 inhibitor.17
We then selected the three cell lines that were susceptible to VSV‐GP killing and had reduced interferon competence, Du145, 22Rv1 and PC‐3M‐Luc, for efficacy testing in xenotransplant mouse models. VSV‐GP was found to selectively replicate in Du145 and 22Rv1 tumors, which induced long‐term complete remission of up to 100% and long‐term survival with no visible toxicity (Fig. 4). Immunofluorescence analysis of tumor sections showed that viral replication co‐localized with the activation of caspase 3, in line with previous data showing that VSV induces apoptosis in the infected cancer cell.33 This impressive potency compares favorably with other oncolytic virus studies investigating VSV to treat of PCa mouse models. Although VSV also targets the tumor34, 35 and reduces tumor size,36, 37 treatment is limited by the potential of fatal viral encephalitis, not seen for VSV‐GP.38 Interestingly, subcutaneous tumors derived from VCaP, a cell line that in vitro appeared to be IFN‐I responsive and less susceptible to VSV‐GP oncolysis, still showed some therapeutic responses when treated with VSV‐GP (Fig. 4). Despite not being a complete remission, the significant delay in tumor growth observed was not expected. One possible explanation might be that while the recombinant interferon used in our in vitro assays shows cross‐species activity, the interferon generated by the mouse host does not.39 Consequently, the modest interferon production in VCaP cells might not suffice in countering the VSV‐GP activity. In addition, lack of correlation between the in vitro and in vivo outcomes has been previously reported in the OV context by other groups,40, 41 indicating that the final therapeutic outcome is not only shaped by the permissivity of the tumor cells but also by the context where they are found. In the same line, treatment of the syngeneic TRAMP‐C1 tumors, which in vitro were highly sensitive to VSV‐GP infection and killing, resulted in a measurable but limited therapeutic response (Fig. 5). Our in vitro screening had revealed that this cell line was capable of efficiently mounting an IFN‐I induced antiviral response. As in the syngeneic context, in contrast to the xenograft models, the tumor can be stimulated by the cytokines produced by the host, we believe that upon VSV‐GP infection of TRAMP‐C1 bearing mice, the virus‐induced IFN‐I protects the tumor from further VSV‐GP replication and killing, limiting the therapeutic response. The predictive value of an active IFN‐I response and the influence on the therapy of the tumor microenvironment in the syngeneic context is an issue that will be addressed in future studies.
Our final objective was to test if VSV‐GP is also effective when used systemically, which, in contrast to intratumoral application, would allow effective targeting of metastatic disease. Using the 22Rv1 model, we showed that a single intravenous dose of VSV‐GP was sufficient to cause remission of localized s.c. tumors in all treated mice (Fig. 4). Furthermore, we studied VSV‐GP efficacy in a well‐established bone metastasis model consisting in the intratibial inoculation of PC‐3M cells that express the firefly luciferase as a marker for imaging.16 A single intravenous injection of VSV‐GP was able to reach the metastatic sites and cause remission of the established tumors within 30 days after treatment initiation (Fig. 6). Again, this impressive potency compared favorably with other oncolytic viruses. An adenovirus‐based oncolytic virus was found to significantly reduce tumor growth rates, but failed to induce complete tumor remission.42, 43 Furthermore, systemic treatment with a recombinant fusogenic herpes simplex virus (HSV) reduced both primary tumor and metastatic burden in a lung metastasis model for PCa,44 but again with no complete remission.
In conclusion, VSV‐GP as a single therapeutic agent was found to be highly effective and safe both in in vitro and in vivo xenograft PCa models upon systemic treatment. Especially, the long‐term complete remission of bone marrow metastases supports the further development of VSV‐GP for the treatment of patients with CRPC, for which current treatment options are limited.
Supporting information
Supporting Information Legends
Supporting InformationFigure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1
Acknowledgements
The authors thank Dr. Georg Schäfer (Department of Pathology, Medical University Innsbruck) for his support in obtaining patient samples for the primary culture of prostate epithelial cells and Dr. Walther Parson (Institute of Legal Medicine, Medical University of Innsbruck) for cell line authentication. They thank Maria Rauth, Bettina Großlercher and Melissa Mayr for their excellent technical assistance. This work was funded by the Division of Virology, Medical University Innsbruck and the Christian Doppler Research Association.
[The copyright line for this article was changed on August 15, 2019, after original publication.]
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Steliarova‐Foucher E, Lortet‐Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur. J Cancer 2013; 49:1374–403. [DOI] [PubMed] [Google Scholar]
- 3. Bourke MG, Salwa S, Harrington KJ, et al. The emerging role of viruses in the treatment of solid tumours. Cancer Treat Rev 2011; 37:618–32. [DOI] [PubMed] [Google Scholar]
- 4. Rajani KR, Vile RG. Harnessing the power of onco‐immunotherapy with checkpoint inhibitors. Viruses 2015; 7:5889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pol J, Buqué A, Aranda F, et al. Trial watch‐oncolytic viruses and cancer therapy. Oncoimmunology 2016; 5:e1117740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14:642–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. United States Food and Drug Administration . FDA approves first‐of‐its‐kind product for the treatment of melanoma. 2015.
- 8. European medicines Agency . Imlygic ‐ EPAR summary for the public. 2015.
- 9. Taguchi S, Fukuhara H, Homma Y, et al. Current status of clinical trials assessing oncolytic virus therapy for urological cancers. Int J Urol 2017; 24:342–51. [DOI] [PubMed] [Google Scholar]
- 10. Tober R, Banki Z, Egerer L, et al. VSV‐GP: a potent viral vaccine vector that boosts the immune response upon repeated applications. J Virol 2014; 88:4897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mead DG, Ramberg FB, Besselsen DG, et al. Transmission of vesicular stomatitis virus from infected to noninfected black flies co‐feeding on nonviremic deer mice. Science 2000; 287:485–7. [DOI] [PubMed] [Google Scholar]
- 12. Stojdl DF, Lichty BD, TenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti‐cancer agents. Cancer Cell 2003; 4:263–75. [DOI] [PubMed] [Google Scholar]
- 13. Belkowski LS, Sen GC. Inhibition of vesicular stomatitis viral mRNA synthesis by interferons. J Virol. 1987; 61:653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor‐specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 2000; 6:821–5. [DOI] [PubMed] [Google Scholar]
- 15. Santer FR, Höschele PPS, Oh SJ, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011; 10:1644–55. [DOI] [PubMed] [Google Scholar]
- 16. Buijs JT, Rentsch CA, van der Horst G, et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol 2007; 171:1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dold C, Rodriguez Urbiola C, Wollmann G, et al. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV‐GP oncolytic viral therapy. Mol Ther Oncolytics 2016; 3:16021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santer FR, Erb HHH, Oh SJ, et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 2015; 6:6105–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mruk DD, Cheng CY. Enhanced chemiluminescence (ECL) for routine immunoblotting: an inexpensive alternative to commercially available kits. Spermatogenesis 2011; 1:121–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 21. Cao W, Henry MD, Borrow P, et al. Identification of alpha‐dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998; 282:2079–81. [DOI] [PubMed] [Google Scholar]
- 22. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43:D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. American Society of Cancer . Cancer facts & figures 2017. Am Cancer Soc. 2017; [Google Scholar]
- 24. Yu N, Puckett S, Antinozzi P. A, et al. Changes in susceptibility to oncolytic vesicular stomatitis virus during progression of prostate cancer. J Virol 2015; 89:5250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peehl DM. Primary cell cultures as models of prostate cancer development. Endocr Relat Cancer 2005; 12:19–47. [DOI] [PubMed] [Google Scholar]
- 26. Bühler P, Wolf P, Katzenwadel A, et al. Primary prostate cancer cultures are models for androgen‐independent transit amplifying cells. Oncol Rep. 2010; 23:465–70. [PubMed] [Google Scholar]
- 27. Goldstein AS, Huang J, Guo C, et al. Identification of a cell of origin for human prostate cancer. Science 2010; 329:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park JW, Lee JK, Phillips JW, et al. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc Natl Acad Sci USA 2016; 113:4482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blackham AU, Northrup S. A, Willingham M, et al. Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma. J Surg Res 2014; 187:412–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Westcott MM, Liu J, Rajani K, et al. Interferon beta and interferon alpha 2a differentially protect head and neck cancer cells from vesicular stomatitis virus‐induced oncolysis. J Virol 2015; 89:7944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dunn GP, Sheehan KCF, Old LJ, et al. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res 2005; 65:3447–53. [DOI] [PubMed] [Google Scholar]
- 32. Carey BL, Ahmed M, Puckett S, et al. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol 2008; 82:12104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Balachandran S, Porosnicu M, Barber GN. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol 2001; 75:3474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moussavi M, Fazli L, Tearle H, et al. Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice. Cancer Res 2010; 70:1367–76. [DOI] [PubMed] [Google Scholar]
- 35. Moussavi M, Tearle H, Fazli L, et al. Targeting and killing of metastatic cells in the transgenic adenocarcinoma of mouse prostate model with vesicular stomatitis virus. Mol Ther 2013; 21:842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao X, Huang S, Luo H, et al. Evaluation of vesicular stomatitis virus mutant as an oncolytic agent against prostate cancer. Int J Clin Exp Med. 2014; 7:1204–13. [PMC free article] [PubMed] [Google Scholar]
- 37. Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 2004; 330:34–49. [DOI] [PubMed] [Google Scholar]
- 38. Muik A, Stubbert LJ, Jahedi RZ, et al. Re‐engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res 2014; 74:3567–78. [DOI] [PubMed] [Google Scholar]
- 39. Veomett MJ, Veomett GE. Species specificity of interferon action: maintenance and establishment of the antiviral state in the presence of a heterospecific nucleus. J Virol. 1979; 31:785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yarde DN, Nace R. A, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti‐myeloma activity in vivo. Exp Hematol. 2013; 41:1038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leddon JL, Chen CY, Currier MA, et al. Oncolytic HSV virotherapy in murine sarcomas differentially triggers an antitumor T‐cell response in the absence of virus permissivity. Mol Ther Oncolytics. 2014;1:14010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu W, Neill T, Yang Y, et al. The systemic delivery of an oncolytic adenovirus expressing decorin inhibits bone metastasis in a mouse model of human prostate cancer. Gene Ther 2015; 22:31–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu Z, Gupta J, Zhang Z, et al. Systemic delivery of oncolytic adenoviruses targeting transforming growth factor‐β inhibits established bone metastasis in a prostate cancer mouse model. Hum Gene Ther 2012; 23:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakamori M, Fu X, Pettaway CA, et al. Potent antitumor activity after systemic delivery of a doubly fusogenic oncolytic herpes simplex virus against metastatic prostate cancer. Prostate 2004; 60:53–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Legends
Supporting InformationFigure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1