

Abstract
Cancer metabolism is currently a hot topic. Since it was first realized that cancer cells rely upon an altered metabolic program to sustain their rapid proliferation, the enzymes that support those metabolic changes have appeared to be good targets for pharmacological intervention. Here, we discuss efforts pertaining to targets in cancer metabolism, focusing upon the tricarboxylic acid cycle and the mechanisms which feed nutrients into it. We describe a broad landscape of small-molecule inhibitors, targeting a dozen different proteins, each implicated in cancer progression. We hope that this will serve as a reference both to the areas being most highly examined today and, relatedly, the areas that are still ripe for novel intervention.
Keywords: : cancer, drug discovery, glutaminase, glutaminolysis, glycolysis, isocitrate dehydrogenase, metabolism, patents, TCA cycle
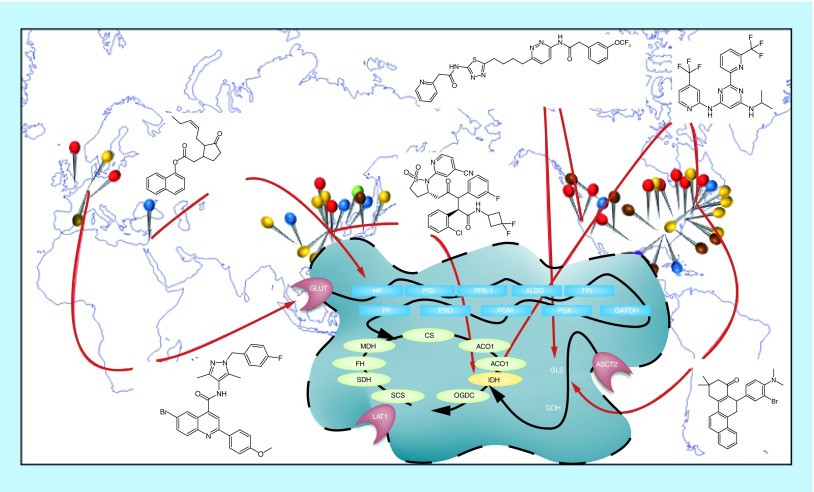
Global solutions to a central problem. Researchers across the globe have patented efforts to target enzymes involved in cancer cell metabolism. Pins on the map represent individual research efforts, and are color coded to pathways or proteins. Several interesting small-molecule inhibitors revealed by these efforts are shown.
In December of 2016, the United States Congress passed the 21st Century Cures Act, which increased funding to the NIH by US$4.8 billion over 10 years. Of this funding, $1.8 billion was earmarked for the Cancer Moonshot, a 7-year program to double the pace of discovery in detection, prevention and cure of cancer. This is just one example of the increasing recognition of the widespread impact of cancer, and of the need to direct new funding to combat this disease. While many current approaches involve new technologies, such as Car-T therapy, others draw instead from older science, to which modern ideas and techniques are now being brought to bear.
In 1956, Warburg reviewed the results of several decades of research, and described the altered metabolic phenotype of cancer cells that would come to be known as the Warburg effect [1]. The observations of Warburg and his contemporaries demonstrated that most tumors do not undergo standard respiration, but instead engage in fermentation of glucose to lactic acid, independent of oxygen availability. More recent studies have shown that this is part of a conserved metabolic program in most rapidly proliferating cells, in which consumed glucose enters the tricarboxylic acid (TCA) cycle in only small amounts, with most metabolic intermediates being redirected into a series of biosynthetic pathways. Most TCA cycle input then comes from assorted anaplerotic inputs, and in particular glutamine, one of the most common amino acids in the bloodstream [2–4].
Because the Warburg effect is exhibited by most cancer cells, a great deal of effort has gone into developing strategies to pharmacologically target metabolic proteins, and interrupt their function to slow tumor growth or to shrink tumors. In this review, we will highlight some of the most interesting patents from the last few years describing inhibitors of proteins involved in nutrient import, glycolysis, anaplerosis and the TCA cycle. Notably, we will be examining only patents reporting novel inhibitors of proteins directly involved in metabolic biosyntheses, and not upstream regulators or downstream targets of these proteins. Furthermore, we limit ourselves to only relatively recent patents (generally starting from 2014) that explicitly investigate some form of cancer, and thus we may fail to consider molecular scaffolds reported in patents which did not explicitly discuss cancer. The patents we do consider are summarized in Table 1. Finally, we do not extensively cover molecules that have advanced to clinical trials, but instead attempt to examine the overall chemical and intellectual landscape surrounding the various pathways examined. We hope this review will help to shed light on the most active areas of cancer metabolism research today, and on targets that are still open for new exploration.
Table 1. . Status of described patents.
| Inventor | Assignee | Examples | Target | #Molcs | Classification? | Legal status | Ref. |
|---|---|---|---|---|---|---|---|
| Mjalli | VTV Therapeutics LLC | 1 | HK2 | ∼150 | N | AF | [13] |
| Andrews | VTV Therapeutics LLC | n.d. | HK2 | ∼60 | N | AF | [14] |
| Behar | Vidac Pharma LTD. | 2, 3 | HK2 | 1 | N | WO: AF, US: pending | [15] |
| Chen | Neonc Technologies, Inc. | 4 | GAPDH | 1 | N | Unknown | [19] |
| Zhang | Shanghai Institute of Materia Medica | 6 | PGK1 | ∼30 | N | Search and examination | [20] |
| Chen | Emory University | 7 | PGM | 5 | N | WO: AF, US2012: active, US2017: pending | [22] |
| Jung | Gwangju Institute of Science and Technology | 8 | ENO | 1 | N | WO: AF, US: active | [23] |
| Cantley | Beth Israel Deaconess Medical Center, Inc. | 9, 10 | PKM2 | ∼10 | N | WO: AF, US: active | [26] |
| He | Guizhou Medical University | 11, 12 | PKM2 | ∼10 | N | Unknown | [27] |
| Kang | Emory University | 13 | GDH | 1 | N | Active | [31] |
| Newcomb | Elan Pharmaceuticals, LLC | 14 | GLS | ∼10 | N | Expired | [35] |
| Lemieux | Agios Pharmaceuticals Inc | n.d. | GLS | ∼290 | Y | Active | [36] |
| Lemieux | Agios Pharmaceuticals Inc. | n.d. | GLS | ∼120 | Y | WO: AF, US2013: active, US2018: pending | [37] |
| Cianchetta | Agios Pharmaceuticals Inc. | n.d. | GLS | ∼10 | Y | WO: AF, US: active | [38] |
| Bennett | Calithera Biosciences, Inc. | 15 | GLS | ∼730 | N | WO: AF, US: abandoned, JP: active | [39] |
| Parlati | Calithera Biosciences, Inc. | n.d. | GLS | 1 | N | WO: AF, US: pending | [40] |
| Finlay | AstraZeneca AB | n.d. | GLS | ∼70 | N | WO: AF, US2015: active, US2018: pending | [41] |
| Finlay | AstraZeneca AB | 17 | GLS | 6 | N | WO: AF, US2016: active, US2019: pending | [42] |
| Bhavar | Rhizen Pharmaceuticals SA | n.d. | GLS | ∼50 | Y | WO: AF, US2015: active, US2017: pending | [43] |
| Bhavar | Rhizen Pharmaceuticals SA | n.d. | GLS | ∼40 | Y | WO: AF, US2015: active, US2017: pending | [44] |
| Ruan | Hangzhou Gamma Biotech Co., LTD. | 18 | GLS | ∼10 | N | WO: AF, US: pending | [45] |
| Liang | Centaurus Biopharma Co., LTD. | n.d. | GLS | ∼20 | Both | Unknown | [46] |
| Piwnica-Worms | Washington University | n.d. | GLS | 1 | N | Abandoned | [47] |
| Di Francesco | University of Texas System | n.d. | GLS | ∼600 | Y | WO: AF, US2015: active, US2017: pending | [48] |
| Di Francesco | University of Texas System | n.d. | GLS | ∼240 | Y | WO: AF, US: abandoned | [49] |
| McDermott | University of Pittsburgh | 16 | GLS | ∼50 | N | WO: AF, US: active | [50] |
| Lewis | University of Texas System | n.d. | GLS | ∼50 | Y | WO: AF, US2016: active, US2018: pending | [51] |
| Slusher | The Johns Hopkins University | 19, 20 | GLS | 1 | N | WO: AF, US: pending | [59] |
| Cerione | Cornell University | 21 | GLS | ∼30 | N | WO: AF, US: pending | [62] |
| Lee | Dongguk University Industry-Academic Cooperation Foundation | 22 | MDH | ∼50 | Y | Unknown | [63] |
| Amatangelo | Celgene Corporation | n.d. | IDH | 1 | N | WO: AF, US: pending | [70] |
| Konteatis | Agios Pharmaceuticals Inc. | 23 | IDH | ∼40 | Y | WO: AF, US: pending, AU: active | [71] |
| Popovici-Muller | Agios Pharmaceuticals Inc. | 24, 25 | IDH | ∼180 | Y | WO: AF, US: active | [72] |
| Travins | Agios Pharmaceuticals Inc. | n.d. | IDH | 1 | Y | WO: AF, US: active | [73] |
| Ashwell | Forma Therapeutics, Inc. | 26 | IDH | ∼60 | Y | WO: AF, US: active | [74] |
| Ashwell | Forma Therapeutics, Inc. | 27 | IDH | ∼30 | Y | WO: AF, US2015: active, US2018: pending | [75] |
| Ashwell | Forma Therapeutics, Inc. | 28 | IDH | ∼80 | Y | WO: AF, US: active | [76] |
| Ashwell | Forma Therapeutics, Inc. | 29 | IDH | ∼100 | Y | WO: AF, US: active | [77] |
| Ashwell | Forma Therapeutics, Inc. | n.d. | IDH | ∼10 | Y | WO: AF, US: active | [78] |
| Ashwell | Forma Therapeutics, Inc. | n.d. | IDH | ∼180 | Y | WO: AF, US2015: active, US2017: pending | [79] |
| Bauer | Eli Lilly and Company | 35 | IDH | ∼10 | N | Unknown | [80] |
| Boxer | USA Department of Health and Human Services | 37 | IDH | ∼150 | Y | WO: AF, CA/AU: pending | [81] |
| Cao | Jiangsu Provincial Institute of Traditional Chinese Medicine | n.d. | IDH | ∼140 | Y | Search and examination | [82] |
| Fischer | Merck Sharp & Dohme Corp. | 36 | IDH | ∼130 | N | WO: AF, US: abandoned, EP: pending | [83] |
| Huang | Neuform Pharmaceuticals, Inc. | n.d. | IDH | 1 | N | WO: AF, US: active | [84] |
| Saito | Daiichi Sankyo Company | 38 | IDH | ∼170 | N | WO: AF, US: active | [85] |
| Schirmer | Bayer Pharma Aktiengesellschaft | n.d. | IDH | 1 | N | WO: AF, US: pending | [86] |
| Wang | Medshine Discovery Inc. | n.d. | IDH | ∼10 | N | Unknown | [87] |
| Wang | Nanjing Sanhome Pharmaceutical Co. | n.d. | IDH | ∼40 | N | WO: AF, CA: pending | [88] |
| Wu | Shanghai Shipu Biotechnology Co. | 39 | IDH | ∼20 | Y | Unknown | [89] |
| Yang | Shanghai Meton Pharmaceutical Co. | n.d. | IDH | ∼10 | Y | Unknown | [90] |
| Zhang | Sichuan University | n.d. | IDH | ∼50 | Y | Search and examination | [91] |
| Zhang | Isocure Biosciences Inc. | 31 | IDH | 2 | N | Unknown | [92] |
| Zhang | Isocure Biosciences Inc. | 30 | IDH | ∼20 | N | Unknown | [93] |
| Zhang | Isocure Biosciences Inc. | 32 | IDH | 10 | N | Unknown | [94] |
| Zhao | Lianyungang Runzhong Pharmaceutical Co. | n.d. | IDH | 8 | N | WO: AF, US: pending | [95] |
| Zhu | Chia Tai Tianqing Pharmaceutical Group Co. | n.d. | IDH | ∼30 | Y | WO: AF, CN: search and examination | [96] |
| Zhu | Chia Tai Tianqing Pharmaceutical Group Co. | 34 | IDH | ∼20 | Y | WO: AF, CN: search and examination | [97] |
| Oprea | STC. UNM | 40 | GLUT5 | 1 | N | WO: AF, US: pending | [103] |
| Shanmugam | Emory University | 41 | GLUT4 | ∼10 | N | Unknown | [104] |
| Jun | The Johns Hopkins University | 42 | GLUT1 | 2 | N | WO: AF, CN: search and examination | [105] |
| Chen | Ohio State Innovation Foundation | 43 | GLUT1 | 30 | N | WO: AF, US: Active | [106] |
| Heisler | Bayer Pharma Aktiengesellschaft | 44 | GLUT1 | ∼190 | N | WO: AF | [107] |
| Heisler | Bayer Pharma Aktiengesellschaft | 45 | GLUT1 | ∼70 | N | WO: AF | [108] |
| Kanai | Osaka University | 46 | LAT1 | ∼10 | N | WO: AF, US: active | [114] |
| Esslinger | The University of Montana | 47 | ASCT2 | ∼10 | N | Active | [115] |
| Manning | Vanderbilt University | 48 | ASCT2 | 7 | N | Unknown | [116] |
The boldface type are compound numbers.
Information from the patents described in this manuscript is presented above. This includes the first listed inventor, the first listed assignee, the reference number to that patent in the bibliography, the specific compounds discussed from that patent (n.d. means no compounds were discussed), the major protein target investigated, the approximate number of compounds described with meaningful biological data (generally less than the total number of molecules directly described), whether the biological data are classification (grouped into potency ranges) or not, and the current legal status of the patent. The number of described molecules was generally rounded down to the nearest ten, and is intended only to provide an idea of how much biological potency data each patent provides for a given series. The legal status was obtained via Google Patents in April 2019, and generally reflects only the WO and US filing status, unless those were unavailable, or substantially different filing statuses were available in a different jurisdiction. ‘Unknown’ means that Google Patents did not have any information recorded for any filing jurisdiction. Most WO listings were AF – application filing.
ASCT2: Neutral amino acid transporter B(0); ENO: Enolase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; GLS: Glutaminase; GLUT: Glucose transporter; HK: Hexokinase; IDH: Isocitrate dehydrogenase; LAT1: Large neutral amino acid transporter; MDH: Malate dehydrogenase; #Molcs: Number of molecules discussed; PGK: Phosphoglycerate kinase; PGM: Phosphoglycerate mutase; PKM2: Pyruvate Kinase isoform M2; TCA: Tricarboxylic acid; TPI: Triosephosphate isomerase.
Glycolysis
Glycolysis is a 10-step series of reactions in which glucose is converted to pyruvate, which then enters into the TCA cycle (Figure 1). Cancer cells depend upon an increase in glycolysis, even while little of the carbon from glucose eventually enters the TCA cycle [4]. Indeed, in the 1920s, Warburg made the observation that glucose uptake was increased in most tumor cells, a process that became known as the Warburg effect [1]. This increased glycolytic rate is coupled to an increase in lactic acid generation, even in oxygen-rich environments, leading to reduced entry of pyruvate into the TCA cycle [5]. Furthermore, many of the various enzymes and metabolic intermediates have been found to have functions outside the glycolytic pathway. Lactate and pyruvate, for example, have been tied to chemoresistance, while enzymes including hexokinase, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and pyruvate kinase (PKM) have been found to be involved in transcriptional regulation [6]. Moreover, pathways such as the pentose-phosphate shunt allow for glycolytic intermediates to be converted into a variety of biological-building blocks. The collective evidence suggests that rapidly dividing cells, such as cancer cells, prioritize metabolite production from glucose over energy generation via the TCA cycle [4,7].
Figure 1. . Glutaminolysis.

Glutaminolysis is a series of ten reactions that convert glutamine to pyruvate, a substrate which is then able to enter the tricarboxylic acid cycle for purposes of energy production. Of the ten enzymes involved in glutaminolysis, six have been targeted by patented drug discovery efforts in recent years.
ALDO: Fructose-bisphosphate aldolase; ENO: Enolase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HK: Hexokinase; PFK-1: Phosphofructokinase; PGI: Phosphoglucose isomerase; PGK: Phosphoglycerate kinase; PGM: Phosphoglycerate mutase; PK: Pyruvate kinase; TCA: Tricarboxylic acid; TPI: Triosephosphate isomerase.
To date, there is little clear evidence for a single ‘switch’, which turns on the Warburg effect in cancer cells. Early evidence suggested that a specific variant of pyruvate kinase, PKM2, might play that role, as it is expressed in many cancer cells but not in healthy tissue, and further has a much lower catalytic activity than the PKM1 variant found in non-transformed cells [5]. However, it is poorly expressed in some cancer cells that still experience Warburg effect metabolism, suggesting that other genes must also be involved in the metabolic reprogramming cancer cells experience [8]. Despite a poor understanding of the causes, the fact remains that many of the enzymes involved in glycolysis are upregulated in assorted cancers, and that this can be exploited for medical purposes. For example, positron emission tomography is widely utilized in conjuction with radio-labeled glucose to monitor tumors in the body [9]. In a related way, the various glycolytic enzymes make tempting targets for drug discovery. Indeed, virtually every step of glycolysis has been targeted during drug discovery efforts at this point. Although to our knowledge there is not yet an approved glycolysis inhibitor in the clinic [10,11], progress in this area is ongoing.
The first step in glycolysis, an essentially irreversible reaction, which consumes one molecule of ATP to convert glucose to glucose-6-phosphate, is initiated by hexokinase. There are four isoforms of hexokinase, with HK2 being overexpressed in many cancers [12], and thus representing a promising drug target. Unsurprisingly, several groups have pursued HK2 inhibitors in recent years. VTV Therapeutics, for example, described a series of glucose-competitive inhibitors based around a ribose/indole scaffold (Table 1) [13,14], such as compound 1 in Figure 1 (all compounds in Figure 1 are summarized in Table 2). The most potent molecules in the series had low-nanomolar IC50 values versus recombinant HK2 (the compound 1 IC50 is 36 nM), and were able to inhibit the growth of SKOV-3 ovarian cancer cells at mid-nanomolar to low-micromolar potency (the compound 1 IC50 is 390 nM). Vidac Pharma has taken an alternate approach and reported inhibitors such as 2 [15], which cause HK2 to detach from the mitochondrial membrane, thus preventing its biological activity without competing with glucose. Compound 2 was shown to be selective for HK2 over HK1 by thermophoresis assay (EC50 ∼ 100 nM vs HK2 thermophoresis), and was demonstrated to inhibit cancer cell growth in 65 different cell lines, xenografts and PDX models with IC50 values between 14 nM and 6.5 μM. Vidac also reported a series of piperazine derivatives such as compound 3 in the same patent, but provided relatively little biological data, and so it is unclear how these compounds performed compared with 2.
Table 2. . Glycolysis inhibitors.
| Inhibitor | Target | IC50 | |
|---|---|---|---|
| Protein | Cell | ||
| 1 | HK | 36 nM | 390 nM |
| 2 | HK | ∼100 nM | 14 nM–6.5 μM |
| 3 | HK | N.D.R. | N.D.R. |
| 4 | GAPDH | 45 μM | 50–400 μM |
| 5 | PGK | 1 nM | N.D.R. |
| 6 | PGK | <50 μM | 1.3 μM |
| 7 | PGM | 13.1 μM | ∼15 μM |
| 8 | ENO | <2.5 μM | 2.5 μM |
| 9 | PKM | 7 μM | 250 μM |
| 10 | PKM | 20 μM | 250 μM |
| 11 | PKM | 13.5 μM | 193 μM |
| 12 | PKM | 5 μM | 69 μM |
Summary of inhibitory potencies of compounds inhibiting steps of the glycolytic pathway. Values marked as approximate (∼) were generally read off a graph, while exact values were generally reported in a table. For values marked as ‘N.D.R.’, no data were reported for that compound. ‘Protein’ IC50 values were generally determined in systems with recombinant or otherwise purified proteins, while ‘Cell’ IC50 values generally apply to the inhibition of cancer cell growth, or a similar cellular readout.
ENO: Enolase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HK: Hexokinase; PGK: Phosphoglycerate kinase; PGM: Phosphoglycerate mutase; PKM: Pyruvate kinase.
Hexokinase is the first of five ‘preparatory phase’ reactions in glycolysis, which are energetically disfavorable but produce necessary synthetic intermediates [6]. The other four enzymes, glucose-6-phosphate isomerase, phosphofructose kinase, fructose bisphosphate aldolase and triosephosphate isomerase, have been the targets of only a small number of patents in the last decade [16–18], and none that we could locate in the past few years. The remaining five enzymes in glycolysis, however, are all part of the so-called ‘payoff phase’, in which a net gain of NADH and ATP molecules are produced, and various attempts have been made to inhibit the activity of each enzyme in this phase. The first of these, GAPDH, has seen more recent activity. Neonc Technologies has described compound 4, a combination of perillyl alcohol and 3-bromo-pyruvate, each of which were known to have some activity against GAPDH [19]. Compound 4 was shown to inhibit the activity of GAPDH with an IC50 of about 45 μM, and the growth of a panel of breast, brain, colon and ovarian cancer cells with IC50 values ranging from 50 to 400 μM. In each case, compound 4 was more potent than the parent molecule 3-bromo-pyruvate.
Patents have also been published recently pertaining to phosphoglycerate kinase (PGK), the second ‘payoff phase’ enzyme in glycolysis. Notably, the Shanghai Institute for Materia Medica (SIMM) has reported a series of 33 quinazoline derivatives that inhibit the activity of PGK [20], and appear to build upon molecules reported by Abbott Laboratories in a 2012 filing [21]. The Abbott molecules were all based around a quinoxaline scaffold, with molecules such as 5 having IC50 values against recombinant PGK as low as 1 nM. SIMM adjusted to a quinazoline scaffold, and added three heterocycles to the compounds, as exemplified by compound 6. SIMM reported only single-point inhibition values at 50 μM for their compounds against recombinantly expressed PGK, so while most of their compounds exhibited well over 50% inhibition at that concentration, it is impossible to directly compare them to the nanomolar compounds reported by Abbott. However, Abbott did not report any inhibition of cancer cell growth. By comparison, the SIMM compounds were shown to inhibit the growth of HepG2 liver cancer cells, with compound 6 having an IC50 value of 1.3 μM.
Interest in the next two enzymes in glycolysis, phosphoglycerate mutase (PGM) and enolase (ENO), has been similarly sparse, with the most recent patent filings for inhibitors of either being from 2014. Emory University reported a number of anthracene dione compounds based off the common dye alizarin, with compound 7 having an IC50 of 13.1 μM against recombinantly expressed PGM [22]. This was reported to be substantially similar to the inhibitory potency of parent compound alizarin. However, the alizarin dyes are much more hydrophilic than compound 7, and indeed 7 was better able to inhibit the proliferation of several cancer cell lines, including H1299 lung cancer cells and KG1a leukemia cells, compared with alizarin. Compound 7 was unable to effect HFF, HaCaT or PIG1 immortalized skin cells, suggesting that it is at least moderately selective for cancer over non-transformed cells. Targeting ENO, the Gwangju Institute of Science and Technology described a series of triazine compounds. Compound 8, the most potent member in the series, was able to inhibit 80% of recombinant ENO activity at a concentration of 2.5 μM [23]. The same concentration of the drug was able to block approximately 50% of HCT116 colorectal cancer cell migration or invasion. At 10 μM, the drug could significantly reduce the growth of tumors in a zebrafish model without significantly impacting embryonic development. However, negative effects on zebrafish biology were seen at 20 μM, suggesting a narrow therapeutic window.
The final reaction in the glycolytic pathway is catalyzed by PKM, which is expressed as several different isoforms. It was at one point proposed that a switch in expression between two of these, PKM1 in healthy cells to PKM2 in transformed cells, was the driving factor behind the Warburg effect [6,24,25]. While this idea has since been found to be less universal than originally proposed, it was none the less a promising lead in understanding the metabolic demands of cancer cells, and inhibitor development has been relatively rich. In 2014, the Beth Israel Deaconess Medical Center filed a patent reporting drugs targeting specifically the PKM2 isoform of PKM, which is highly overexpressed compared with PKM1 in many cancer cells [26]. The patent revealed eight different chemical families, with compounds such as 9 and 10 having IC50 values versus recombinant PKM2 of 7 and 20 μM, respectively. Both compounds were selective for PKM2, with 50 μM of either inhibiting PKM1 by only approximately 15%. However, neither compound was particularly potent against cancer cell growth, with each having an IC50 value of 250 μM against H1299 lung cancer cells. More recently, the Guizhou Medical University filed a claim on a series of biotinylated flavones based on the natural product scutellarin [27]. Compound 11, for example, bound primarily to PKM2 as determined in immunoprecipitation assays, exhibited an IC50 versus recombinant PKM2 of 13.5 μM, and was eightfold more selective for PKM2 versus PKM1. However, cell potency was lackluster, with an IC50 of 193 μM against HeLa cervical cancer cell growth. Non-biotinylated compounds such as 12 showed a sharply improved potency, with an IC50 versus PKM2 of 5 μM, 18-fold selectivity for PKM2 over PKM1, and an IC50 against HeLa cell growth of 69 μM.
Anaplerosis
Glycolysis is the major pathway by which a common nutrient, glucose, is converted into pyruvate, then enters the TCA cycle via conversion to acetyl-CoA by pyruvate dehydrogenase (PDH). However, in most cancer cells, glucose is primarily diverted into the formation lactate, or various biosynthetic materials, and thus little glucose carbon enters the TCA cycle [28]. To make up for the reduced entry of acetyl-CoA into the TCA cycle, cancer cells rely upon a variety of anaplerotic reactions to introduce intermediate materials into other points in the cycle. There are five major anaplerotic reactions in human cells, which consume pyruvate, aspartate, glutamate, fatty acids and adenylosuccinate (Figure 2). The most commonly utilized pathways are those involving glutamate, pyruvate and fatty acids [29]. Of these substrates, the most readily available is glutamate, via the common serum component glutamine.
Figure 2. . Anaplerosis.

In healthy cells, the tricarboxylic acid cycle is fueled primarily by pyruvate, which is converted to acetyl-coA by pyruvate dehydrogenase. However, in most cancer cells, there is insufficient pyruvate entering the tricarboxylic acid cycle to fuel metabolic demands. Thus, cancer cells make heavy use of anaplerotic reactions. The most commonly utilized anaplerotic inputs to mammalian cells are shown. Of these, the most used in cancer cells is glutamine conversion to α-ketoglutarate via the combined reactions of glutaminase (GLS) and glutamate dehydrogenase (GDH). Only GLS and GDH have been targeted by recent patented drug discovery efforts.
αKG: α-Ketoglutarate; βFA: Beta-fatty acids; ADS: Adenylosuccinate; ADSL: Adenylosuccinate lyase; Asp: Aspartate; AST: Aspartate transaminase; Cit: Citrate; Fum: Fumarate; GDH: Glutamate dehydrogenase; Gln: Glutamine; GLS: Glutaminase; MCM: Methylmalonyl-CoA mutase; Oxa: Oxaloacetate; PC: Pyruvate carboxylase; PDH: Pyruvate dehydrogenase; Pyr: Pyruvate; Suc: Succinate; TCA: Tricarboxylic acid.
Pyruvate is able to enter the TCA cycle via two pathways: PDH, which is the canonical capstone of glycolysis, and pyruvate carboxylase (PC), which converts pyruvate to acetyl-CoA. Of the two, neither has been significant targeted for cancer intervention in recent years. The same is true of adenylosuccinate lyase, which converts adenylosuccinate to fumarate, and methylmalonyl-CoA mutase, which is a point of convergence for several anaplerotic pathways and generates succinyl-CoA [29]. Aspartate transaminase has also been largely ignored. However, two key enzymes in glutamine metabolism, glutaminase (GLS) and glutamate dehydrogenase (GDH), have been heavily examined.
Glutamine is the most common circulating amino acid, and can be converted by GLS in the mitochondria to glutamate, which is then converted by GDH to α-ketoglutarate, a TCA cycle intermediate [30]. GDH has been directly targeted by Emory University (Table 1), which described anthracenedione compounds such as 13 (Table 3) [31]. Similar to their approach with compound 7, 13 was derived from the dye molecule purpurin, which inhibits GDH with a Ki of 1.9 μM, but is cell impermeable. Addition of the allyl group of 13 lowered the inhibitory potency, with a resultant Ki of 28.6 μM. However, the compound exhibited cell permeability and inhibited the viability of both H1299 lung cancer and MDA-MB-231 breast cancer cells with an IC50 of about 20 μM. It was further able to inhibit the growth of H1299 xenograft tumors in mice, but only when millimolar concentrations of the compound were administered.
Table 3. . Anaplerosis inhibitors.
| Inhibitor | Target | IC50 | |
|---|---|---|---|
| Protein | Cell | ||
| 13 | GDH | 28.6 μM† | 20 μM |
| 14 | GLS | ∼100 nM | ∼300 nM |
| 15 | GLS | 5 nM | 30 nM |
| 16 | GLS | 31 nM | 140 nM |
| 17 | GLS | 25 nM | 9 nM |
| 18 | GLS | N.D.R. | 10–2000 nM |
| 19 | GLS | N.D.R. | N.D.R. |
| 20 | GLS | N.D.R. | N.D.R. |
| 21 | GLS | 2.5 μM | 5 μM |
Summary of inhibitory potencies of compounds inhibiting anaplerosis to the tricarboxylic acid cycle. Values marked as approximate (∼) were generally read off a graph, while exact values were generally reported in a table.
Reported as a Ki rather than an IC50. For values marked as ‘N.D.R.’, no data were reported for that compound. ‘Protein’ IC50 values were generally determined in systems with recombinant or otherwise purified proteins, while ‘Cell’ IC50 values generally apply to the inhibition of cancer cell growth, or a similar cellular readout.
GDH: Glutamate dehydrogenase; GLS: Glutaminase.
GLS, which acts as a gatekeeper to GDH, has been far more extensively investigated. Indeed, it is one of the most heavily investigated metabolic enzymes in cancer. The existence of GLS was first proposed by Krebs in 1935, and over the next several decades assorted efforts helped to characterize the enzyme as being a tetrameric protein, activated by inorganic phosphate, which localized to the mitochondria [4,32]. There are two isozymes of glutaminase: GLS (the kidney-type enzyme, expressed in all tissues), and GLS2 (the liver-type enzyme, most highly expressed in liver) [4]. Of these, data from the cancer genome atlas [33] show that the vast majority of cancers highly express at least one of the two isozymes. While GLS2 has been poorly studied in connection with cancer, GLS is known to be important to the growth and survival of a number of aggressive cancers, including leukemia, breast cancer, colorectal cancer, kidney cancer, lung cancer, melanoma and pancreatic cancer [4]. Such cancers are now sometimes referred to as ‘glutamine addicted’, due to their dependence upon heavy consumption of glutamine [32].
Of the cancers which overexpress GLS, many have a mean 5-year survival time of under 50% (e.g., pancreatic, lung, brain or kidney cancer [34]), suggesting that targeting GLS could be of immediate therapeutic benefit to high-need patients. It is then unsurprising that well over 2000 small-molecule inhibitors having been reported to date [4]. As was the case for glycolysis inhibitors, no GLS inhibitors are yet approved for use in the clinic [10,11], but research is ongoing. The vast majority of GLS inhibitors have been derived from compound 14, first reported in 2002 [35]. Since then, molecules have been described by Agios Pharmaceuticals [36–38], Calithera Biosciences [39,40], AstraZeneca [41,42], Rhizen Pharmaceuticals [43,44], Hangzhou Gamma Biotech [45], and Centaurus Biopharma [46], as well as by Washington University, the University of Texas, Cornell University and the University of Pittsburgh [47–51]. This class of molecules is thought to bind to an allosteric regulatory site on the glutaminase enzyme, and has selectivity for GLS over the second glutaminase isozyme, GLS2, in the range of tenfold or greater [52–54]. Given the many excellent reviews already dedicated to this class of inhibitors [4,55–57], we will describe only a few highlights here.
The founding member of this class of inhibitors, compound 14, inhibits a splice variant of GLS highly expressed in a number of cancer cells with a reported IC50 of approximately 100 nM, and was shown to lower glutamate levels in Caco2 colon cancer cells with an IC50 of approximately 300 nM [35]. Numerous reports since the initial description have examined 14, and shown it to inhibit the proliferation of several different types of cancer [4,54]. Of the many reported derivatives of 14, the most heavily studied at this point is 15, reported by Calithera Biosciences [39]. Compound 15 improves upon the properties of 14, with reported IC50 values of 5 nM against GLS, and 30 nM against the proliferation of P493-6 B-cells transformed with oncogenic c-myc. Compound 15 has been shown to be particularly effective in triple-negative breast cancer cells [53], and has been the subject of multiple clinical trials since its initial description. A number of other compounds, such as 16 [50], from the University of Pittsburgh and Cornell University, have been described that integrate heterocyclic scaffolds to replace the flexible regions of 14 or 15. Compound 16 inhibits GLS with an IC50 value of 31 nM, and blocks the proliferation of MDA-MB-231 cells with an IC50 value of 140 nM. Compound 15 had IC50 values of 180 nM and 33 nM in the same respective assays. More recently, a number of scaffolds have been described that diverge more noticeably from 14, but which most likely bind to the same allosteric site on GLS. AstraZeneca described a number of bis-pyridazine compounds such as 17 [42] that resemble heterocyclic compounds like 16 but lack the thiadiazole rings common to most compounds in this class. Compound 17 inhibits GLS with an IC50 of 25 nM, and inhibits the proliferation of NCI-H1703 lung cancer cells with an IC50 of 9 nM, revealing a similar potency to compounds such as 15 and 16.
The most novel compounds in the class may be a series reported by Hangzhou Gamma Biotech, and based on the organoselenium compound Ebselen [45]. Ebselen is a glutathione peroxidase inhibitor, but has broad off-target activity, which was at one point thought to include GLS. Researchers at Hangzhou Gamma Biotech showed that Ebselen did not bind to GLS, and then chose to modify the scaffold to effect binding. The result was a series of compounds such as 18, which are thought to bind to the same allosteric region as 14. Compound 18 was shown to bind to GLS via a direct binding readout, and to inhibit the growth of a panel of four different human cancer cell lines, with T24 bladder cancer cells being the most sensitive (IC50: 10 nM) and PC3 prostate cancer cells being most resistant (IC50: 2000 nM). PC12 rat adrenal gland cancer cells were less sensitive than any human cancer cells tested (IC50: 5000 nM), and healthy L2 rat lung cells were unaffected by concentrations as high as 10 μM. The parent compound Ebselen was unable to inhibit the growth of any tested cell line at 10 μM, suggesting the GLS targeting compounds were more effective. However, the compound also inhibited the proliferation of JC82 cells, a strain of actinobacteria, suggesting that the selectivity problems present with ebselen may have been maintained in 18.
Other classes of GLS inhibitor have also been reported. One recently described set of molecules was based around 6-diazo-5-oxo-L-norleucine (DON), one of the oldest known inhibitors of GLS [4]. DON is an irreversible substrate-competitive compound, and is generally toxic, with a number of verified targets, and so even though it has excellent efficacy in cell culture it is unsuitable for use in live subjects [4,58]. However, by preparing a series of prodrugs which would be cleaved to DON in cancer cells, researchers from the Johns Hopkins University demonstrated that compounds such as 19 had similar tissue distribution to DON upon injection in mice, but resulted in much lower plasma concentrations of active DON, suggesting it would have reduced off-target effects [59]. They then showed that a related compound, 20, could inhibit the growth of 4T1 triple-negative mouse breast cancer cells more potently than 15 in vivo, and also inhibited tumor growth in several other xenograft models. Compound 20, however, was shown to have very poor stability in plasma, and to be rapidly degraded in the presence of liver microsomes. No antitumor effect was demonstrated for 19, while antitumor effects were shown for molecules unstable in plasma, so it is unclear if the most stable compounds in this series had any antitumor efficacy of note.
Another series of molecules was recently reported by Cornell University. These molecules have been proposed to bind to an allosteric region of GLS distinct from that occupied by 14, but this has not yet been verified by crystallographic analysis [60]. Still, the inhibitors are structurally distinct from those described thus far, and unlike compounds such as 14, they are thought to have nearly equal potency against both GLS and GLS2 [61]. Since GLS2 has been shown to be important in some cancers [4], this becomes an interesting property, although it has not yet been robustly investigated. Still, compound 21, for example, has an IC50 value of 2.5 μM against recombinantly expressed GLS, and an IC50 value of approximately 5 μM against the proliferation of MDA-MB-231 triple-negative breast cancer cells [62].
TCA cycle
The TCA cycle is a series of nine reactions, driven by eight enzymes, and is conserved in all aerobic organisms (Figure 3). These reactions create the key intermediates used to feed the electron transport chain, which is the major source of ATP in most cells. The crucial nature of the TCA cycle to the survival of aerobic cells makes these enzymes tricky to target, however, and only one of the eight enzymes directly involved in TCA cycle metabolism, malate dehydrogenase (MDH), has been targeted in connection with cancer in recent years. MDH was examined in a joint filing by the Dongguk University Industry-Academic Cooperation Foundation and the Korea Research Institute of Bioscience and Biotechnology (Table 1) [63]. The investigators described a series of molecules which acted as dual inhibitors of isozymes MDH1 and MDH2. The MDH enzymes catalyze the oxidation of malate to oxaloacetate, by reducing the cofactor NAD+ to NADH. Molecules such as 22 (Table 4) compete with NAD+ for binding to both MDH1 and MDH2, with a reported IC50 value for each enzyme of 1–5 μM. The molecule was also shown to be able to inhibit the growth of over a dozen cancer cell lines, with lung, colon and liver cancer cells being especially sensitive. Finally, the molecule was able to block the growth of HCT-116 colon cancer xenografts in mice, showing that dual inhibition of MDH1 and MDH2 could be a promising therapeutic approach. However, a much more active field of research has formed around a second TCA cycle enzyme, isocitrate dehydrogenase (IDH).
Figure 3. . The tricarboxylic acid cycle.

The tricarboxylic acid cycle is the heart of energy production in mammalian cells. It comprises nine reactions, catalyzed by eight enzymes, which create metabolites to enter the electron transport chain in order to produce the so-called ‘high-energy’ molecules ATP and NADH. In recent years, malate dehydrogenase has been directly targeted by patented drug-discovery efforts. Mutant forms of isocitrate dehydrogenase have been much more widely examined.
ACO1: Aconitase; CS: Citrate synthase; FH: Fumarase; IDH: Isocitrate dehydrogenase; MDH: Malate dehydrogenase; OGDC: α-Ketoglutarate dehydrogenase; PDH: Pyruvate dehydrogenase; SCS: Succinyl-coA synthetase; SDH: Succinate dehydrogenase; TCA: Tricarboxylic acid.
Table 4. . Tricarboxylic acid cycle inhibitors.
| Inhibitor | Target | IC50 | |
|---|---|---|---|
| Protein | Cell | ||
| 22 | MDH1/2 | 1–5 μM | 1–20 μM |
| 23 | mIDH | <50 nM | <50 nM |
| 24 | mIDH | <100 nM | N.D.R. |
| 25 | mIDH | <100 nM | N.D.R. |
| 26 | mIDH | <100 nM | <100 nM |
| 27 | mIDH | <10 nM | <10 nM |
| 28 | mIDH | <10 nM | <100 nM |
| 29 | mIDH | <100 nM | <10 nM |
| 30 | mIDH | 0.6 nM | N.D.R. |
| 31 | mIDH | 536 nM | N.D.R. |
| 32 | mIDH | 1–2 nM | N.D.R. |
| 33 | mIDH | 12 nM | 8 nM |
| 34 | mIDH | <20 nM | 6.7 nM |
| 35 | mIDH | 5.7 nM | 0.2 nM |
| 36 | mIDH | 3 nM | N.D.R. |
| 37 | mIDH | <300 nM | N.D.R. |
| 38 | mIDH | 7 nM | ∼20 nM |
| 39 | mIDH | <10 μM | ∼20 μM |
Summary of inhibitory potencies of compounds inhibiting steps in the tricarboxylic acid cycle. Values marked as approximate (∼) were generally read off a graph, while exact values were generally reported in a table. For values marked as ‘N.D.R.’, no data were reported for that compound. ‘Protein’ IC50 values were generally determined in systems with recombinant or otherwise purified proteins, while ‘Cell’ IC50 values generally apply to the inhibition of cancer cell growth, or a similar cellular readout.
MDH: Malate dehydrogenase; mIDH: Mutant isocitrate dehydrogenase.
Three IDH enzymes exist in humans: IDH1 and IDH2 share substantial sequence homology and are NADP+-dependent enzymes that catalyze a reversible reaction interconverting isocitrate and α-ketoglutarate [64]. IDH1 is located in the cytosol, and has roles in glucose sensing and lipid metabolism, while IDH2, in the mitochondria, is primarily involved in the regulation of the TCA cycle. IDH3 is a distinct enzyme with different regulating factors than IDH1/2, and catalyzes an irreversible conversion of isocitrate to α-ketoglutarate as part of the TCA cycle. Interestingly, mutant forms of IDH1 and IDH2 have been noted in several cancers. The mutation of IDH1 was first noted in colorectal cancer in 2007, and since then various genomics studies have found heavy IDH1 and IDH2 mutational burdens in glioma, acute myeloid leukemia, chondrosarcomas, cholangiocarcinomas and thyroid carcinomas [65]. More recent studies have shown that approximately 70% of grade II–III gliomas, and secondary glioblastomas, express mutant forms of IDH1 or IDH2, while only approximately 17% of acute myeloid leukemias express the mutant enzyme, and other indications have even lower expression levels [66].
IDH1 and IDH2 mutations occur almost exclusively at Arg 132 in the enzymatic active sites, and lead to major functional consequences, namely the partial catalysis of α-ketoglutarate to 2-hydroxyglutarate, and intracellular accumulation of the latter molecule [65]. 2-Hydroxyglutarate, in turn, interferes with α-ketoglutarate-binding proteins such as histone demethylases, leading to oncogenic epigenetic changes [67]. Despite relatively few indications expressing mutant IDH enzymes [66], targeting IDH neoenzymatic activity in cancer has shown great promise, particularly against acute myeloid leukemia, with several IDH1 and IDH2 inhibitors progressing through clinical trials for that indication [65]. Indeed, unlike the case of every other enzyme discussed in this review, there is a mutant IDH2 inhibitor approved for use in acute myeloid leukemia, enasidenib, with three other molecules currently in clinical trials [11], proving that targeting mutant IDH has great potential benefit to some patients. However, as these inhibitors target the mutant enzymes specifically, they would not be expected to have any benefit to patients whose tumors expressed wild-type IDH. As for other molecules in this review, we will show only highlights from each series investigated. However, a number of excellent reviews discuss many of these molecules in greater depth, and we would encourage interested readers to consult those [68,69].
As was the case for inhibitors of GLS, inhibitors of IDH enzymes have been reported by a large number of organizations, with recent reports originating with groups such as the Celgene Corporation [70], Agios Pharmaceuticals [71–73], Forma Therapeutics [74–79], Eli Lily [80], the University of North Carolina [81], the Jiangsu Provincial Institute of Traditional Chinese Medicine [82], Merck [83], Neuform Pharmaceuticals [84], the Daiichi Sankyo Company [85], Bayer Pharma [86], Medshine Discovery Inc. [87], Nanjing Sanhome Pharmaceutical Co. [88], Shanghai Shipu Biotechnology Co. [89], Shanghai Meton Pharmaceutical Co. [90], Sichuan University [91], Isocure Biosciences [92–94] and the Chia Tai Tianquing Pharmaceutical Group [95–97]. However, unlike the case of GLS inhibitors, a far greater degree of chemical space has been investigated in the pursuit of IDH inhibitors. Agios Pharmaceuticals has historically been one of the leaders in this area, and their most recent filings describe a series of heterocyclic inhibitors of mutant IDH2 [71,73], and a series of aryl-urea-based inhibitors of mutant IDH1 and IDH2 [72]. In the first case, two separate patents describe pyrimidine [71] and 1,3,5-triazine [73] compounds of otherwise similar structure. In total, approximately 1400 compounds were reported, although biological data were provided for only a small number of those compounds. Of these, however, compound 23 had a reported IC50 value of less than 50 nM against both the IDH2 R140Q mutant in enzymatic assays and against the proliferation of U87MG brain cancer cells stably expressing IDH2 R140Q [71]. A highly related compound, enasidenib, has since been examined clinically in collaboration with Celgene Corporation [70]. In the second series reported, compound 24 was shown to have an IC50 <100 nM against IDH R132H, while compound 25 exhibited a similar IC50 against IDH R140Q. Unfortunately, no cell results were reported for these molecules, so it is difficult to know the cell permeability of most members of the series. Similarly, each of the approximately 350 compounds reported had inhibitory potency described for either IDH R140Q or IDH R132H, and not both, and so it is not possible to determine their specificity for one mutant form over the other from the data provided.
Forma Therapeutics has been another major player in developing inhibitors of mutant IDH enzymes. In 2016, Forma filed a barrage of six patents, each examining one variety of quinolinone-based inhibitors [74–79]. The first series, exemplified by 26, generally combined the quinolinone with a fused bicyclic pyridine or pyrimidine [74]. Compound 26 had IC50 values less than 100 nM against the enzymatic activity of the IDH1 mutants R132H and R132C, as well as against the proliferation of HCT-116 colon cancer cells stably expressing either mutant isoform of IDH1. Two other compound series converted the fused-pyrimidine ring to a thiazole or thiadiazole [78], or a nonfused pyrimdine ring [79], with the best molecules in each series having reported efficacy values in the four test systems similar to 26. Interestingly, in the three other series tested, an additional potency classification was created for select assays: molecules with IC50 values under 10 nM. In one series, the fused pyrimidine ring was converted to 1,2-hydropyridine [75], and molecule 27 was the most potent in the series, with an IC50 less than 100 nM against the enzymatic activity of IDH1 R132C, and IC50 values less than 10 nM against the enzymatic activity of IDH1 R132H, as well as the proliferation of HCT-116 cells stably expressing either IDH1 mutant. Compound 28, from a series of pyridine containing compounds, had an IC50 value less than 10 nM against the proliferation of IDH1 R132C expressing cells, but less than 100 nM for the three other metrics [76]. The final series of compounds each contained a benzene ring in place of the pyrimidine [77]. Very few of the compounds had results reported for all four experimental conditions, but 29 was substantially more potent against IDH1 R132H than IDH1 R132C, with cells stably expressing either mutant being inhibited with an IC50 of less than 10 nM, or with IC50 values ranging from 100 nM to 1 μM, respectively. The molecule was more potent against the enzymatic activity of IDH1 R132H than IDH1 R132C as well, showing that even though most of the reported compounds across these series had similar potency against both mutants, it is possible to design selectivity for one mutant over the other.
Other groups have also filed for multiple patents in the last few years. One of these, Isocure Biosciences, has reported three different scaffolds that inhibit mutant IDH enzymes. The first of these examined tetrahydropyrazolopyradine compounds such as 30 [93]. These molecules were meant to improve upon GSK321, a tetrahydropyrazolopyradine reported by GlaxoSmithKline. GSK321 is an allosteric regulator of mutant IDH1, which locks it into a catalytically inactive conformation, with IC50 values of 3–5 nM against the various R132 mutants of IDH1 [98]. However, it was also moderately active against wild-type IDH1, with an IC50 value of 46 nM. A related compound reported by Bayer had an IC50 value of 20 nM against mutant enzyme, with no reported value for wild-type IDH1 [86]. While most of the compounds reported by Isocure were less potent against mutant IDH1 than was GSK321 or the related Bayer compound, 30 inhibited the R132H mutant at 0.6 nM, and the R132C mutant at 0.8 nM. Moreover, it required over 5 μM compound to inhibit the wild-type enzyme [93]. A series of structurally related tetrahydroimidazopyrazine compounds were described in a second series, many of which were designed to bind irreversibly to IDH1, but even the most potent reported compound, 31, had IC50 values for either mutant enzyme over 500 nM [92]. Compound 32, on the other hand, was derived from a different series, but one which was also meant for irreversible binding. In the case of 32, IC50 values of 1–2 nM were reached for IDH1 R132H and R132C, while wild-type IDH1 was inhibited with an IC50 of 9.5 μM [94]. None of the compounds reported by Isocure had cell data associated with them, but it is clear that they were able to obtain excellent selectivity for mutant versus wild-type IDH enzymes.
Another group that has made multiple patent filings pertaining to IDH inhibitors involves a collaboration between the Chia Tai Tianquing Pharmaceutical Group, Lianyungang Runzhong Pharmaceutical Co. and Centaurus Biopharma Co. In one reported series, the group investigated triazine compounds similar to Agios compound 23, with nearly identical structures and biological efficacies [95]. The other two series are derivatives of an older class of Agios compounds, which we have not discussed thus far, and includes molecules such as ivosidenib (compound 33) [69]. Compound 33 itself is a 12-nM inhibitor with excellent pharmacokinetic parameters, which is in several clinical trials [69,99]. Compounds such as 34 were able to modestly improve upon the potency of 33 (6.7 nM IC50 for 34 vs 46 nM IC50 for 33 in U87MG glioblastoma cells), but more importantly, it had superior pharmacokinetic values, with an oral dose of 5 mg/kg compound in mice resulting in a plasma half-life of 10.7 h for 34 compared with 3.3 h for 33 [96,97]. Several other groups have taken comparable approaches, exploring structures similar to 23 [84,88,90,91], 24 and 25 [82], and 30 and 31 [87], and obtained similar effects: small improvements to potency, selectivity and/or pharmacokinetic parameters compared with the compounds already discussed. However, a few other unique scaffolds have been reported.
Eli Lily filed a claim for compounds modestly similar to those developed by Forma Therapeutics (26–29), but predominantly exploring the structure/activity relationship at the opposite end of the ring system. Compounds such as 35 form irreversible covalent bonds to IDH1 or IDH2 [80]. Compound 35 inhibits IDH1 R132H enzymatic activity with an IC50 value of 5.7 nM, wild-type IDH1 with an IC50 value of 85.4 nM and inhibited the production of 2-hydroxyglutarate but not α-ketoglutarate in HT1080 fibrosarcoma cells expressing IDH1 R132C or IDH1 R132H (IC50: 0.2 nM), further suggesting specificity for mutant IDH enzymes. It was among several compounds in the series reported to inhibit 2-hydroxyglutarate in xenograft models of TB08 glioblastoma tumors expressing IDH1 R132H. Describing an even more novel molecular series, Merck identified over 100 compounds featuring a 6-7-6-fused ring system such as 36 [83]. IC50 values versus IDH1 R132H for the series ranged from approximately 40 μM for the least potent compounds to 3 nM for compounds such as 36. The Daiichi Sankyo Company and the University of Virginia nearly simultaneously reported two series of similar compounds, based on isoxazoles [85] or thiazoles [81], respectively. Thiazole compounds such as 37 inhibited 2-hydroxyglutarate production by IDH1 R132H with an IC50 value under 300 nM, while isoxazole 38 had an IC50 value of 7 nM, and inhibited 2-hydroxyglutarate production by IDH1 R132H in cell lines at approximately 20 nM IC50. Shanghai Shipu Biotechnology recently reported a wide variety of imidazole or triazole containing compounds, but even their most potent inhibitors, such as 39, were only tested at the level of 10 μM, so it is difficult to assess their potency compared with series from other sources [89]. Still, these various compounds help to demonstrate the vast degree of chemical space that has been examined in attempting to inhibit mutant IDH activity.
Nutrient import
Thus far, we have considered the pathways which feed into the TCA cycle, and the cycle itself, as drug targets to combat tumor growth. However, one other related mechanism remains to be considered: targeting the transmembrane receptors that facilitate nutrient entry into cells. Because glucose and glutamine are the major nutrients used in the pathways discussed, we will focus our attention here on glucose transporters (GLUTs) and amino acid transporters (Figure 4).
Figure 4. . Nutrient transporters.

Before being used in the tricarboxylic acid cycle, nutrients must first enter the cell. Major transporters include the GLUTs, and the transporters of assorted amino acids. The GLUTs, LAT1 and ASCT2 have all been targeted by patented drug discovery efforts recently.
ASCT2: Neutral amino acid transporter B(0); GLUT: Glucose transporter; LAT1: Large neutral amino acid transporter; TCA: Tricarboxylic acid.
The GLUT family of enzymes are responsible for bringing glucose, or closely related molecules, into cells. As they are the primary entry point for glucose, they are intimately linked with the increased glucose metabolism noted by Warburg [1]. Fourteen different GLUT proteins have been discovered in humans [100]. GLUT1–4 are well-characterized transporters for glucose, GLUT5 functions to transport fructose, and the remaining GLUTs are more recently discovered and less well studied [101]. Given the increase in glucose consumption in cancer, it is of little surprise that several GLUTs have been found to be overexpressed in cancer, particularly GLUT1 and GLUT3, which have a higher affinity for glucose than the other GLUT proteins. GLUT expression tends to vary with tumor and tissue type. For example, lymphomas tend to express GLUT3 but not GLUT1, but about half of epithelioid sarcomas and uterine sarcomas have high GLUT1 expression. Similarly, while GLUT2 expression is rarer across all cancers, it is expressed in almost all gastric cancers, and in about 30% of breast, colon or liver cancers [100]. Notably, GLUT expression is one of the driving forces that allows positron emission tomography scans to reveal tumor locations [9,102]. Despite their obvious importance to cancer, no inhibitors of the GLUTs have yet advanced to clinical approval, but at least one molecule (ritonavir) is currently undergoing clinical trials for prostate cancer and multiple myeloma [10,11].
Researchers from STC UNM and Rosalind Franklin University recently reported a series of molecules targeting the fructose transporter GLUT5 (Table 1), arguing that fructose was not regulated by insulin, but given that fructose consumption had increased dramatically in recent years, its transporter represented an ideal target for drug discovery [103]. They further noted that GLUT5 was overexpressed in approximately 27% of tumors examined, including breast cancer cell lines where normal, healthy tissue had no detectable GLUT5 expression, thus suggesting that any inhibitor could have relatively broad applicability. They described 14 compounds, with compound 40 (Table 5) having an IC50 value for GLUT5 of 100 μM, whereas concentrations as high as 2 mM did not effect transport by GLUT1–4. Similarly, a collaborative effort between Emory University, Northwestern University and Washington University uncovered inhibitors of GLUT4. Arguing that multiple myeloma had a significantly higher expression of GLUT4 compared with other GLUTs, they designed a series of 22 compounds, with molecules such as 41 inhibiting 2-deoxyglucose transport by GLUT4 overexpressed in HEK cells, with an IC50 value of 18.2 μM, while having an IC50 >100 μM against GLUT1 or GLUT8 when similarly overexpressed in HEK cells [104]. However, the molecule was only moderately selective when applied to cancer cell growth: it was able to inhibit the growth of KMSl 1 multiple myeloma cells ectopically expressing a GFP control with an IC50 value of 13.4 μM. However, this IC50 value increased to only 21.8 μM when the cells instead ectopically expressed GLUT1, suggesting either off-targets for the molecule, or that GLUT4 plays a role substantially different than GLUT1 in the case of multiple myeloma.
Table 5. . Nutrient import inhibitors.
| Inhibitor | Target | IC50 | |
|---|---|---|---|
| Protein | Cell | ||
| 40 | GLUT5 | 100 μM | N.D.R. |
| 41 | GLUT4 | 18.2 μM | 13.4 μM |
| 42 | GLUT1 | 25–40 nM | 80–700 nM |
| 43 | GLUT1 | ∼3 μM | 1–8 μM |
| 44 | GLUT1 | 336 nM | N.D.R. |
| 45 | GLUT1 | 37 nM | N.D.R. |
| 46 | LAT1 | 36–131 μM | 110 nM |
| 47 | ASCT2 | 240–310 nM† | N.D.R. |
| 48 | ASCT2 | 9.6 μM | ∼10–50 μM |
Summary of inhibitory potencies of compounds inhibiting steps in the tricarboxylic acid cycle. Values marked as approximate (∼) were generally read off a graph, while exact values were generally reported in a table.
Reported as a Ki rather than an IC50. For values marked as ‘N.D.R.’, no data were reported for that compound. ‘Protein’ IC50 values were generally determined against the ability of the transmembrane protein to transport its target molecule into a cell, while ‘Cell’ IC50 values generally apply to the inhibition of cancer cell growth, or a similar cellular readout.
More commonly, GLUT1 has been targeted. Recently, the Johns Hopkins University claimed a series of macroclactones derived from rapamycin. Compound 42 was able to inhibit 2-deoxyglucose and 3-O-methylglucose uptake into A549 lung cancer cells with IC50 values of 25.7 and 38.2 nM, respectively [105]. The molecule had no effect on glucose transport in GLUT1 knockout DLD-1 colon cancer cells, but maintained activity in wild-type cells, even while the GLUT1–4 inhibitor cytochalasin B still negated glucose transport, suggesting specificity for GLUT1. The molecule was similarly able to inhibit the proliferation of various cancer cells with IC50 values between 80 and 700 nM. The Ohio State Innovation Foundation took a similar approach, but derived molecules from the smaller precursor ciglitazone, a PPAR-γ inhibitor that inhibits glucose import totally independent of its PPAR-γ-related effects. The resulting thiazolidinedione inhibitors were able to directly and potently inhibit GLUTs, with compound 43, for example, inhibiting GLUT1 with an IC50 of 2 μM, and inhibiting the growth of prostate, breast and pancreatic cancer cells at similar potencies [106]. The compound had little effect on non-transformed cell lines.
Bayer has also recently reported two series of novel GLUT1 inhibitors. In the first series, Bayer described over 200 N-pyrazolyl carboxamides. Compounds such as 44 inhibited GLUT1 (as measured by quantification of intracellular ATP following glucose treatment) with an IC50 value of 336 nM, but inhibited GLUT2 (as measured by ATP quantification following fructose treatment) with an IC50 value of 35.2 μM, a 100-fold increase [107]. Similar selectivity in the two experiments was observed for several other compounds in the series. The second series of compounds contained a smaller number of structurally similar molecules, such as 45. These compounds generally had somewhat better selectivity for GLUT1 or GLUT2 compared with the first series, with 45 having IC50 values of 37 nM and 51.2 μM against the two GLUTs, respectively [108]. Compounds from either series were used to treat a variety of cancer cells to determine their antiproliferative effects, according to the patents, but no data were reported for either series describing their efficacy in that application.
Like glucose, a variety of amino acids are consumed by cells to fuel their metabolic function. In cancer, two of the most important amino acid transporters are LAT1 and ASCT2 [109]. LAT1, the L-type amino acid transporter 1, is a sodium-independent transporter with a high affinity for leucine, but which also transports other large, neutral amino acids across the cell membrane. ASCT2 is a sodium-dependent transporter with a low affinity (∼300 μM Km) for LAT1 substrates, but with a very high affinity (Km ∼20 μM) for glutamine. Furthermore, LAT1 and ASCT2 expressions are upregulated from twofold to 100-fold across different types of cancer, and play essential roles in cancer growth [109]. The transporters have been shown to be important in a number of different diseases: LAT1 has been shown to predict a poor prognosis in pancreatic ductal carcinomas [110] and in non-small-cell lung cancers [111], ASCT2 has been shown to be crucial for the progression of prostate tumors [112], and both LAT1 and ASCT2 predict worse outcomes in tongue cancer [113]. These transporters are thus attractive targets for drug development. As yet no LAT1 or ASCT2 inhibitor has entered clinical trials, or been approved for clinical use [10,11], but a number of studies have made progress toward changing that.
While a large number of LAT1 inhibitors have been described, patents have been filed on relatively few in recent years. However, one patent from Osaka University describes a large series of compounds targeting LAT1 and its homolog LAT2. The authors prepared a series of small, flexible molecules, prepared as fumaric acid salts [114]. Compound 46 was able to inhibit the amino acid uptake by HEK293T cells stably expressing either LAT1 or LAT2 with IC50 values of 36.6 μM and 131.3 μM, respectively. The compound also inhibited the proliferation of Mia-PaCa-2 pancreatic cancer cells, with an IC50 value of 110 nM. Other compounds in the series were similarly much more potent at inhibiting Mia-PaCa-2 proliferation than at inhibiting amino acid uptake. It is unclear if this is due to off-target effects, or due to Mia-PaCa-2 cells being especially sensitive to LAT inhibition. The compound was also able to inhibit the growth of a Mia-PaCa-2 xenograft by 90% when orally administered at a daily dose of 1 mg/kg, however, showing that it is orally bioavailable.
ASCT2 has seen somewhat more activity lately. The University of Montana has reported a series of hydroxyl-proline analogs, which are dual inhibitors of ASCT2 and ASCT1. Some compounds, such as 47, had substantial potency against either transporter, with Ki values of 310 and 240 nM for ASCT1 and ASCT2, respectively, as measured by two-electrode voltage clamping of stage V–VI oocytes microinjected with cRNA for either gene [115]. Other compounds in the series were similarly unselective between the two ASCTs. The patent claims the molecules can be transformed into radioligands for use in diagnostic imaging, but does not provide any evidence of the efficacy of such reagents compared with any other radioimaging reagent. Similarly, no evidence is provided that the compounds can actually inhibit cancer progression. More recently, Vanderbilt University described a series of compounds that culminated in 48, which was able to inhibit glutamine uptake by HEK-293 cells with an IC50 value of 9.6 μM [116]. It was further able to inhibit the growth of a variety of lung, breast and colon cancer cell lines, with colorectal cancer cells being particularly sensitive. Indeed, DLD1 and HT29 cells were more sensitive than any other examined in the 29 cell line panel, and in the cases of HCT-116, HT29, Colo-205 and RKO colon cancer cells, 48 was a more effective inhibitor than glutaminase inhibitor 15, suggesting that for at least some cancers, preventing nutrient entry might be more effective than preventing nutrient utilization once it is already in the cell. Furthermore, the compound was able to inhibit the growth of colon cancer xenografts or PDX models, but only with a relatively high daily dose (75 mg/kg) administered by intraperitoneal injection. Nonetheless, while amino acid transport inhibition has been a less studied target than GLUT inhibition, compounds 46, 47 and 48 show that there are potential benefits when inhibiting LAT1 and ASCT2.
Conclusion
As discussed above, we have attempted to lay out a landscape of pharmaceutical space, examining a variety of patented efforts to interfere in cancer cell metabolism as a mechanism of inhibiting tumor growth. We have specifically focused upon the TCA cycle, the catalytic process at the heart of energy generation in all mammalian cells, and the processes which most directly feed into the cycle. Such inhibitors are of particular interest due to fundamental metabolic changes that occur in cancer cells, which help them sustain rapid proliferation [3,117]. However, the TCA cycle, glycolysis and anaplerosis are just some of the many metabolic processes in cells. Other pathways, such as the pentose-phosphate shunt, nucleotide biosynthesis, fatty acid synthesis and polyamine synthesis are all critically implicated in cellular metabolism, and many of these have also been the target of significant drug discovery efforts [24,118]. Moreover, we have limited ourselves primarily to a consideration of small molecules that directly act on the targets of interest. We encountered numerous reports of biologics, antibodies or interfering RNAs, which act directly on the targets discussed, while preparing this report. Similarly, the regulatory pathways for many of these targets have been extensively investigated. Thus, it should be understood that the molecular landscape presented here was limited by the scope of our investigation, and not by the imagination or efforts of the drug discovery community.
Of the topics we were able to consider, several trends are immediately evident. Most notably, research is not currently evenly divided among metabolic targets. The TCA cycle itself, for instance, is necessary for the survival of all human cells, and targeting any enzyme in the cycle could thus have highly toxic side effects. While there may be a valuable therapy targeting such enzymes waiting to be discovered, that process will likely require a painstaking undertaking. We also suspect that the strategies being emphasized reflect the momentum that has gathered within each particular aspect of cell metabolism. There has been great success targeting enzymes such as GLS, and mutant isoforms of IDH1 and IDH2, molecules targeting each of which are involved in a number of clinical trials [118].
Overall, we do feel there is substantial hope to be found in the area of cancer metabolism. Some of the earliest developed anticancer drugs were the antimetabolites, molecules such as 5-fluorouracil, which mimics nucleotide metabolites and thus interfere with their biosynthesis [24]. Many of these molecules, discovered near the advent of chemotherapy, are still in use today against a variety of indications. This strongly suggests that metabolism represents an area of vulnerability for cancer cells. Furthermore, while many of the patents we have discussed lack key information pertaining to selectivity, efficacy in whole tumor models, or even the ability to inhibit the growth of cultured cancer cells, they all share one thing in common: they demonstrate that it is possible to inhibit the target enzyme with a small molecule. If medicinal chemists, from industry to academia, have shown us nothing else in the last few decades, it is that they can make a molecule cell permeable, selective and potent.
Future perspective
Over the next 3–5 years, we would not be surprised to see another inhibitor of mutant IDH enzymes enter the clinic. The research in that field is well advanced, with numerous scaffolds currently under consideration in clinical trials, and we suspect that ultimately the correct molecule will be identified, and find its way from the bench to the bedside. We suspect that a GLS inhibitor will not be too far behind, and might even emerge by the end of the next decade. GLS inhibition appears to be as promising as mutant IDH inhibition, with a similar ability to inhibit cancer cell growth while only minimally affecting healthy cells. However, while IDH inhibitors have been examined in a variety of scaffolds, GLS inhibitors have been almost exclusively limited to derivatives and analogs of compound 14. We suspect that in the near future, GLS inhibitor research will begin to explore new classes of compounds with different scaffolds. Indeed, the academic literature already shows this trend, with inhibitors having been described for at least three different allosteric regulatory sites on GLS, in addition to its catalytic pocket [4].
We expect that the most surprising growth over the next decade, however, will be in glycolysis. While none of the inhibitors in Figure 1 have yet shown great success, they demonstrate that no fewer than 60% of the enzymes involved in glycolysis can currently be targeted. Furthermore, each enzyme is relatively poorly investigated from the drug discovery perspective, leaving the field wide open for novel approaches. Given that most cancer cells are heavily dependent on glucose, coupled with the unique microenvironment of a tumor, it might be easier to tune a glycolysis inhibitor to a level that harms the tumor but not the surrounding healthy tissue, than it would be to modulate a comparable GLUT inhibitor. While we expect IDH and GLS research to have the greatest payoff in the near future, inhibitors of glycolysis may be the most exciting to watch over the next decade, with the greatest chance for huge advancements. Regardless of where the advance comes from, however, we strongly feel that clinical relevance for novel metabolic inhibitors is a question of when, and not if, and that these molecules may be of benefit to patients in the relatively near future.
Executive summary.
Cancer metabolism
Increased uptake of glucose and glutamine.
Glucose converted primarily to lactate, glutamine used to fuel tricarboxylic acid (TCA) cycle.
Glycolysis
Six of ten enzymes targeted in recent discovery efforts.
Only a few scaffolds described, open field for novel discovery efforts.
Anaplerosis
Almost all efforts target glutamine anaplerosis, through glutaminase.
Most efforts look at just one drug scaffold.
Tricarboxylic acid (TCA) cycle
Very little effort to target enzymes directly involved in TCA cycle metabolism.
Mutant forms of isocitrate dehydrogenase catalyze a new reaction, and form a new metabolite that drives cancer growth.
Many different scaffolds target mutant isocitrate dehydrogenase enzymes.
Nutrient uptake
Major transmembrane receptors: glucose transporters, large neutral amino acid transporter and neutral amino acid transporter B(0).
Several efforts target glucose transporters. Fewer efforts target large neutral amino acid transporter or neutral amino acid transporter B(0).
Acknowledgments
The authors thank C Westmiller for her skilled assistance in preparing this manuscript. The authors also thank KS Green, M Endo and YH Hur for assistance in translating several of the patents cited in this review, and R Li for proofreading assistance. Furthermore, the authors sincerely apologize to any researcher whose work could not be included in this review due to space or scope constraints.
Footnotes
Financial & competing interests disclosure
Funding for this study was provided by grants from the NIH to RA Cerione (GM122575 and CA201402). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Warburg O. On the origin of cancer cells. Science 123(3191), 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 2.Lu W, Pelicano H, Huang P. Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell 18(3), 199–200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29(3), 313–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katt WP, Lukey MJ, Cerione RA. A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 9(2), 223–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akins NS, Nielson TC, Le HV. Inhibition of glycolysis and glutaminolysis: an emerging drug discovery approach to combat cancer. Curr. Top. Med. Chem. 18(6), 494–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganapathy-Kanniappan S, Geschwind J-FH. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol. Cancer 12(1), 152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930), 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akhenblit PJ, Pagel MD. Recent advances in targeting tumor energy metabolism with tumor acidosis as a biomarker of drug efficacy. J. Cancer Sci. Ther. 8(1), 20–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momcilovic M, Shackelford DB. Imaging cancer metabolism. Biomol. Ther. (Seoul). 26(1), 81–92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Lartigue J. Hallmark tumor metabolism becomes a validated therapeutic target. Hematol. Oncol. 16(1), e47–e52 (2018). [Google Scholar]
- 11.Muthu M, Nordström A. Current status and future prospects of clinically exploiting cancer-specific metabolism – why is tumor metabolism not more extensively translated into clinical targets and biomarkers? Int. J. Mol. Sci. 20(6), 1385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 19(1), 17–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VTV Therapeutics LLC: WO2016196890A1 (2016).
- 14.VTV Therapeutics LLC: WO2018009539A1 (2018).
- 15.Vidac Pharma Ltd.: WO2018083705A1 (2018).
- 16.Exelixis, Inc.: WO2012149528A1 (2012).
- 17.Aurelium BioPharma, Inc.: US20050026231A1 (2005).
- 18.Universite Paul Sabatier Toulouse III; Centre National de la Recherche Scientifique CNRS; Universite de Montreal: FR2857012A1 (2005).
- 19.NeOnc Technologies, Inc.: WO2018102412A1 (2018).
- 20.Shanghai Institute of Materia Medica, Chinese Academy of Sciences; Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences: CN107814792A (2018).
- 21.Abbott Laboratories; Abbott Laboratories Trading Shanghai Company, Ltd.: WO2012045196A1 (2012).
- 22.Emory University: US20140294818A1 (2014).
- 23.Gwangju Institute of Science and Technology: WO2014065572A1 (2014).
- 24.Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem. Biol. 24(9), 1161–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christofk HR, Vander Heiden MG, Harris MH. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452(7184), 230–233 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Beth Israel Deaconess Medical Center, Inc.: US8877791B2 (2014).
- 27.Guizhou Medical University: CN107266466A (2017).
- 28.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 41(3), 211–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunengraber H, Roe CR. Anaplerotic molecules: current and future. J. Inherit. Metab. Dis. 29(2), 327–331 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 22(5), 796–804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emory University: US20160228466A1 (2016).
- 32.Wang JB, Erickson JW, Fuji R. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18(3), 207–219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45(10), 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quaresma M, Coleman MP, Rachet B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: a population-based study. Lancet 385(9974), 1206–1218 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Elan Pharmaceuticals LLC: US20020115698A1 (2002).
- 36.Agios Pharmaceuticals, Inc.: US20140142081A1 (2014).
- 37.Agios Pharmaceuticals, Inc.: US20140142146A1 (2014).
- 38.Agios Pharmaceuticals, Inc.: WO2015143340A1 (2015).
- 39.Calithera Biosciences, Inc.: WO2014089048 (2014).
- 40.Calithera Biosciences, Inc.: US20160287585A1 (2016).
- 41.AstraZeneca AB; Cancer Research Technology Limited: WO2015181539A1 (2015).
- 42.AstraZeneca AB; Cancer Research Technology Limited: WO2017089587A1 (2017).
- 43.Rhizen Pharmaceuticals SA: WO2015101958A2 (2015).
- 44.Rhizen Pharmaceuticals SA: WO2015101957A2 (2015).
- 45.Hangzhou Gamma Biotech Co., Ltd.: WO2017084598A1 (2017).
- 46.Centaurus Biopharma Co., Ltd.: CN107474024A (2017).
- 47.Washington University; Board of Regents, The University of Texas System: US20150273088A1 (2015).
- 48.Board of Regents, The University of Texas System: WO2016004404A2 (2016).
- 49.The University of Texas System: US20160002248A1 (2016).
- 50.University of Pittsburgh - of the Commonwealth System of Higher Education; Cornell University: WO2016054388A1 (2016).
- 51.Board of Regents, University of Texas System: WO2017004359A1 (2017).
- 52.Huang Q, Stalnecker C, Zhang C. et al. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 293(10), 3535–3545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gross MI, Demo SD, Dennison JB. et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13(4), 890–901 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Robinson MM, McBryant SJ, Tsukamoto T. et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406(3), 407–414 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu X, Meng Y, Li L. et al. Overview of the development of glutaminase inhibitors: achievements and future directions. J. Med. Chem. 62(3), 1096–1115 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Wu C, Chen L, Jin S, Li H. Glutaminase inhibitors: a patent review. Expert Opin. Ther. Pat. 28(11), 823–835 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Zimmermann SC, Duvall B, Tsukamoto T. Recent progress in the discovery of allosteric inhibitors of kidney-type glutaminase. J. Med. Chem. 62(1), 46–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tenora L, Alt J, Dash RP. et al. Tumor-targeted delivery of 6-diazo-5-oxo-l-norleucine (DON) using substituted acetylated lysine prodrugs. J. Med. Chem. 62(7), 3524–3538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johns Hopkins University: US20180221395A1 (2018).
- 60.Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol. Cancer Ther. 11(6), 1269–1278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59(2), 298–308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cornell University; Ithaca College: WO2016090350A1 (2016).
- 63.Dongguk University Industry-Academic Cooperation Foundation; Korea Research Institute of Bioscience and Biotechnology: WO2018164549A1 (2018).
- 64.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J. Natl Cancer Inst. 102(13), 932–941 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 34(2), 186–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 29, 6409 (2010). [DOI] [PubMed] [Google Scholar]
- 67.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci. Adv. 2(5), e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen J, Yang J, Cao P. The evolving landscape in the development of isocitrate dehydrogenase mutant inhibitors. Mini Rev. Med. Chem. 16(16), 1344–1358 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Ma T, Zou F, Pusch S, Xu Y, von Deimling A, Zha X. Inhibitors of mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2): an update and perspective. J. Med. Chem. 61(20), 8981–9003 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Celgene Corporation; Agios Pharmaceuticals, Inc.: WO2017146794A1 (2017).
- 71.Agios Pharmaceuticals, Inc.: WO2015006591A1 (2015).
- 72.Agios Pharmaceuticals, Inc.: WO2014062511A1 (2014).
- 73.Agios Pharmaceuticals, Inc.: WO2015006592A1 (2015).
- 74.Forma Therapeutics, Inc.: WO2016171755A1 (2016).
- 75.Forma Therapeutics, Inc.: WO2016044789A1 (2016).
- 76.Forma Therapeutics, Inc.: WO2016044787A1 (2016).
- 77.Forma Therapeutics, Inc.: WO2016044782A1 (2016).
- 78.Forma Therapeutics, Inc.: WO2016171756A1 (2016).
- 79.FORMA Therapeutics, Inc.: WO2016044781A1 (2016).
- 80.Eli Lilly and Company: WO2018111707A1 (2018).
- 81.United States Dept. of Health and Human Services; The University of North Carolina at Chapel Hill: WO2017223202A1 (2017).
- 82.Jiangsu Provincial Institute of Traditional Chinese Medicine: CN106810512A (2017).
- 83.Merck Sharp & Dohme Corp.; Yu Yang. WO2016089830A1 (2016).
- 84.Neuform Pharmaceuticals, Inc.: WO2017069878A1 (2017).
- 85.Daiichi Sankyo Company, Limited; National Cancer Center: WO2016052697A1 (2016).
- 86.Bayer Pharma Aktiengesellschaft: WO2017016992A1 (2017).
- 87.Medshine Discovery Inc.: WO2019015672A1 (2019).
- 88.Nanjing Sanhome Pharmaceutical Co., Ltd.: WO2018014852A1 (2018).
- 89.Shanghai Shipu Biotechnology Co., Ltd.: CN108403696A (2018).
- 90.Shanghai Meton Pharmaceutical Co., Ltd.: WO2018095344A1 (2018).
- 91.Sichuan University: CN107382840A (2017).
- 92.Isocure Biosciences Inc.: WO2019023165A1 (2019).
- 93.Isocure Biosciences Inc.: WO2018071404A1 (2018).
- 94.Isocure Biosciences Inc.: WO2018118793A1 (2018).
- 95.Chia Tai Tianqing Pharmaceutical Group Co., Ltd.; Lianyungang Runzhong Pharmaceutical Co., Ltd.; Centaurus Biopharma Co., Ltd.: WO2017016513A1 (2017).
- 96.Chia Tai Tianqing Pharmaceutical Group Co., Ltd.; Lianyungang Runzhong Pharmaceutical Co., Ltd.; Centaurus Biopharma Co., Ltd.: WO2017162156A1 (2017).
- 97.Chia Tai Tianqing Pharmaceutical Group Co., Ltd.; Lianyungang Runzhong Pharmaceutical Co., Ltd.; Centaurus Biopharma Co., Ltd.: WO2017162157A1 (2017).
- 98.Okoye-Okafor UC, Bartholdy B, Cartier J. et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 11, 878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Popovici-Muller J, Lemieux RM, Artin E. et al. Discovery of AG-120 (ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 9(4), 300–305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Szablewski L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta - Rev. Cancer 1835(2), 164–169 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 202(3), 654–662 (2005). [DOI] [PubMed] [Google Scholar]
- 102.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891 (2004). [DOI] [PubMed] [Google Scholar]
- 103.STC.UNM; Rosalind Franklin University: WO2016201214A1 (2016).
- 104.Emory University; Northwestern University; Washington University: WO2018125968A1 (2018).
- 105.The Johns Hopkins University: WO2017136731A1 (2017).
- 106.Ohio State Innovation Foundation: US9174951B2 (2015).
- 107.Bayer Pharma Aktiengesellschaft: WO2016202898A1 (2016).
- 108.Bayer Pharma Aktiengesellschaft: WO2016202935A1 (2016).
- 109.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin. Cancer Biol. 15(4), 254–266 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Yanagisawa N, Ichinoe M, Mikami T. et al. High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J. Clin. Pathol. 65(11), 1019–1023 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Kaira K, Oriuchi N, Imai H. et al. Prognostic significance of L-type amino acid transporter 1 expression in resectable stage I–III nonsmall cell lung cancer. Br. J. Cancer 98, 742 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang Q, Hardie R-A, Hoy AJ. et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 236(3), 278–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Toyoda M, Kaira K, Ohshima Y. et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 110, 2506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Osaka University: WO2014112646A1 (2014).
- 115.The University of Montana: US20150056138A1 (2015).
- 116.Vanderbilt University: WO2018107173A1 (2018).
- 117.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144(5), 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Tripathi SC, Fahrmann JF, Vykoukal JV, Dennison JB, Hanash SM. Targeting metabolic vulnerabilities of cancer: small molecule inhibitors in clinic. Cancer Rep. 2(1), e1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]