

Abstract
Water, despite being a driving force in biochemical processes, has an elusively complex microscopic behavior. While water can increase its local density near amphiphilic protein surfaces, water is also thought to evaporate from hydrophobic surfaces and cavities, an effect known as “dewetting”. The existence and extent of dewetting effects remains elusive due to the difficulty in observing clear “drying” transitions in experiments or simulations. Here, we use explicit solvent molecular dynamics (MD) simulations to study the molecular solvation at the binding interfaces of two distinctive molecular complexes: the highly hydrophilic barnase–barstar and the highly hydrophobic MDM2–p53. Our simulations, in conjunction with simple volumetric analyses, reveal a strikingly different water behavior at the binding interfaces of these two molecular complexes. In both complexes, we observe significant changes in the water local density as the two proteins approach, supporting the existence of a clear dewetting transition in the case of MDM2–p53, with an onset distance of 5.6–7.6 Å. Furthermore, the solvation analysis reported herein is a valuable tool to capture and quantify persistent or transient dewetting events in future explicit solvent MD simulations.
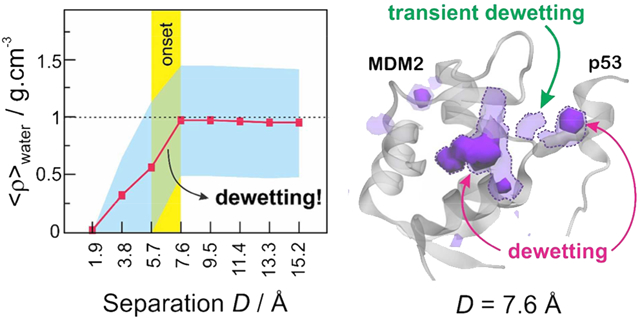
Graphical Abstract
INTRODUCTION
Water, the ubiquitous solvent of life, has a mysteriously flexible nature. Despite its undeniable importance as one of the driving forces for essentially all biological processes,1-4 to understand water’s microscopic behavior remains a challenging task. In the collapse of purely hydrophobic systems5-8 or in complex problems involving protein folding,9-14 aggregation,15,16 and binding,4,17-19 many experimental and theoretical studies have attempted to decipher the exact role of water, at a molecular level. The molecular solvation picture, however, remains elusive, as solvation effects are often subtler than what we can rationally anticipate.20
As a highly polar solvent, water has an extraordinary ability to buffer and mediate electrostatic interactions, which play a major role in the biomolecular world.21 Proteins, for instance, typically have a highly ordered solvation shell, with a density 10–20% higher than that exhibited by bulk water.22,23 These water molecules help in stabilizing the protein fold and might even exert some control over the protein dynamics (and function).24-27
On the other hand, water is also thought to assist in hydrophobic associations by decreasing its density near hydrophobic surfaces and pockets. Such “dewetting” transitions—typically rationalized as water locally behaving as a “vapor bubble”—could play an important role in accelerating hydrophobic associations,2,6,7 driving the folding of protein hydrophobic cores,10 or favoring the binding of hydrophobic ligands.28-32 However appealing, the existence, frequency, and extent of “dewetting” effects remains elusive, especially in topologically complex systems such as proteins, mainly because of the difficulty in observing clear drying transitions in experiments or computer simulations.
In particular, explicit solvent molecular dynamics (MD) simulations are a valuable tool to provide microscopic physical details not directly available in experiments and as such have been recently used to investigate the existence of “dewetting” effects, mainly in the folding scenario.7,9,13-16 While a clear dewetting transition was found to facilitate the collapse of the melittin tetramer,15 most of the studies could not find conclusive evidence of the expected “dewetting” effects. In the case of the association between BphC domains, dewetting was only observed when the electrostatic properties of the domains were turned off,13 suggesting that, even in the hydrophobic core of proteins, van der Waals and electrostatic attractions with the solvent are still too strong to allow collective dewetting. In another study of the aggregation of amyloid β 16–22 protofilaments implicated in Alzheimer’s disease, the authors observed a clear drying transition only when the protein–water 1/r6 Lennard–Jones term was decreased by 10%.16 It is not impossible, then, that current force fields hinder the observation of dewetting transitions by slightly overestimating water–solute attractive potentials. Additionally, it is likely that, although existent, dewetting effects are subtler and more transient than the collective drying that would be required to create a large and easily identifiable “vapor bubble”.
Here, we use explicit solvent MD to investigate the molecular solvation at the binding interfaces of two distinctive protein complexes—the barstar–barnase complex and MDM2 protein bound to p53 transactivation peptide—at varying separation distances. While barstar and barnase have been extensively studied on account of their extremely fast kinetics of binding due to optimal electrostatic complementarity,33-36 the MDM2–p53 complex has received significant attention due to its particularly hydrophobic character,37 besides being a potential target for new anticancer therapies.38-40 Figure 1 displays the binding interfaces of these two protein complexes, with residues colored as charged (blue), polar (green), or apolar (gray). Clearly, most of the binding interfaces in barstar and barnase are comprised of polar or charged residues (Figure 1A). On the other hand, the entire bottom of the concave binding region in MDM2 is composed of apolar residues, flanked by charged and polar at the edges (Figure 1B).
Figure 1.

Binding surfaces (A) in the hydrophilic barnase–barstar complex and (B) in the hydrophobic MDM2–p53 transactivation peptide. Residues are colored as apolar (gray), polar (green), or charged (blue). In each complex, the center of the receptor binding surface is highlighted, as well as the most protruding residues in their binding partners (Asp39 in barnase, Trp23 in p53). The atomic coordinates were obtained from the PDB (IDs 1BRS and 1YCR).
By performing explicit solvent MD simulations in conjunction with simple volumetric analysis, we reveal a strikingly different water behavior near the binding interfaces of these two protein complexes. Interestingly, the approach between protein pairs in each molecular complex promotes opposite variations in the water local density, consistent with the nature of each system. In the MDM2 case, we observe a clear dewetting transition induced by the approach of p53, with a critical dewetting distance of about 5.6–7.6 Å. We additionally detect localized dewetting fluctuations closer to the binding partner, p53, which likely prelude the dewetting transition. Furthermore, the simple solvation analysis proposed herein could be used in future MD studies not only to capture but also to quantify persistent or transient dewetting effects in other protein–protein complexes.
METHODS
MD Simulations.
Atomic coordinates of the two molecular complexes were obtained from the Protein Data Bank,41 with PDB IDs 1BRS (barnase–barstar) and 1YCR (MDM2 bound to p53 transactivation peptide). To generate a realistic encounter pathway for each complex, the protein pairs were manually pulled away along their binding axis and configurations were saved every 1.9 Å, with a separation distance (D) ranging from 0 Å (native complex) up to 15.2 Å (separated proteins). To define the binding axes, we used the vector connecting the center of residue Asp39 in barstar to the geometrical center of barnase and the vector connecting the center of Trp23 in p53 to the geometrical center of MDM2.
The 18 resulting systems (9 describing the encounter pathway between barnase and barstar and 9 describing the encounter pathway between p53 and MDM2) were solvated with TIP3P water molecules,42 in rectangular boxes of dimensions of 56 × 64 × [78 + D] Å (barnase + barstar) or 56 × 64 × [58 + D] Å (MDM2 + p53), with the z-axis aligned along the binding vector. To each box, Na+ or Cl− ions were added to make the systems electrically neutral. Protonation states were estimated with H++.43,44
Simulations were performed with NAMD 2.12,45 using the CHARMM36 force field.46,47 Equilibration consisted of 1000 steps of energy minimization of water and ions, followed by 5 ps of NVT at 310 K and 500 ps of NPT simulations, at 1 bar and 310 K, to equilibrate the density. After that, 40 ns of production NPT simulations were run, at 310 K and 1 bar. In both equilibration and production simulations, the protein conformations were completely fixed, allowing only solvent and ions to move, so as to not add any noise to the solvation analysis.
Solvation Analysis.
To analyze the water behavior in the protein encounter pathways, we used two different approaches (Figure 2). In the first (Figure 2A), we focused on the binding regions of the receptor proteins (barnase and MDM2), as defined by a sphere of radius = 6 Å located at the center of the binding region. The center of the binding region was defined as the position occupied by the most protruding heavy atom from the binding partner (Trp23 in p53 or Asp39 in barstar), in the bound complexes. These positions are highlighted by red spheres in Figure 1. To estimate the average local density in the binding regions, we calculated
| (1) |
where ⟨Nwater⟩ is the average number of water molecules obtained from the simulations, 18.016 g·mol−1 is the molecular mass of water, 6.02204 × 1023 is the Avogadro number, and V is the water-accessible volume of the binding regions. The second line in eq 1 is based on the fact that 1 Å3 = 10−24 cm3 and allows one to obtain the local density (in g/cm3) using volumes in the more adequate units of Å3. For each separation distance, D, the water-accessible volume in the binding regions was estimated with POVME,48-50 after manual deletion of all water molecules, using a grid resolution of 1 Å and a DistanceCutoff = 1.2 Å (Supporting Figure S1 and Table S1). This approach is referred to as “solvation in the binding interface”.
Figure 2.

(A) First approach to analyze water behavior consisting of counting the number of water molecules near the binding surface of the receptor. (B) Second approach consisting of measuring the empty (i.e., dry) volume near the binding surface and in the interdomain gap.
In the second approach, we used POVME to estimate the vacated (that is, desolvated) volume in certain regions of the solvent throughout the simulations. For this approach, again we defined spheres centered at the binding regions of the receptor proteins (barnase or MDM2), but we used spheres of two different sizes: a smaller one focusing on the binding region (radius = 6 Å) and a larger one that extends the analysis into the interdomain gap between the two binding proteins (radius = 18 Å). Prior to POVME calculations, all hydrogen atoms were deleted from the trajectories to facilitate the analysis. The empty volumes within each sphere were computed for every 50th frame of the trajectories (total of 800 frames), using a grid resolution of 1 Å and a DistanceCutoff = 1.2 Å. Using the VolMap plugin in VMD,51 the resulting volumes were then accumulated into “vacancy maps”, which reflect the probability of these regions to be found empty (i.e., desolvated) during the course of the simulations (Supporting Figure S2). This approach is referred to as “desolvation in the binding region/interdomain gap”. It allows one not only to quantify dewetting effects but also to localize transient solvent fluctuations.
RESULTS
Solvation in the Binding Regions of Barnase and MDM2.
As a first approach to investigate the solvation behavior near the binding surfaces of the receptor proteins (Figure 3A), we counted the number of water molecules lying within these regions throughout the MD trajectories. As a result, Figure 3B shows the average number of water molecules solvating the binding regions of barnase (left) and MDM2 (right) as a function of the separation distance (D) to their respective binding partners. In agreement with the character of their binding regions, barnase displays a significantly higher number of water molecules surrounding its binding interface (~12) as compared to the MDM2 binding cavity (~3). In both systems, the number of water molecules sharply decreases as the binding partners approach, but while barnase continues to display a well solvated binding surface, the number of water molecules in the MDM2 binding pocket drops to zero at D = 1.9 Å (Figure 3B). In the case of barnase, the discrete water expulsion observed between 3.8 and 5.7 Å is likely a simple consequence of the beginning of a steric hindrance from the approaching barstar protein. In the MDM2 protein, however, the expulsion of water molecules starts at a larger separation distance (between 5.7 and 7.6 Å), suggesting the occurrence of a hydrophobic-driven dewetting transition. The latter is confirmed when we relate the average number of water molecules to the water-accessible volume in each binding region, as indicated in eq 1, obtaining the average local density, ⟨ρ⟩water (Figure 3C). In the case of barnase, we observe a sharp “wetting” of the binding region, which starts when its binding partner, barstar, is less than 5.7 Å away. Interestingly, the barnase binding region is strongly solvated even when barstar is very close (1.9 Å). Contrariwise, the approach of the p53 transactivation peptide induces a clear “dewetting” of the MDM2 binding region, whose onset distance (5.6–7.6 Å) is large enough to accommodate 2–3 times the diameter of a water molecule. It should be noticed that the local densities displayed in Figure 3C are only semiquantitative, since they are based on discrete estimates of local volumes, which are not exact. They do provide, however, a good picture of the strikingly different molecular solvation occurring at the binding interfaces of barnase–barstar versus MDM2–p53.
Figure 3.

Solvation in the binding regions of barnase (left) and MDM2 (right). (A) Definition of the binding regions. (B) Average number of water molecules in the binding region as a function of the distance (D) to the binding partners. (C) Water local density in the binding regions, as calculated in eq 1, as a function of the distance (D) to the binding partners. In panels B and C, the critical distance at which the approach of the binding partners promotes heterogeneous solvation at the binding regions is highlighted by a vertical yellow bar. The standard deviation is shown in cyan.
Desolvation in the Binding Regions and Intedomain Gap.
In this second approach, we used POVME to calculate desolvated volumes within a cutoff of 6 or 18 Å from the center of the binding surfaces in the receptor proteins (Figure 4A). As a result, Figure 4B shows the distribution of desolvated volumes at different separation distances, D, for the binding region (left) and interdomain gap (right) of MDM2–p53 (top) and barnase–barstar (bottom). The percentage of dry volume, Vdry (%), was calculated as
| (2) |
where Vvoid is the desolvated volume measured by POVME for each frame of the trajectories and Vtotal is the total water-accessible volume within each sphere, also computed with POVME after manual deletion of water molecules (see Supporting Figure S1 and Table S1).
Figure 4.

Desolvation in the binding region and interdomain gap. (A) Regions considered in the analysis, defined by spheres of radius 6 Å (red) or 18 Å (orange), illustrated for D = 9.5 Å. (B) Distribution of “desolvated” volumes, calculated as in eq 2, for MDM2–p53 (top) or barnase–barstar (bottom), in the binding regions (left) or in the interdomain gap (right).
In the case of MDM2, we observe significant desolvation of the binding region (r = 6 Å) at separation distances of D < 7.6 Å, as revealed by the shift of the probability distribution of dry volumes toward greater values (Figure 4B, top left panel). In fact, 50–80% of the water-accessible volume in the binding region of MDM2 is often vacated when p53 is less than 7.6 Å away. When we extend the analysis into the interdomain gap (Figure 4B, top right panel), we see that the approach of p53 below 7.6 Å also increases the desolvated volume, suggesting that the dewetting persists in the interdomain region, at least to some extent. Moreover, the significantly shifted distribution of dry volumes for separation distances of D < 7.6 Å confirms that the onset distance for dewetting effects lies in between 5.7 and 7.6 Å, in good agreement with results obtained in our previous solvation analysis (Figure 3).
In contrast, we observe no evidence of any desolvation near barnase, regardless of the separation distance to its binding partner, barstar (Figure 4B, bottom panels). In fact, the insignificant desolvated volume estimated in the binding region of barnase (<3%) is in very good agreement with a strongly solvated (or “wet”) binding surface, as predicted by our previous solvation analysis (Figure 3). Also, when the desolvation analysis is projected into the interdomain gap, we find that the interdomain region between barnase and barstar is more solvated (smaller “dry volumes”) as compared to the region between MDM2 and p53, regardless of the separation distance, D (Figure 4, right panels).
We further accumulated the “desolvated” volumes in the solvent region to generate “water vacancy maps”, which allows one to visualize the regions that are more frequently empty (i.e., desolvated) during the simulations. As a result, Figure 5 contours the water vacancy maps at levels of 0.10 and 0.25, for separation distances ranging from 3.8 to 9.5 Å. To facilitate the analysis, regions that are found empty at least 25% of the simulation time (solid blobs) are referred to as “often dry” and those empty at least 10% of the simulation time (dashed blobs) are referred to as “transiently dry”. The maps were calculated within a cutoff radius of 18 Å from the center of the MDM2 binding region, as illustrated in Figure 4A. We found that the inner part of the MDM2 binding pocket is often dry regardless of the distance from p53, while the outer parts of the binding region are often dry only when p53 is less than 7.6 Å away (blue arrows in Figure 5). We also found an often dewetting located close to Trp23 in the p53 (pink arrows), regardless of the separation distance to MDM2, which could help destabilize the water molecules in the interdomain gap as the two proteins approach. Interestingly, there is evidence of transient dewetting in the interdomain region between MDM2 and p53 for separation distances up to 7.6 Å (green arrow). Although subtle, such transient dewetting clearly preludes the dewetting transition observed at 5.7 Å and could thus be regarded as the initial solvent fluctuations leading to the nucleation of a small vapor bubble. Noteworthy, no such frequent or even transient dewetting effects were observed in the vacancy maps computed for barstar and barnase (Supporting Figure S3).
Figure 5.

Water vacancy maps contoured at 0.25 (solid blobs) and 0.10 (dashed blobs), representing regions of the solvent that are often empty (desolvated) during the simulations of MDM2 and p53, at separation distances of 3.8–9.5 Å. These maps were calculated within a cutoff radius of 9.5 Å from the center of the MDM2 binding region. The arrows indicate regions of MDM2 often dry when p53 is less than 7.6 Å away (in blue), an often dry region near Trp23 in p53 (in pink), and a transient dewetting in the interdomain gap (in green).
DISCUSSION AND CONCLUSIONS
We performed explicit solvent MD simulations to investigate the molecular solvation details that take part in the bindings between MDM2 and p53 and between barstar and barnase. We selected these two strikingly different complexes—the first highly hydrophobic and the second highly hydrophilic—to guarantee that our methodology can describe a large range of solvation behavior.
By using two different approaches combined—the first based on discretely counting water molecules lying within the binding regions and the second based on measuring “vacated” volumes in the solvent near the binding interfaces—we could capture important heterogeneous solvation in the binding regions of MDM2 and barnase, induced by the approach of their respective binding partners (p53 and barstar, respectively). These two approaches produced complementary and consistent results, allowing us to identify (i) wetting versus dewetting transitions in barnase–barstar versus MDM2–p53, (ii) the critical interprotein distance at which heterogeneous solvation starts to occur, (iii) the extent of dewetting effects (in %), and (iv) the exact location of important solvent fluctuations preluding the drying transition in MDM2.
The strikingly different molecular solvation observed in the binding of barstar–barnase and MDM2–p53 is likely influenced not only by their distribution of amino acid residues—many charged residues in barnase/barstar versus many hydrophobic residues in MDM2/p53—but also by the shape of their binding interfaces. While barnase displays a relatively flat binding surface, MDM2 has a significantly concave binding cavity to host p53’s protruding Trp23 residue. At close distances, the combination of a concave cavity with a protruding binding partner makes the interdomain gap between MDM2 and p53 resemble a tunnel, a geometry that has been argued to disrupt water hydrogen bonds, making water presence less favorable.15 Therefore, the relatively long-ranged dewetting transition observed in MDM2 at the approach of p53 is likely a consequence not only of a high content of apolar amino acid residues but also of the highly negative curvature of MDM2’s binding region. Moreover, the onset dewetting distance of 5.6–7.6 Å is large enough to accommodate 2–3 times the diameter of a water molecule, confirming that the dewetting transition observed in MDM2 is not caused by simple steric hindrance with the approaching p53 (i.e., “water expulsion”).
In sharp contrast, in all of our analyses, we did not observe any evidence of a dewetting transition in the highly hydrophilic barstar–barnase complex, not even solvent fluctuations that could lead to local and transient dewetting effects. Instead, we found that barstar induces a sharp “wetting” of the binding surface of barnase, which persists even at very close separation distances (1.9 Å). This indicates that the eventual water expulsion necessary for binding occurs at the very last stage of complex formation, in contrast with the “partial desolvation” mechanism that has been suggested to “steer” the binding of barstar and barnase at a distance of ~3 Å.34,36 On the other hand, these results are in good agreement with previous calculations with the variational implicit solvent method (VISM),52-55 which predicted that water was strongly favored in between barnase and barstar as the two proteins approach.56
Overall, our results highlight the fascinating flexibility of water, which can assist the association of very distinctive protein complexes by displaying strikingly different behaviors: either increasing its local density prior to steric expulsion in the binding of barstar and barnase or gradually disappearing from the MDM2 binding pocket at the approach of p53. Therefore, while the association between barnase and barstar appears to follow the “water expulsion” mechanism,9 as proposed in the folding scenario, the desolvation detected in the binding of MDM2 and p53 provides further support to the “dewetting” mechanism, as proposed by ten Wolde and Chandler,10 further extending its occurrence to the context of protein–protein interactions. According to this mechanism, the drying transition between MDM2 and p53 could increase the attraction between the hydrophobic interfaces of the proteins, accelerating their binding after they reach a critical distance of 7.6 Å.
Finally, we propose that the simple solvation analysis reported herein could be easily applied in other explicit solvent MD studies to capture, quantify, and localize transient solvent fluctuations leading up to dewetting effects, in the context of protein–protein interactions. One particularly interesting direction would be to combine this analysis with methods such as grid inhomogeneous solvation theory (GIST),57,58 to get additional information on the thermodynamic driving forces that are at play in heterogeneous water behavior.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Bo Li and Prof. Joachim Dzubiella for stimulating discussions. This work was supported in part by the National Institutes of Health (NIH), the National Biomedical Computation Resource (NBCR), and the San Diego Supercomputer Center (SDSC).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b07773.
Figures and table describing (i) the methodology used to calculate solvent accessible volumes and desolvated volumes and (ii) desolvation results for barnase–barstar complex (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Ball P Water as an active constituent in the cell biology. Chem. Rev 2008, 108, 74–108. [DOI] [PubMed] [Google Scholar]
- (2).Chandler D Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [DOI] [PubMed] [Google Scholar]
- (3).Levy Y; Onuchic JN Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 389–415. [DOI] [PubMed] [Google Scholar]
- (4).Hummer G Molecular binding: Under water’s influence. Nat. Chem 2010, 2, 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Parker JL; Claesson PM; Attard PJ Bubbles, cavities, and the long-ranged attraction between hydrophobic surfaces. J. Phys. Chem 1994, 98, 8468–8480. [Google Scholar]
- (6).Lum K; Chandler D; Weeks JD Hydrophobicity at small and large scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar]
- (7).Huang X; Margulis CJ; Berne BJ Dewetting-induced collapse of hydrophobic particles. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 11953–11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huang X; Zhou R; Berne BJ Drying and hydrophobic collapse of paraffin plates. J. Phys. Chem. B 2005, 109, 3546–3552. [DOI] [PubMed] [Google Scholar]
- (9).Cheung MS; Garcia AE; Onuchic JN Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).ten Wolde PR; Chandler D Drying-induced hydrophobic polymer collapse. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 6539–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Miller TF; Vanden-Eijnden E; Chandler D Solvent coarse-graining and the string method applied to the hydrophobic collapse of a hydrated chain. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 14559–14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang Q; Ding SW; Hua CY; Yang HC; Chen CL A computer simulation study of water drying at the interface of protein chains. J. Chem. Phys 2004, 121, 1969–77. [DOI] [PubMed] [Google Scholar]
- (13).Zhou R; Huang X; Margulis CJ; Berne BJ Hydrophobic collapse in multidomain protein folding. Science 2004, 305, 1605–1609. [DOI] [PubMed] [Google Scholar]
- (14).Rhee YM; Sorin EJ; Jayachandran G; Lindahl E; Pande VS Simulations on the role of water in the protein-folding mechanism. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 6456–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu P; Huang X; Zhou R; Berne BJ Observation of a dewetting transition in the collapse of the melittin tetramer. Nature 2005, 437, 159–162. [DOI] [PubMed] [Google Scholar]
- (16).Krone MG; Hua L; Soto P; Zhou R; Berne BJ; Shea J-E Role of water in mediating the assembly of alzheimer amyloid β 16–22 protofilaments. J. Am. Chem. Soc 2008, 130, 11066–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Young T; Abel R; Kim B; Berne BJ; Friesner RA Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ahmad M; Gu W; Helms V Mechanism of fast peptide recognition by SH3 domains. Angew. Chem., Int. Ed 2008, 47, 7626–7630. [DOI] [PubMed] [Google Scholar]
- (19).Wang L; Berne BJ; Friesner RA Ligand binding to protein-binding pockets with wet and dry regions. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Berne BJ; Weeks JD; Zhou R Dewetting and hydrophobic interaction in physical and biological systems. Annu. Rev. Phys. Chem 2009, 60, 85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Davis ME; McCammon JA Electrostatics in biomolecular structure and dynamics. Chem. Rev 1990, 90, 509–521. [Google Scholar]
- (22).Cheng Y-K; Rossky PJ Surface topography dependence of biomolecular hydrophobic hydration. Nature 1998, 392, 696–699. [DOI] [PubMed] [Google Scholar]
- (23).Makarov V; Pettitt BM; Feig M Solvation and hydration of proteins and nucleic acids: a theoretical view of simulation and experiment. Acc. Chem. Res 2002, 35, 376–384. [DOI] [PubMed] [Google Scholar]
- (24).Wong CF; Zheng C; McCammon JA Glass transition in SPC/E water and in a protein solution: a molecular dynamics simulation study. Chem. Phys. Lett 1989, 154, 151–154. [Google Scholar]
- (25).Ansari A; Jones CM; Henry ER; Hofrichter J; Eaton WA The role of solvent viscosity in the dynamics of protein conformational changes. Science 1992, 256, 1796–1798. [DOI] [PubMed] [Google Scholar]
- (26).Fenimore PW; Frauenfelder H; McMahon BH; Parak FG Slaving: solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16047–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mattos C Protein-water interactions in a dynamic world. Trends Biochem. Sci 2002, 27, 203–208. [DOI] [PubMed] [Google Scholar]
- (28).Qvist J; Davidovic M; Hamelberg D; Halle B A dry ligand-binding cavity in a solvated protein. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 6296–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Baron R; Setny P; McCammon JA Water in cavity-ligand recognition. J. Am. Chem. Soc 2010, 132, 12091–12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Setny P; Baron R; McCammon JA How can hydrophobic association be enthalpy driven? J. Chem. Theory Comput 2010, 6, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang L; Berne BJ; Friesner RA Ligand binding to protein-binding pockets with wet and dry regions. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Krimmer SG; Cramer J; Schiebel J; Heine A; Klebe G How nothing boosts affinity: hydrophobic ligand binding to the virtually vacated S1’ pocket of thermolysin. J. Am. Chem. Soc 2017, 139, 10419–10431. [DOI] [PubMed] [Google Scholar]
- (33).Gabdoulline RR; Wade RC Simulation of the diffusional association of barnase and barstar. Biophys. J 1997, 72, 1917–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gabdoulline RR; Wade RC. On the protein-protein diffusional encounter complex. J. Mol. Recognit 1999, 12, 226–234. [DOI] [PubMed] [Google Scholar]
- (35).Lee L-P; Tidor B Barstar is electrostatically optimized for tight binding to barnase. Nat. Struct. Biol 2001, 8, 73–76. [DOI] [PubMed] [Google Scholar]
- (36).Hoefling M; Gottschalk KE Barnase-barstar: from first encounter to final complex. J. Struct. Biol 2010, 171, 52–63. [DOI] [PubMed] [Google Scholar]
- (37).Guo Z; Li B; Dzubiella J; Cheng L-T; McCammon JA; Che J Heterogeneous hydration of p53/MDM2 complex. J. Chem. Theory Comput 2014, 10, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chène P Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [DOI] [PubMed] [Google Scholar]
- (39).Chène P Inhibition of the p53-MDM2 interaction: targeting a protein-protein interface. Mol. Cancer Res 2004, 2, 20–28. [PubMed] [Google Scholar]
- (40).Zhao Y; Bernard D; Wang S Small molecule inhibitors of MDM2-p53 and MDMX-p53 interaction as new cancer therapeutics. BioDiscovery 2013, 8, e8950. [Google Scholar]
- (41).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (43).Gordon JC; Myers JB; Folta T; Shoja V; Heath LS; Onufriev A H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Anandakrishnan R; Aguillar B; Onufriev AV H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic modeling and simulations. Nucleic Acids Res 2012, 40, W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Huang J; MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Durrant JD; de Oliveira CA; McCammon JA POVME: an algorithm for measuring binding-pocket volumes. J. Mol. Graphics Modell 2011, 29, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Durrant JD; Votapka L; Sorensen J; Amaro RE POVME 2.0: an enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput 2014, 10, 5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wagner JR; Sorensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: software for mapping binding pocket flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Humphrey W; Dalke A; Schulten K VMD – Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (52).Dzubiella J; Swanson JMJ; McCammon JA Coupling hydrophobicity, dispersion, and electrostatics in continuum solvent models. Phys. Rev. Lett 2006, 96, 087802. [DOI] [PubMed] [Google Scholar]
- (53).Dzubiella J; Swanson JMJ; McCammon JA Coupling nonpolar and polar solvation free energies in implicit solvent models. J. Chem. Phys 2006, 124, 084905. [DOI] [PubMed] [Google Scholar]
- (54).Cheng L-T; Dzubiella J; McCammon JA; Li B Application of the level-set method to the implicit solvation of nonpolar molecules. J. Chem. Phys 2007, 127, 084503. [DOI] [PubMed] [Google Scholar]
- (55).Cheng L-T; Xie Y; Dzubiella J; McCammon JA; Che J; Li B Coupling the level-set method with molecular mechanics for variational implicit solvation of nonpolar molecules. J. Chem. Theory Comput 2009, 5, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ricci CG; Li B; Cheng L-T; Dzubiella J; McCammon JA Martinizing” the variational implicit solvent method (VISM): Solvation free energy for coarse-grained proteins. J. Phys. Chem. B 2017, 121, 6538–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Nguyen CN; Young TK; Gilson MK Grid inhomogeneous solvation theory: hydration and thermodynamics of the miniature receptor cucurbit[7]uril. J. Chem. Phys 2012, 137, 044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ramsey S; Nguyen C; Salomon-Ferrer R; Walker RC; Gilson MK; Kurtzman T Solvation thermodynamic mapping of molecular surfaces in AmberTools: GIST. J. Comput. Chem 2016, 37, 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.