

SUMMARY
Mechanisms coordinating pancreatic β cell metabolism with insulin secretion are essential for glucose homeostasis. One key mechanism of β cell nutrient sensing uses the mitochondrial GTP (mtGTP) cycle. In this cycle, mtGTP synthesized by succinyl-CoA synthetase (SCS) is hydrolyzed via mitochondrial PEPCK (PEPCK-M) to make phosphoenolpyruvate, a high-energy metabolite that integrates TCA cycling and anaplerosis with glucose-stimulated insulin secretion (GSIS). Several strategies, including xenotopic overexpression of yeast mitochondrial GTP/GDP exchanger (GGC1) and human ATP and GTP-specific SCS isoforms, demonstrated the importance of the mtGTP cycle. These studies confirmed that mtGTP triggers and amplifies normal GSIS and rescues defects in GSIS both in vitro and in vivo. Increased mtGTP synthesis enhanced calcium oscillations during GSIS. mtGTP also augmented mitochondrial mass, increased insulin granule number, and membrane proximity without triggering de-differentiation or metabolic fragility. These data highlight the importance of the mtGTP signal in nutrient sensing, insulin secretion, mitochondrial maintenance, and β cell health.
Graphical Abstract
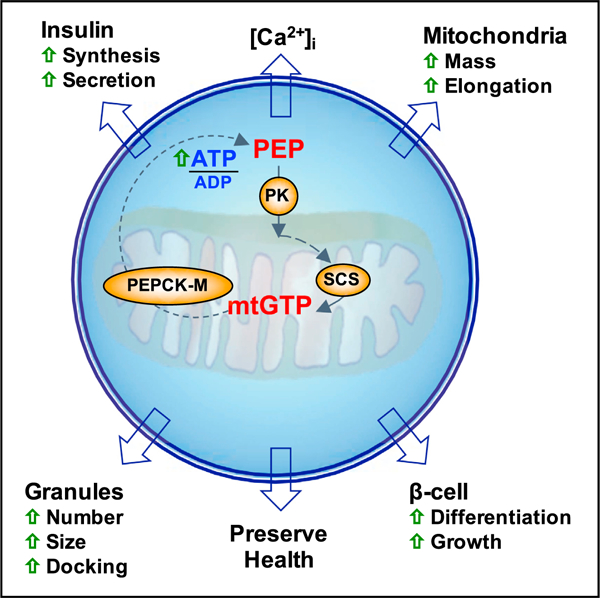
In Brief
Jesinkey et al. report that mitochondrial GTP (mtGTP) is an integrative nutrient sentinel regulating b cell function. Signaling from mtGTP raises calcium independent of oxidative phosphorylation to promote insulin secretion. Without overworking the β cell, mtGTP cycling potentiates insulin secretion, nutrient sensing, and mitochondrial expansion alongside promoting health and increasing insulin reserves.
INTRODUCTION
Metabolic signals intrinsically regulate coherent glucose sensing and are crucial for β cell function. Such signaling metabolites must reflect ambient nutrient concentrations and transmit this information to downstream targets as diverse as ion channels, cytoskeleton, vesicle membranes, protein synthesis, and transcription factors. Mitochondrial guanosine triphosphate (mtGTP) is an essential signaling intermediate for insulin secretion, which is clinically apparent in hyperinsulinemia hyperammonemia (HI/HA), a human inborn error in glutamate metabolism that is associated with protein-induced hypoglycemia (Kibbey et al., 2007, 2014; MacMullen et al., 2001).
mtGTP hydrolysis is coupled to the mitochondrial synthesis of phosphoenolpyruvate (PEP) and requires three-way collaboration between GTP-specific succinyl-coenzyme A synthetase (SCS-GTP; SUCLG2), mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M), and pyruvate carboxylase (PC) and/or glutamate dehydrogenase (GDH) (Figure S1A; Cline et al., 2004; Lu et al., 2002; Stark and Kibbey, 2014; Stark et al., 2009). Tricarboxylic acid (TCA) cycling generates mtGTP via SCS-GTP independently of oxidative phosphorylation (OxPhos) and distinct from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH)-generating substrate cycles (Kibbey et al., 2007). Since mammalian mitochondria lack a GTP transporter, mtGTP is effectively trapped in the mitochondrial matrix (McKee et al., 1999, 2000; Vozza et al., 2004). The mitochondrial GTPase PEPCK-M biochemically transduces the mtGTP signal out to the cytosol by synthesizing PEP, which exits the mitochondria via the citrate isocitrate carrier (CIC) (Drahota et al., 1983). In the cytosol, PEP can lower ADP via pyruvate kinase (PK) driven by the large favorable free energy drop (equivalent to two ATPs). This makes the mtGTP-PEP cycle uniquely poised to reflect cellular energy potential.
Substrate-level mtGTP synthesis is catalyzed via SCS-GTP. Since a parallel ADP-specific isoform (SCS-ATP) competes for the substrate succinyl-coenzyme A (succinyl-CoA), the ratio of SCS-GTP to SCS-ATP and the matrix concentrations of the respective di- and triphosphate nucleotides influence mtGTP synthesis rates. Previously, we silenced SCS-ATP (to increase mtGTP) or SCS-GTP (to decrease mtGTP) in vitro and demonstrated a link between mtGTP synthesis and insulin secretion (Kibbey et al., 2007). Here, we confirm and further characterize the mtGTP-PEP cycle signal via xenotopic enzyme and transporter expression in vitro and in vivo.
RESULTS
Transient Overexpression of Human SCS-ATP and SCS-GTP
SCS is a heterodimer with a shared catalytic α-subunit and either an ATP or a GTP nucleotide-specific β-subunit. Plasmid constructs were generated for the expression of GFP fused to human ATP-specific (hSCS-ATP) or GTP-specific (hSCS-GTP) SCS β-subunit, with mitochondria-targeted GFP (MitoGFP) as the control (Figure 1A). Transient transfection of INS-1 832/13 rat insulinoma cells was confirmed by robust human SCS β-sub-unit mRNA expression (Figures S1B and S1C). Overexpression of the β-subunits did not alter endogenous α-subunit levels (Figure S1D). GFP reporter fluorescence co-localized with mitochondria (Figure S1E). Consistent with prior reports, ~70% of total SCS activity was SCS-GTP in non-transfected INS-1 832/13 cell lysates (Figure S1F) (Kibbey et al., 2007; Kowluru, 2001). Overexpression of hSCS-ATP reduced the GTP-specific reaction to 5% of total activity, while overexpression of hSCS-GTP increased the GTP-specific reaction to ~98% of total activity (Figure S1F). Increased hSCS-ATP virtually abolished glucose-stimulated insulin secretion (GSIS), but hSCS-GTP overexpression potently increased GSIS (Figure 1A). In this acute transgenic system, the correlation between GSIS and SCS-GTP expression was consistent with prior knockdown studies (Kibbey et al., 2007) arguing against off-target effects.
Figure 1. Overexpression of hSCS-GTP Enhances Insulin Secretion.

(A–C) Transgenic overexpression of hSCS-ATP or hSCS-GTP via transient (A), constitutive (B), and inducible (C) systems. Insulin release from transiently transfected INS-1 832/13 cells, and clonal cells lines with constitutive or tet-inducible overexpression at basal (gray bars) versus stimulatory (black bars) glucose. Basal was 3 mM glucose (A and B) and 2.5 mM glucose (C); stimulatory was 15 mM (A and B) and 16.7 mM glucose (C).
(D) Western blot of the V5 epitope of human SCS β-subunits in two clones of the inducible cell lines and insulin secretion in response to 16.7 mM glucose from these cells after 0.2 mg/mL DOX treatment.
(E) Insulin secretion from control (black), constitutive hSCS-ATP (blue), and constitutive hSCS-GTP (red) cells. Control, INS-1 832/13; mtGFP, mitochondria-targeted green fluorescent protein.
The data are means ± SEMs (A)–(E) secretion, n = 6 per group; (D) V5-tagged protein expression, n = 3 per group. Statistical significance is *p < 0.05, ***p < 0.001, and ****p < 0.0001 by two-way ANOVA with Sidak’s correction (A), two-way ANOVA with Tukey’s correction (B), two-way ANOVA with two-stage linear step-up (C), Student’s unpaired two-tailed t test (D), and two-way ANOVA with Dunnett’s multiple comparisons (E).
Constitutive Overexpression of Human SCS-ATP and SCS-GTP
Clonal cell lines were made with constitutive overexpression of either V5-tagged hSCS-ATP or hSCS-GTP (Figure 1B). Endogenous rat SCS β-subunit mRNA expression was largely unaltered in the constitutive cell lines, except that rat SCS-GTP was slightly higher in hSCS-GTP clones, although a difference in protein expression was not detected (Figures S2A and S2B). Transgenic expression of V5-tagged hSCS β-subunit protein (Figure S2B, top panel) and mRNA (Figures S2C and S2D) were both elevated. Endogenous rat α-subunit mRNA and protein levels were unchanged (Figures S2B and S2E). As with transient expression, the clonal cell line expressing hSCS-ATP showed decreased GSIS, while cells with constitutive hSCS-GTP expression had elevated basal insulin secretion and GSIS (Figure 1B).
Inducible Expression of Human SCS-ATP and SCS-GTP
Doxycycline (DOX)-inducible (tet-on) clonal cell lines were also generated with each V5-tagged SCS transgene under the control of a reverse tet-repressor system (rtTA) (Figure 1C). DOX induced robust increases in mRNA and mitochondrial protein expression, which was maximal 24 h after treatment and persisted for 48 h after DOX washout (Figures S2F–S2L). Induction and clearance rates of both trans-proteins were similar. Transgene induction (1 day induced, 2 days washout) decreased GSIS in hSCS-ATP cells and increased it in hSCS-GTP cells (Figure 1C). Clones expressing each hSCS β-subunit were selected based on ‘‘low’’ and ‘‘high’’ trans-protein expression; again, insulin secretion was further attenuated in clones with higher hSCS-ATP expression, whereas clones with higher hSCS-GTP expression displayed higher insulin secretion (Figure 1D).
Glucose Sensing Is SCS Isoform Dependent
To assess whether the increase in GSIS observed with enhanced mtGTP production is detrimental to substrate sensitivity, GSIS was measured across a range of glucose concentrations (Figure 1E). The hSCS-ATP cells had diminished GSIS across the entire glucose range. In contrast, hSCS-GTP cells exhibited enhanced insulin release at all glucose concentrations, with peak insulin levels reaching 180% over control at 25 mM glucose and a greater rate of increase in GSIS from 7 to 25 mM. These data suggest that mtGTP-mediated signaling increases sensitivity and responsiveness to glucose.
Tetracycline-Regulated b Cell Succinyl-Coenzyme A Synthetase (TaBaSCo) Mice
Two inducible transgenic mouse models, tetracycline-regulated β cell succinyl-CoA synthetase (TaBaSCo), were made (Figure 2A), with transgenes expressing GFP-tagged human SCS β isoforms under the regulation of the tet-operon. Heterozygous hSCS-ATP and hSCS-GTP lines were crossed with rat insulin promoter (RIP)-tTA (tet-off) mice to generate double transgenic offspring (TaBaSCo-ATP = hSCS-ATP 3 RIP-tTA and TaBaSCo-GTP = hSCS-GTP 3 RIP-tTA). The RIP-tTA and WT mice on 50% C57BL6/J, 50% CD-1 background had the same weight, fasting plasma glucose, or insulin and were combined as controls (Figures S3A–S3C). TaBaSCo-ATP or -GTP body weights did not differ significantly from controls (Figures S3D–S3F), and the pancreas had a normal histologic appearance (Figure 2B). GFP-tagged hSCS-GTP and hSCS-ATP expression was islet specific, co-localized with insulin, and suppressed by DOX (Figure 2C). Overnight-fasted TaBaSCo-GTP mice were hypoglycemic relative to controls, whereas TaBaSCo-ATP mice exhibited fasting hyperglycemia (Figure 2D). Meanwhile, fasting plasma insulin concentrations were significantly decreased in TaBaSCo-ATP mice relative to controls, and fasting plasma insulin was elevated in TaBaSCo-GTP mice compared to controls and nearly 10-fold greater than concentrations in TaBaSCo-ATP mice, which is consistent with a primary b cell effect (Figure 2E). The mirroring effects on plasma glucose and insulin seen with hSCS-ATP and hSCS-GTP overexpression were observed in both male and female mice (Figures S3G–S3J). DOX in the drinking water of TaBaSCo-ATP mice (+DOX) reversed fasting hyperglycemia and increased plasma insulin (Figures 2F and 2G). Perifusion of isolated islets from both TaBaSCo-GTP and -ATP mice showed basal insulin secretion similar to that from control islets (Figures 2H–2J). However, both first- and second-phase GSIS was markedly decreased from TaBaSCo-ATP islets (Figure 2H) and significantly higher from TaBaSCo-GTP islets (Figure 2I). DOX treatment of TaBaSCo-ATP islets improved both first- and second-phase GSIS (Figure 2J), suggesting that the β cell transgenes were responsible for the differences in whole-body glucose homeostasis.
Figure 2. Metabolic Characteristics of Pre-backcrossed tet-off TaBaSCo Mice.

(A) TaBaSCo transgenic mice with tet-off islet overexpression of hSCS-ATP or hSCS-GTP.
(B) Representative H&E staining of pancreatic sections from wild-type (WT), TaB-ATP – DOX, and TaB-GTP ± DOX mice.
(C) Representative islet immunofluorescence of GFP-tag and co-localization with insulin from male TaB-ATP mice ± DOX.
(D and E) Plasma glucose (D) and insulin (E) from overnight fasted mice (littermates n = 8, TaB-ATP n = 7, littermates n = 5, TaB-GTP n = 3 individual mice).
(F and G) Plasma glucose (F) and insulin (G) from overnight fasted TaB-ATP + DOX and TaB-ATP – DOX mice (n = 4 mice per group).
(H–J) Perifused islet insulin secretions as indicated (H and I: n = 3, J: n = 5 technical replicates).
The data are means ± SEMs. TaBaSCo-ATP, TaB-ATP; TaBaSCo-GTP, TaB-GTP. Scale bars (B and C), 10 mm. Statistical significance is *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by Welch’s unpaired two-tailed t test (D and E), Student’s unpaired two- tailed t test (F and G), and unpaired two-tailed t test with Holm-Sidak’s correction (H–J).
Unfortunately, fully backcrossing the mice to C57BL/6J resulted in low-level mosaic transgene expression for both isoforms with a consistent, albeit diminished phenotype (Figures S3K–S3M and S4A–S4E). Instead of tet-off, an attempt to rescue expression using backcrossed tet-on mice also led to poor transgene expres-sion, suggesting an intrinsic issue with the operon in β cells of C57BL/6J mice (Figures S4F–S4P). TaBaSCo-GTP female mice had significantly increased fasting serum insulin concentrations compared to TaBaSCo-ATP mice (Figure S4M), but no differences in fasting serum glucose were observed between the two isoforms (Figures S4G and S4L). Although TaBaSCo-GTP males had significantly increased glucose tolerance during an intraperitoneal glucose tolerance test (GTT), serum insulin was not comparatively increased, whereas the opposite was true for females (Figures S4I, S4J, S4N, and S4O). Furthermore, isolated islets from female tet-on TaBaSCo-ATP mice treated with +DOX showed decreased GSIS relative to untreated islets (Figure S4P), which is consistent with the isolated islet GSIS data from pre-backcrossed mice. Weak, mosaic expression dampened the phenotype in congenic mice and limited additional in vivo studies. Nevertheless, together with the findings from the cell lines and pre-backcrossed mice, these studies support a role for the mtGTP-generating pathway in modulating insulin secretion and glucose homeostasis in vivo.
Inner Mitochondrial Membrane GTP Permeability Enhances Insulin Secretion
Unlike mammalian cells, yeast do not express the GTP-specific isoform of SCS. Consequentially, they have a GTP/GDP exchanger (GGC1) in the inner mitochondrial membrane (Vozza et al., 2004) that exchanges cytosolic GTP for mtGDP (Gordon et al., 2006; Przybyla-Zawislak et al., 1998). Therefore, we evaluated whether xenotopic expression of GGC1 in INS-1 cells could enhance insulin secretion by promoting the mitochondrial uptake of GTP, thereby increasing mtGTP, as does hSCS-GTP overexpression (Figure 3A). To assess this possibility, clonal INS-1 832/13 cells expressing yeast GGC1 tagged with a C-terminal GFP were generated. Expression of GGC1 and its GFP tag mRNA and GFP-tagged GGC1 protein in the mitochondria were confirmed (Figures 3B and 3D). GTP/GDP exchange transport by GGC1 was confirmed using GDH oxidation of NADH as a reporter. GDH is allosterically inhibited by mtGTP, so decreased Vmax of GDH activity correlates with increased mtGTP concentration. GGC1 overexpressing cells had decreased GDH Vmax compared to control INS-1 cells, indicating increased GTP transport into the mitochondria by GGC1 (Figure 3E). GGC1 enhanced basal insulin secretion and GSIS compared to controls (Figure 3F). Since hSCS-ATP cells have diminished mtGTP synthesis, GGC1 was expressed in these clonal cell lines to determine whether GGC1 can increase insulin secretion by restoring mtGTP levels (Figure 3G). GGC1 expression in constitutive hSCS-ATP cells (Figures 3H–3J) increased both basal insulin secretion and GSIS in spite of increased hSCS-ATP expression (Figure 3K). These data suggest that increased mtGTP concentration is sufficient to enhance insulin secretion via a mechanism that is independent of SCS-GTP activity.
Figure 3. The yeast GTP/GDP Exchanger Increases mtGTP Permeability and Insulin Secretion.

(A–C) Expression of yeast GGC1, a GTP/GDP exchanger, exchanges cytosolic GTP down its concentration gradient for matrix GDP. mtGTP is also generated by endogenous SCS-GTP (red circle). PEPCK-M uses mtGTP to metabolize anaplerotic OAA into cataplerotic PEP (A). mRNA of GFP (B) and GGC1 (C) from cells transiently transfected with GGC1-GFP versus empty vector (control).
(D) Western blot of anti-GFP and anti-voltage-dependent anion channel (VDAC) as loading control in whole-cell lysates, mitochondrial (mito), and cytosolic (cyto) fractions of INS-1 cells transfected with GGC1-GFP (GGC1+) or empty vector (control).
(E) NADH oxidation via GDH in isolated mitochondria as a function of GTP concentration.
(F) Insulin secretion from INS-1 cells overexpressing control (empty vector), mtGFP, and GGC1 after 45 min of exposure to basal 3 mM (gray bars) or stimulatory 15 mM (black bars) glucose.
(G) Mitochondria from cells overexpressing hSCS-ATP (blue circles) have decreased mtGTP, making GGC1 the principal source of matrix GTP.
(H–J) mRNA of (H) GFP, (I) GGC1, and (J) hSCS-ATP from cells transiently transfected with GGC1-GFP versus empty vector (control).
(K) Insulin secretion from constitutive hSCS-ATP cells (solid bars) and hSCS-ATP cells transfected with empty vector (blue striped) or GGC1-GFP vector (red striped) after 45 min exposure to basal 2.5 mM or stimulatory 16.7 mM glucose.
The data are means ± SEMs (for B, C, and H–J: n = 2 per group; for E: n = 7; for F: n = 6 per group; for K: basal glucose hSCS-ATP n = 6, control hSCS- ATP n = 25, GGC1 hSCS-ATP n = 6, stimulatory glucose hSCS-ATP n = 6, control hSCS- ATP n = 23, GGC1 hSCS-ATP n = 5; technical replicates for all panels). Statistical significance is *p < 0.05, ***p < 0.001, and ****p < 0.0001 by unpaired two-tailed t test (B, C, and E), two-way ANOVA with Tukey’s multiple comparisons test (F and K), one-way ANOVA with Tukey’s multiple comparisons test (H and J), and one-way ANOVA with Holm-Sidak’s multiple comparisons test (I).
mtGTP Enhances Insulin Biosynthesis and Content
Continuous insulin hypersecretion has been implicated in the demise of β cells, a phenomenon referred to as ‘‘β cell exhaustion.’’ Since basal insulin secretion and GSIS are both augmented by increased mtGTP synthesis, the significant increased demand for insulin could exceed biosynthetic capacity leading to b cell exhaustion. Insulin protein and mRNA levels were significantly increased in hSCS-GTP cells (Figures 4A and 4B) despite continuously higher insulin secretion. Conversely, although insulin mRNA levels were slightly elevated, hSCS-ATP cells had decreased insulin protein. Notably, hSCS-GTP cells had increased insulin granule number, size, and proximity to the plasma membrane visualized by transmission electron microscopy (TEM) (Figures 4C–4F and S5). These findings suggest that increased mtGTP synthesis not only increases insulin secretion and sensitivity to glucose but also synergizes insulin production and vesicle localization with the metabolic advantage of higher mtGTP turnover.
Figure 4. Insulin Content, Synthesis, and Granule Localization in Constitutive hSCS-ATP and hSCS-GTP Cells.

(A and B) Total insulin protein content (A) and insulin I and II mRNA (B) from control, hSCS-ATP, and hSCS-GTP constitutive cell lines.
(C) TEM images of control, hSCS-ATP, and hSCS-GTP cells. The arrows identify the insulin granules. Scale bar, 2 μm.
(D) Ratio of area of all insulin granules relative to cytoplasmic cross-sections for each cell line.
(E and F) Average size (E) and distance (F) from the plasma membrane of insulin granules. Control is the parental INS-1 cell line.
The data are means ± SEMs (for A: n = 4 per group; for B: n = 6 per group; for D: number of insulin granules counted toward area for ratio to cytoplasmic area was control n = 243, hSCS-ATP n = 94, hSCS-GTP n = 241; for E and F: control n = 243, hSCS- ATP n = 94, hSCS-GTP n = 241; technical replicates for A and B; replicates are individual insulin granules for D–F). Statistical significance is *p < 0.05 and ****p < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test (A, E, and F) and two-way ANOVA with Tukey’s multiple comparisons test (B).
hSCS-GTP Cells Maintain Metabolic Fitness Despite Increased Secretory Burden
Overworked b cells exhibit increased redox stress and endoplasmic reticulum (ER) stress, among other complications, leading to functional impairment and eventually apoptosis (Swisa et al., 2017; Vetere et al., 2014). Furthermore, loss of glucose responsiveness with chronic exposure to glucolipotoxicity (GLT) has been associated with de-differentiation away from a mature β cell phenotype (Kim and Yoon, 2011; Weir and Bonner-Weir, 2004). Therefore, we hypothesized that the burden of increased insulin synthesis and secretion would injure hSCS-GTP cells. hSCS-GTP cells had a 20% higher proliferation rate than hSCS-ATP cells, despite higher secretory demands (Figure 5A). mRNA levels of key β cell maturity markers such as insulin, PDX1, MafB, and FOXO1 were also elevated in hSCS-GTP cells relative to hSCS-ATP cells (Figures 5B and 5C). mRNA expression levels of PK isoform M2 (PK-M2), the principal PK isoform in β cells that has active and inactive states, was decreased in hSCS-GTP cells (Ashizawa et al., 1991; MacDonald and Chang, 1985; Figure 5B). There was a perhaps compensatory increase of PEPCK-M mRNA in hSCS-GTP cells (Figure 5D), suggesting a functional link to mtGTP (Figure S1A).
Figure 5. Constitutive hSCS-GTP Expression Promotes Cellular Growth, Differentiation, and Health.

(A) Cell proliferation assessed by bromodeoxyuridine (BrdU) incorporation in control, hSCS-ATP, and hSCS-GTP cells.
(B) Heatmap of mRNA expression (ΔΔCt fold change relative to average ΔCt) for indicated gene (n = 6 per group).
(C and D) Individual mRNA expression of (C) cellular differentiation markers and (D) PEPCK-M under normal culture conditions.
(E and F) Effect of hyperglycemia (20 mM glucose) and glucolipotoxicity (GLT; 20 mM glucose + 0.3 mM palmitate and 0.6 mM oleate [P/O]) on the ratio of endogenous rat SCS-GTPβ:SCS-ATPβ mRNA expression (E) and PEPCK-M mRNA expression in INS-1 cells cultured overnight under indicated conditions (n = 4 per group; F). (G) Basal (2.5 mM glucose) and stimulated (16.7 mM glucose) insulin secretion from indicated cell lines cultured overnight in standard media or GLT (20 mM glucose and 0.4 mM palmitate) conditions.
(H) Change in mRNA expression in response to GLT exposure overnight. Gene expression is relative to the control cells grown at 2.5 mM glucose overnight. All of the mRNA data were normalized to actin. The data are means ± SEMs (for A: control n = 20, hSCS-ATP n = 9, hSCS-GTP n = 9; for B: n = 6 per group; for C: n = 6 per group; for D: n = 6 per group; for E: n = 4 per group; for F: n = 4 per group; for G: n = 6 for all treatment groups, except n = 5 for hSCS-ATP 16.7 mM glucose + GLT; for H: n = 4 per group). Significance is *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by Brown-Forsythe and Welch’s one-way ANOVA with Games-Howell’s multiple comparisons test (A), Holm-Sidak’s multiple two-tailed t tests (C), unpaired two-tailed t-test (D), one-way ANOVA with Tukey’s multiple comparisons test (E and F), two-way ANOVA with Tukey’s multiple comparisons test (G), and two-way ANOVA with Dunnett’s multiple comparisons test (H).
GLT impairs islet function, decreases insulin content, and induces β cell apoptosis (Poitout and Robertson, 2008). Exposing hSCS-GTP cells to GLT conditions could reveal underlying metabolic fragility, rendering these cells incapable of handling metabolic stress. Cells were exposed to high glucose with palmitate, or palmitate and oleate, overnight. In control cells, GLT led to an increase in the mRNA expression of endogenous SCS-GTP relative to SCS-ATP, but diminished PEPCK-M expression and impaired GSIS (Figures 5E and 5F). GLT further impaired the already dampened GSIS response from hSCS-ATP cells, while hSCS-GTP cells sustained strong insulin secretion that was comparable to that of healthy control cells (Figure 5G). This resilience was concurrent with mRNA levels of insulin, Nkx6.1, and PEPCK-M, which were better maintained in hSCS-GTP cells exposed to GLT than in control cells (Figure 5H). The ratio of endogenous SCS-GTP to SCS-ATP mRNA expression was also higher in hSCS-GTP cells, suggesting that increasing SCS-GTP relative to total SCS could be a protective adaptation against GLT in β cells. These unexpected observations suggest that mtGTP promotes metabolic fitness, survival, differentiation, and protection from GLT.
mtGTP Shapes Mitochondria
Confocal images of cells transiently expressing hSCS-GTP (Figure S1E) had longer, more fused mitochondria. Similarly, hSCS-GTP mitochondria visualized by TEM were more elongated than controls and more fragmented in hSCS-ATP cells (Figure 6A). Relative to hSCS-ATP cells, hSCS-GTP mitochondria were ~30% larger and the mitochondria:cytoplasmic cross-sectional area was 25% greater in hSCS-GTP cells (Figures 6A, 6B, and S6A). hSCS-GTP mitochondria had a larger aspect ratio and form factor, both indicative of longer, more interconnected mitochondria (Figures 6C and 6D). At high and low glucose concentrations, hSCS-GTP cells exhibited a 50% decrease in the mRNA of outer mitochondrial membrane fusion protein mitofusin-2 (MFN-2; Figure S6B). Conversely, mRNA transcript levels of mitochondrial fission protein DRP-1 were increased by 28% in hSCS-GTP cells relative to hSCS-ATP cells exposed to hyperglycemia. No differences in OPA-1 (the inner mitochondrial membrane fusion protein) mRNA were observed. Changes in mitochondrial mass, morphology, and expression of genes regulating mitochondrial fission and fusion suggest that mtGTP may adaptively modulate mitochondrial dynamics.
Figure 6. Expression of hSCS-GTP Modifies Mitochondrial Morphology and Metabolic Regulation of Insulin Secretion.

(A) Representative TEM images of indicated constitutive expression cell lines grown under normal culture conditions. Scale bar, 1 μm.
(B) Average size of mitochondrion for each cell line.
(C) Average aspect ratio of mitochondria analyzed via TEM imaging in each cell line.
(D) Form factor values for the cell lines.
(E) Mitochondrial oxygen consumption rate (OCR) (antimycin and rotenone subtracted) measured using the Agilent Seahorse ‘‘mito stress test’’ protocol following glucose stimulation (16.7 mM glucose), oligomycin (oligo), and FCCP in indicated constitutive overexpression cell lines. ATP = (OCRglucose – OCRantimycin/rotenone) – OCRoligomycin; Leak = OCRoligomycin – OCRantimycin/rotenone.
(F–J) Simultaneous monitoring of insulin secretion (F), OCR (G), NAD(P)H (H), Ca2+ (I), and extracellular lactate (J) from perifusion of hSCS-ATP and hSCS-GTP cells at basal and upon exposure to high glucose (16.7 mM) or amino acids (10 mM L-leucine, 4 mM L-glutamine) (n = 5).
(K) Insulin secretion in response to 2.5 mM glucose (G2.5) and indicated stimuli (BCH, 2- aminobicyclo[2,2,1]heptane-2-carboxylic acid; SAME, succinate methyl ester, propionate) (n = 6 per group).
(L) Insulin secretion in response to incubations with G2.5, 9 mM glucose (G9.0), and G9.0 + pharmacological agents (TBT, tolbutamide; Ex-4, exendin-4; GKa, glucokinase activator).
(M) Insulin secretion in response to 2.5 mM glucose (G2.5) 10 mM L-arginine (L-Arg), 10 mM L-leucine (L-Leu), 10 mM L-pyruvate (L-Pyr), and 10 mM ketoiso-caproate (α-KIC) (n = 6 per group).
The data are means ± SEMs (for B: control n = 299, hSCS-ATP n = 322, hSCS-GTP n = 232; for C: control n = 448, hSCS-ATP n = 440, hSCS-GTP n = 324; for D: control n = 43, hSCS-ATP n = 29, hSCS-GTP n = 35; for E: control and hSCS-GTP n = 8 per group, hSCS- ATP n = 7 per group; for F and H–J: n = 3 separate experiments; for G: n = 5 per group; for K–M: n = 6 per group; technical replicates for all panels). Statistical significance is *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test (B–D), two-way ANOVA with Tukey’s multiple comparisons test (E), multiple two-tailed t tests with the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli (F–H), Holm-Sidak’s multiple two-tailed t tests (I and J), and two-way ANOVA with Dunnett’s multiple comparisons test (K–M).
Real-Time mtGTP-Regulated Metabolism and Insulin Secretion
Previous work silenced SCS-ATP to increase mtGTP, which increased the glucose-stimulated O2 consumption rate (OCR) without increasing mitochondrial ATP synthesis (Kibbey et al., 2007). In the present study, stimulatory glucose increased OCR across all cell lines with no difference in ATP-dependent and -independent respiration (Figures 6E and S6C). Despite the large difference in insulin secretion at high glucose, maximal respiration capacity (Trifluoromethoxy carbonylcyanide phenylhydrazone [FCCP] triggered OCR) was not significantly increased in hSCS-GTP cells and only slightly, albeit significantly, decreased in hSCS-ATP cells (Figure 6E). The extracellular acidification rate (ECAR), a measurement that integrates multiple features of metabolism with net extracellular proton transport, more than doubled with glucose stimulation in control and hSCS-ATP cells, but increased an additional 10% in hSCS-GTP cells (Figures S6D and S6E). Unlike primary β cells, insulinoma cells express the monocarboxylate transporter that co-transports lactate with a proton. In addition to lactate, CO2 generated from the pentose phosphate pathway (PPP) or from mitochondrial oxidation also contributes to ECAR. Strong suppression of the electron transport chain (ETC) by antimycin and rotenone indicates that mitochondria-generated bicarbonate accounts for 70% of ECAR (Figure S6F), and the small additional increase in ECAR seen in hSCS-GTP could be attributed to either PPP or lactate.
To better assess dynamic mtGTP-dependent coordination of cellular and mitochondrial metabolism with insulin secretion, hSCS-ATP and hSCS-GTP cells were perifused during simultaneous measurement of insulin secretion, O2 consumption, NAD(P)H autofluorescence, cytosolic calcium (Ca2+), and lactate release (Figures 6F–6J). Basal, first- and second-phase responses to glucose and L-glutamine and L-leucine were highest in hSCS-GTP cells, while hSCS-ATP cells had a weak response to glucose and were unresponsive to the two amino acids (Figure 6F). Both hSCS-GTP and hSCS-ATP cells had a similar OCR with high glucose or amino acid exposure (Figure 6G), ruling out any major differences in oxidative metabolism. With glucose treatment, hSCS-GTP cells had lower mitochondrial membrane potential and cellular NAD(P)H autofluorescence (Figures 6H and S6G). In hSCS-ATP cells, NAD(P)H levels were higher with stimulatory glucose treatment but slightly lower with amino acid treatment compared to hSCS-GTP cells (Figure 6H).
The amino acid and GSIS response paralleled cytosolic Ca2+ (Figure 6I). While glucose did trigger a short-lived, first-phase Ca2+ response in hSCS-ATP cells, this increase was not sustained and was not triggered by amino acids, correlating with the pattern of impaired insulin secretion observed in these cells. Ca2+ response was robust in the hSCS-GTP cells, matching the insulin secretion profile observed in these cells. Lactate production, indicative of PEP synthesis from glycolysis and/or PEPCK-M, was 2-fold greater in hSCS-GTP cells and consistent with the ECAR results (Figure 6J). This also indicates greater PK activity, which would lower cytosolic ADP to support Ca2+-dependent insulin release.
mtGTP Influences Nutrient, Hormonal, and Pharmacological Insulin Secretion
Hydrolysis of mtGTP by PEPCK-M depends on anaplerosis from either PC or GDH. The non-metabolizable compound 2-aminobi-cyclo[2,2,1]heptane-2-carboxylic acid (BCH) activates GDH by binding to its L-leucine allosteric site, increasing insulin secretion by contributing to mitochondrial α-ketoglutarate (Bertrand et al., 2002), and as a result, increasing succinyl-CoA to promote mtGTP synthesis from SCS-GTP. In hSCS-GTP cells, BCH stimulated insulin secretion by an additional 35% relative to control (Figure 6K). Similarly, anaplerotic succinate (SAME) boosted insulin secretion from hSCS-GTP cells by an extra 33% (Figure 6K), potentially by expanding the mitochondrial oxaloacetate (OAA) pool to enhance PEPCK-M flux and/or increased mtGTP cycling (Figure S1A). While anaplerotic propionate activates PC via a propionyl-CoA allosteric mechanism, it inhibits insulin secretion at high concentrations (Perry et al., 2016; Ximenes et al., 2007). Propionate decreased basal hSCS-GTP insulin secretion to control levels (Figure 6K). Since propionate treatment did not decrease insulin secretion from control cells, the effect is specific to a metabolic context in which mtGTP cycling is increased. In contrast to hSCS-GTP cells, none of the tested anaplerotic substrates were able to promote insulin secretion from hSCS-ATP cells.
Sulfonylurea tolbutamide, GLP-1 receptor agonism, and glucokinase activation increased insulin secretion from control cells and further augmented insulin secretion from hSCS-GTP cells (Figure 6L). Of these agents, sulfonylurea and GLP-1 receptor agonism modestly increased GSIS from the hSCS-ATP cells, although these cells had significantly depressed insulin secretion compared with the other two cell lines. The nutrient secretagogues L-arginine, L-leucine, pyruvate, and α-ketoisocaproate (aKIC) enhanced insulin secretion above basal glucose in hSCS-GTP cells relative to control (Figure 6M). Of these, only L-arginine improved insulin secretion in hSCS-ATP to levels comparable to control. This would suggest that mtGTP broadly interacts with both KATP-dependent and -independent mechanisms to influence insulin secretion.
mtGTP Regulates Cataplerosis
Precision central carbon metabolic flux measurements were performed to directly assess the role of mtGTP in anaplerotic and/or cataplerotic metabolism. Glucose-stimulated metabolite concentrations were nearly identical, with the exception of higher pyruvate and lower aspartate and glutamate in hSCS-GTP cells (Figure S7A). Metabolite concentrations are poor indicators of metabolic flux, so quantitative kinetic (ν) and steady-state (ϕ) isotope labeling studies were both performed using mass isoto-pomeric multiordinate spectral analyses (MIMOSA) (Figure S7; Alves et al., 2015). At steady state, the fractional contribution of glucose oxidation to the TCA cycle (VPDH/VCS) was similar in both SCS cell lines (Figure 7A). Consistent with the OCR data, the kinetic analysis showed no difference in citrate synthase velocity (VCS) (Figures 7F and S7B) between cell lines. In fact, there was a small but statistically significant reduction in the estimates of glycolysis (Vglycolysis) and pyruvate dehydrogenase (PDH) flux (Figure 7F) that more likely reflects the higher sensitivity of the dynamic analysis to the larger distribution of acetyl-CoA enrichments (Figure S7C), rather than an actual physiological effect. This assessment is supported by the kinetic modeled VPDH/VCS, which showed no difference between the cell lines (Figure S7D).
Figure 7. Impact of mtGTP Levels on Metabolic Fluxes.

(A–D) Fractional flux through pyruvate dehydrogenase (VPDH; A), pyruvate carboxylase (VPC; B), phosphoenolpyruvate carboxykinase (VPCK; C), and ME (VME; D) obtained from the steady-state analysis of enrichments following incubation with [U-13C6]glucose.
(E) Analysis of VPCK/VME from steady-state data.
(F) Absolute fluxes calculated by the mathematical analysis of the time-dependent accumulation of 13C-label into the glycolytic and mitochondrial intermediates.
(G) Analysis of VPCK/VME from the calculated absolute fluxes.
The data are means ± SEMs, except the modeled data (F and G), which are least-square fit ± SD of the distribution obtained from the Monte Carlo simulations (n = 100). For (A)–(D): n = 6 per group; for (E): hSCS-ATP n = 6, hSCS-GTP n = 5. Statistical significance is *p < 0.05 and **p < 0.005 by unpaired two-tailed t test (A–D) and unpaired two-tailed t test with robust regression and outlier removal (ROUT) test for outliers (E). Statistical analysis of (F) and (G) was performed using a statistical tool built into CWave.
The rates of PC relative to CS flux (VPC/VCS) calculated at (ϕ) steady state (Figure 7B) as well as kinetic (ν) measurement of VPC (Figures 7F and S7E) were similar between cell lines. Thus, contrary to our hypothesis predicting the dependence of PEPCK-M-mediated mtGTP hydrolysis on enhanced PC flux, there was no difference in anaplerosis between the cell lines. VPDH/VCS and VPC/VCS were similar (Figures 7A and 7B) to reported measurements from the parental cell line INS-1 maintained under similar conditions (Alves et al., 2015). PEPCK-M hydrolyzes mtGTP to convert OAA into PEP that cataplerotically exits the TCA. Consistent with this mtGTP reliance, hSCS-GTP cells had a higher fractional PEPCK-M flux (VPCK) (Figures 7C and S7F). Of all of the fluxes measured, VPCK showed the greatest fold increase (Figure 7F), suggesting that mtGTP finely regulates this flux. When compared to independent measurements of malic enzyme (ME) flux (VME; Figures 7D and 7F), mtGTP tripled the ratio of VPCK/VME in both steady-state and kinetic experiments (Figures 7E and 7G–7I). These results suggest that even with unchanged anaplerosis, the principal mitochondrial metabolic change associated with the salutary mtGTP responses is a diver-sion of carbon flux from ME to PEPCK-M (Figures 7E and 7G–7I).
DISCUSSION
The fidelity of glucose sensing is crucial for survival. The uniform concordance between mtGTP synthesis rates and insulin secretion in the multiple in vivo and in vitro models tested confirms a rele-vant and important physiological role for mtGTP signaling. The concept of mtGTP signaling emerged from an inborn error in metabolism affecting b cell function. Specifically, mutations in the GTP-binding domain of GDH associate with hypoglycemia in HI/HA due to insulin hypersecretion and concomitant suppression of counter-regulatory glucagon release (Kibbey et al., 2014). The present study provides additional strong, consistent evidence implicating mtGTP and PEP metabolism in the regulation of insulin secretion. Several different systems were used to toggle mtGTP synthesis rates and help circumvent potential off-target effects (e.g., clonal selection, chronic adaptive responses, variable transfection efficiency). The importance of mtGTP itself (rather than SCS) was validated by xenotopic GGC1 expression that increased the permeability of the mitochondria to GTP. In vivo studies and perifused islet studies from TaBaSCo mice establish the relevance of the mtGTP signal for whole-body physiology as an amplifier and sentinel of β cell glucose sensing.
An unexpected additional observation is that mtGTP appears to provide resilience to metabolic stresses such as GLT and favors a mature, differentiated β cell that includes increased PEPCK-M expression (van der Meulen et al., 2017). Significant secondary adaptive responses in PEPCK-M expression, insulin biosynthesis, and other transcription and metabolic factors will require future mechanistic delineation. ER stress from high insulin biosynthetic demand is proposed to cause b cell failure. Results from the hSCS-GTP cells provide a very optimistic model in which in the context of increased mtGTP synthesis, increased insulin mRNA transcription and biosynthesis co-exist with enhanced secretion, nutrient sensitivity, β cell differentiation, and health. The extent to which this pathway determines β cell differentiation and is responsible for islet dysfunction in the progression toward diabetes remains to be ascertained. Similarly, the mechanisms by which mtGTP may directly or indirectly influence mitochondrial morphology and mass are not clear. While many of the fusion and fission proteins hydrolyze GTP to perform their functions, the GTPase domains of these proteins are located outside the matrix where mtGTP is generated.
Changes in the ATP:ADP ratio have long been correlated with insulin secretion. Mounting evidence implicates other non-oxidative metabolic pathways for this function. These pathways include anaplerosis via PC and GDH; cataplerosis via ME and PEPCK-M; or cytosolic NADPH production via cytosolic ME (ME1), isocitrate dehydrogenase 1 (IDH1), and the PPP (Prentki et al., 2013). Of these, only anaplerosis by GDH generating mtGTP and OAA that supports cataplerotic PEP synthesis by PEPCK-M correlates with the metabolic defect associated with human HI/HA.
The association of this anaplerotic-cataplerotic mtGTP-PEP cycle with physiologic insulin secretion can be observed with other inborn errors of metabolism. For instance, HNF4α (the gene mutated in MODY1) regulates HNF1α (MODY3) to strongly modulate PEPCK-M and PK expression (Pongratz et al., 2009; Servitja et al., 2009). More recently, hyperinsulinemic hypoglycemia was associated with dominant human mutations in UCP2 (Ferrara et al., 2017). The role of UCP2 as a strict proton uncoupler may have initially been overstated, as it can catalyze proton-coupled mitochondrial transport that can deplete matrix OAA in exchange for Pi (Vozza et al., 2014). Although GDP inhibits UCP2 (Berardi and Chou, 2014), no difference in proton leak was noted in the SCS cell lines (Figure 6E). UCP2 loss-of-function could preserve OAA pools for mitochondrial PEP syn-thesis, promoting insulin secretion. Therefore, mutations in GDH, HNF4α, HNF1α, and UCP2 suggest the consequences of a disrupted mtGTP and PEP pathway.
In INS cells, mitochondrial acetyl-CoA is almost entirely of glucose origin (Alves et al., 2015). The similarity between basal and glucose-stimulated OCR in the hSCS-GTP cells argues against OxPhos as a component of the mtGTP-dependent mechanism. Anaplerosis through ME and/or IDH1 and PPP may generate NADPH (Prentki et al., 2013). With the exception of propionate, anaplerotic stimuli enhanced the mtGTP-mediated amplification of insulin secretion. What is surprising is that the mtGTP effect does not correlate with NAD(P)H, PC, or ME. Rather, diversion of anaplerosis from ME into PEPCK-M was important to initiate and sustain the increased cytosolic Ca2+ and insulin release associated with elevated mtGTP synthesis.
Because alterations in neither OxPhos nor NAD(P)H can explain the mtGTP-mediated regulation of insulin secretion, we propose an alternative model based on the requirement of cytosolic ADP lowering to close KATP channels. Mitochondrial metabolism couples the proton motive force (PMF) used by ATP synthase to form the γ-phosphate bond between Pi and ADP. The ATP:ADP ratio reaches a maximum as it approaches the mitochondrial phosphorylation potential. Once this ‘‘state IV’’-like equilibrium is reached, OxPhos cannot further increase the ATP:ADP ratio. It is counterintuitive, then, how PEP synthesis in the mitochondria, which actually consumes two nucleotide triphophates, could actually raise the ATP:ADP ratio further. A potential explanation considers that PEP contains the highest free energy of any biologic phosphoester bond, equivalent to approximately two ATPs because of the spontaneous enol-to-keto tautomerization during pyruvate formation. Since the substrate-level PEP potential exceeds the mitochondrial phosphorylation potential, PK can lower cytosolic ADP further, effectively increasing the ATP:ADP ratio. In this manner, mitochondrial PEP synthesis collaborates with glycolysis to maximize cytosolic ADP lowering via PK. Our stable isotope techniques assessing PK flux only account for pyruvate metabolized by mitochondria and do not measure the fraction of pyruvate diverted into lactate production. Since lactate production rates were higher in hSCS-GTP cells, our measurements likely underestimate mtGTP-dependent PEPCK-M flux.
A mechanism centered on PEP hydrolysis does not exclude contributions from OxPhos, other second messengers (e.g., NADPH), or homeostatic metabolic pathways (e.g., glutathione reduction) (Ferdaoussi et al., 2015; Ivarsson et al., 2005; Prentki et al., 2013). Theoretically, there are advantages that an mtGTP and PEP pathway can confer to metabolic efficiency, as it is coupled to fluctuations in cytosolic metabolism and Ca2+ oscillations (Merrins et al., 2013) and is also localized to KATP channels and the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA), where PEP and/or ADP have known regulatory activ-ities (Ashcroft and Rorsman, 2013; Dhar-Chowdhury et al., 2005; Ho et al., 2015; Xu and Becker, 1998; Zima et al., 2006). PEP has a pivotal position in carbohydrate metabolism, and its levels are controlled by the tightly regulated PK reaction. The importance of PEP is further illustrated by a recent report identifying that PK-dependent PEP metabolism elicits insulin secretion in K+-permeabilized islets independently of OxPhos (Pizarro-Delgado et al., 2016). Given that PC flux is associated with insulin secretion, that PEPCK-M flux accounts for the majority of PC flux, and that PEPCK-M cycling rates are at least as high as 30%–40% of the glycolytic rate (Stark et al., 2009), mtGTP in coordination with PEP is well poised to signal energy levels in β cells. Thus, SCS-generated mtGTP provides a powerful signal that integrates TCA cycle flux and anaplerosis to amplify PEP and regulate insulin secretion. It is a key mediator whose metabolism could be targeted to develop novel therapeutic interventions.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Kibbey (Richard.kibbey@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models & Breeding
C57BL6/J mouse lines transgenic for either a GFP-tagged hSCS-ATP or hSCS-GTP expression cassette were bred with a CD-1 mouse line transgenic for the rat insulin promoter tetracycline transactivator (JAX, NOD.Cg-Tg(Ins2-tTA)1Doi/DoiJ, Stock No: 004937) ‘‘tet-off’’ regulatory cassette (RIP-tTA) to generate doubly hemizygous RIP-tTA/ hSCS-ATP or RIP-tTA/ hSCS-GTP mice, in which pancreatic β-cell specific expression of the respective human SCS subunits is repressed in the presence of doxycycline (Luco et al., 2006; Shockett et al., 1995; Weir et al., 1996). The nomenclature used to describe the double transgenic offspring for the RIP-tTA; SCS-ATP line is ‘‘TaBaSCo-ATP’’ and for the RIP-tTA; SCS-GTP is ‘‘TaBaSCo-GTP.’’ The initial phenotyping experiments were performed in these mice that were on a mixed 50% C57BL6/J, 50% CD-1 background. These mice are referred to as ‘‘pre-backcrossed’’ throughout the manuscript. Subsequent experiments in ‘‘backcrossed’’ mice were performed in mice that were crossed to the C57BL6/J for at least nine generations.
To construct the tet-on mouse models, C57BL/6 mice strains transgenic for either GFP-tagged human SCSATP or SCSGTP expression cassettes were bred with a CD-1 transgenic mouse strain expressing the rat insulin promoter reverse tetracycline trans-activator (RIP-rtTA) to produce the ‘‘tet-on’’ mouse lines, where pancreatic beta-cell specific expression of the respective human SCS β-subunits is induced in the presence of doxycycline. Congenic mice were backcrossed for at least seven generations to C57BL/6. The nomenclature describing the double transgenic offspring for the RIP-rtTA; SCSATP line is referred to as tet-on-’’TaBaSCo-ATP’’ and the RIP-rtTA; SCSGTP is termed tet-on ‘‘TaBaSCo-GTP.’’
All mice were genotyped by using tail genomic DNA isolated via the QIAGEN Blood & Tissue Kit. Mice were genotyped by PCR using genotyping primers (listed in Key Resources Table, Oligonucleotides).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Invitrogen mouse anti-V5 | Thermo Fisher Scientific | Cat# R960–25; RRID: AB_2556564 |
| Mouse anti-beta actin | Abcam | Cat# ab8226; RRID: AB_306371 |
| Rabbit anti-GFP | Abcam | Cat# ab6556; RRID: AB_305564 |
| Goat anti-VDAC (N18) | Santa Cruz Biotechnology | Cat# sc-8828; RRID: AB_793935 |
| Primary rabbit polyclonal antibodies against rodent SCSα (peptide sequence: DAAKKAVASVAKK) | This paper | N/A |
| Primary rabbit polyclonal antibodies against rodent SCS-ATPβ (peptide sequence: KEAHVDVKFQLPI) | This paper | N/A |
| Primary rabbit polyclonal antibodies against rodent SCS-GTPβ (peptide sequence: DAAKKAVASVAKK) | This paper | N/A |
| Guinea pig anti-insulin | Sigma | discontinued |
| secondary Alexa Fluor 555 goat anti-guinea pig IgG | Sigma | Cat # SAB4600297 |
| secondary Alexa Fluor 488 donkey anti-rabbit (H+L) | Abcam | Cat # ab150073; RRID: AB_2636877 |
| Bacterial and Virus Strains | ||
| Invitrogen One Shot® OmniMAX 2-T1R Chemically Competent E. coli | Thermo Fisher Scientific | C854003 |
| Biological Samples | ||
| TaB-ATP pancreatic islets | This paper | N/A |
| TaB-GTP pancreatic islets | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 (Life Technologies) | Thermo Fisher Scientific | 11668019 |
| DMEM-Base Cell culture media | Sigma Aldrich | D5030 |
| RPMI 1640 Cell culture media | Sigma Aldrich | SLM-240 |
| Quantitect reverse transcriptase | QIAGEN | 205311 |
| SYBR PCR reagent | BioRad | 1708880 |
| OptiMEM I | Thermo Fisher Scientific | 31985–062 |
| complete mini EDTA-free protease inhibitor cocktail (Roche) | Sigma Aldrich | 11836170001 |
| Exendin-4 | Tocris | 1933 |
| tolbutamide | Fluka Analytical | 46968 |
| glucokinase activator | Merck | 346021 |
| TMRE | Molecular Probes | T669 |
| MitoTracker Green | Molecular Probes | M7514 |
| [U-13C6]glucose | Cambridge Isotope Laboratories | 110187–42-3 |
| 2H4-taurine | CDN Isotopes | D-1971 |
| Doxycycline Slow Release Pellets | Innovative Research of America | B-168 |
| Critical Commercial Assays | ||
| Micro BCA protein assay kit | Thermo Fisher Scientific | 23235 |
| Rat High Range ELISA | ALPCO | 80-INSRTH-E01 |
| Mouse Ultrasensitive ELISA | ALPCO | 80-INSMSU-E01 |
| RNeasy Mini Kit | QIAGEN | 74106 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69504 |
| BrdU Cell Proliferation Assay | Chemicon International, Millipore | 2750 |
| HIGH range rodent insulin ELISA assay kit (colorimetric) | ALPCO | Discontinued |
| Invitrogen Quant-iT PicoGreen dsDNA Reagent | Thermo Fisher Scientific | P7581 |
| Qiaquick Gel Extraction Kit | QIAGEN | 28704 |
| QIAGEN Plasmid Maxi Kit | QIAGEN | 12163 |
| Experimental Models: Cell Lines | ||
| Clonal INS-1 832/13 cell line overexpressing the human insulin gene (INS-1) | C.B. Newgard (Duke University School of Medicine) | N/A |
| hSCS-ATP constitutive cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human ATP-specific SCSβ subunit) | This paper | N/A |
| hSCS-GTP constitutive cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human GTP-specific SCSβ subunit) | This paper | N/A |
| hSCS-ATP inducible ‘‘tet-on’’ cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human ATP-specific SCSβ subunit and the reverse tet-repressor system (rtTA)) | This paper | N/A |
| hSCS-GTP inducible ‘‘tet-on’’ cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human GTP-specific SCSβ subunit and the reverse tet-repressor system (rtTA)) | This paper | N/A |
| 293FT Cell line (Invitrogen) | Thermo Fisher Scientific | R70007 |
| GGC1 Overexpressing stable cell line | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| ‘‘TaBaSCo-ATP’’ or TaB-ATP mice double transgenic for the RIP-tTA and human SCS-ATP beta-subunit over-expression; tet-off | This paper | N/A |
| ‘‘TaBaSCo-GTP’’ or TaB-GTP mice double transgenic for the RIP-tTA and human SCS-GTP beta-subunit over-expression; tet-off | This paper | N/A |
| ‘‘TaBaSCo-ATP’’ or TaB-ATP mice double transgenic for the RIP-rtTA and human SCS-ATP beta-subunit over-expression; tet-on | This paper | N/A |
| ‘‘TaBaSCo-GTP’’ or TaB-GTP mice double transgenic for the RIP-rtTA and human SCS-GTP beta-subunit over-expression; tet-on | This paper | N/A |
| RIP-tTA Mouse line (tet-off) | The Jackson Laboratory | NOD.Cg-Tg(Ins2-tTA)1Doi/DoiJ, Stock No: 004937 |
| RIP-rtTA Mouse Line (tet-on) | The Jackson Laboratory | Tg(Ins2-rtTA)2Efr/J Stock No: 008250 |
| Oligonucleotides | ||
| Genotyping primer tTA FWD: AAC AAC CCG TAA ACT CGCC | This paper | N/A |
| Genotyping primer tTA REV: AAA TCT TGC CAG CTT TCC | This paper | N/A |
| Genotyping primer human SCSATP FWD: GCA AGA AGC TGG TGT CTC CGT TCC | This paper | N/A |
| Genotyping primer human SCSATP REV: GCT TGA CCA TGT TTT CTG CTG C | This paper | N/A |
| Genotyping primer GFP FWD: GCA CGA CTT CTT CAA GTC CGC CAT GCC | This paper | N/A |
| Genotyping primer GFP REV: GCG GAT CTT GAA GTT CAC CTT GAT GCC | This paper | N/A |
| RT-qPCR Primers (Listed in Table S1) | This paper | N/A |
| Recombinant DNA | ||
| ViraPower T-Rex Lentiviral Expression System (Invitrogen) | Thermo Fisher Scientific | Thermo Fisher Scientific |
| Transient GFP-tagged human SCS-ATP beta-subunit overexpression vector pEXP-hSCSATP | This paper | This paper |
| Transient GFP-tagged human SCS-ATP beta-subunit overexpression vector pEXP-hSCSGTP | This paper | N/A |
| Transient yeast GFP-tagged GGC1 overexpression vector pEXP-GGC1 | This paper | N/A |
| Transient mitochondrial GFP overexpression vector pEXP-mtGFP | This paper | N/A |
| PLHCX lentiviral vector for overexpression of GGC1 with C-terminal GFP tag | This paper | N/A |
| PLHCX lentiviral vector for overexpression GFP | This paper | N/A |
| Human SCS-GTPβ (NCBI sequence NM_003848.2) | Origene | SC317700 |
| and human SCS-ATPβ (NCBI sequence NM_003850.1) | Origene | SC117714 |
| vector pET21b-YHM1 expressing GGC1 (NCBI Sequence NM_001180258) | Debkumar Pain laboratory | N/A |
| Retro-X Universal Expression System | Clontech | 631530 |
| Software and Algorithms | ||
| MetaMorph Image Analysis Software | Molecular Devices | N/A |
| iTEM imaging software | EMSIS GmbH | N/A |
| ImageJ software | (NIH; https://imagej.nih.gov/ij/) | N/A |
| Metabolic modeling was performed using CWave software, version 4.0 (Mason et al., 2003) running in MATLAB (Natick, MA, USA) version R2017b with OS × version 10.13.6. | Alves et al., 2015 | N/A |
| Monte-Carlo analysis with 100 repetitions | Patel et al., 2010 | N/A |
| El-Maven and Polly MS Processing Platforms | Elucidata, Cambridge MA | N/A |
| GraphPad Prism 7 software | GraphPad Software | N/A |
| BD CellQuest Pro FACS Analysis Software | BD Biosciences | N/A |
| Other | ||
| Seahorse Bioscience XF24 Analyzer | Agilent | N/A |
| FEI Tecnai transmission electron microscope | ThermoFisher Scientific | N/A |
| FlexStation 3 | Molecular Devices | N/A |
| Olympus Morada CCD camera | Olympus Life Science | N/A |
| Beckman Glucose Analyzer II | Beckman Instruments, Fullerton, CA | N/A |
| Bio-Rep Perifusion Instrument | Bio-Rep | N/A |
| Olympus fluorescent microscope | Olympus Life Science | N/A |
| SCIEX 5500 QTRAP equipped with a SelexION for differential mobility separation (DMS) | SCIEX | N/A |
| Hypercarb column (3 μm particle size, 3×150 mm) | Thermo Fisher Scientific | N/A |
| BD FACSCalibur Flow Cytometer | BD Biosciences | N/A |
For experiments where doxycycline treatment (DOX) was used, mice between 8–12 weeks of age were administered DOX for 2 weeks via a subcutaneous slow release pellet (35 mg/pellet for 21 days; Innovative Research of America, Sarasota, FL) or via the drinking water at 1.125 g doxycycline/L supplemented with 37.5% sucrose to circumvent dosing and dehydration issues associated with taste aversion for doxycycline water. Water was provided in light-protected bottles and changed every 4 days.
All studies were conducted in mice aged 8–15 weeks; males or females were studies as indicated in the individual figure legends corresponding to the data from that study.
INS-1 832/13 Clonal Cell Line
Initial stocks of the clonal INS-1 832/13 cell line overexpressing the human insulin gene were a generous gift from C.B. Newgard (Duke University School of Medicine) (Hohmeier et al., 2000). These cells were cultured as monolayers in RPMI-1640 supplemented with 11.1 mM D-glucose, 10% (v/v) fetal bovine serum, 10,000 units/mL penicillin and 10 mg/mL streptomycin, 10 mM HEPES, 2 mM L-glutamine (GlutaMAX, Invitrogen), 1 mM sodium pyruvate, and 60 mM sodium bicarbonate. Cells were incubated at 37°C in 5% CO2, 95% air. All experiments used cells harvested between passages 10 and 40.
Constitutive hSCS Overexpression Cell Lines
Clonal insulinoma cell lines were generated with overexpression of either the hSCS-ATP or hSCS-GTP tagged with the V5 epitope and driven by a CMV promoter under the influence of the tet operon (Figure 1B) that is constitutively active in the absence of the tet-repressor. INS-1 832/13 cells were transduced to overexpress either the human ATP or GTP-specific SCSβ subunit using the ViraPower T-Rex Lentiviral Expression System (Invitrogen). pLenti4/TO/V5-DEST vectors expressing the human SCS-ATP sequence (GenBank: NM_003850.1) or the human SCS-GTP sequence (GenBank: NM_003848.2) with a C-terminal V5 tag were constructed. 293FT cells at passage 4 were transfected with either of these constructs and the ViraPower Packaging Mix (Invitrogen) using Lip-ofectamine 2000 (Life Technologies) in OptiMEM I with serum to produce lentivirus. Virus-containing media was collected 72 hours post-transfection. INS-1 832/13 cells at passage 10 were then transduced with the human SCS-ATPb or human SCS-GTPβ lentivirus. Positive colonies were selected by resistance to 50 mg/mL Zeocin (Invitrogen) and expanded in RPMI-1640 media supplemented with 10% tetracycline-free FBS. To obtain tetracycline-regulated expression of SCSβ (inducible tet-on system), the INS-1 832/13 cells were co-transduced with either of the human SCSβ lentiviruses and the tetracycline-repressor (TR) lentivirus. Positive colonies were selected using 50 mg/mL Zeocin (Invitrogen) and 5 mg/mL Blasticidin (Invitrogen). Expression of human SCSβ from the constitutive cell lines and the inducible tet-on cell lines was determined by western blotting for the V5 tag and qPCR to amplify the human SCS-ATP and -GTP sequences.
Inducible hSCS Overexpression Cell Lines
INS-1 832/13 cells were transfected with a reverse tet-repressor system (rtTA) expressing human SCS-ATP (GenBank: NM_003850.1) or human SCS-GTP (GenBank: NM_003848.2) with a C-terminal V5 tag. Prior to use in the cell culture assays, doxy-cycline hydrochloride, a tetracycline derivative (Sigma) was added to the culture media at a concentration of 0.2 ug/ml (this concentration is referred to as DOX) for 24 hours followed by a 48 hour wash out period. Positive clone screening for each transgenic line was initially performed via Dot-Blot for expression of the V5 tag in the presence of DOX. Subsequent confirmation of positive expression of the transgene and DOX response was performed by RT-qPCR for V5 tag and hSCS, and western blotting for the V5 tag.
METHOD DETAILS
RT-qPCR
Total RNA was extracted using RNeasy with RNase-Free DNase (QIAGEN) from 6-well plates or batches of 15–20 medium sized islets following 2 days of transfection. Reverse transcriptase reactions were performed separately on a PTC-100 Thermocycler (BioRad, Hercules, CA) using Quantitect reverse transcriptase (QIAGEN) prior to real time quantitative PCR analysis on an Opticon 2 DNA En-gine (Applied Bioscience) using SYBR PCR reagent (BioRad). All reactions confirmed a single product of the expected size by agarose gel electrophoresis. Reaction efficiencies for actin, SCS-ATP and SCS-GTP (rat and human) were greater than 1.90. Δ SCS isoform mRNA levels were expressed as percent of actin (%Actin = ESCS C(t)SCS /EActin C(t)Actin*100) or as the ΔΔ C(t) normalized to actin, where E is efficiency of the steady state reaction and C(t) is the cycle threshold. Primers were synthesized at the Yale School of Medicine HHMI/IDT facility (Table S1).
Western Blotting
Cells were lysed in RIPA lysis buffer (1% NP-40, 0.15 M NaCl, 0.01 M sodium phosphate, 1 mM EDTA) containing complete mini EDTA-free protease inhibitor cocktail (Roche). Lysate protein concentration was measured using Micro BCA protein assay kit (23235, Thermo Fisher Scientific).
30 μg of total protein lysate was loaded on a 4%–12% or 12% Tris-glycine gel (BioRad) and transferred to a PVDF membrane (Immobilon-P 0.45 mm, BioRad). Antibodies used for western blotting were supplied by the following vendors: Invitrogen mouse anti-V5 (R960–25, Thermo Fisher Scientific), mouse anti-beta actin (ab8226, Abcam), rabbit anti-GFP (ab6556, Abcam), and goat anti-VDAC (N18) (sc-8828, Santa Cruz Biotechnology). Primary rabbit polyclonal antibodies were raised (Invitrogen) that were capable of recognizing rodent epitopes for SCSα (peptide sequence: DAAKKAVASVAKK), SCS-ATPβ (peptide sequence: KEAHVDVKFQLPI), and SCS-GTPβ (peptide sequence: DAAKKAVASVAKK).
hSCS Transient Overexpression Vectors
Expression vectors for transient overexpression of the hSCS-GTP and hSCS-ATP β-subunits and a mitochondria-targeted GFP in cells were constructed using the Gateway Clonase II System by Invitrogen. Human SCS-GTPb (GenBank: NM_003848.2, cDNA clone obtained from Origene, Cat. No. SC317700) and human SCS-ATPβ (GenBank: NM_003850.1, cDNA clone obtained from Origene, Cat. No. SC117714) were PCR amplified with primers containing 5′ and 3′ attB recombination sites. Primer sequences are:
attB_SCSGTP_fwd
5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGGCGTCCCCCGTAGCAGCGCAG-3′, attB_SCSGTP_rev
5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTTCTTGGCCACACTGGCCAC-3′, attB_SCSATP_fwd
5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGGCGGCCTCCATG-3′, attB_SCSATP_rev
5′- GGGGACCACTTTGTACAAGAAAGCTGGGTCTATTGGCAACTGAAA-3′
where primer sequence in italics corresponds to the attB recombination sites. Resulting PCR products were gel-purified using Qiaquick Gel Extraction Kit (QIAGEN) and recombined into donor vector pDONR 221 (Invitrogen) using BP Clonase II enzyme mix, a combination of bacteriophage λ Integrase (Int) and E. coli Integration Host Factor (IHF) that catalyze strand exchanges between the attB sites of the amplified cDNA with the attP sites of the donor vector to produce entry vectors pENTR-SCS-ATP and pENTR-SCS-GTP. A 1 µL aliquot of the recombination reaction was used to transform 50 µL of One Shot® OmniMAX 2-T1R Chem-ically Competent E. coli (Invitrogen) that was plated on 50 μg/mL kanamycin LB-agar. Colonies were then propagated in selective media and entry vectors were isolated using QIAGEN Plasmid Maxi Kit. Purified entry vectors were then recombined with destination vector Vivid Colors pcDNA6.2/C-EmGFP-DEST Gateway® Vector (Invitrogen) using LR Clonase II enzyme mix consisting of bacteriophage λ Int and Excisionase (Xis), as well as E. coli IHF, that insert the gene sequence flanked by the recombination sites on the entry vector between the sites on the destination vector in frame with a C-terminal GFP sequence. 1 μL of this recombination reaction was used to transform 50 μL of One Shot® OmniMAX E. coli that was plated on 100 μg/mL ampicillin LB-agar. Bacterial colonies were grown and final expression vectors pEXP-hSCSATP and pEXP-hSCSGTP were isolated and sequenced by GeneWiz, Inc. To synthesize the control mitochondria-targeted GFP vector, the mitochondria-targeting sequence was amplified from pEYFP-mito (BD Biosciences Clontech, cat. no. 6115–1) using primers attB-mitoF 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAACCATGGATGTCCGTCCTGACGCCG-3′ and attB-mitoR 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCGATCTTGGCGCGCGGCAC-3′. The resulting PCR product was recombined to finally produce pEXP-mitoGFP as described. Cells transfected with pEXP-hSCSATP are referred to as hSCS-ATP; cells transfected with pEXP-hSCSGTP are hSCS-GTP cells and cells transfected with pEXP-mitoGFP are MitoGFP control in the manuscript.
GGC1 Overexpression
The vector pET21b-YHM1 expressing GGC1 (NCBI Sequence NM_001180258) was a generous gift from the Debkumar Pain laboratory. The GGC1 gene was amplified from this vector template using primers containing attB viral integration sites. The PCR product was recombined into a pDONR vector to produce an entry vector using the BP reaction (Invitrogen; Gateway Clonase II system). The pENTR-GGC1 was recombined with the pDEST vector to obtain an expression vector pEXP-GGC1 expressing GGC1 with a C-terminal GFP fusion tag. INS-1 832/13 cells were transfected with the pEXP-GGC1 vector, which was validated by sequencing and confirmation of clones positive for the complete GGC1 gene in frame with the GFP tag.
GGC1 overexpressing cells were made using the Retro-X Universal Expression System (Clontech; #631530). Briefly, GP2–293 cells (HEK293 based packaging cell line) were co-transfected with the a PLHCX retroviral expression vector containing the GFP tagged GGC1 expression cassette, and the pEco (ecotropic) packaging vector containing the viral envelope gene for infection. Viruses were harvested from cells and used to transform INS-1 cells to produce the stable GGC1 overexpressing cell line. These cells were used to confirm insulin secretion studies and to generate the data for mitochondrial GTP concentration (measured indirectly by GDH assay). Proper vector construction was validated by the sequencing (Genewiz, Inc.) of clones for the complete GGC1 gene in frame with the GFP tag. Vector function was further confirmed by western blot analysis for expression of the GFP protein and by RT-qPCR to detect GFP and GGC1 mRNA transcripts.
Vector Transfection
INS-1 832/13 cells or the constitutive hSCS cell lines were transfected with the various transient expression vectors as indicated using Lipofectamine 2000 (Life Technologies) in OptiMEM I with serum. Ratio of vector DNA to lipofectamine was determined as recommended by the product guidelines. Successful transfection and expression of vector in cells was assessed by RT-qPCR and western blotting as shown in the corresponding data for the studies described.
Mitochondrial Isolation
Cells from four 10 cm2 dishes per condition were washed with PBS prior to being scraped in ice-cold mitochondrial isolation buffer (65 mM sucrose, 215 mM mannitol, 5 mM KH2PO4, 5 mM KHCO3, 3 mM MgCl2, 5 mM HEPES [pH 7.4]). Cells were centrifuged at 800 rcf. for 5 min. After centrifugation, the supernatant was aspirated and the remaining pellet was resuspended in 1 mL of ice cold 1X mitochondrial buffer, transferred to a 2 mL glass dounce homogenizer and passed 50 times with a type B pestle. Lysates were centrifuged for 3.5 min at 1,800 rcf., and the supernatant was centrifuged at 8,500 rcf. for an additional 5 min. The mitochondria-enriched pellet was again resuspended in isolation buffer.
SCS Enzyme Assay
Cells were transfected with either pEXP-hSCSATP, pEXP-hSCSGTP or pEXP-mitoGFP control overexpression constructs in 6-well culture plates 24 hours prior to the assay. 200 μL of extraction buffer (20 nM KPO4, 0.4% CHAPS, pH 7.3) containing complete mini EDTA-free protease inhibitor cocktail (Roche) was added to each well and cells were scraped into eppendorfs. Cell samples were centrifuged at 2000 rpm at 4°C for 5 min to remove precipitate. Cell lysates were assayed immediately. 10 μL cell lysate and 165 μL assay buffer (50 mM KPO4, 10 mM MgCl2, pH 7.3) were added per well in a clear flat-bottom 96-well plate (BD Falcon). Reaction was started by adding 75 μL reagent mixture (1.05 mM DTNB, 1.07 mM succinyl-coA in assay buffer) containing 5.33 mM ADP to measure SCS-ATP activity, or 5.33 mM GDP to measure SCS-GTP activity. SCS enzyme activity was measured as NTB2− release from the reaction of DTNB with the sulfhydryl group of the coenzyme A released upon synthesis of succinate by SCS-ATP or SCS-GTP. NTB2− levels were measured by absorbance at 405 nm after a 300 s reaction time using Spectramax, and normalized to total lysate protein. Reactions were performed at room temperature.
GTP Uptake Assay
The principle of this assay was to use endogenous matrix GDH activity as a sensor of mitochondrial GTP transport. GTP is an allosteric inhibitor of the GDH reaction so increased permeability to GTP/GDP exchange would be expected to produce a crossover response (no exchange, or uptake, means low matrix GTP and more GDH activity while high exchange means high matrix GTP and decreased GDH activity). Mitochondria were isolated from cells transfected with control vector, and cells transfected with pEXP-GGC1. In a 96-well plate containing reaction buffer (45 mM TE, 1.9 mM EDTA, 87.5 mM NH4OAc, 87.5 mM ADP, 175 mM NADPH, 9 mM alpha-ketoglutarate) GTP was added in the following log concentrations [mM]: 1, 0, −1, −1.301, −1.62, −2, −2.301, −2.602, −3, −4, −5. The plate background was read at λ 340/540 nm on a Spectramax. 25 μL of isolated mitochondrial preparation or 10 μL of GDH enzyme (USB) (positive control) were subsequently added, and NADH oxidation reaction rate was read at λ 340/540 for 1 hour. Vmax was determined from NADH oxidation at each GTP concentration and normalized to protein concentration.
Insulin Secretion Assays
GSIS assays were performed by preincubating cells in DMEM-base (Sigma) supplemented with 2.5 or 3 mM glucose and 22 mM NaHCO3 for 1.5 h followed by a 45 min incubation in DMEM-base with either 2.5 or 3 mM glucose for basal secretion and either 15 or 16.7 mM glucose for stimulated secretion as indicated. Media samples were centrifuged at 1800 rcf. for 5 min at 4°C, of which 200 μL supernatant was collected to assay for insulin concentration using the Rat High Range ELISA (80-INSRTH-E01, ALPCO). Cells were washed in 1x PBS and lysed in 1 mL 0.1% Triton X-100. Insulin levels were normalized to total protein measured by Micro BCA protein assay kit (23235, Thermo Fisher Scientific).
Glucolipotoxicity (GLT) was evaluated in cells incubated overnight in RPMI media containing 0.4 mM palmitate and 20 mM glucose, or 0.4 mM palmitate, 0.6 mM oleate (P/O) and 20 mM glucose. BCH (10 mM), SAME (10 mM), propionate (10 mM) were added acutely to cells in RPMI media containing 2.5 mM glucose and insulin secretion was measured after 2h incubation. Exendin-4 (Tocris, Cat. No. 1933) was added acutely to INS-1 832/13 cells at 10 nM, tolbutamide (Fluka Analytical, Cat. No. 46968) at 100 μM and glucokinase activator (Merck, Cat. No. 346021) at 100 nM in RPMI media containing 2.5 mM glucose and insulin secretion was measured after 2h incubation. 10 mM L-Arginine (L-Arg), 10 mM L-leucine (L-Leu), 10 mM L-Pyruvate (L-Pyr) and 10 mM α-ketoisocaproate (α-KIC) were also added acutely in RPMI media containing 2.5 mM glucose and insulin secretion was measured after 2h incubation.
Cell Proliferation Rate
Constitutively Hscsβ-overexpressing clones and parental INS-1 832/13 cells were seeded in clear, flat-bottomed 96-well Corning plates at 1 × 105 cells/mL media, 100 µL per well. Wells containing cell culture media supernatant without cells were used as blanks, and cells grown without BrdU were used to calculate background. Multiplication rates were determined by total BrdU incorporation as detected by an anti-BrdU antibody and goat anti-mouse IgG peroxidase conjugate secondary using the BrdU Cell Proliferation Assay (Chemicon International, Millipore). BrdU levels were measured by absorption at 450/550 nm dual wavelength on Flexstation 3.
Cell Perifusion
Perifusion studies that simultaneously quantified insulin secretion, lactate production and OCR, or imaged NAD(P)H or cytosolic Ca2+ in INS-1 832/13 and constitutively expressing hSCS-ATP or hSCS-GTP cells were performed by the laboratory of Ian Sweet as previously described (Gilbert et al., 2008; Sweet et al., 2002). For imaging, INS-1 cells were harvested and plated on coverslips the day before the experiment. NAD(P)H levels were measured by autofluorescence using an excitation wavelength of 360 nm and reading emission at 460 nm. To normalize relative fluorescence units (RFU), steady state RFU was determined at the end of each experiment by adding potassium cyanide (KCN) followed by FCCP. Normalized NAD(P)H fluorescence was calculated as described previously (Gilbert et al., 2008). Cytosolic Ca2+ was measured via Fura-2 AM fluorescence (Invitrogen) after cells were treated with 2 µm Fura-2 AM and 0.02% pluronic acid for 40 min at 37°C. For measurements of insulin secretion, lactate production and oxygen consumption, the cells were harvested (about 5 million/chamber) and loaded in the perifusion chambers the same day as the experiment. OCR was determined by measuring oxygen tension in the inflow and outflow perfusate by electrode; insulin secretion was measured using insulin ELISA (Mercodia) and lactate production enzymatically using lactate dehydrogenase and measurement of NADH fluorescence (Sweet et al., 2002).
Oxygen Consumption & Extracellular Acidification
INS-1 cells were plated in 24-well Seahorse XF24 cell culture microplates in growth media prior to each study on the Seahorse Bioscience XF24 Analyzer. Cells were pre-incubated in assay media (DMEM base with 2.5 mM glucose, and no sodium bicarbonate, no serum) for 1 hour prior to studies. All reagents were diluted in assay media and loaded into the ports of the flux plate (port A: 16.7 mM glucose; port B: 5 µM oligomycin; port C: 1 µM FCCP; port D: 5 µM antimycin A and 10 µM rotenone.
TMRE/MitoTracker Green Imaging
INS-1 cells constitutively overexpressing the human SCS-ATP and SCS-GTP β-subunit isoforms were plated on MatTek Glass Bottom Culture Dishes (Part No.: P35G-0–10-C) and, following overnight incubation, were stained with the mitochondrial membrane-potential dye TMRE (15 nM; Molecular Probes, T669) and MitoTracker Green (MTG; 200 nM; Molecular Probes, M7514) and incubated for 1 hr. The cells were then washed 3x with media (RPMI-1640; Catalog #: 31800–022) containing TMRE but not MTG. Subsequently, live-cell imaging was performed on the mitochondria using a Zeiss LSM 710 Confocal Microscope. Relative mitochondrial membrane potential was quantified by taking the ratio of TMRE (574 nm) to MTG (510 nm) fluorescence intensity. Mitochondrial morphological parameters (e.g., form factor and aspect ratio) were assessed with MetaMorph Image Analysis Software (Molecular Devices).
Electron microscopy
The cultured cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), then post-fixed in 1% OsO4 in the same cacodylate buffer at room temperature for 1 hour. After staining en bloc with 2% aqueous uranyl acetate for 30 min, cells were dehydrated in a graded series of ethanol to 100% and finally embedded in EMbed 812 resin. Blocks were then polymerized in a 60°C oven for 24 hr. Thin sections (60 nm) were cut by a Leica ultramicrotome and post-stained with 2% uranyl acetate and lead citrate. Sample grids were examined with a FEI Tecnai transmission electron microscope at 80 kV of accelerating voltage, and digital images were recorded with an Olympus Morada CCD camera and iTEM imaging software (EMSIS GmbH).
TEM Images Analysis
By using transmission electron microscopy (TEM) we imaged the constitutive cell lines to evaluate various structural properties of mitochondria and insulin granules. TEM images were captured for the INS-1 832/13 and constitutively-expressing hSCS-GTP and hSCS-ATP cell lines. Since imaging is limited to the field of view and not the entire cell population, we captured 40 images/group, which allowed us to examine large numbers of mitochondria and insulin granules to extrapolate our findings to the true population. Furthermore, to reduce bias, the acquisition of images was randomized. Given that these images were to be subjected to comparative analysis between each cell line, it was important to determine that they were indeed comparable. Therefore, we determined the total number of slides used for analysis and the cytoplasmic area captured. Since all images do not contain mitochondria and insulin granules, these calculations were performed separately for the mitochondrial and insulin granule analyses. TEM images were analyzed using ImageJ software (NIH; https://imagej.nih.gov/ij/) with the scale set to 600.01 pixels/mm.
Mouse Metabolic Phenotyping
Glucose tolerance tests were performed after an overnight fast. Mice were injected intraperitoneally with 1 g/kg glucose, and blood was collected by tail bleed at 0, 15, 30, 45, 60, 90, and 120 min for plasma insulin and glucose measurements. Plasma glucose concentrations were measured using the glucose oxidase method on a Beckman Glucose Analyzer II (Beckman Instruments, Fullerton, CA). Serum insulin was measured via Mouse Ultrasensitive ELISA (80-INSMSU-E01, ALPCO). Fasting serum glucose and insulin concentrations were also measured using the same methods.
Isolated Pancreatic Islet Perifusions
Pancreata were excised from anesthetized mice and islets were isolated by collagenase P digestion followed by centrifugation with Histopaque 1100 solution (made by combining Histopaque 1077 (10771, Sigma) and Histopaque 1119 (11191, Sigma) at a ratio of 1:1.2) for density separation of pure islets. Islets were then hand-picked under a light microscope using a flame smoothed glass pipette to ensure that the islets were free of visible exocrine contamination (Kibbey et al., 2014). Islets were allowed to recover for 48 h. When necessary, doxycycline (5 µg/mL) treatment was done during this recovery time period and is indicated in the manuscript when used. Approximately 50–80 islets were then layered between a slurry of acrylamide gel column beads (Bio-Gel P4G; BioRad 150–412) and perifusion buffer DMEM (D-5030, Sigma) prepared as per the manufacturer’s instructions and supplemented with 2.5 mM glucose, 10 mM HEPES, 2 mM glutamine and 0.2% fatty acid free BSA. The islets were perifused at 100 µl/min for a 1hr equilibration period using a Bio-Rep Perifusion Instrument (Miami, FL, USA) that maintains precise temperature, gas (5% CO2/95% air) and flow control. After the stabilization period, the islets were perifused with 2.5 mM glucose for 10 minutes followed by stimulatory 16.7 mM glucose for 45 minutes. During the perifusion, outflow was collected into a 96-well plate format and secreted insulin concentrations were measured by a high range rodent insulin ELISA assay kit (ALPCO; this specific colorimetric kit detecting both rat and mouse insulin is discontinued) and normalized to islet DNA which was measured using a Invitrogen Quant-iT PicoGreen dsDNA Reagent (P7581, Fisher Scientific).
Immunofluorescent Staining
Whole islets were isolated, as described above – see Method Details section on ‘‘isolated pancreatic islet perifusions’’ – fixed with 4% paraformaldehyde, mounted on paraffin embedded slides by the Yale Pathology Tissue Services, and later de-paraffinized in a staining chamber at 56°C overnight. The following day the slides were removed and subjected to the following series of washes: Histoclear (Thermo Fisher Scientific) for 15 min, Histoclear for 15 min, 100% ethanol for 10 min, 95% ethanol for 10 min, 70% ethanol for 10 min, and water for 5 min. Antigen retrieval was performed by placing samples in a pressure cooker with 7 mM citrate buffer, pH 6, for 1 minute. Slides were then soaked in 1x PBS for 5 minutes and the blocked with 2% BSA in 1x PBS for 30 minutes. After blocking, the slides were incubated overnight at 4°C with primary antibodies at a 1:1000 dilution in 2% BSA in 1x PBS. After overnight incubation, the slides were washed 3x for 10 min with 1x PBS, and then incubated for 30 min at room temperature with the fluorescent secondary antibody (1:1000). Coverslips were mounted using Vectashield Mounting Media (Vector Labs) and slides were imaged at 200x using the Olympus BX50 microscope equipped with a Diagnostic Instruments, Inc model: 8.2 color mosiac digital camera.
Primary antibodies used were guinea pig anti-insulin (Sigma; discontinued) with secondary Alexa Fluor 555 goat anti-guinea pig IgG (SAB4600297, Sigma) and primary rabbit anti-GFP (ab6556, Abcam) with secondary Alexa Fluor 488 donkey anti-rabbit (H+L) (ab150073, Abcam).
FACS Flow Cytometry
Isolated islets from TaB-ATP and TaB-GTP mice backcrossed at least nine generations to C57BL/6 were first dispersed, centrifuged at 250 × g and filtered through 40 µm cell strainers. Cells were diluted to 1x106 - 5x106 cells/mL. Cells were fixed for 15 min at room temperature with 4% paraformaldehyde in 1x PBS. Cells were then permeabilized by incubating in 0.3% Triton X-100 in 1x PBS for 15 min. Cells were then blocked for 30 min with 10% BSA in PBS. Cells were incubated with 1:200 primary antibody guinea pig anti-insulin (Sigma; discontinued) in 3% BSA in 1x PBS overnight at 4°C. Cells were then washed three times with 1x PBS, and incubated in 1:200 secondary goat Alexa 555 conjugated anti-guinea pig antibody (Sigma; SAB4600297) in 3% BSA in 1x PBS for 1 hour at room temperature. Cells were washed three more times with 1x PBS and re-suspended in ice cold ~1x PBS at 1x106 cells/mL. Cells were sorted on a BD FACSCalibur Flow Cytometer, and data analysis was performed on BD CellQuest Pro Analysis software. Flow rate was set at a few hundred cells per second; instruments settings were FSC: voltage E-1, amplitude gain 7.04, linear mode; SSC: voltage 333, amplitude gain 1.49, linear mode; FL1: voltage 638, logarithmic mode; FL2: voltage 491, logarithmic mode. Compen-sation was set for FL1 at 0.8% FL2, and for FL2 at 31.8% FL1.
13C-Isotopic Labeling
SCS-ATP and SCS-GTP cells were cultured as monolayers in RPMI-1640 complete medium as previously described (Kibbey et al., 2007). 13C-Isotopic labeling studies were performed in DMEM medium (D5030, Sigma-Aldrich) supplemented with glucose (9mM), glutamine (4mM), pyruvate (0.05mM) and lactate (0.45mM). Cells were preincubated in this media for 2h until a metabolic steady state was reached at which time unlabeled glucose was replaced with [U-13C6]glucose (110187–42-3, Cambridge Isotope Laboratories). Following the addition of 13C-label, cells were quenched at t = 0, 5, 15, 30, 60, 120 and 240min (n = 6 per time point). Cell quenching involved a rapid wash with ice-cold 1x PBS and metabolite extraction in 150 µL of an ice-cold solution containing 20% methanol, 0.1% formic acid, 1mM phenylalanine, 3mM NaF, 100uM EDTA and 10 µM 2H4-taurine (CDN Isotopes, D-1971) as a load control. All the samples were lyophilized and resuspended in 50 µL of water prior the LC-MS/MS analysis.
LC-MS/MS Analysis
Metabolite concentrations and 13C-enrichments were determined by mass spectrometry using a SCIEX 5500 QTRAP equipped with a SelexION for differential mobility separation (DMS). Samples were injected onto a Hypercarb column (3 µm particle size, 3x150 mm, Thermo Fisher Scientific) at a flow rate of 1 mL/min. Metabolites were eluted with a combination of aqueous (A: 15mM ammonium formate and 10uM EDTA) and organic mobile phase (B: 60% acetonitrile, 35% isopropanol and 15mM ammonium formate) according to the following gradient: t = 0min, B = 0%; t = 0.5min, B = 0%, t = 1min, B = 40%; t = 1.5min, B = 40%; t = 2min, B = 0%; t = 6min, B = 0%. Metabolite detection was based on multiple reaction monitoring (MRM) in negative mode using the following source parameters: CUR: 30, CAD: high, IS: –1500, TEM: 625, GS1: 50 and GS2: 55. DMS parameters were DT: low, MD: 2-propanol, MDC: low, DMO: 3 and DR: off, while Separation Voltage (SV) and Compensation Voltage (CoV) were optimized individually for each metabolite in order to maximize signal intensity and isobar resolution. The individual MRM transition pairs (Q1/Q3) are listed in Table S2.
Retention times were confirmed with known standards and peaks integrated using El-Maven (Elucidata). The atomic percent excess (APE) was calculated using Polly™ interface (Elucidata) and corrected for background noise and for natural abundance. Endogenous taurine, an intracellular osmolyte, was used as internal control for cell density as previously validated (Kibbey et al., 2007).
13C-Isotopomer Analysis
The calculation of citrate and OAA isotopomers was performed using the MRM Q1/Q3 = 191/111. This MRM detects the citrate fragments resulting from the loss of carbon C1 or C5. The deconvolution of citrate isotopomers followed the same principles of citrate symmetry, citrate families and exchange through isocitrate dehydrogenase as previously described (Alves et al., 2015). The comparison of isotopomer enrichments using Q1/Q3 = 191/111 or Q1/Q3 = 191/67 is shown in Figure S7. The poor S/N obtained from Q1/Q3 = 191/67 supported the choice of Q1/Q3 = 191/111 for the analysis of citrate enrichments.
Steady-State Flux Ratio
VPDH/VCS and VPC/VCS were calculated as previously described (Alves et al., 2015). The fractional fluxes through VPCK and VME were calculated according to Equations 1 and 2, respectively.
| (Eq. 1) |
| (2) |
The derivation of the equations for steady-state flux ratios is as follows:
Derivation of Equation 1
While the fractional enrichment of [M+3] PEP,f[M+3]PEP, can be genereted from glycolysis and mitochondrial reactions, f[M+2]PEP only arises from the latter. The mathemetical representation of the variation of f[M+2]PEP is:
Where fMalatePrecursor is the sum of the malate enrichments capable of generating [M+2]PEP:
By definition, at the steady – state : , which leads to :
Rearranging this equation reveals:
Derivation of Equation 2
The principles applied in the derivation of Equation 1 can also be used to study the variation of f[M+2]Pyruvate such that:
By definition, at steady state: and VPK = VGlycolysis + VPCK · In addition, anaplerosis must equal cataplerosis in orter to maintain the concentration of the TCA cycle intermediates constant. Therefore, in the absence of a significant source of glutamate - driven anaplerosis VPC = VPCK + VME · A consequence of this relationship and the law of mass balance is that VGlycalysis = VPDH · After substituting all these terms:
Rearranging this equation reveals:
Modeling of 13C-labeled time courses
The integrated analysis of 13C labeling time courses was performed using a mathematical model of the TCA cycle similar to the one described previously (Alves et al., 2015). The model describes the transfer of label through the distal portion of glycolysis and the TCA cycle using [U-13C3]DHAP as a driving function. The label is distributed through all possible isotopomers for PEP, pyruvate, citrate, αKG, glutamate, succinate, malate and OAA and it is used to measure the flux through glycolysis, pyruvate kinase, pyruvate dehydrogenase, pyruvate carboxylase, citrate synthase, reversed isocitrate dehydrogenase and β-oxidation. Isotopomers were grouped in combination pools based on the number and/or position of labeled carbons and used to fit target data. The quality of fits is shown in Figure S7. Metabolic modeling was performed using CWave software, version 4.0 (Mason et al., 2003) running in MATLAB (Natick, MA, USA) version R2017b with OS × version 10.13.6. The distributions of uncertainty were calculated using a Monte-Carlo analysis with 100 repetitions (Patel et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are reported as the mean ± SEM. Comparisons between groups were made using unpaired, two-tailed Student t tests. For comparison between two groups with multiple time points multiple t tests were used, and one- or two-way ANOVA was used for comparison between three or more groups when appropriate and as indicated. For the glucose tolerance tests and serum insulin data, t tests were used to discern differences between the area under the curve (AUC) values. p < 0.05 was considered significant. P value ranges, exact statistical tests used and n-values per group are all reported in the main and supplementary figure legends. No statistical tests were used to pre-determine sample sizes, though the sample sizes used are comparable to what is used in the field. Statistical analyses were performed using GraphPad Prism 7 software.
Supplementary Material
Highlights.
Enhanced mitochondrial GTP (mtGTP) production amplifies insulin secretion
mtGTP regulates PEPCK-M and cytosolic Ca2+, not OxPhos, to trigger insulin secretion
Exuberant secretion and enhanced insulin biosynthesis can coexist without toxicity
mtGTP turnover enhances insulin content, granule docking, and mitochondrial mass
ACKNOWLEDGMENTS
This work was supported by US Public Health Service grants R01 DK-092606, R01 DK-110181, and K08 DK-080142 (to R.G.K.), and Clinical and Translational Science Awards (CTSA) UL 1 RR-0024139, Diabetes Research Centers (DRC) P30 DK-045735, Mouse Metabolic Phenotyping Center (MMPC) U24 DK-059635, and the DRC Cell Functional Analysis Core DK-17047 (University of Washington). We would like to thank Dr. Gerald Shulman (Yale University) for assistance with rodent phenotyping and technical, instrumental, and moral support; Dr. Xiao-Bing Gao (Yale University) for histologic analysis of hypothalamic tissues; Dr. Debkumar Pain (University of Medicine and Dentistry of New Jersey) for the GGC1 construct; and Dr. Matthew Merrins (University of Wisconsin) for critically reading the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.058.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
The modeling of the LC-MS/MS datasets in C-Wave are available from the corresponding author on request.
REFERENCES
- Alves TC, Pongratz RL, Zhao X, Yarborough O, Sereda S, Shirihai O, Cline GW, Mason G, and Kibbey RG (2015). Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab 22, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, and Rorsman P (2013). K(ATP) channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol 9, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa K, Willingham MC, Liang CM, and Cheng SY (1991). In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem 266, 16842–16846. [PubMed] [Google Scholar]
- Berardi MJ, and Chou JJ (2014). Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab 20, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand G, Ishiyama N, Nenquin M, Ravier MA, and Henquin JC (2002). The elevation of glutamate content and the amplification of insulin secretion in glucose-stimulated pancreatic islets are not causally related. J. Biol. Chem 277, 32883–32891. [DOI] [PubMed] [Google Scholar]
- Cline GW, Lepine RL, Papas KK, Kibbey RG, and Shulman GI (2004). 13C NMR isotopomer analysis of anaplerotic pathways in INS-1 cells. J. Biol. Chem 279, 44370–44375. [DOI] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, and Coetzee WA (2005). The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J. Biol. Chem 280, 38464–38470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drahota Z, Rauchová H, Miková M, Kaul P, and Bass A (1983). Phospho-enolpyruvate shuttle–transport of energy from mitochondria to cytosol. FEBS Lett 157, 347–349. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional b cells. J. Clin. Invest 125, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara CT, Boodhansingh KE, Paradies E, Fiermonte G, Steinkrauss LJ, Topor LS, Quintos JB, Ganguly A, De Leon DD, Palmieri F, and Stanley CA (2017). Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab 102, 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert M, Jung SR, Reed BJ, and Sweet IR (2008). Islet oxygen consumption and insulin secretion tightly coupled to calcium derived from L-type calcium channels but not from the endoplasmic reticulum. J. Biol. Chem 283, 24334–24342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DM, Lyver ER, Lesuisse E, Dancis A, and Pain D (2006). GTP in the mitochondrial matrix plays a crucial role in organellar iron homoeostasis. Biochem. J 400, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, and Newgard CB (2000). Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430. [DOI] [PubMed] [Google Scholar]
- Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, in ‘t Veld P, Renström E, and Schuit FC (2005). Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54, 2132–2142. [DOI] [PubMed] [Google Scholar]
- Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, and Shulman GI (2007). Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab 5, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbey RG, Choi CS, Lee HY, Cabrera O, Pongratz RL, Zhao X, Birkenfeld AL, Li C, Berggren PO, Stanley C, and Shulman GI (2014). Mitochondrial GTP insensitivity contributes to hypoglycemia in hyperinsulinemia hyperammonemia by inhibiting glucagon release. Diabetes 63, 4218–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, and Yoon KH (2011). Glucolipotoxicity in Pancreatic β-Cells. Diabetes Metab. J 35, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A (2001). Adenine and guanine nucleotide-specific succinyl-CoA synthetases in the clonal beta-cell mitochondria: implications in the beta-cell high-energy phosphate metabolism in relation to physiological insulin secretion. Diabetologia 44, 89–94. [DOI] [PubMed] [Google Scholar]
- Lu D, Mulder H, Zhao P, Burgess SC, Jensen MV, Kamzolova S, Newgard CB, and Sherry AD (2002). 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc. Natl. Acad. Sci. USA 99, 2708–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Maestro MA, del Pozo N, Philbrick WM, de la Ossa PP, and Ferrer J (2006). A conditional model reveals that induction of hepatocyte nuclear factor-1alpha in Hnf1alpha-null mutant beta-cells can activate silenced genes postnatally, whereas overexpression is deleterious. Diabetes 55, 2202–2211. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, and Chang CM (1985). Pancreatic islets contain the M2 isoenzyme of pyruvate kinase. Its phosphorylation has no effect on enzyme activity. Mol. Cell. Biochem 68, 115–120. [DOI] [PubMed] [Google Scholar]
- MacMullen C, Fang J, Hsu BY, Kelly A, de Lonlay-Debeney P, Saudubray JM, Ganguly A, Smith TJ, and Stanley CA; Hyperinsulinism/hyperammonemia Contributing Investigators (2001). Hyperinsulinism/hyperammonemia syndrome in children with regulatory mutations in the inhibitory guanosine triphosphate-binding domain of glutamate dehydrogenase. J. Clin. Endocrinol. Metab 86, 1782–1787. [DOI] [PubMed] [Google Scholar]
- Mason GF, Falk Petersen K, de Graaf RA, Kanamatsu T, Otsuki T, Shul-man GI, and Rothman DL (2003). A comparison of (13)C NMR measurements of the rates of glutamine synthesis and the tricarboxylic acid cycle during oral and intravenous administration of [1-(13)C]glucose. Brain Res. Brain Res. Protoc 10, 181–190. [DOI] [PubMed] [Google Scholar]
- McKee EE, Bentley AT, Smith RM Jr., and Ciaccio CE (1999). Origin of guanine nucleotides in isolated heart mitochondria. Biochem. Biophys. Res. Commun 257, 466–472. [DOI] [PubMed] [Google Scholar]
- McKee EE, Bentley AT, Smith RM Jr., Kraas JR, and Ciaccio CE (2000). Guanine nucleotide transport by atractyloside-sensitive and -insensitive carriers in isolated heart mitochondria. Am. J. Physiol. Cell Physiol 279, C1870–C1879. [DOI] [PubMed] [Google Scholar]
- Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA, and Satin LS (2013). Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 activity. J. Biol. Chem 288, 33312–33322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Scheetz MH, Drusano GL, and Lodise TP (2010). Identification of optimal renal dosage adjustments for traditional and extended-infusion piperacillin-tazobactam dosing regimens in hospitalized patients. Antimicrob. Agents Chemother 54, 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Borders CB, Cline GW, Zhang XM, Alves TC, Petersen KF, Rothman DL, Kibbey RG, and Shulman GI (2016). Propionate Increases Hepatic Pyruvate Cycling and Anaplerosis and Alters Mitochondrial Metabolism. J. Biol. Chem 291, 12161–12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Delgado J, Deeney JT, Corkey BE, and Tamarit-Rodriguez J (2016). Direct Stimulation of Islet Insulin Secretion by Glycolytic and Mitochondrial Metabolites in KCl-Depolarized Islets. PLoS One 11, e0166111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitout V, and Robertson RP (2008). Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr. Rev 29, 351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongratz RL, Kibbey RG, Kirkpatrick CL, Zhao X, Pontoglio M, Yaniv M, Wollheim CB, Shulman GI, and Cline GW (2009). Mitochondrial dysfunction contributes to impaired insulin secretion in INS-1 cells with dominant-negative mutations of HNF-1alpha and in HNF-1alpha-deficient islets. J. Biol. Chem 284, 16808–16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, and Madiraju SR (2013). Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18, 162–185. [DOI] [PubMed] [Google Scholar]
- Przybyla-Zawislak B, Dennis RA, Zakharkin SO, and McCammon MT (1998). Genes of succinyl-CoA ligase from Saccharomyces cerevisiae. Eur. J. Biochem 258, 736–743. [DOI] [PubMed] [Google Scholar]
- Servitja JM, Pignatelli M, Maestro MA, Cardalda C, Boj SF, Lozano J, Blanco E, Lafuente A, McCarthy MI, Sumoy L, et al. (2009). Hnf1alpha (MODY3) controls tissue-specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol. Cell. Biol 29, 2945–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockett P, Difilippantonio M, Hellman N, and Schatz DG (1995). A modified tetracycline-regulated system provides autoregulatory, inducible gene expression in cultured cells and transgenic mice. Proc. Natl. Acad. Sci. USA 92, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Kibbey RG (2014). The mitochondrial isoform of phosphoenol-pyruvate carboxykinase (PEPCK-M) and glucose homeostasis: has it been overlooked? Biochim. Biophys. Acta 1840, 1313–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shul-man GI, and Kibbey RG (2009). Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem 284, 26578–26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet IR, Cook DL, Wiseman RW, Greenbaum CJ, Lernmark A, Matsumoto S, Teague JC, and Krohn KA (2002). Dynamic perifusion to maintain and assess isolated pancreatic islets. Diabetes Technol. Ther 4, 67–76. [DOI] [PubMed] [Google Scholar]
- Swisa A, Glaser B, and Dor Y (2017). Metabolic Stress and Compromised Identity of Pancreatic Beta Cells. Front. Genet 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, Liu S, Ackermann AM, Caceres E, Hunter AE, et al. (2017). Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab 25, 911–926.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetere A, Choudhary A, Burns SM, and Wagner BK (2014). Targeting the pancreatic beta-cell to treat diabetes. Nat. Rev. Drug Discov 13, 278–289. [DOI] [PubMed] [Google Scholar]
- Vozza A, Blanco E, Palmieri L, and Palmieri F (2004). Identification of the mitochondrial GTP/GDP transporter in Saccharomyces cerevisiae. J. Biol. Chem 279, 20850–20857. [DOI] [PubMed] [Google Scholar]
- Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, et al. (2014). UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 111, 960–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, and Bonner-Weir S (2004). Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53 (Suppl 3), S16–S21. [DOI] [PubMed] [Google Scholar]
- Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, and Broadus AE (1996). Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA 93, 10240–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ximenes HM, Hirata AE, Rocha MS, Curi R, and Carpinelli AR (2007). Propionate inhibits glucose-induced insulin secretion in isolated rat pancreatic islets. Cell Biochem. Funct 25, 173–178. [DOI] [PubMed] [Google Scholar]
- Xu KY, and Becker LC (1998). Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum vesticles. J. Histochem. Cytochem 46, 419–427. [DOI] [PubMed] [Google Scholar]
- Zima AV, Kockskamper J, and Blatter LA (2006). Cytosolic energy reserves determine the effect of glycolytic sugar phosphates on sarcoplasmic reticulum Ca2+ release in cat ventricular myocytes. J. Physiol 577, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.