

Abstract
The associative memory, water-mediated, structure and energy model (AWSEM) has been successfully used to study protein folding, binding, and aggregation problems. In this work, we introduce AWSEM-IDP, a new AWSEM branch for simulating intrinsically disordered proteins (IDPs), where the weights of the potentials determining secondary structure formation have been finely tuned, and a novel potential is introduced that helps to precisely control both the average extent of protein chain collapse and the chain’s fluctuations in size. AWSEM-IDP can efficiently sample large conformational spaces, while retaining sufficient molecular accuracy to realistically model proteins. We applied this new model to two IDPs, demonstrating that AWSEM-IDP can reasonably well reproduce higher-resolution reference data, thus providing the foundation for a transferable IDP force field. Finally, we used thermodynamic perturbation theory to show that, in general, the conformational ensembles of IDPs are highly sensitive to fine-tuning of force field parameters.
Graphical Abstract
1. INTRODUCTION
Intrinsically disordered proteins (IDPs) and structured proteins containing intrinsically disordered regions (IDRs) are ubiquitously found in the proteomes of higher organisms. These elements carry out a variety of critical biological functions despite their structurally disordered nature.1–7 Extensive progress in investigating IDPs in the past 20 years has highlighted the limits of the classical fixed structure−function paradigm of molecular biology,8 suggesting a number of new mechanisms, including “coupled folding and binding” along with others.9–14 While many studies treat IDPs as conformational ensembles having no well-defined secondary and tertiary structures, it has more and more been recognized that certain types of structural order are in fact encoded in the overall disordered state.15–20 Hence, numerous experimental studies, employing solution-based techniques, such as nuclear magnetic resonance (NMR)21 and small-angle X-ray scattering (SAXS),12 have investigated various classes of IDPs. Generally, these methods provide only ensemble averaged measurements, although more recently complete distributions of structural variables have become available through single-molecule observations based on Förster resonance energy transfer (FRET).22 Nevertheless, it remains challenging to capture experimentally the detailed structural dynamics of IDPs. To address this blind spot, and also help guide further experiments, computational approaches have come to play an increasingly important role, in illuminating the molecular nature of IDPs’ conformational ensembles.16,17,23,24
Different computational methodologies have been used to study IDPs, including models that require specific experimental inputs as well as de novo molecular dynamics (MD) simulations at the all-atom level which do not use experimental data.25–27 Methods such as the energy-minima mapping and weighting method (EMW),28 ASTEROIDS,21 and the ENSEMBLE program29 usually take experimental constraints to generate a best-fitted IDP ensemble through back-calculations from specific experiments on each system. These methods almost exclusively rely on fitting the experimental data, using only relatively simple energy functions to describe the chain. In contrast, traditional atomistic MD simulations attempt to model IDPs at the same high structural resolution employed in developing X-ray crystal structures. De novo approaches provide the possibility of discovering new conformations of completely unexpected types. Yet, obtaining adequate statistical sampling of the configuration space of IDPs by means of purely atomistic MD simulations is quite challenging, requiring very long runs at high computational expense. These studies have also already given the impression that atomistic force field inaccuracies are significant with regard to modeling IDPs.30–33 Coarse-grained (CG) models, on the other hand, replace atomistic details with a coarser description that can be more rapidly simulated. By having a higher computational efficiency, coarse-grained models dramatically broaden the exploration of the conformational space of IDPs and IDRs.34–40 Most of the earlier CG models used for IDP simulations have employed generic polymer physics approaches and have been neither systematically benchmarked against experiments reporting on the properties of the structural ensembles sampled by IDPs nor, alternatively, benchmarked against comprehensive atomistic simulations. Therefore, it is desirable to develop a transferable CG force field for IDPs that aims to reproduce the salient structural features of IDPs, namely, the extended geometry of the chain and the nature of their conformational disorder, in particular, by taking into account sequence-specific effects.
We might ask: Why has it been so hard to model “intrinsically disordered proteins”? There are two fundamental reasons arising from their statistical physics:
First, “intrinsic disorder” implies, by its definition, large fluctuations in structure. If one were to use thermodynamic perturbation theory to treat any errors in individual terms in the force field as perturbations on the perfect model, one would see that the sensitivity of any average structural feature to small errors in the potential directly depends on the correlated fluctuations in the unperturbed ensemble of those structural features to be monitored and the energetic error terms (which are themselves structural variables!). Thus, ipso facto, intrinsic disorder with its large structural fluctuations then implies there will be high sensitivity of the average structure of an IDP to modeling errors.
Second, sequence disordered polymers exhibit many phase transitions that all meet near multiple points in the phase diagram: collapse, folding, liquid crystal order, etc.41 Making a small error in the force field that fails to locate properly which part of the diagram the molecule actually is in thus has big consequences. This difficulty is unlike what happens for simulations of well-ordered globular proteins where it can be assumed, at the start, that the system is weakly fluctuating in the fully ordered part of the phase diagram and thus in the most insensitive part of the phase diagram! Simulations can be started in near native conformations, biasing the system to fluctuate less for example. Remember that Pauling was led to the main themes of secondary structure in well-ordered proteins without even considering such important forces as hydrophobicity! His force field was poor, but his structures were excellent.
At the same time that we see that the structure of an intrinsically disordered protein must be sensitive to the details of the force field owing to the big fluctuations of IDPs, likewise thermodynamic perturbation theory by the same token also suggests that the thermodynamic consequences of making these structural errors are small because the system is soft! In other words, there will be entropy/energy compensation. In this way we see that very often less than perfect structural simulations will still get the global mechanism right due to compensating contributions. Biology and thermodynamics can forgive modest modeling errors.
The associative-memory, water-mediated, structure and energy model (AWSEM),42 which has been successfully applied to study globular protein folding,42 protein recognition and binding,20,35 aggregation,43 membrane proteins,44,45 protein−DNA association, and functional transitions,46–48 provides a promising opportunity for predictive simulations of IDPs. It is a coarse-grained model that has been developed using concepts from the energy funnel theory of folding of globular proteins and structural data on well-folded proteins. It contains both physics-based potentials and bioinformatics-motivated local structural biasing terms.49 The synergy among the biophysical and bioinformatic potentials provides the needed flexibility for AWSEM’s further development to simulate IDPs. For instance, specific potential terms can be tuned to regulate the formation of protein secondary or tertiary structures. The local structure biasing term can be drawn from diverse data sources including experiments42 or from in silico simulations of more elaborate fully atomistic models.50,51
In this article, we introduce “AWSEM-IDP”, a new coarse-grained model specialized for simulating IDPs. It is based on the standard AWSEM, but three major changes have been made: (1) the weights of the hydrogen bonding potentials have been modified to reflect the reduced propensities for secondary structure formation characteristic of IDPs; (2) the local fragment library is derived from either IDP experiments or structural ensembles obtained from atomistic simulations; (3) a novel radius of gyration (Rg) term is added into the AWSEM Hamiltonian to regulate finely the collapse of the chain, enabling delicate control of its size fluctuations. We tested the performance of AWSEM-IDP on two examples: the H4 histone tail (H4 tail) and ParE2-associated antitoxin 2 (PaaA2). H4 tail is 26-residues long and largely lacks any secondary structure. PaaA2 is 71-residues long but has clear secondary structural elements in an extended chain geometry. Both sets of simulations show significant agreement between the AWSEM-IDP generated ensembles and the corresponding experimental measurements or atomistic simulations. We also carried out energy landscape analysis of these IDPs, comparing the energy distributions with those found for globular proteins. Finally, we used thermodynamic perturbation theory to calculate how IDPs’ structural propensities depend on the details of the potential, finding the responses of structural variables to force field perturbations are at least an order of magnitude larger than the same responses for globular proteins.
All together, this study introduces AWSEM-IDP as a transferable model for simulating various types of IDPs, whose computational efficiency allows broad, well-converged sampling of the disordered ensemble. It should be particularly useful for simulating mixed biomolecular complexes that contain IDPs or IDRs along with well-folded structural segments.
2. METHODS
2.1. AWSEM-IDP Hamiltonian.
AWSEM-IDP is a specialization of AWSEM,42 a coarse-grained protein force field, where each amino acid is represented by the positions of Cα, Cβ (H for glycine), and O atoms. The coordinates of other heavy atoms are calculated following the ideal peptide geometry. The total Hamiltonian of AWSEM-IDP, which largely coincides with that of AWSEM, is given below
| (1) |
where Vbackbone ensures a proteinlike backbone connectivity and stereochemistry. Vcontact and Vburial describe water- and protein-mediated tertiary interactions and also the preferences for each amino acid to be buried or exposed. Detailed definitions of the first five terms are provided in the references.42,49 In this work, we report on tuning the parameters for the and terms for IDP simulations, hence denoting these terms with a single prime notation. We also introduce here a new term, which allows for the control of the collapse and the size fluctuations of an IDP chain. In the following subsections, these three terms are introduced in greater detail.
2.2. Hydrogen Bonding Potential.
is a sum of three hydrogen bonding terms, as shown in eq 2
| (2) |
where Vβ favors formation of well-structured hydrogen bonding networks in β-sheets, VP−AP enables a protein chain to adopt approximate parallel or antiparallel β-sheet conformations before more detailed hydrogen bonds are fully formed, and Vhelical controls the formation of hydrogen bonds in α-helices. , and indicate the corresponding weights of these potentials. These terms have been described in detail else-where.42,49
IDPs show a lesser propensity to form secondary structure elements than do globular proteins. Collapse itself tends to increase secondary structural content.41 Doubtless, the tendency to form secondary structure is also reinforced by minimally frustrated correlation between secondary and tertiary inter-actions.41,52–54 In our test simulations, we found that, with the default VHbond setup, IDPs already tend to form more stable secondary structures than are seen in experiments. Therefore, while for AWSEM-IDP we kept the functional forms of these hydrogen bonding terms, we have recalibrated the relative weights of these terms, namely, , and , such that the resulting α-helix and β-sheet propensities are more appropriate for IDPs and IDRs (see Supporting Information for further details of this calibration).
2.3. Fragment Memory Potential.
is a bioinformatics fragment memory (“FM”) potential that structurally biases short fragments of the protein chain, typically 3−9 residues at a time, toward conformations that are based on “memory” structures. In AWSEM the latter memory terms have been selected by matching the fragment sequence to sequences of proteins in the globular protein structural database, usually selected from the (see Figure 1)
| (3) |
Figure 1.
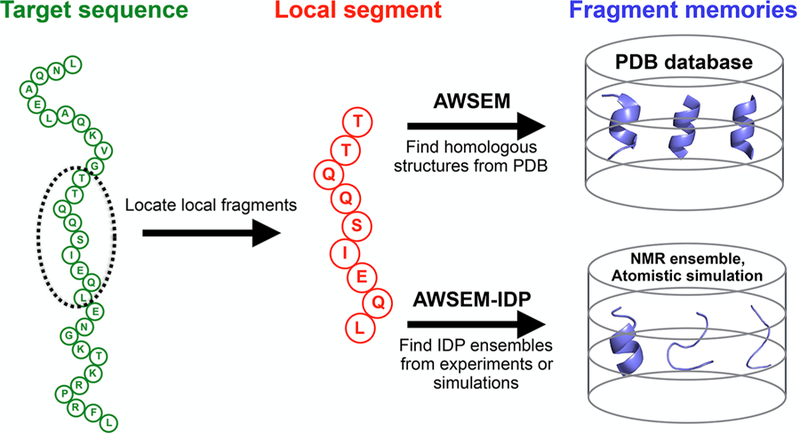
Schematic diagram of the fragment memory terms in AWSEM and AWSEM-IDP. In both AWSEM and AWSEM-IDP, the target sequence (green) is assigned into short local segments (red). Then structural fragments called “memories” (blue) are chosen to bias the local segment. The original forms of AWSEM search for fragment memories from the PDB database, while AWSEM-IDP utilizes NMR ensembles or atomistic simulation trajectories to construct the fragment library. The example sequence shown here is the amino-terminal domain of phage 434 repressor (PDB ID: 1R69).
In eq 3, the outer summation is carried out over aligned fragment memories, while the inner summations are carried out over all possible pairs of Cα and Cβ that are separated by two or more residues. rij is the distance between the ith Cα and jth Cβ atom in the target sequence, and is the corresponding distance in the memories. σij=|i − j|0.15 is the tolerance factor for gauging similarity between two distances. λFM sets the overall weight of the FM term.
As shown in Figure 1, the fragment memory library in the standard form of AWSEM is constructed from structures in the PDB database, with the specific memory conformations selected on the basis of the similarity between the target sequence and the individual memory sequences. This approach is not optimally suited for studying IDPs or the disordered regions in globular proteins,42 because most structures in the PDB, which serve as templates for potential fragment memories, belong to globular proteins having a significant amount of secondary structure that has been partially induced by the supporting tertiary structure. This bias, in turn, typically will result in overestimation of secondary structure formation in IDPs or IDRs. Therefore, in AWSEM-IDP we have decided to rely instead on taking fragment memories either from the representative snapshots of the target protein carried out using atomistic simulations, similar in spirit to the way it was done in atomistic AWSEM,51,55 or by taking them from the experimentally obtained structural ensembles for these peptide fragments, since these ensembles are expected to describe more accurately the realistic conformational details.56
2.4. Rg Potential.
The standard AWSEM can accurately predict the size of globular proteins. However, for some disordered proteins highly extended in physiological conditions, the AWSEM Hamiltonian tends to overcollapse the IDP chain, especially for longer ones. To remedy this deficiency, we propose a new term in AWSEM-IDP in the following expression
| (4) |
where N is the number of residues in the target sequence and is the desired value for the average of the radius of gyration, which typically can be determined by related experiments such as FRET or SAXS. α and β modulate the width of the curve, thus modulating the degree of allowed fluctuation in degree of collapse. The depth of the potential is controlled by D is a scaling factor to fine tune the sought after average compaction.
The major advantage of this potential over the more commonly used alternatives, such as harmonic or Morse potentials for the radius of gyration, is the ability of this term to sculpt more flexibly the potential profile (Figure 2). In particular, this potential allows the simulated chain to overcome the unrealistically large energy barriers for expansion of the chain that arise from the harmonic well much in the way the Morse potential does. The chain collapse potential therefore allows accessing extended chain conformations characteristic of IDPs, while separately controlling the extent of the fluctuations at the bottom of potential profile. In this way, eq 4 goes beyond what can be done with the Morse potential. The width, depth, and slope of the can all be carefully adjusted to regulate both the general collapse and the distribution of accessible conformations of the IDP chain. Hence, this potential could be useful not only in IDP simulations, but also in computational studies of various other biological and artificial polymer chains, where more precise control of collapse dynamics is needed.
Figure 2.
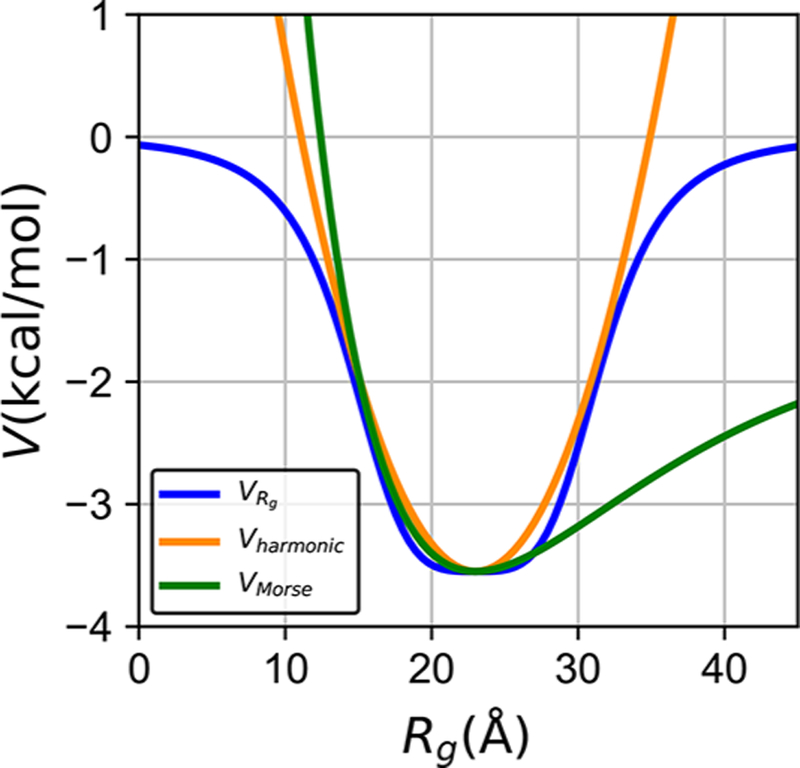
potential introduced in this work allows for more powerful control of chain fluctuations of IDPs than the harmonic and Morse Rg potentials. The harmonic potential (orange) and Morse potential (green) tend to restrain Rg in a narrow energy well with a steep energy barrier away from the ideal Rg value. In comparison, (blue) shows a shallow bottom and allows the chain to escape the restraint.
2.5. Force Field Parametrization.
We used a two-step protocol to calibrate the modified and new parameters in AWSEM-IDP. Since and both account for local structure, these two terms were parametrized first. After local secondary structures were reproduced sufficiently and faithfully close to the targets from atomistic simulations or experimentally determined structural ensembles, the parameters in were subsequently optimized. The parameters obtained for the , and terms in the current model are listed in Table 1. A detailed description of the parametrization procedure is provided in the Supporting Information.
Table 1.
Typical IDP Parameters Used in AWSEM-IDP
| term | param | value | unit |
|---|---|---|---|
| 1.0 | kcal/mol | ||
| 1.0 | kcal/mol | ||
| 1.2 | kcal/mol | ||
| |i – j|min | 3 | ||
| |i – j|max | 12 | ||
| 0.001–0.002 | kcal/mol | ||
| D | −0.2 to −0.8 | kcal/mol | |
| α | 0.001 | kcal/mol Å−2 | |
| β | 0.0005–0.003 | Å−4 | |
| γ | 1.1−1.2 |
2.6. Testing Models.
The wild-type N-terminal H4 histone tail (H4 tail) and ParE2-associated antitoxin 2 (PaaA2) are both well-studied IDPs with important biological functions in regulating eukaryotic chromatin folding57,58 and prokaryotic cell growth and death,59,60 respectively (Figure 3). These two IDPs were chosen as the test systems to evaluate the performance of AWSEM-IDP because they have quite distinct chain lengths, with rather different characteristics of their respective conformational ensembles. The H4 tail is relatively short, with a small fraction of secondary structure elements, while PaaA2, on the other hand, is a longer IDP and is more extended with two preformed α-helices. The specific parameters for these targets are listed in the Supporting Information.
Figure 3.
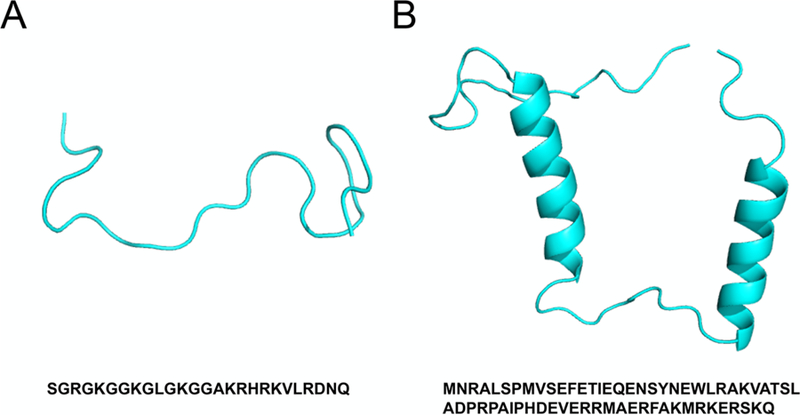
Experimental structures and sequences of H4 tail and PaaA2. (A) X-ray crystallography structure of H4 tail in the context of the encompassing nucleosomal particle (PDB ID: 1KX5). (B) NMR ensemble structure of PaaA2 (PDB ID: 3ZBE). Amino acid sequences are shown under the corresponding structures.
2.7. Simulation Details.
We performed all molecular dynamics simulations using the open-source simulation package LAMMPS (Feb 2016 version), in which both the original AWSEM and AWSEM-IDP codes have been implemented.42 We used nonperiodic shrink-wrapped boundary conditions and the Nose−Hoover thermostat. The simulation time step was set at 2 fs. We unfolded the initial structure at 800 K to generate a random peptide chain as the initial conformation and then slowly cooled down the system from 800 to 300 K over 5 × 105 time steps. Then, we ran 10 production simulations at 300 K for 1.5 × 107 time steps, recording snapshots every 1000 time steps. The first 5 × 106 time steps of trajectories were discarded as the equilibration phase. All the analyses reported below are based on the final 1 × 107 time steps. The convergence of all simulations was confirmed by the root-mean-square inner product analysis61 (see Supporting Information, including Figure S1, for details).
2.8. Analyses.
Since AWSEM is based on a coarse-grained (CG) representation of amino acids, we converted the CG beads into more elaborate atomistic representation based on ideal peptide backbone geometry.42 We determined secondary structure assignments in simulations by STRIDE62 implemented in VMD (version 1.9.2). We also calculated the radius of gyration (Rg) and end-to-end distance (De2e) of structures in the ensembles as global structural metrics using Cα atom coordinates.
Particularly for PaaA2, we compared our simulations with the NMR and SAXS experimental results that are available online.63 We determined the secondary structure of PaaA2 from NMR chemical shift data from the BioMagResBank database64 (BMRB entry: 18841) with the δ2D method,65 which translates a set of chemical shifts into probabilities of secondary structure elements. We also computed theoretical SAXS intensities from simulations with CRYSOL (version 2.8.2)66 and compared these with experimental results.
To measure the heterogeneity of ensembles, we employed the distribution of pairwise structural overlap values: q. This pairwise q quantifies the structural similarity between any two conformations, and the formula for pairwise q between structure i and j is given by
| (5) |
where represents the Cα distance between residues a and b for structure i, σab = (1+ |a − b|)0.15 is the resolution of this metric, and Npairs is the number of a and b pairs summed for all possible choices. The range of pairwise q is from 0 to 1, with the higher values indicating stronger structural similarity between conformations. Hence, the shape of the pairwise q distribution reflects the heterogeneity of the corresponding structural ensemble. Pairwise q distributions have been used to elucidate the intrinsic conformational preferences and the structural heterogeneities of histone tail conformation ensembles in previous simulation studies.16,17
3. RESULTS AND DISCUSSION
We first describe the results of AWSEM-IDP calculations for the two test systems, H4 tail (Section 3.1) and PaaA2 (Section 3.2), comparing our results with either atomistic simulations or experimental data. In the third subsection, we then characterize the secondary and tertiary structural properties of the H4 tail and PaaA2 and some well-folded globular proteins from the energy landscapes perspective (Section 3.3).
3.1. Coarse-Grained Simulations of H4 Tail.
We first applied AWSEM-IDP to the H4 histone tail, which is 26-residues long, and has no prominent secondary structure elements (Figure 3). Winogradoff et al.18 previously performed atomistic replica-exchange molecular dynamic (REMD)67 simulations of the H4 tail at 300 K for 6 μs in total, using the amber99SB*68 and ions9469 force fields with the TIP3P water model. We randomly selected 100 conformational snapshots from those atomistic simulation trajectories to construct the fragment memory database for AWSEM-IDP.
We characterized the distribution of the chain sizes as measured by the radius of gyration, Rg, of the H4 tail, calculated from both the atomistic18 and the AWSEM-IDP simulations (Figure 4A). The average Rg value from AWSEM-IDP (8.2 ± 0.8 Å) reproduces its atomistic simulation counterpart (8.6 ± 1.4 Å) well. Furthermore, the Rg probability distributions from atomistic and AWSEM-IDP simulations significantly overlap. Both distributions exhibit long tails stretching toward larger Rg values that correspond to extended chain conformations. Note that the Rg biasing potential, introduced in this work, provides enough flexibility to control the complete Rg distribution, not only the average value of the radius of gyration, enabling more accurate modeling of the H4 tail in more extended conformations. Interestingly, histone tails are known to change their degree of chain condensation throughout the cell cycle.16–18,70–72 By tuning the Rg potential, we can thus nudge histone tails to explore specific regions of chain extension (Figure S2), providing a basis for more accurate coarse-grained modeling of polynucleosomal arrays in future studies.
Figure 4.
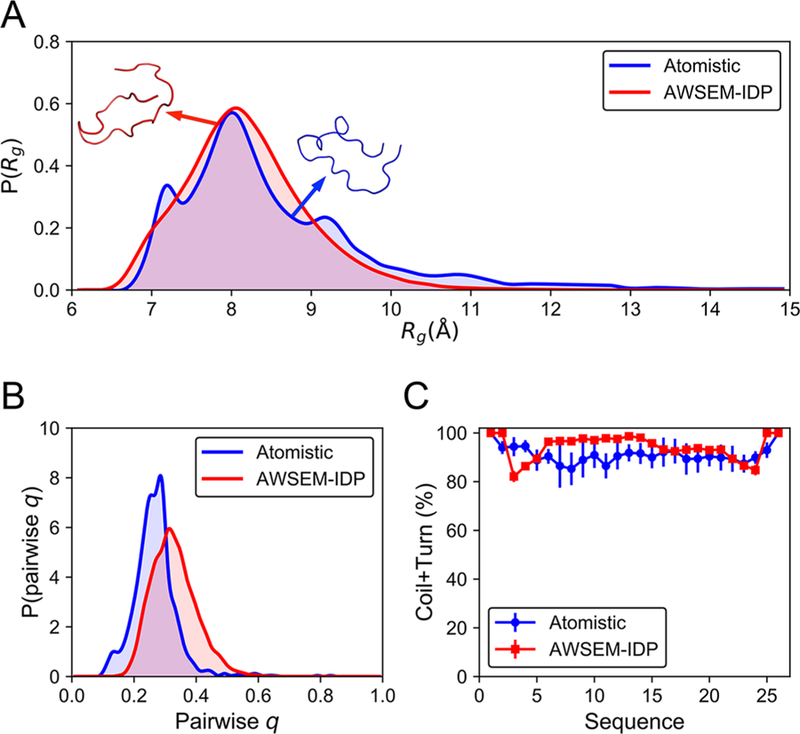
AWSEM-IDP captures reasonably well the structural features of the H4 tail obtained from atomistic simulations. (A) The probability distributions of Rg indicate similar global overall properties of AWSEM-IDP (red) and atomistic (blue) simulated ensembles. Representative snapshots at the average Rg values of the corresponding distributions are displayed for the atomistic (blue) and AWSEM-IDP (red) simulations. (B) The probability distributions of pairwise q demonstrate a somewhat shifted, but roughly similar, level of structural heterogeneity in AWSEM-IDP and atomistic MD. (C) Local disordered secondary structural propensities (coil + turn) from atomistic and AWSEM-IDP results are close.
We examined the heterogeneity of the structures sampled in AWSEM-IDP and all-atom MD simulations by measuring the distributions of the pairwise q (Figure 4B). A similar level of structural heterogeneity was found in both the atomistic and the AWSEM-IDP simulations. The average pairwise q obtained from AWSEM-IDP (0.33 ± 0.07), however, is slightly larger than that found from atomistic MD (0.27 ± 0.07), possibly resulting from AWSEM’s tendency to overstructure protein chains.73
In addition to comparing the global characteristics of chain conformations, we also analyzed the local propensities for the secondary structure formation. Because the H4 tail is intrinsically disordered, lacking well-defined secondary structure,16–18 we used the combination of coil and turn probabilities as a metric of local structural disorder and heterogeneity (Figure 4C). This comparison indicates that AWSEM-IDP replicates the amount of flickering secondary structures observed in atomistic simulations with a relatively high fidelity. In both atomistic and AWSEM-IDP simulations, the coil + turn probabilities fluctuate around 90%. This particularly high level of disorder is not surprising because of the high proportion of positively charged (lysine and arginine) and flexible (glycine) residues in the H4 tail amino acid sequence (Figure 3A). Overall, these comparisons reveal robust agreement between the conformational ensembles sampled by atomistic simulations and those sampled by AWSEM-IDP simulations. In particular, the results obtained from AWSEM-IDP simulations of the H4 histone tail more faithfully reflect the atomistic results than do those found using the standard AWSEM force field (Figure S3).
3.2. Coarse-Grained Simulations of PaaA2.
We also tested the performance of AWSEM-IDP on another disordered protein, PaaA2. PaaA2 is relatively longer (71 residues) than H4 histone tails and has more stable secondary structural elements, namely, two α-helices (Figure 3B). Sterckx et al.63 calculated a PaaA2 ensemble based on NMR and SAXS experimental results. We used all 50 structures from their ensemble as the fragment memory library in our subsequent simulations.
We first analyze the AWSEM-IDP sampled ensemble by projecting the conformational space onto two collective variables, Rg and end-to-end distance (De2e) (Figure 5A). The resulting two-dimensional landscape topography reveals three well-connected conformational basins (labeled as i, ii, and iii in Figure 5A), with moderate energy barriers of ∼2 kBT, suggesting high conformational lability.
Figure 5.
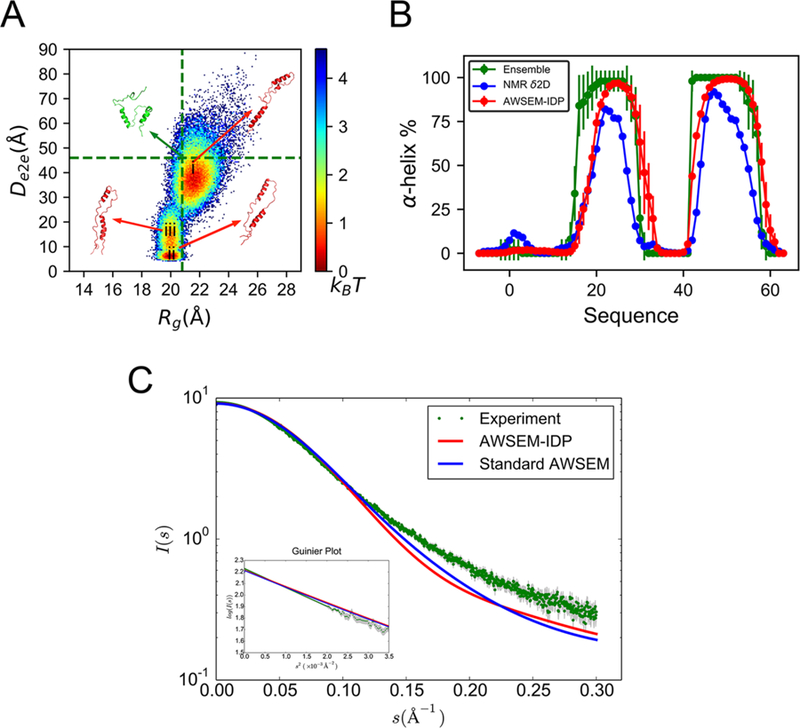
AWSEM-IDP simulations agree well with experiments in the global and local structures of PaaA2. (A) The free energy landscape of PaaA2 is projected on the coordinates of Rg and De2e. The vertical and horizontal lines in the figure are the average Rg and De2e from the experimental ensemble calculated on the basis of NMR and SAXS data in Sterckx et al.63 Representative structures are shown for the experimental ensemble (green) and for different basins in AWSEM-IDP simulations (red). (B) The two helical structures in both experimental ensemble (green) and δ2D calculation from NMR chemical shifts data (blue) are well-replicated by AWSEM-IDP simulations (red), with similar positions and probabilities. (C) The AWSEM predicted SAXS curves and the related Guinier plot (inset figure). Experimental errors are labeled in gray.
To quantify further the simulation results, we compare the locations of the free energy basins to those inferred from theexperimentally guided structural ensemble.63 All three free energy basins are located along the average Rg (20.8 ± 3.2 Å) of the latter ensemble (the vertical green dotted line in Figure 5A), showing consistency between the chain dimensions of the simulated and experimental ensembles. The experimentally guided structural ensemble gives the range 30−62 Å for the end-to-end distance De2e, covering the largest free energy basin explored by AWSEM-IDP. The average value 46.0 Å is marked with a horizontal green dotted line in Figure 5A. Notice that the two other free energy basins have lower De2e values than the experimental reference. This again could arise from the tendency of AWSEM to overcollapse, or it represents subpopulations that are too small for experiments to see.
Beyond global analyses of the chain conformations, we also looked into the local structural details of ensemble members. The PaaA2 experimental ensemble63 indicates two prominent α-helices, connected by a highly flexible loop (Figure 3B). This topology is important for carrying out some of the significant biological functions of PaaA2, such as the molecular recognition driving toxin inhibition.63,74 To analyze this structural feature, we calculated the average α-helical tendency of all the PaaA2 residues along the simulation trajectory. As seen in Figure 5B, PaaA2 has two well-defined α-helices in AWSEM-IDP simulations (shown in red). Moreover, both the positions and structural probabilities of these helices are quantitatively consistent with those in the experimental ensemble (green), as well as with the helical probabilities calculated directly from the NMR chemical shifts data63 by the δ2D method65 (blue). This agreement suggests that AWSEM-IDP can reproduce reasonably well the local structural details obtained from experimental measurements. By contrast, in the standard AWSEM simulations, the first helix comes out as too long in comparison to the NMR determination (Figure S5B). Reducing the weight of the helical structure formation term in AWSEM-IDP is apparently necessary to improve the modeling of secondary structures of disordered proteins. Besides the experiment-guided ensemble and NMR chemical shifts signal, we also compared the simulation results to the SAXS experimental data63 (Figure 5C). The experimental and simulated curve overlap with high precision for s < 0.10 Å−1. For greater s, the small deviations indicate less extended structures in simulations than those found in experiments. The slopes of the Guinier plot (log(I(s)) versus s2) show similar global structures in experimental and simulated ensembles with close Rg values. This comparison indicates both ensembles have similar global size.
3.3. Analyzing IDPs from the AWSEM-Specific Energy Landscapes Perspective.
We discuss and quantify in this section the role of those AWSEM-IDP energy terms that provide the most important contributions to the formation of secondary and tertiary structure. In the AWSEM-IDP Hamiltonian, the formation of protein secondary structures primarily results from the effects of two potential terms, VHbond and Vrama. VFM also commonly contributes to local structure formation, but we must remember the fragment memories for IDPs do not necessarily carry directly the signals for conventionally well-defined helical or extended secondary structure. Residual tertiary interactions in IDPs, on the other hand, arise largely from the terms Vcontact and Vburial, where Vcontact indicates water-mediated or protein-mediated interactions between pairs of amino acids distant in sequence, and Vburial governs the burial preference a particular residue. We define the secondary and tertiary average energies per residue using eqs 6 and 7 as
| (6) |
| (7) |
Following these definitions, we calculated ⟨Esecondary⟩ and ⟨Etertiary⟩ for H4 histone tail and PaaA2, along with these analyses for two globular proteins (PDB: 1R69, 1UBQ) and one mostly globular protein with a disordered tail (PDB: 1UZC). These data are plotted in Figure 6. As one could have anticipated, both the H4 histone tail and PaaA2 have higher ⟨Esecondary⟩ and ⟨Etertiary⟩ than the three ordered proteins. Between the two IDPs, PaaA2 has lower average secondary structure energy compared with that of the H4 tail. The average tertiary energy of the H4 tail is approximately equal to PaaA2, however, with similar level of fluctuations. This comparison suggests that PaaA2 and H4 tail may potentially belong to two different classes of IDPs: PaaA2 has stable secondary structural elements but is lacking tertiary organization, while H4 tail may be relatively collapsed but lacks stable secondary structures. 1R69 and 1UBQ, which are well-folded globular proteins, are characterized by lower secondary and tertiary structure energies than IDPs have, as expected. 1UZC, which has unstructured segments, shows correspondingly an intermediate behavior: low secondary structure energy but destabilized tertiary structure energy. Table S1 and Figure S6 elaborate on additional term-by-term contributions from other terms of the AWSEM Hamiltonian and also temporal evolution of Esecondary and Etertiary during MD runs.
Figure 6.
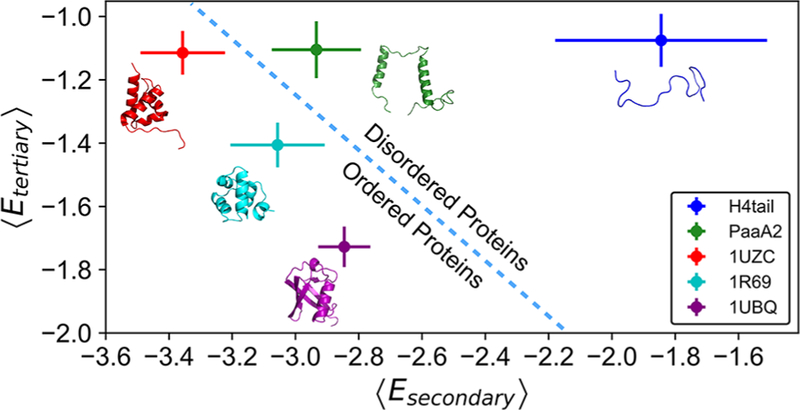
Means and variances of AWSEM-specific energies corresponding to secondary and tertiary structures can efficiently demarcate protein disorder. The average energies and corresponding standard deviations for secondary and tertiary structures are shown for all simulated proteins (H4 tail, blue; PaaA2, green; 1UZC, red; 1R69, cyan; 1UBQ, purple). Initial conformations of each protein are illustrated accordingly. The dashed line serves as a qualitative border between the ordered and disordered proteins. All energies are in the units of kcal/mol.
We see that the analyses more or less correspond to our intuitions about the differences between IDPs and well-structured globular proteins. Still more telling differences can be seen in the fluctuations and corresponding modeling sensitivities as monitored by susceptibilities or response functions. We calculate the sensitivities of the radius of gyration and the helical occupation probabilities. Both of these, as we have seen, can be monitored experimentally. The susceptibility of Rg to potential variation is computed as
| (8) |
while the sensitivity of helical occupations along the sequence
| (9) |
can be computed on an individual residue basis (where Ph,i is an indicator function, being 1 if the residue i is found in helical conformation, and 0 otherwise). Here, the AWSEM potential is assumed to have the following form
| (10) |
where the Vk terms represent various types of interactions, and γk parameters indicate the corresponding weights. We see in Figure 7A that the Rg modeling sensitivities for the IDPs studied are more than an order of magnitude larger than those are for globular proteins. This seems to trace back to considerable sensitivity of secondary structure occupation to the model terms as shown in Figure 7B. We see that the fraying ends of helices in PaaA2 are especially sensitive to energy modeling errors. This is where the structure of this IDP fluctuates most strongly.
Figure 7.
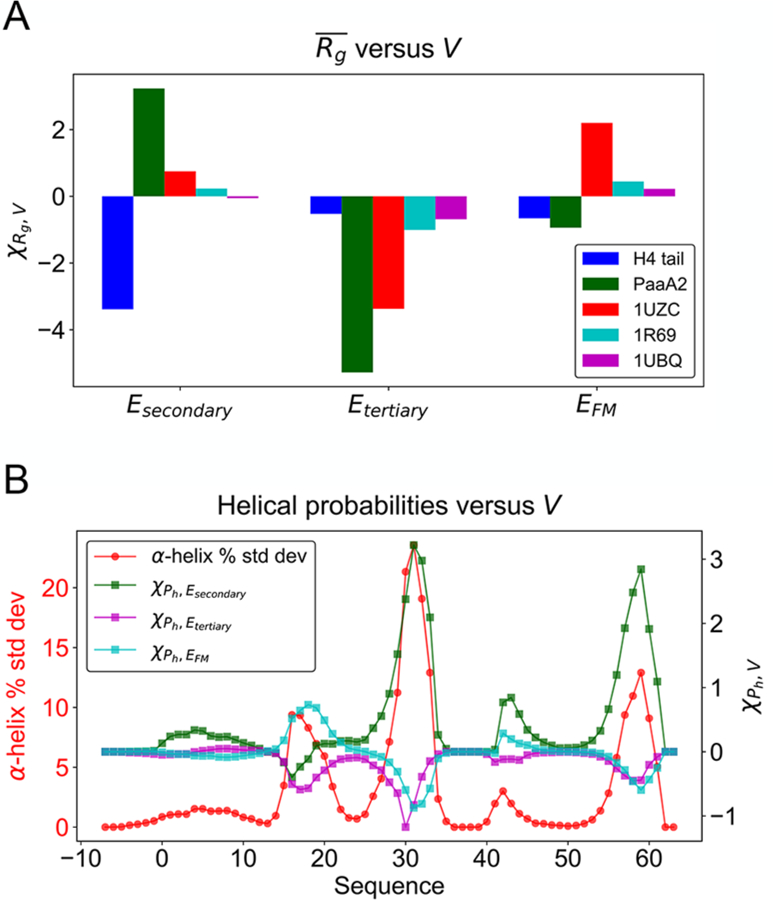
Structural features of IDPs are highly sensitive to the force field potential variation compared with globular proteins. (A) The susceptibilities of Rg to variation of the potential for IDPs and globular proteins are shown. (B) The susceptibilities of helical occupations along the PaaA2 sequence are primarily determined by the covariance with Esecondary (green squares), whose peaks coincide with the locations where the helical probability fluctuation (red circles) reaches a maximum. A similar plot with the raw value of helical probability as reference is given in Figure S7.
4. CONCLUSIONS
In this paper, we introduce AWSEM-IDP, a coarse-grained model tailored for modeling intrinsically disordered proteins. Two terms from the standard AWSEM Hamiltonian, VHbond and VFM, were modified, and one new term, , was added. Lowering the weight of VHbond diminishes secondary structure formation, thereby better representing the amount of secondary structure observed in IDPs. The VFM term can be constructed and tuned using the structural ensembles obtained from either experiments or long time scale atomistic simulations, allowing AWSEM-IDP to replicate accurately the known structural features of any given IDP. Finally, the new term provides fine control over the chain’s global fluctuations, being important for reproducing the average chain radius as well as variance and tails of the Rg distribution that may be known either from experiments or atomistic simulations.
The quality of predictions from AWSEM-IDP will depend on the quality of the available experimental input data or the accuracy of atomistically generated ensembles. Experimental databases for IDPs, such as pE-DB,75 are rapidly evolving and will become more useful. We must bear in mind however that obtaining accurate descriptions of IDPs by atomistic MD simulations alone will remain a challenge, both due to the intrinsic sensitivity of IDP structure to force field error and the incomplete samplings of fully atomistic landscapes. In particular, our calculations of such sensitivities indicate an order-of-magnitude amplification of errors compared to globular proteins, which has profound implications in the context of recent attempts to improve atomistic force fields to better model IDPs and unfolded protein chains.
In summary, AWSEM-IDP enables the exploration of large conformational spaces of IDPs while still maintaining sufficient chemical accuracy. The present work should provide the foundation for simulating large protein complexes that include both ordered and disordered protein segments, such as nucleosomes.
Supplementary Material
ACKNOWLEDGMENTS
G.A.P.’s efforts were supported by the Amazon AWS Machine Learning Research Award. P.G.W.’s work is supported by NIH Grant R01 GM44557 and the Bullard−Welch Chair at Rice University Grant C-0016. Computational resources are provided by the Deepthought2 HPC cluster at University of Maryland. We would like to thank David Winogradoff for providing the H4 tail all-atom simulation data and the insightful discussions. We also thank Haiqing Zhao for helpful suggestions and discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b05791.
Detailed descrition on the parametrization procedure of the modified and new terms in AWSEM-IDP, and the RMSIP calculation for evaluating the convergence of simulations; table to represent the values of all terms in the AWSEM Hamiltonian for all the simulated proteins; and supplementary figures of the RMSIP analysis results of H4 tail and PaaA2 simulations, the effect of different parameters, the results obtained from the standard AWSEM simulations, the predicted SAXS curves, the time evolutions of Esecondary and Etertiary, and the susceptibilities of helical occupations (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Dunker AK; Lawson JD; Brown CJ; Williams RM; Romero P; Oh JS; Oldfield CJ; Campen AM; Ratliff CM; Hipps KW; et al. Intrinsically Disordered Protein. J. Mol. Graphics Modell 2001, 19, 26–59. [DOI] [PubMed] [Google Scholar]
- (2).Uversky VN; Dunker AK Understanding Protein Non-Folding. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mao AH; Crick SL; Vitalis A; Chicoine CL; Pappu RV Net Charge Per Residue Modulates Conformational Ensembles of Intrinsically Disordered Proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Babu MM; Kriwacki RW; Pappu RV Versatility from Protein Disorder. Science 2012, 337, 1460–1461. [DOI] [PubMed] [Google Scholar]
- (5).Van Der Lee R; Buljan M; Lang B; Weatheritt RJ; Daughdrill GW; Dunker AK; Fuxreiter M; Gough J; Gsponer J; Jones DT; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Habchi J; Tompa P; Longhi S; Uversky VN Introducing Protein Intrinsic Disorder. Chem. Rev 2014, 114, 6561–6588. [DOI] [PubMed] [Google Scholar]
- (7).Wright PE; Dyson HJ Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Papoian GA Proteins with Weakly Funneled Energy Landscapes Challenge the Classical Structure-Function Paradigm. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 14237–14238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Papoian GA; Wolynes PG The Physics and Bioinformatics of Binding and Folding - an Energy Landscape Perspective. Biopolymers 2003, 68, 333–349. [DOI] [PubMed] [Google Scholar]
- (10).Wright PE; Dyson HJ Linking Folding and Binding. Curr. Opin. Struct. Biol 2009, 19, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tompa P Intrinsically Disordered Proteins: a 10-Year Recap. Trends Biochem. Sci 2012, 37, 509–516. [DOI] [PubMed] [Google Scholar]
- (12).Bernadó P; Svergun DI Structural Analysis of Intrinsically Disordered Proteins by Small-Angle X-Ray Scattering. Mol. BioSyst 2012, 8, 151–167. [DOI] [PubMed] [Google Scholar]
- (13).Jensen MR; Ruigrok RW; Blackledge M Describing Intrinsically Disordered Proteins at Atomic Resolution by NMR. Curr. Opin. Struct. Biol 2013, 23, 426–435. [DOI] [PubMed] [Google Scholar]
- (14).Baker CM; Best RB Insights into the Binding of Intrinsically Disordered Proteins from Molecular Dynamics Simulation. WIRES Comput. Mol. Sci 2014, 4, 182–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Materese CK; Savelyev A; Papoian GA Counterion Atmosphere and Hydration Patterns Near a Nucleosome Core Particle. J. Am. Chem. Soc 2009, 131, 15005–15013. [DOI] [PubMed] [Google Scholar]
- (16).Potoyan DA; Papoian GA Energy Landscape Analyses of Disordered Histone Tails Reveal Special Organization of Their Conformational Dynamics. J. Am. Chem. Soc 2011, 133, 7405–7415. [DOI] [PubMed] [Google Scholar]
- (17).Potoyan DA; Papoian GA Regulation of the H4 Tail Binding and Folding Landscapes via Lys-16 Acetylation. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 17857–17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Winogradoff D; Echeverria I; Potoyan DA; Papoian GA The Acetylation Landscape of the H4 Histone Tail: Disentangling the Interplay between the Specific and Cumulative Effects. J. Am. Chem. Soc 2015, 137, 6245–6253. [DOI] [PubMed] [Google Scholar]
- (19).Winogradoff D; Zhao H; Dalal Y; Papoian GA Shearing of the CENP-A Dimerization Interface Mediates Plasticity in the Octameric Centromeric Nucleosome. Sci. Rep 2015, 5, 17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhao H; Winogradoff D; Bui M; Dalal Y; Papoian GA Promiscuous Histone Mis-Assembly is Actively Prevented by Chaperones. J. Am. Chem. Soc 2016, 138, 13207–13218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schneider R; Huang J.-r.; Yao M; Communie G; Ozenne V; Mollica L; Salmon L; Jensen MR; Blackledge M Towards a Robust Description of Intrinsic Protein Disorder Using Nuclear Magnetic Resonance Spectroscopy. Mol. BioSyst 2012, 8, 58–68. [DOI] [PubMed] [Google Scholar]
- (22).Schuler B; Soranno A; Hofmann H; Nettels D Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys 2016, 45, 207–231. [DOI] [PubMed] [Google Scholar]
- (23).Burger VM; Gurry T; Stultz CM Intrinsically Disordered Proteins: Where Computation Meets Experiment. Polymers 2014, 6, 2684–2719. [Google Scholar]
- (24).Bhowmick A; Brookes DH; Yost SR; Dyson HJ; Forman-Kay JD; Gunter D; Head-Gordon M; Hura GL; Pande VS; Wemmer DE; et al. Finding Our Way in the Dark Proteome. J. Am. Chem. Soc 2016, 138, 9730–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang W; Ye W; Jiang C; Luo R; Chen H-F New Force Field on Modeling Intrinsically Disordered Proteins. Chem. Biol. Drug Des 2014, 84, 253–269. [DOI] [PubMed] [Google Scholar]
- (26).Song D; Luo R; Chen H-F The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model 2017, 57, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Robustelli P; Piana S; Shaw DE Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang A; Stultz CM The Effect of a ΔK280 Mutation on the Unfolded State of a Microtubule-Binding Repeat in Tau. PLoS Comput. Biol 2008, 4, e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Marsh JA; Forman-Kay JD Ensemble Modeling of Protein Disordered States: Experimental Restraint Contributions and Validation. Proteins: Struct., Funct., Bioinf 2012, 80, 556–572. [DOI] [PubMed] [Google Scholar]
- (30).Best RB; Zheng W; Mittal J Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Henriques J. a.; Cragnell C; Skepö M Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput 2015, 11, 3420–3431. [DOI] [PubMed] [Google Scholar]
- (32).Rauscher S; Gapsys V; Gajda MJ; Zweckstetter M; de Groot BL; Grubmüller H Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: a Comparison to Experiment. J. Chem. Theory Comput 2015, 11, 5513–5524. [DOI] [PubMed] [Google Scholar]
- (33).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell A CHARMM36m: an Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kumar S; Showalter SA; Noid WG Native-Based Simulations of the Binding Interaction Between RAP74 and the Disordered FCP1 Peptide. J. Phys. Chem. B 2013, 117, 3074–3085. [DOI] [PubMed] [Google Scholar]
- (35).Zheng W; Schafer NP; Davtyan A; Papoian GA; Wolynes PG Predictive Energy Landscapes for Protein-Protein Association. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 19244–19249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Knott M; Best RB Discriminating Binding Mechanisms of an Intrinsically Disordered Protein via a Multi-State Coarse-Grained Model. J. Chem. Phys 2014, 140, 175102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kurcinski M; Kolinski A; Kmiecik S Mechanism of Folding and Binding of an Intrinsically Disordered Protein as Revealed by Ab Initio Simulations. J. Chem. Theory Comput 2014, 10, 2224–2231. [DOI] [PubMed] [Google Scholar]
- (38).Emperador A; Sfriso P; Villarreal MA; Gelpi JL; Orozco M PACSAB: Coarse-Grained Force Field for the Study of Protein-Protein Interactions and Conformational Sampling in Multiprotein Systems. J. Chem. Theory Comput 2015, 11, 5929–5938. [DOI] [PubMed] [Google Scholar]
- (39).Kmiecik S; Gront D; Kolinski M; Wieteska L; Dawid AE; Kolinski A Coarse-Grained Protein Models and Their Applications. Chem. Rev 2016, 116, 7898–7936. [DOI] [PubMed] [Google Scholar]
- (40).Lee KH; Chen J Multiscale Enhanced Sampling of Intrinsically Disordered Protein Conformations. J. Comput. Chem 2016, 37, 550–557. [DOI] [PubMed] [Google Scholar]
- (41).Luthey-Schulten Z; Ramirez BE; Wolynes PG Helix-Coil, Liquid Crystal, and Spin Glass Transitions of a Collapsed Heteropolymer. J. Phys. Chem 1995, 99, 2177–2185. [Google Scholar]
- (42).Davtyan A; Schafer NP; Zheng W; Clementi C; Wolynes PG; Papoian GA AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zheng W; Schafer NP; Wolynes PG Free Energy Landscapes for Initiation and Branching of Protein Aggregation. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 20515–20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kim BL; Schafer NP; Wolynes PG Predictive Energy Landscapes for Folding α-Helical Transmembrane Proteins. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Truong HH; Kim BL; Schafer NP; Wolynes PG Predictive Energy Landscapes for Folding Membrane Protein Assemblies. J. Chem. Phys 2015, 143, 243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Potoyan DA; Zheng W; Komives EA; Wolynes PG Molecular Stripping in the NF-κB/IκB/DNA Genetic Regulatory Network. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhang B; Zheng W; Papoian GA; Wolynes PG Exploring the Free Energy Landscape of Nucleosomes. J. Am. Chem. Soc 2016, 138, 8126–8133. [DOI] [PubMed] [Google Scholar]
- (48).Tsai M; Zhang B; Zheng W; Wolynes PG The Molecular Mechanism of Facilitated Dissociation of Fis Protein from DNA. J. Am. Chem. Soc 2016, 138, 13497–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Papoian GA; Wolynes PG In Coarse-Grained Modeling of Biomolecules; Papoian GA, Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, 2017; Chapter 4, pp 121–189. [Google Scholar]
- (50).Kwac K-J; Wolynes PG Protein Structure Prediction Using an Associated Memory Hamiltonian and All-Atom Molecular Dynamics Simulations. Bull. Korean Chem. Soc 2008, 29, 2172–2182. [Google Scholar]
- (51).Chen M; Lin X; Zheng W; Onuchic JN; Wolynes PG Protein Folding and Structure Prediction from the Ground Up: The Atomistic Associative Memory, Water Mediated, Structure and Energy Model. J. Phys. Chem. B 2016, 120, 8557–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gō N The Consistency Principle in Protein Structure and Pathways of Folding. Adv. Biophys 1984, 18, 149–164. [DOI] [PubMed] [Google Scholar]
- (53).Bryngelson JD; Wolynes PG Spin Glasses and the Statistical Mechanics of Protein Folding. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 7524–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Saven JG; Wolynes PG Local Conformational Signals and the Statistical Thermodynamics of Collapsed Helical Proteins. J. Mol. Biol 1996, 257, 199–216. [DOI] [PubMed] [Google Scholar]
- (55).Chen M; Lin X; Lu W; Onuchic JN; Wolynes PG Protein Folding and Structure Prediction from the Ground Up II: AAWSEM for α/β Proteins. J. Phys. Chem. B 2017, 121, 3473–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Allison JR; Varnai P; Dobson CM; Vendruscolo M Determination of the Free Energy Landscape of α-Synuclein Using Spin Label Nuclear Magnetic Resonance Measurements. J. Am. Chem. Soc 2009, 131, 18314–18326. [DOI] [PubMed] [Google Scholar]
- (57).Strahl BD; Allis CD The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (58).Jenuwein T; Allis CD Translating the Histone Code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- (59).Hayes F; Van Melderen L Toxins-Antitoxins: Diversity, Evolution and Function. Crit. Rev. Biochem. Mol. Biol 2011, 46, 386–408. [DOI] [PubMed] [Google Scholar]
- (60).Hallez R; Geeraerts D; Sterckx Y; Mine N; Loris R; Van Melderen L New Toxins Homologous to ParE Belonging to Three-Component Toxin-Antitoxin Systems in Escherichia Coli O157: H7. Mol. Microbiol 2010, 76, 719–732. [DOI] [PubMed] [Google Scholar]
- (61).Amadei A; Ceruso MA; Di Nola A On the Convergence of the Conformational Coordinates Basis Set Obtained by the Essential Dynamics Analysis of Proteins’ Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet 1999, 36, 419–424. [PubMed] [Google Scholar]
- (62).Frishman D; Argos P Knowledge-Based Protein Secondary Structure Assignment. Proteins: Struct., Funct., Genet 1995, 23, 566–579. [DOI] [PubMed] [Google Scholar]
- (63).Sterckx YG; Volkov AN; Vranken WF; Kragelj J; Jensen MR; Buts L; Garcia-Pino A; Jové T; Van Melderen L; Blackledge M; et al. Small-Angle X-Ray Scattering- and Nuclear Magnetic Resonance-Derived Conformational Ensemble of the Highly Flexible Antitoxin PaaA2. Structure 2014, 22, 854–865. [DOI] [PubMed] [Google Scholar]
- (64).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; et al. BioMagResBank. Nucleic Acids Res 2007, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Camilloni C; De Simone A; Vranken WF; Vendruscolo M Determination of Secondary Structure Populations in Disordered States of Proteins Using Nuclear Magnetic Resonance Chemical Shifts. Biochemistry 2012, 51, 2224–2231. [DOI] [PubMed] [Google Scholar]
- (66).Svergun D; Barberato C; Koch MH CRYSOL - a Program to Evaluate X-Ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr 1995, 28, 768–773. [Google Scholar]
- (67).Sugita Y; Okamoto Y Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (68).Best RB; Hummer G Optimized Molecular Dynamics Force Fields Applied to the Helix-Coil Transition of Polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (70).Gao M; Nadaud PS; Bernier MW; North JA; Hammel PC; Poirier MG; Jaroniec CP Histone H3 and H4 N-Terminal Tails in Nucleosome Arrays at Cellular Concentrations Probed by Magic Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc 2013, 135, 15278–15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Erler J; Zhang R; Petridis L; Cheng X; Smith JC; Langowski J The Role of Histone Tails in the Nucleosome: a Computational Study. Biophys. J 2014, 107, 2911–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kenzaki H; Takada S Partial Unwrapping and Histone Tail Dynamics in Nucleosome Revealed by Coarse-Grained Molecular Simulations. PLoS Comput. Biol 2015, 11, e1004443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Schafer NP; Kim BL; Zheng W; Wolynes PG Learning to Fold Proteins Using Energy Landscape Theory. Isr. J. Chem 2014, 54, 1311–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Sterckx YG-J; Jové T; Shkumatov AV; Garcia-Pino A; Geerts L; De Kerpel M; Lah J; De Greve H; Van Melderen L; Loris R A Unique Hetero-Hexadecameric Architecture Displayed by the Escherichia Coli O157 PaaA2-ParE2 Antitoxin-Toxin Complex. J. Mol. Biol 2016, 428, 1589–1603. [DOI] [PubMed] [Google Scholar]
- (75).Varadi M; Kosol S; Lebrun P; Valentini E; Blackledge M; Dunker AK; Felli IC; Forman-Kay JD; Kriwacki RW; Pierattelli R; et al. pE-DB: a Database of Structural Ensembles of Intrinsically Disordered and of Unfolded Proteins. Nucleic Acids Res 2014, 42, D326–D335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.