

Abstract
Non-Hodgkin lymphoma (NHL) is one of the most common types of hematologic malignancies. Pretargeted radioimmunotherapy (PRIT), the sequential administration of a bispecific antibody-based primary tumor-targeting component followed by a radionucleotide-labeled treatment effector, has been developed to improve the treatment efficacy and to reduce the side effects of conventional RIT. Despite the preclinical success of PRIT, clinical trials revealed that the immunogenicity of the bispecific antibody as well as the presence of competing endogenous effector molecules often compromised the treatment. One strategy to improve PRIT is to utilize bio-orthogonal ligation reactions to minimize immunogenicity and improve targeting. Herein, we report a translatable pretargeted nanoradioimmunotherapy strategy for the treatment of NHL. This pretargeting system is composed of a dibenzylcyclooctyne (DBCO)-functionalized anti-CD20 antibody (α-CD20) tumor-targeting component and an azide- and yttrium-90-(90Y) dual-functionalized dendrimer. The physicochemical properties of both pretargeting components have been extensively studied. We demonstrated that an optimized dual-functionalized dendrimer can undergo rapid strain-promoted azide–alkyne cycloaddition with the DBCO-functionalized α-CD20 at the physiological conditions. The treatment effector in our pretargeting system can not only selectively deliver radionucleotides to the target tumor cells but also increase the complement-dependent cytotoxicity of α-CD20 and thus enhance the antitumor effects, as justified by comprehensive in vitro and in vivo studies in mouse NHL xenograft and disseminated models.
Keywords: non-Hodgkin lymphoma, pretargeted radioimmunotherapy, antibody, dendrimer, bioorthogonal ligation reaction
Graphical Abstract
Non-Hodgkin lymphoma (NHL) is one of the most common types of hematological malignancies, with over 70,000 NHL cases diagnosed annually in the USA.1 In contrast to solid tumors that localize in only several organs, malignant lymphoid cells commonly travel through the lymphatic system to disseminate tumors throughout the body. The current standard of care for NHL involves the combination of antibody-based immunotherapy, cytotoxic chemotherapy, and/or radiotherapy.2,3 Rituximab, a chimeric anti-CD20 antibody (α-CD20) that targets lymphoma cells expressing the CD20 antigen, is the most widely used antibody in NHL immunotherapy.4,5 Although the long-term survival rate of aggressive NHL, such as diffuse large B-cell lymphoma, has reached up to 70%, many NHL patients suffer long-term side effects associated with cytotoxic chemotherapy and high-grade whole-body radiotherapy well after their treatments. In addition, more than half of relapsed/refractory B-cell NHL patients do not respond to conventional α-CD20 treatment6 because their immune effector cells (e.g., natural killer cells) cannot cross-link the cancer cell membrane-bound α-CD20 to activate the complement-dependent cytotoxicity (CDC) mechanism.6–8 Therefore, developing NHL treatment strategies is necessary to overcome these therapeutic challenges.
One strategy for treating relapsed/refractory NHL is to deliver radiation at the site of tumor since lymphoma cells usually remain sensitive to ionizing radiation even after becoming resistant to chemoimmunotherapy.9,10 Radioimmunotherapy (RIT) has been developed to deliver ionizing radiation directly to malignant lymphoid cells through a radionucleotide-labeled (e.g., 90Y) antibody that binds specifically to the overexpressed antigen on the surface of lymphoma cells.11,12 Studies have found RIT is more efficient at treating NHL than the combination of immunotherapy and external-beam radiotherapy. Early stage clinical trials have revealed that approximately 75% of relapsed B-cell NHL patients responded to a single dose of 90Y-labeled α-CD20 treatment.12 However, the direct administration of radiolabeled antibody prolongs radiation exposure to radiosensitive organs (e.g., bone marrow) because of the slow accumulation of radiolabeled antibody in the tumor and the slow elimination of antibody from the circulation system.13,14 The slow accumulation of antibody in the tumor also reduces the potency of radionuclides because of isotopic decay.13,14 In recent years, several multistep consecutive pretargeting strategies have been proposed to overcome these challenges.14–18 Most pretargeting systems are composed of three components: a primary tumor-targeting component, a clearing agent component, and an effector component. The primary tumor-targeting component, usually a non-internalizing bispecific antibody, is a tumor-targeting vehicle that binds specifically to the target antigen on the cancer cells. It often contains a tag or ligand that can bind selectively to the effector component through strong noncovalent interactions (e.g., biotin-avidin interaction and hapten-antihapten interaction). A clearing agent is usually administrated 24–48 h after the initial treatment to remove the primary tumor-targeting component from the circulation system. The effector, which contains tag/ligand that binds specifically to the primary tumor-targeting component and delivers cargo (e.g., radionuclides), is then delivered to kill the target tumor cells. Ideally, the unbound effector component can be rapidly cleared from the body after systemic administration. Although preclinical studies have successfully demonstrated the use of a pretargeted radioimmunotherapy (PRIT) strategy to improve the tumor uptake of radionucleotides, some clinical trials have found that the immunogenicity of bispecific antibodies as well as the competitive binding by the endogenous effector molecules (e.g., biotin) and/or clearing agents often compromised treatments.16
Recent advances in bioorthogonal chemistry and polymer chemistry provide opportunities for pretargeted therapy.19 In contrast to the early generation pretargeting systems, bioorthogonal ligation reactions, such as tetrazine-alkene ligation and strain-promoted azide-alkyne cycloaddition (SPAAC), do not require the use of an immunogenic protein tag and a clearing agent for selective binding and clearing. Well-defined, low-molecular-weight branched polymers, like dendrimers (e.g., poly(amidoamine) (PAMAM)), are particularly attractive for use as effectors in pretargeted cancer imaging or RIT because their surfaces can be easily engineered to modulate specific toxicity and biological properties.20,21 In addition, low-molecular-weight polymers can be cleared rapidly by the kidneys after systemic administration.22 Recent preclinical studies and clinical trials have demonstrated the use of dendrimers for cancer immunotherapy and chemotherapy.20,21,23 However, the translation of a bioorthogonal click chemistry-based pretargeting system is hindered by the poor chemical stability of the first-generation bioorthogonal ligands, such as trans-cyclooctene (TCO)24 and the slow reaction kinetics of the second-generation bioorthogonal ligation reactions (e.g., SPAAC between azide and dibenzylcyclooctyne (DBCO)).24,25
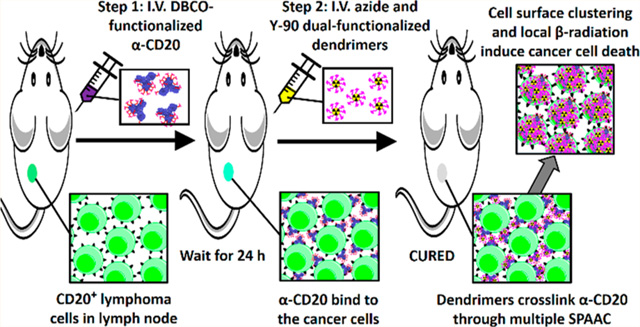
Herein, we present a translatable, two-step, two-component, pretargeting strategy for the treatment of NHL. This next-generation pretargeting system is composed of a DBCO-functionalized rituximab as the primary tumor-targeting component and an azide-functionalized 90Y-labeled PAMAM as the antitumor effector (see Figure 1). We hypothesized that an optimized azide-functionalized dendrimer can undergo rapid multiple SPAAC with DBCO-functionalized α-CD20 through multimeric interactions. The treatment not only selectively delivers radionucleotides to the target tumor cells but also increases CDC by forming a hyper-cross-linked membrane attack complex with membrane-bound α-CD20 that kills the cancer cell without effector-immune cells (see Figure 1). The physiochemical properties and SPAAC kinetics of the pretargeting system have been extensively investigated in the present study. In vitro binding assay and toxicity studies were performed in CD20 antigen-overexpressing (CD20+) Raji B-cell lymphoma cells and CD20 antigen-negative (CD20−) Karpas 299 (K299) T-lymphoblastic lymphoma cancer cells to evaluate the antitumor effects of the pretargeting system. In vivo tumor uptake and biodistribution of both pretargeting components have been investigated in the Raji and K299 dual-xenograft tumor model. Finally, the therapeutic efficacy of this PRIT approach was further evaluated in Raji xenograft and disseminated lymphoma murine models. The antitumor mechanism was investigated through comprehensive histopa-thological studies.
Figure 1.

Scheme illustrating the two-step pretargeted radioimmunotherapy strategy for the treatment of non-Hodgkin lymphoma.
RESULTS AND DISCUSSION
Design, Fabrication, and Characterization of Pretargeting Components for Pretargeted Immunotherapy and Pretargeted Radioimmunotherapy.
DBCO-functionalized α-CD20 (α-CD20(DBCO)f, where f is the degree of functionalization (DOF)) and dual-functionalized PAMAM (PAMAM(D-Y)8(M)m(N)n, where D-Y is yttrium(III)-coordinated DOTA and m and n are the numbers of conjugated methoxyl- and azide-functionalized oligoethylene glycol) were prepared using well-established bioconjugation techniques (see Figures 2a and 3a). The target DBCO to α-CD20 molar ratios were 5:1, 10:1, 20:1, 30:1, and 40:1. The actual target degrees of functionalization of α-CD20 were 2, 5, 10, 15, and 18 (see Figure S1), respectively, as determined by UV spectroscopy and matrix-assisted laser desorption ionization-time-of-flight mass spectroscopy (MALDI-TOF MS). The DBCO-functionalized α-CD20 was further labeled with Alexa Fluor 488 (A488), either through SPAAC with the terminal DBCO or through amine-NHS coupling reaction (see Figures S2 and S3), for in vitro binding and in vivo biodistribution studies. A family of dual-functionalized PAMAM was prepared with an average of 8 Y3+-coordinated DOTA and approximately 30 methoxyl- and azide-functionalized oligoethylene glycol in different molar ratios. The tailor-made dendrimers have been characterized by complementary characterization techniques (see Figures S4–S16). The chelation of nonradioactive 89Y3+ was confirmed by X-ray photoelectron spectroscopy (see Figure S14), and the chelation of radioactive 90Y3+ was quantified by a Geiger counter. The number-average molecular weight and hydro-dynamic diameter of the dual-functionalized PAMAM were 30–31 kDa (see Figure S4) and approximately 7 nm (see Figures S15 and 16), respectively, as determined by MALDI-TOF MS and light-scattering methods. Selected dual-functionalized PAMAM was further functionalized with rhodamine (see Figure S13) for in vitro binding and in vivo biodistribution studies.
Figure 2.

Functionalization and characterization of rituximab (α-CD20). (a) Functionalization of rituximab for pretargeting. The target degrees of functionalization were 5, 10, 20, 30, and 40, and the actual degrees of functionalization (f) were 2, 5, 10, 15, and 18, respectively.(b) Representative flow cytometry histograms of CD20-antigen overexpressing Raji cells after incubation with different concentrations of (i) FITC-labeled rituximab, (ii) α-CD20(DBCO)1(A488)1, (iii) α-CD20(DBCO)4(A488)6, and (ii) α-CD20(DBCO)7(A488)11. (c)(i) The plot of normalized mean-fluorescence intensities (MFIs) of Raji cells after staining with different concentrations of α-CD20. (ii) Microscopic dissociation constants (Kd,micro) of FITC-labeled rituximab and different DBCO-functionalized α-CD20 with different degrees of functionalization, as determined by the FACS binding assay in the CD20-overexpressing Raji cell line. The microscopic dissociation constant (Kd,micro) is equivalent to the concentration of antibody required to achieve half-saturated binding.
Figure 3.

Functionalization and evaluation of azide and yttrium dual-functionalized PAMAM G4 for pretargeted immunotherapy and pretargeted radioimmunotherapy. (a) Functionalization azide and yttrium dual-functionalized PAMAM G4. (b) In vitro evaluation of SPAAC between the different dual-functionalized PAMAM and the selected α-CD20(DBCO)10. (i–iv) Time-dependent UV spectra of 3.13 μM (final concentration) of α-CD20(DBCO)10 recorded before and after incubation with 3.13 μM of dual-functionalized PAMAM at 37 °C for 5 to 180 min. (v, vi) Time-dependent UV absorbance at (v) 280 nm and (vi) 310 nm of α-CD20(DBCO)10 after incubation with an equivalent amount of different dual-functionalized PAMAM. (vii) The plot of the pseudo-first-order rate constants (kobs,280 nm and kobs,310 nm) of SPAAC between different dual-functionalized dendrimers and α-CD20(DBCO)10. (c)(i, ii) Time-dependent UV spectra of 3.13 μM (final concentration) of α-CD20(DBCO)10 recorded before and after incubation with (i) 1.56, and (ii) 6.25 μM of dual-functionalized PAMAM(D-89Y)8(N)29 at 37 °C for 5–180 min. (iii, iv) Time-dependent UV absorbance at (iii) 280 nm and (iv) 310 nm of α-CD20(DBCO)10 after incubation with different molar ratios of PAMAM(D-89Y)8(N)29. The concentration-dependent second-order rate constant was calculated to be about 230 M−1 s−1 (see Figure S22). (NB. * represents p < 0.05; n.s. represents statistically insignificant (i.e., p > 0.05).)
The binding affinities and specificities of different DBCO-functionalized α-CD20 were evaluated using fluorescence-activated cell sorting (FACS) binding assays in CD20+ Raji and CD20– K299 cell lines (see Figure S17). To prevent dye labeling from introducing further steric hindrances to the antibodies, α-CD20 were labeled through SPAAC between azide-functionalized A488 and part of the terminal DBCO (see Figure S2). At low DBCO loading (DOF ≤ 10), the microscopic dissociation constants (Kd,micro) of the functionalized α-CD20 (5–6 nM, see Figure 2b,c) were comparable with that of FITC-labeled rituximab (4.82 ± 0.74 nM, see Figures S18 an 2b(i)) and the reported Kd,micro (Kd = 4.96 ± 0.21 nM).26 The CD20 binding affinity significantly decreased when the DOF increased to above 15. The Kd,micro of α-CD20(DBCO)15 and α-CD20(DBCO)18 were 65% and 140% higher than that of FITC-labeled α-CD20 (see Figure 2a,b) because of the steric hindrances introduced by the bulky DBCO ligand. The negative control study carried out in the K299 cells confirmed that the functionalization of α-CD20 does not induce nonspecific binding even at high DBCO loading (see Figure S19). Given that increasing the number of terminal DBCO increased the probability of the α-CD20 reacting with the azide-functionalized PAMAM, the remaining part of this study focused on the use of α-CD20(DBCO)10 (Kd,micro = 6.1 ± 0.7 nM) as the primary tumor-targeting component.
The kinetics of SPAAC between α-CD20(DBCO)10 and different azide-functionalized PAMAM were investigated using the spectroscopic method. Figures 3b(i–iv), S20, and S21 show the time-dependent UV absorption spectra of α-CD20-(DBCO)10 after incubation with an equivalent amount of azide-functionalized PAMAM and the corresponding time-dependent absorbance at 280 and 310 nm. In general, the absorbance at 280 and 310 nm from DBCO gradually decreased after incubation with azide-functionalized PAMAM because of the structural change of cyclooctyne into triazole upon cycloaddition (see Figure 3b). The absorbance of α-CD20(DBCO)10 at 280 and 310 nm reached plateaus after incubation with PAMAM(D-89Y)8(N)29 for 60 min (see Figure 3b(v,vi)), suggesting the cycloaddition was completed and all DBCO was converted into triazole. The absorbance at 310 nm never dropped to zero because the triazole product retained some absorbance.27 The rate of change of absorbance at 280 and 310 nm followed pseudo-first-order kinetics with an observed pseudo-first-order rate constant (kobs) of 39.1 × 10−3 ± 2.6 × 10−3 min−1 (see Figure 3b(vii)). The reaction between α-CD20(DBCO)10 and PAMAM(D-89Y)8(M)7(N)23 followed a similar pattern, but the rate of cycloaddition was slightly slower (kobs = 26.6 × 10−3 ± 2.1 × 10−3 min−1) than that of PAMAM(D-89Y)8(N)29. The rates of SPAAC between α-CD20(DBCO)10, PAMAM(D-89Y)8(M)16(N)16, and PAMAM-(D-89Y)8(M)24(N)8 were significantly slower than those of the previous two azide-rich dendrimers because of lower local concentrations of terminal azide in the later PAMAM (see Figure 1c(iv)). Further concentration-dependent kinetic studies were performed to obtain a better insight into the rate of cycloaddition between α-CD20(DBCO)10 and PAMAM-(D)8(N)28. As shown in Figures 3b(iv), 3c and S22, the rate of cycloaddition (and thus the observed pseudo-first-order rate constants) increased with the equilibrium concentrations of PAMAM(D-89Y)8(N)28. The second-order rate constant (k2) of the reaction pair was found to be 232 M−1 s−1 (or k2 = 8 M−1 s−1) for each terminal azide group in the dendrimer, Figure S22, which is more than 100 times faster than its small moleculae analogues (k2 = 2.1 ± 0.2 M−1 s−1).25 The kinetics of this optimized bioorthogonal reaction pair is comparable with that of the first-generation tetrazine-TCO ligation reaction (k2 = 210 M−1 s−1), in which the ligation ligands have limited chemical stabilities in physiologic conditions.25
Hyper-cross-linked rituximab has shown to induce apoptosis (programmed cell death) in lymphoma cells;7,8,28,29 therefore, the cross-linking efficiencies of different azide-functionalized PAMAM were further investigated using size exclusion chromatography-differential refractometry (SEC-dRI, see Figure 4). Figure 4a(ii,iii) shows 40 and 47 wt % of the α-CD20(DBCO)10 were unbound after incubation with limited amounts of PAMAM(D-89Y)8(M)24(N)8 and PAMAM-(D-89Y)8(M)16(N)16, respectively. The predominant products obtained in both studies were low-molecular-weight antibody-dendrimer nanoaggregates composed of 2–4 dendrimers and 2–4 antibodies. On the other hand, PAMAM-(D-89Y)8(M)7(N)28 and PAMAM(D-89Y)8(N)29 effectively cross-linked with all α-CD20(DBCO)10 under identical physiological conditions (see Figure 4a(iv,v). Quantitative chromatographic studies indicated that approximately 50 wt % of the conjugates have a molecular weight above 1.5 × 106 Da. In situ differential refractometry studies indicated the apparent diameter of these mixtures was between 30 and 50 nm, suggesting the formation of nanoclusters upon SPAAC. The comprehensive SEC-dRI study indicated that increasing the number of terminal azide groups (and thus avidity) in the PAMAM enhances its ability to form a hyper-cross-linked premixture with α-CD20(DBCO)10. Therefore, PAMAM-(D-89Y)8(N)29 was selected for further in vitro and in vivo studies.
Figure 4.

Cross-linking efficiencies of different dual-functionalized PAMAM after incubated with α-CD20(DBCO)10 at the physiological conditions. (a) SEC-MALLS analysis of α-CD20(DBCO)10, different dual-functionalized PAMAM, and their 1:1 mixture after being incubated at 37 °C for 30 min. (b) SEC-MALLS analysis of α-CD20(DBCO)10, PAMAM(D-89Y)8(N)29, and their mixtures obtained after incubating with different molar ratios of PAMAM(D-89Y)8(N)29 at 37 °C for 30 min. The SEC-MALLS analysis indicates that PAMAM(D-89Y)8(N)29 prefers to form high molecular-weight nanoclusters with α-CD20(DBCO)10 rather than form well-defined antibody-dendrimer conjugates.
In Vitro Evaluation of the Two-Step Nanopretargeting Strategy.
A FACS binding assay was utilized to validate the two-step pretargeting strategy. The FACS study confirmed dose-dependent binding of the azide-functionalized A488 to the α-CD20(DBCO)10-treated Raji cells due to SPAAC (see Figure 5a(i)). The mean fluorescence intensity (MFI) of the Raji cells increased with the increasing concentration of azide-functionalized A488, but it never reached a plateau (see Figure 5a(iii)). By contrast, no binding was observed between a high concentration of azide-functionalized A488 and preblocked α-CD20(DBCO)10-stained Raji cells (in which the terminal DBCO was inactivated by sodium azide (NaN3) pretreatment; see Figure S23) as well as the negative control K299 cells treated under the same experimental conditions (see Figure S24). This result confirms that α-CD20(DBCO)10 undergoes SPAAC with azide-functionalized A488 after specially binding to the CD20 antigen on the Raji cells. Further FACS study confirmed the binding of fluorescent PAMAM-(D-89Y)8(N)29(Rhod)8 to the Raji cells (see Figure 5a(ii)). The MFI of the α-CD20(DBCO)10-treated Raji cells increased with the rising concentration of PAMAM-(D-89Y)8(N)29(Rhod)8. In contrast to azide-functionalized A488, saturated binding was observed when the dendrimer concentration increased to 10 nM (see Figure 5a(iii)). This phenomenon can be explained by the higher avidity of PAMAM(D-89Y)8(N)29(Rhod)8, which amplified the binding affinity.30 Control studies confirmed that the bioorthogonal reaction between PAMAM(D-89Y)8(N)29(Rhod)8 and the DBCO-labeled Raji cells was highly selective (see Figure S23). The premixture prepared from comparable amounts of α-CD20(DBCO)10 (0.6 μg/million cells) and α-CD20(DBCO)10 (800 nM/million cells) bound to the Raji cells, but the MFI of the direct labeling cells (MFI = 68) was considerably weaker than that of the pretargeting method (MFI = 117; see Figure S25). No binding was observed between PAMAM-(D-89Y)8(N)29(Rhod)8 and preblocked α-CD20(DBCO)10- stained Raji cells and K299 cells. The selective labeling of Raji cells through the two-step pretargeting method was further confirmed by confocal laser scanning microscopy (CLSM, see Figure 5b). Strong rhodamine fluorescence colocalizing with A488 fluorescence on the surface of Raji cells was observed after briefly incubating the α-CD20(DBCO)10(A488)2-pretreated cells with PAMAM(D-89Y)8(N)29(Rhod)2 in physiologic conditions. The antibody-dendrimer premixture also selectively labeled the Raji cell membrane, but their fluorescence intensities were weaker, and uneven distribution was observed. No rhodamine fluorescence was observed in the preblocked α-CD20(DBCO)10(A488)2-pretreated cells. This result confirms the observed Rhod fluorescence in the earlier pretargeted labeling can be attributed to SPAAC between membrane-bound α-CD20 and dendrimers. Significant internalization of dendrimers and antibodies was observed after incubating the stained cells in physiological conditions for another 6 h. This phenomenon can be explained by the rapid cell surface hyper-cross-linking (clustering) triggering clathrin-medicated endocytosis,30 despite rituximab being a slow internalizing antibody.
Figure 5.

In vitro evaluation of the α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29(Rhod)8-based pretargeted system. (a) Flow cytometry histograms of α-CD20(DBCO)10-pretreated Raji cells after incubation with different concentrations of (i) azide-functionalized A488 and (ii) PAMAM(D-89Y)8(N)29(Rhod)8 containing 11.6–23,220 nM of terminal azide. (iii) The plot of MFIs of α-CD20(DBCO)10-pretreated Raji cells after being incubated with different concentrations of azide-functionalized A488 and PAMAM(D-89Y)8(N)29(Rhod)8, as determined by the flow cytometry method. (b) Representative CLSM images of (i) unstained Raji cells (control); (ii) Raji cells stained with α-CD20(DBCO)10(A488)2 for 1 h, washed, with and without further incubation at 37 °C for another 6 h; (iii) Raji cells pretreated with α-CD20(DBCO)10(A488)2 for 1 h before staining with PAMAM(D-89Y)8(N)29(Rhod)8 for another 1 or 6 h; (iv) Raji cells incubated with the antibody-dendrimer premixture for 1 or 6 h; and (v) Raji cells incubated with preblocked α-CD20(DBCO)10(A488)2 for 1 h before being incubated with PAMAM(D-89Y)8(N)29(Rhod)8 for another 1 h.
The mechanisms of actions by rituximab are governed by antibody-dependent cellular cytotoxicity (ADCC) and CDC in the presence of immune effector cells.6,7 The direct anticancer activities of rituximab alone are limited in vitro.6,7 Hyper-crosslinking of membrane-bound α-CD20 has been found to increase the CDC through caspase activation pathways.28,29 Caspase 3 activation plays a central role in early stage apoptosis.31 The early stage apoptotic activity induced by PAMAM(D-89Y)8(N)29 after being cross-linked with the membrane-bound α-CD20(DBCO)10 through the pretargeting strategy was quantified by caspase 3 (active) red-DEVD-FMK assay (see Figure 6a). Raji cells treated with unmodified rituximab and α-CD20(DBCO)10 did not significantly enhance the caspase 3 activities compared with cells in the negative control group. Raji cells treated with moderate concentrations of PAMAM(D-89Y)8(N)29 also did not induce apoptosis. However, PAMAM(D-89Y)8(N)29 induced apoptosis in a dose-dependent manner in the α-CD20(DBCO)10-pretreated Raji cells. The caspase 3 activities of Raji cells that received the pretargeted treatment with α-CD20(DBCO)10 were approximately 10 times higher than that of untreated or α-CD20(DBCO)10-treated Raji cells without PAMAM-(D-89Y)8(N)29. The high apoptotic activities were likely due to the hyper-cross-linking of membrane-bound α-CD20 that induces apoptosis through calcium influx-dependent and mitochondrial signal pathways.32 By contrast, the apoptotic activity induced by an equivalent amount of antibody-dendrimer premixture was approximately 65% weaker than that when treated through the two-step pretargeting method. The lower apoptotic activities were felt to be secondary by the weaker antibody-antigen binding capacity that has been observed in FACS and time-dependent CSLM studies. Given the absence of antibody-antigen binding, the K299 cells did not show any significant apoptotic activities after being treated with different α-CD20 and dendrimer formulations.
Figure 6.

In vitro toxicities of direct and pretargeted treatments with α-CD20(DBCO)10 and nonradioactive PAMAM-(D-89Y)8(N)29/radioactive PAMAM(D-90Y)8(N)29 in CD20− K299 and CD20+ Raji cell lines. (a) Caspase 3 activities of Raji and K299 cells recorded 20 h after direct or sequential treatment with α-CD20(DBCO)10(A488)2 and/or PAMAM(D-89Y)8(N)29(Rhod)8. High caspase 3 activities indicate early stage apoptosis (i.e., programmed cell death). (b) Proliferation of K299 and Raji cells recorded 4 days after direct or sequential treatment with α-CD20(DBCO)10(A488)2 and/or PAMAM(D-89Y)8(N)29(Rhod)8, as determined by trypan blue exclusion assay. (c) Proliferation of Raji-Luc cells after direct or sequential treatment with α-CD20-(DBCO)10(A488)2 and/or nonradioactive PAMAM-(D-89Y)8(N)29 (Rhod)8/radioactive PAMAM(D-89Y)8(N)29(Rhod)8, as determined by the luciferase-based bioluminescence assay recorded 4 days after the initial treatment. The inserted number in the histograms represent the relative cell viabilities. (NB. * represents p < 0.05; n.s. represents statistically insignificant (i.e., p > 0.05).)
The proliferation of Raji and K299 cells after receiving different direct or pretargeted treatments was quantified by the trypan blue exclusion assay (see Figure 6b). Similar to previous in vitro toxicity studies, a saturated concentration (0.6 μg/106 cells) of unmodified rituximab and α-CD20(DBCO)10 showed relatively low cytotoxicities (cell viabilities ≈85%) in Raji cells and negligible cytotoxic effects in K299 cells (viability >95%). In vitro treatment with 200 nM of PAMAM(D-89Y)8(N)29 alone showed very low cytotoxic effects in both cell lines. The cell viability of the α-CD20(DBCO)10-pretreated Raji cells was significantly reduced after being treated with 50 nM of PAMAM(D-89Y)8(N)29 (viability = 73 ± 5%), whereas no cytotoxicity was observed in K299 cells. The viability of Raji cells progressively decreased when the concentration of PAMAM(D-89Y)8(N)29 increased. The viability of Raji cells progressively decreased as the concentration of PAMAM-(D-89Y)8(N)29 increased, with the viability rate of 21 ± 3% at 200 nM of PAMAM(D-89Y)8(N)29. By contrast, one-step direct treatment with premixture prepared from a comparable amount of α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29 only showed moderate cytotoxicity (42 ± 4%). The lower in vitro cytotoxicity was again consistent with the low binding efficiency of the antibody-dendrimer premixture that was observed in the FACS binding assay and time-dependent CSLM study.
A luciferase assay was employed to investigate the effect of β-radiation from 90Y-labeled PAMAM on the proliferation of luciferase-labeled Raji cells (Raji-Luc) in vitro (see Figures 6c and S26). Similar to the trypan blue exclusion assay, the bioluminescence intensities (and thus viabilities) of Raji-Luc cells dropped by 57% after being treated with the pretargeted α-CD20(DBCO)10 followed by PAMAM(D-89Y)8(N)29 treatment compared to 45% in Raji cells treated with the antibody-dendrimer premixture. We then proceeded to show that Raji-Luc cells were highly sensitive to β-radiation. In vitro treatment with 100 nM of radiolabeled PAMAM(D-90Y)8(N)29 alone reduced the bioluminescence intensity of Raji cells by 69%. PRIT with α-CD20(DBCO)10 followed by PAMAM-(D-90Y)8(N)29 effectively inhibited the proliferation of Raji-Luc cells by 95%. By contrast, direct treatment with the radiolabeled premixture only reduced the proliferation by approximately 80% (3.3 times weaker than the DBCO/PAMAM pretargeted approach).
In Vivo Evaluations of the Pretargeted Nanoimmuno-therapy and Nanoradioimmunotherapy Strategies.
The DBCO/PAMAM pretargeting strategy was further evaluated in the Raji and K299 dual-xenograft tumor models in mice. The time-dependent in vivo fluorescence imaging study confirmed PAMAM(D-89Y)8(N)29(Rhod)2 selectively accumulated in the Raji xenograft tumor, but not the K299 xenograft tumor, when mice were pretreated with α-CD20(DBCO)10(A488)2 (see Figure 7a). The saturated rhodamine epifluorescence signal was recorded 4 h after intravenous (i.v.) administration of the dendrimer and remained relatively constant afterward. Direct targeting with the antibody-dendrimer premixture also selectively accumulated in the Raji xenograft tumor, but the average epifluorescence signal was approximately three times lower than that recorded for the pretargeting group, and the epifluorescence intensities gradually decreased after reaching a plateau at 2 h after the i.v. administration. In the control study, no significant epifluorescence was observed in tumor-bearing mice administrated with preblocked α-CD20-(DBCO)10(A488)2 before PAMAM(D-89Y)8(N)29(Rhod)2 (p = 0.0920) compared with the nontreatment control group, suggesting the enhanced permeability and retention effect would not facilitate the accumulation of dendrimers in both xenograft tumors. Further ex vivo biodistribution studies confirmed the α-CD20(DBCO)10(A488)2 selectively accumulated in the Raji xenograft tumor with approximately 21%ID/g of the i.v.-administered antibody being accumulated in the Raji tumor 48 h after the administration (see Figures 7b, S27, and S28). Similar to the in vitro binding studies, an insignificant amount of the α-CD20(DBCO)10(A488)2 was accumulated in the K299 tumor. Further biodistribution studies indicated approximately 20%ID/g of the i.v.-administered α-CD20 accumulated in the Raji tumor, but apparently <4%ID/g of the α-CD20(DBCO)10(A488)2 that was accumulated in the Raji tumor in the pretargeting group (see Figures 7b and S28). This phenomenon can be explained by that Förster resonance energy transfer (FRET) from A488 in α-CD20 to rhodamine in the dendrimer upon SPAAC (see Figures S29–S32). The selective accumulation of the α-CD20(DBCO)10(A488)2 and PAMAM(D-89Y)8(N)29(Rhod)2 in the xenograft tumors was further confirmed by CLSM in ex vivo tumor sections recorded under the compensation mode (see Figure 7c).
Figure 7.

Biodistribution of rhodamine-labeled dual-functionalized PAMAM administrated via direct-targeting or pretargeting methods in K299 and Raji dual-exnograft tumor bearing mice. (a)(i) Representative time-dependent in vivo fluorescence images (λex = 570 ± 20 nm, λem = 620 ± 20 nm) of the Raji and K299 dual-xenograft tumor-bearing mice recorded after i.v. administration of α-CD20(DBCO)10(A488)2 (7.5 mg/kg) and PAMAM(D-89Y)8(N)29(Rhod)8 (25 mg/kg). The α-CD20 and dendrimer were either administrated as a premixture at day 11 (after tumor inoculation) or sequentially administrated as α-CD20 (or preblocked α-CD20) at day 10 and dendrimer at day 11 (24 h after the administration of α-CD20). The in vivo imaging study recorded the fluorescence emitted from the systemically administrated rhodamine-labeled dendrimer. The inserts show the treatment schedule and locations of the xenograft tumors. (ii) The plot of time-dependent average radiances at the regions of interest (ROI, i.e., xenograft tumor sites) of different imaging-group mice (n = 5 per group). (b) “Apparent” biodistributions of (i) α-CD20(DBCO)10(A488)2 and (ii) PAMAM(D-89Y)8(N)29(Rhod)8 determined by the ex vivo fluorescence imaging technique. The inserted cartoon shows rhodamine in the dual-functionalized dendrimer quenches the fluorescence emitted from the A488 in α-CD20 upon SPAAC because of FRET. (c) Representative CLSM images and the fluorescence intensities of A488 and rhodamine channels of the ex vivo Raji xenograft tumor sessions collected from different imaging-group mice. The confocal microscope was operated under the compensation mode to allow concurrent observations of fluorescence emitted from A488 and rhodamine. (NB. * denotes p < 0.05, and n.s. denotes insignificant.)
Comprehensive hematological, renal, and hepatic studies indicated that healthy CD1 mice tolerated therapeutic doses of α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29 without any significant complications (see Table S1). The Raji xenograft tumor model in mice was utilized to evaluate the antitumor efficacies of the DBCO/PAMAM pretargeted immunotherapy and RIT strategies (see Figure 8a). The antitumor effects of α-CD20 and dual-functionalized PAMAM alone were limited. While rituximab and α-CD20(DBCO)10 (7.5 mg/kg) slightly increased the median survival by approximately 10% (see Figures 8b(i–iii), 8c, and S33a; p = 0.0139 and 0.0294 compared with the nontreatment group, respectively), PAMAM(D-89Y)8(N)29 (25 mg/kg) did not show any anticancer activity by itself (see Figures 8b(iv), 8c, and S33a; p = 0.1685 compared with the nontreatment group). The administration of radiolabeled PAMAM(D-90Y)8(N)29 (25 mg/kg, 7.5 mCi/kg) slightly improved the efficacy and increased the median survival by 17% (see Figures 8b(v), 8c, S33b; p =0.0016 compared with the nontreatment group) likely because of the systemic nonspecific exposure to β-radiation. Pretargeted immunotherapeutic approach through consecutive administration of α-CD20(DBCO)10 followed by PAMAM(D-89Y)8(N)29 significantly increased the average time to progression to approximately 16 days and median survival to approximately 44 days (see Figure 8b(vii)). At the study end point, 33% of the treated mice achieved a prolonged survival with significant tumor regression (see Figures 8b(vi) and 8c). Treatment with the premixture slightly increased the median survival by 22% compared with the nontreatment group (see Figures 8b(vii) and S33a; p = 0.0054 compared with the nontreatment group and p = 0.0461 compared with the pretargeted immunotherapy group), consistent with poor tumor uptake of the antibody-dendrimer conjugates, as demonstrated by the in vivo biodistribution studies. The antitumor effect of the control pretargeted immunotherapy group that was sequentially treated by the preblocked α-CD20(DBCO)10 and PAMAM-(D-89Y)8(N)29 was similar to that of the α-CD20(DBCO)10- treated group without PAMAM(D-89Y)8(N)29 (see Figures 8b(viii), 8c, and S33a; p = 0.7389), demonstrating that hyper-cross-linking on the surface of tumor cells through SPAAC between the cell membrane-bound α-CD20 and the dendrimer is essential to inducing tumor cell killing. PRIT through consecutive administration of α-CD20(DBCO)10 followed by radiolabeled PAMAM(D-90Y)8(N)29 showed even stronger antitumor effects than what was seen in the pretargeted immunotherapy group (see Figures 8b(ix), 8c, and S33b,c; p =0.0373 compared with pretargeted immunotherapy group). Regression of xenograft tumor was observed in all mice treated with PRIT. At the study end point, 67% of the treated mice were tumor-free, and all remaining mice had tumors smaller than baseline measurements (see Figure 8b(ix)). Although the antitumor effects of the radiolabeled premixture were stronger than those of the nonradioactive premixture, only 16% of the mice had complete response to the treatment (see Figures 8b(x), 8c, and S33b). Given the poor retention of PAMAM-(D-90Y)8(N)29 in the tumor, PRIT with preblocked α-CD20(DBCO)10 and PAMAM(D-90Y)8(N)29 showed limited antitumor activities (see Figures 8b(xi), 8c, and S33b). Overall, the xenograft tumor-bearing mice tolerated the systemic treatment with the radioactive dendrimer (see Figure S34).
Figure 8.

In vivo antilymphoma efficacy study in Raji xenograft tumor-bearing mice. (a) Treatment schedule. Xenograft tumor was inoculated by s.c. injection of 2 × 106 Raji cells in the left flank at day 0. Xenograft tumor-bearing mice received treatment at day 12. Therapeutics were either i.v. administrated once at day 12 or consecutively administrated α-CD20 at day 12, followed by dendrimer at day 13 (24 h apart). (b) Individual tumor growth curves of nontreatment group mice and mice treated with α-CD20(DBCO)10 (7.5 mg/kg) and/or PAMAM(D-89Y)8(N)29 (25 mg/kg)/PAMAM(D-89Y)8(N)29 (25 mg/kg, 7.5 mCi/kg). (c) Kaplan–Meier survival curves of Raji xenograft tumor-bearing mice (without tumor larger than 1000 mm3) after receiving different treatments. (n = 6 per group; * denotes p < 0.05; M.S.T. denotes median survival time.) (d) Representative CLSM images of anti-PCNA-, anticaspase 3-, and anti-γ-H2AX-stained Raji xenograft tumor section collected after receiving different direct or pretargeted treatments. In the anti-PCNA-stained tumor sections, all nuclei were stained with DAPI (blue fluorescence). The intense red fluorescence nuclei were PCNA-positive proliferating nuclei. In the anticaspase 3-stained tumor sections, nuclei were stained with DAPI (blue fluorescence). The strong red fluorescence were caspase 3-positive cells. In the anti-γ-H2AX-stained tumor sections, nuclei were stained with DAPI (blue fluorescence). The strong red fluorescence foci labeled the DNA double-strand break.
Further histopathologic studies were performed in the Raji xenograft tumors to investigate the underlying mechanisms of the DBCO/PAMAM pretargeted immunotherapy and RIT strategies. Proliferating cell nuclear antigen (PCNA) immunohistochemistry stain indicated the majority (>90%) of cells in the xenograft tumor treated with PRIT were nonproliferating (see Figures 8d(i) and S35).33 Treatments with pretargeted immunotherapy with α-CD20(DBCO)10 and PAMAM-(D-89Y)8(N)29 and direct RIT with the radiolabeled premixture also effectively decreased the number of proliferating cells by 40% and 50%, respectively, whereas other treatments had no significant effect on the cell proliferation. The ex vivo results were consistent with the data from the in vitro studies where the Luc expression of Raji-Luc treated with PRIT showed very low Luc expression (≈5% compared with the nontreatment control group). Caspase 3 immunohistochemistry stain was used to quantify the early stage apoptosis in xenograft tumor (see Figures 8d(ii) and S36).31 The average number of cells expressing cleaved caspase 3 in the PRIT group was approximately 80% more than that recorded in the pretargeted immunotherapy. The higher apoptotic activities recorded in both pretargeted immunotherapy and PRIT groups were consistent with the in vitro data in which hyper-cross-linking enhanced the CDC of α-CD20. Treatments with an β particle-emitting PAMAM(D-90Y)8(N)29 significantly increased the incidences of DSBs in xenograft tumor. Systemic administration of PAMAM(D-90Y)8(N)29 alone increased the number of γ-H2AX foci (proportional to the number of DSBs) per cell by 300%, compared with the nontreatment control group (<1 focus per cell, see Figures 8d(iii) and S37).34 Single administration of the radiolabeled antibody-dendrimer pre-mixture significantly increased the γ-H2AX expression (24 γ-H2AX foci per cell). Given the improved tumor uptake and retention of PAMAM(D-90Y)8(N)29, PRIT further increased the number of γ-H2AX foci to approximately 45 foci per cell and more significant DNA damages. Therefore, we demonstrated the strong antitumor activities observed in the PRIT group were due to the reduction of proliferation, induction of apoptosis, and increased DNA damages caused by the improved uptake of the dual-functionalized dendrimer.
The antitumor effects of the pretargeted immunotherapy and RIT strategies were further evaluated in a more aggressive disseminated lymphoma model through tail-vein injection of Raji-Luc cells. Based on previous studies, the injected lymphoma cells engraft and form tumors in the lymph nodes, liver, lung, spinal cord, hind-limb joints, and brain.29,35 Mice with diseminated lymphoma received 2 cycles of systemic treatments 5 and 12 days after the xenotransplantation of Raji-Luc cells (see Figure 9a). The progression of disease was monitored by time-dependent full-body bioluminescence imaging. As shown in Figures 9b(i–iv) and S38–S41, the bioluminescence signals from viable Raji-Luc cells progressively increased after treatment with α-CD20 and nonradioactive PAMAM(D-89Y)8(N)29. These treatments failed to increase the paralysis-free survival time (0% paralysis-free survival; p = 0.0920–0.4929 compared with the nontreatment group, see Figure 9c). Treatment with PAMAM(D-90Y)8(N)29 slightly delayed the propagation of lymphoma and increased the median survival by approximately 20 days (12.5% paralysis-free survival, p = 0.0795 compared with the nontreatment group, see Figure 9c) likely because of systemic nonspecific exposure to β-radiation. Pretargeted immunotherapy with α-CD20-(DBCO)10 followed by nonradioactive PAMAM(D-89Y)8(N)29 significantly slowed down the progression of the disease (see Figure 9b(vi)) and nearly doubled the median paralysis-free survival time to 81 days (12.5% paralysis-free survival, p = 0.0090 compared with the nontreatment group; see Figure 9c), whereas the treatment with the antibody-dendrimer premixture only slightly increased the media paralysis-free survival time by approximately 33% (0% paralysis-free survival, p = 0.1354 compared with the nontreatment group; see Figure 9c). Similar to the efficacy study in the xenograft tumor model, PRIT with α-CD20(DBCO)10 followed by PAMAM(D-90Y)8(N)29 showed the most superior therapeutic activities. The bio-luminescence intensities of xenotransplanted mice recorded 46 days after the inoculation were similar to those without the tumor (see Figure 9b(ix)), suggesting the pretargeted treatment completely eradicated the disseminated Raji-Luc cells. All mice that received PRIT survived free of paralysis at the study end point (100% paralysis-free survival, p = 0.0002 compared with the nontreatment arm, p = 0.0098 compared with the pretargeted immunotherapy group; see Figure 9c). One-step treatment with the radiolabeled premixture also improved the survival. The bioluminescence intensities recorded 35 days after xenotransplant were similar to those recorded before inoculation (see Figure 9b(x)), indicating the treatment effectively inhibited the proliferation of transplanted Raji-Luc cells. However, the bioluminescence intensities in 62.5% of the treated mice significantly increased after day 32. The median survival was 83% longer than that of the nontreatment group (p = 0.0029 compared with the nontreatment group, see Figure 9c). At the study end point, 37.5% of the mice that received treatment with the radiolabeled premixture survived without paralysis (see Figure 9c). The inferior survival in the mice treated with one-step RIT (p = 0.0100 compared with the PRIT group) was related to less radioactivity accumulated at the site of tumor in the direct RIT group (3.4 times; p = 0.0004 compared with the PRIT group), as indicated by Cerenkov luminescence imaging recorded 5 days after the treatment completion (see Figure S42). Although both RITs used a systemically administrated therapeutic dose of Y-90 (2 × 7.5 mCi/kg), no significant radiotoxicities (e.g., weight loss or skin hemorrhage) were observed (see Figure S43) because unbound 90Y-labeled dendrimers were excreted from the body, as indicated by the Cerenkov luminescence imaging (see Figure S42).36
Figure 9.

In vivo anticancer activity study in the disseminated B-cell NHL xenotransplant model. (a) Treatment schedule. B-cell NHL was induced by i.v. tail-vein injection of 2 × 106 Raji-Luc cells at day 0. Lymphoma mice received two direct/pretargeted treatments with α-CD20(DBCO)10 (7.5 mg/kg) and/or PAMAM(D-89Y)8(N)29 (25 mg/kg)/PAMAM(D-89Y)8(N)29 (25 mg/kg, 7.5 mCi/kg) at days 5 and 12. For pretargeted therapies, the dendrimer was administrated 24 h after the administration of α-CD20. Progression of disease was monitored by in vivo bioluminescence imaging on days 0, 5, 11, 19, 25, 32, and 46. Mice were intraperitoneally administrated with 3 mg firefly D-luciferin 15 min prior to each imaging session. Cerenkov bioluminescence imaging studies were performed at days 11 and 19 prior to the administration of firefly D-luciferin to quantify the amount of Y-90-labeled dendrimers accumulated in the body. (b) Time-dependent whole-body bioluminescence intensities of individual lymphoma mice recorded before and after different treatments. The inserts show in vivo bioluminescence images recorded 46 days after the xenotransplantation. (c) Kaplan–Meier paralysis-free survival curves of Raji-Luc xenotransplanted mice after receiving different treatments. (n = 7–8 per group; * denotes p < 0.05; M.S.T. denotes median survival time.)
CONCLUSIONS
In conclusion, we have developed a pretargeting strategy for the treatment of NHL. This strategy is based on an improved multiple-bioorthogonal ligation reaction between a DBCO-functionalized antibody pretargeting component and an azide-dual-functionalized dendrimer. Our pretargeting strategy not only improved the delivery of radionucleotides to the target cells but also enhanced the CDC of antibody through hyper-cross-linking with the dual-functionalized dendrimer, which effectively induced apoptosis and inhibited the proliferation of tumor cells. Comprehensive in vivo efficacy studies in xenograft as well as aggressive disseminated lymphoma murine models demonstrated this bioorthogonal pretargeting strategy significantly improved the antitumor effects of antibody therapy without incurring significant systemic toxicities. With a large number of antibodies against various tumor antigens, such as CD19, CD30, CD40, and CD79b, currently in clinical development, this robust pretargeting strategy has broad applicability as a diagnostic and therapeutic tool and may be adopted to treat various solid tumors and hematologic malignancies.
MATERIALS AND METHODS
Materials.
Poly(amidoamine) dendrimer (ethylenediamine core, generation 4.0, PAMAM G4; 10 wt % in methanol; 64 terminal primary amine group per dendrimer; molecular weight = 14,214), azide-fluor 488 (azide A488, ≥90%), dimethyl sulfoxide (anhydrous, ≥99.9%), yttrium(III) chloride hexahydrate (89YCl3, 99.9%), sodium bicarbonate (BioReagent), ammonium acetate (Molecular Biology, ≥98%), deuterium oxide (99.9 atom % D), bovine serum albumin (fraction V lyophilized powder), and sterilized Whatman polyvinyli-dene fluoride (PVDF) syringe filters (0.2 μm pore size) were purchased from Sigma-Aldrich (St. Louis, MO). 1,4,7,10-Tetraazacy-clododecane-1,4,7,10-tetraacetic acid N-hydroxysuccinimide ester (DOTA NHS ester, ≥90%), methyl oligo(ethylene glycol) N-hydroxysuccinimide ester (mEG8-NHS), rhodamine NHS ester, Alexa Fluor 488 NHS ester (A488 NHS ester), sodium azide (Laboratory grade), PE-conjugated anti-CD20 (clone: 2H7; 0.04 mg/mL; molecular weight ≈390 kDa), double deionized water (HPLC grade, submicron filtered), Pierce BCA Protin Assay Kit, and the Slide-A-Lyzer M Dialysis Device (10K MWCO) were purchased from Fischer Scientific (Hampton, NH). Fluorescein isothiocyanate (isomer 1, 95%) was purchased from Alfa Aesar (Haverhill, MA). DBCO-EG13-NHS ester was purchased from BroadPharm (San Diego, CA). Azide-functionalized oligo(ethylene glycol) N-hydroxysuccinimide ester (azido-PEG8-NHS) was purchased from Conju-Probe, LLC (San Diego, CA). Radioactive 90yttrium(III) trichloride (90YCl3) in 0.1 M hydrochloric acid was purchased from PerkinElmer (Shelton, CT). The antihuman CD20 antibody, rituximab, was provided by Genentech, Inc. (San Francisco, CA) under a Material Transfer Agreement. Rituximab was supplied at a concentration of 10 mg/mL(69.5 μM) in an aqueous buffer containing 0.7 mg/mL of polysorbate 80, 9 mg/mL of sodium chloride, and 7.35 mg/mL of sodium citrate dehydrate. Rituximab was used without further purification.
Functionalization and Characterization of Antibodies.
Functionalization of Rituximab with DBCO Ligand.
An antihuman CD20 antibody, rituximab, was derivatized via amine-NHS ester coupling reaction between NHS-ester activated DBCO ligand and the primary amines in the antibody. For a typical functionalization of 10 mg of α-CD20, 1 mL of unmodified α-CD20 (10 mg/mL, pH 6.5;69.5 nmol) was first diluted with 1 mL of 0.2 M sodium bicarbonate buffer (pH 8.5) to adjust the pH to about 8.0 before it was mixed with a 5, 10, 20, 30, or 40 mol equiv of DBCO-EG13-NHS (24.65 mM in DMSO; for target α-CD20:DBCO molar ratios of 1:5, 1:10, 1:20, 1:30, and 1:40 respectively). The mixture was gently vortex mixed (200 rpm) at 20 °C for 1.5 h. The DBCO-functionalized antibodies were purified via dialysis against PBS at 5 °C for 3 days (6 cycles) via 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes. The purified antibodies were sterilized via passing through a PVDF syringe filter (0.2 μm pore size) and stored at 5 °C for further studies.
Functionalization of the DBCO-Conjugated anti-CD20 Antibody with Azide-Fluor 488.
To avoid dye labeling and to further increase the steric hindrance of different DBCO-functionalized α-CD20 as well as to affect the binding affinities of antibodies for the in vitro selection study, azide-A488 was conjugated to α-CD20 through SPAAC with the terminal DBCO in the antibody. Briefly, DBCO-functionalized α-CD20 (2 mg/mL in PBS) was incubated with calculated amounts of azide Fluor 488 (50 mg/mL in DMSO) at 20 °C for 1 h (in the dark). The terminal DBCO to azide A488 molar ratio was 1:1. The dye-labeled antibodies were first purified via prewashed Zaba spin desalting columns (7,000 MWCO, ThermoFisher). The Alexa Fluor 488-labeled antibodies were further purified via equilibrium dialysis against PBS for 3 days at 5 °C (in the dark) via 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes. The purified antibodies were sterilized via passing through a PVDF syringe filter (0.2 μm pore size) and stored at 5 °C (in the dark) before further experiments took place. The concentrations of the Alexa Fluor 488-labeled antibodies were quantified with the BCA protein assay (Pierce) according to the manufacturer’s instructions.
Functionalization of the Selected DBCO-Functionalized anti-CD20 Antibody with Alexa 488 for in Vitro and in Vivo Studies.
Selected α-CD20, α-CD20(DBCO)10, was directly labeled with Alexa 488 via an amine-NHS coupling reaction for further in vitro and in vivo studies. For a typical functionalization of 2 mg of α-CD20(DBCO)10, 2 mL of unmodified α-CD20 (2 mg/mL, pH 7.4; 13.3 nmol) was first diluted with 0.5 mL of sodium bicarbonate buffer (0.2M, pH 8.5) to adjust the pH to about 8.0 before being mixed gently with a 5 mol equiv of A488 NHS ester (15.6 mM in DMSO, 4.3 μL) via vortex mixing (200 rpm) at 20 °C for 1.5 h (in the dark). The A488-labeled antibody was purified via dialysis against PBS at 5 °C for 3 days (6 cycles) via 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes. The purified antibodies were sterilized through a PVDF syringe filter (0.2 μm pore size) and stored at 5 °C before further experiments took place. The concentrations of the Alexa Fluor 488-labeled antibodies were quantified with the BCA protein assay (Pierce) according to the manufacturer’s instructions.
Preblocking DBCO-Functionalized anti-CD20 Antibody.
The terminal DBCO in α-CD20(DBCO)10 were inactivated via treatment with a large excess of sodium azide (NaN3) for the control studies. “Preblocked” α-CD20(DBCO)10 was obtained from incubation of a 2 mg/mL of α-CD20(DBCO)10 in 1× PBS containing 0.1% NaN3 at 4 °C for 24 h. The terminal DBCO undergoes SPAAC with NaN3 to form a stable triazole that cannot undergo further SPAAC. The preblocked α-CD20 was used without further purification.
Characterization of Functionalized Antibodies.
The degrees of functionalization (DOF, i.e., the number of DBCO conjugated to each antibody) of different DBCO-functionalized α-CD20 were determined via UV–vis absorption spectroscopy and MALDI-TOF MS methods. Briefly, the concentrations and degrees of the DBCO incorporation of different purified DBCO-conjugated anti-CD20 were determined spectroscopically using an absorption coefficient of DBCO at 310 nm (εDBCO,310 nm) = 12,000 M−1 L cm−1, an absorption coefficient of anti-CD20 (rituximab) at 280 nm (εrituximab,280 nm) = 1.7 mg−1 mL cm−1 = 245,000 M−1 L cm−1, and a DBCO correction factor at 280 nm (CFDBCO,280 nm) = 1.089 according to the manufacturer’s instructions. The mean number-average molecular weights (mean Mn) of different functionalized anti-CD20 were determined via an AB Sciex 5800 MALDI-TOF mass spectrometer in the UNC Michael Hooker Proteomics Center at the UNC School of Medicine. Before the mass spectroscopy analysis took place, all antibody samples were desalted via dialysis against deionized water at 5 °C for 24 h (3 cycles) using 2 kDa cutoff Slide-A-Lyzer Dialysis cassettes. Desalted samples(0.5 μL of 5 mg/mL) were first loaded on a Teflon-coated plate using a “dried drop” method with sinapinic acid as the matrix. Samples were laser-irradiated with 500 shots, and the corresponding molecular weight was determined by following the standard MALDI-TOF mass spectroscopy method using the peptide calibration mixture 4700 (ABSciex) and BSA as the calibration standards.
Functionalization and Characterization of Dendrimers.
Synthesis of DOTA-Functionalized PAMAM G4 Dendrimer, PAMAM-(D)8.
DOTA was conjugated to the PAMAM G4 dendrimer via the amine-NHS ester coupling reaction. The target PAMAM G4 to DOTA molar ratio was 1:15. A typical procedure for the synthesis of a DOTA-functionalized PAMAM dendrimer was as follows. DOTA-NHS ester (402 mg, 528 μmol) was first dissolved in 1 mL of a cold NaHCO3 buffer (0.05 M, pH 8) before adding to a methanolic solution of the PAMAM G4 dendrimer (5 g, 10 wt % of G4 PAMAM in methanol; 500 mg PAMAM G4, 35.2 μmol). The mixture was stirred at 20 °C in the dark for 24 h. The dendrimer solution was dialyzed against deionized water (4 L) at 20 °C for 3 days (6 cycles) in 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes. The purified dendrimer was sterilized by passing through PVDF syringe filter (0.2 μm pore size) before lyophilized to yield 491 mg (80%) of a white solid. The lyophilized dendrimer was store at −20 °C in the dark before further experiments took place.
Synthesis of DOTA and Azide Dual-Functionalized PAMAM, PAMAM(D)8(M)x(N)y (x + y ≈ 30).
DOTA-functionalized PAMAM G4, PAMAM(D)8, was further functionalized with different target molar ratios of methoxy and azide-terminated oligoethylene glycol NHS esters (either 1:0, 3:1, 1:1, 1:3, or 0:1) via a standard amine-NHS ester coupling reaction. The target dendrimer to oligoethylene glycol molar ratio was 1:50. In a typical reaction, 30 mg of PAMAM(D)8 (1.7 μmol) was first dissolved in 750 μL of a 0.1 M NaHCO3 buffer (pH 8) before being vortex mixed with 340 μL of 0.25 M methoxy or azide-functionalized PEG9-NHS ester (85 μmol, in DMSO) at 20 °C for 5 h. The functionalized dendrimer was purified via equilibrium dialysis against deionized water (4 days, 8 cycles) using 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes. The purified dendrimer was sterilized via passing through a PVDF syringe filter (0.2 μm pore size) before being lyophilized to yield 22–25 mg (≈ 43%) of a white solid. The lyophilized dendrimer was stored at −20 °C in the dark before further experiments took place.
Loading Nonradioactive 89Yttrium(III) into DOTA and Azide Dual-Functionalized Dendrimer, PAMAM(D-89Y)8(M)x(N)y (x + y ≈ 30).
Nonradioactive yttrium(III) (89Y3+)-labeled dual-functionalized PAMAM G4 was prepared under optimized chelation conditions. Standard 1 M yttrium(III) chloride (89YCl3) solution was prepared by dissolving a calculated amount of 89YCl3·6H2O into 0.05 M hydrochloric acid. In a typical loading reaction, 10.4 μL of the 1 M 89YCl3 solution (10.4 μmol, 2 mol equiv per conjugated DOTA) were first neutralized with 190 μL of 1 M of ammonium acetate solution before being added to an aqueous solution containing 20 mg of PAMAM(D)8(M)x(N)y (≈650 nmol, dissolved in 200 μL of deionized water). The mixture was incubated at 37 °C for 4 h. The 89yttrium-(III)-loaded PAMAM was purified via equilibrium dialysis against deionized water using 10 kDa cutoff Slide-A-Lyzer Dialysis cassette for 24 h (3 cycles). The purified dendrimer was sterilized by passing through PVDF syringe filter (0.2 μm pore size) before lyophilized to yield 18 to 20 mg (≈95%) of a white solid. The lyophilized dendrimer was stored at −20 °C in the dark before further experiments took place.
Loading Radioactive 90Yttrium(III) into DOTA and Azide Dual-Functionalized Dendrimer, PAMAM(D-90Y)8(M)x(N)y (x + y ≈ 30).
Radioactive 90Y3+ was loaded into a selective DOTA-functionalized dendrimer via an optimized chelation method. Briefly, radioactive 90YCl3 in 0.1 M hydrochloric acid was neutralized in situ before being loaded into the dual-functionalized dendrimer. For a typical preparation of 30 mg of 90Y3+-labeled PAMAM (≈ 4.5 mCi), 200 μL of 90YCl3 (≈ 20 mCi) in 0.1 M HCl was first added slowly into 400 μL of 1:1 v/v of 3 M of ammonium acetate: 0.2 M of sodium hydrocarboncate before mixed with 1.2 mL of a dendrimer solution (25 mg/mL in H2O). The mixture was stirred at 37 °C for 3 h. After cooling down to 20 °C, the 90Y3+-labeled dendrimer was purified via equilibrium dialysis against deionized water used a 10 kDa cutoff Slide-A-Lyzer Dialysis cassette at 20 °C for 48 h (5 cycles). The purified dendrimer was sterilized through a PVDF syringe filter (0.2 μm pore size) before further experiments took place. The radioactivity of the 90Y3+-labeled dendrimer was quantified with a Geiger counter. The labeling efficiency was about 90%.
Labeling Y(III)-DOTA and Azide Dual-Functionalized PAMAM with Rhodamine, PAMAM(D-Y)8(M)x(N)y(Rhod)8 (x + y ≈ 30).
Selected yttrium(III)-DOTA and azide dual-functionalized PAMAM G4 was further functionalized with rhodamine through an amine-NHS ester coupling reaction with rhodamine NHS ester. The target dendrimer to rhodamine molar ratio was 1:15. For the labeling of PAMAM(D-89Y)8(N)29, PAMAM(D-89Y)8(N)29 (10 mg, 330 nmol) was first dissolved in a 200 μL NaHCO3 buffer (0.1 M, pH 8.0) before being vortex mixed with 25 mg/mL (47 mM in DMSO) of rhodamine-NHS (105 μL, 4.9 μmol) at 20 °C in the dark for 5 h. The rhodamine-labeled PAMAM was purified by dialysis against deionized water using 10 kDa cutoff Slide-A-Lyzer Dialysis cassettes for 3 days (6 cycles). The purified dendrimer was sterilized via passing through a PVDF syringe filter (0.2 μm pore size) before being lyophilized to yield about 8 mg (≈70%) of a bright red solid. The lyophilized dendrimer was stored at −20 °C in the dark before further experiments took place.
Dendrimer Characterization.
The mean number-average diameter (Dn), and mean zeta potential (ζ) of differently modified dendrimers were determined by dynamic light scattering and an aqueous electrophoresis method using a Zetasizer Nano ZS Instrument (Malvern, Inc.). All measurements were based on the average of three separate measurements. Infrared spectroscopy was conducted using a Nicolet 380 Fourier transform infrared (FTIR) spectrometer (Thermo Scientific) equipped with a Smart iTX attenuated total reflectance (ATR) accessory in the Nanomedicines Characterization Core Facility at the UNC School of Medicine. The spectral resolution was 4 cm−1, and 128 scans were averaged per spectrum. 1H NMR spectroscopy was conducted using a 400 MHz 400MR NMR spectrometer (Agilent Technologies) at the UNC Eshelman School of Pharmacy operated at 298 K with 1024 scans being averaged per spectrum, and all chemical shifts were reported in ppm. Recorded spectra were processed using MestReNova (version 10.0.2). UV–vis absorption spectra were collected in a NanoDrop ND-1000 UV–vis spectrophotometer (Thermo Scientific).
Kinetics Study of SPAAC among DBCO-Functionalized anti-CD20 Antibody, α-CD20(DBCO)10, and Azide-Functionalized PMAMA under Physiological Conditions.
The kinetics of strain-promoted 1,3-dipolar cycloaddition between α-CD20(DBCO)10 and different azide-functionalized PAMAM under the physiological conditions (1X PBS, pH 7, 37 °C) were evaluated via time-dependent UV–vis spectroscopy. In a typical study, 0.5 mL of 1 mg/mL (6.3 μM in 0.1 M PBS) of α-CD20(DBCO)10 and 0.5 mL of 195 μg/mL (6.3 μM in 0.1 M PBS) of azide-functionalized PAMAM were first incubated at 37 °C for 15 min. The PAMAM solution was then transferred to the α-CD20(DBCO)10 solution. Time-dependent UV–vis spectra were recorded immediately (within 15 s) after mixing in a PerkinElmer Lambda 20 UV–vis spectrometer operated at 37 °C. The time-dependent absorbance at 280 and 310 nm were plotted versus time, fitted to a first-order exponential decay curve <MI> to determine the pseudo-first-order rate constant (kobs = 1/t1) via the Origin 2017 (OriginLab Corporation) software package. The study between α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29 was repeated using two other concentrations of PAMAM(D-89Y)8(N)29 (1.58 μM and 6.30 μM), and the pseudo-first-order rate constants from all three experiments were plotted versus the dendrimer concentration and fitted to a straight line. The gradient of the fitted line corresponded to the concentration-dependent second-order rate constant.
Size Exclusion Chromatography-Differential Refractometry (SEC-dRI).
SEC-dRI, also known as size-exclusion chromatography-multi angle laser scattering (SEC-MALS), was employed to monitor the cross-linking between the α-CD20(DBCO)10 and dual-functionalized dendrimers under the physiological conditions. In the initial selection study, different dual-functionalized dendrimers (126 nM) were incubated with a large excess of α-CD20(DBCO)10 (3.15 μM, 25 mol equiv) at 37 °C for 30 min before the SEC-dRI study was conducted via an SEC-dRI instrument composed of a Wyatt DAWN EOS light scattering instrument interfaced with an Amersham Biosciences Akta FPLC, Wyatt Optilab refractometer, and Wyatt dynamic light scattering module at the UNC Macromolecular Interaction Facility. A further concentration-dependent study was performed on the selected dendrimer, PAMAM(D-89Y)8(N)29, in which the dendrimer was incubated with 0.5, 1.0, and 2.0 mol equiv of α-CD20(DBCO)10 at 37 °C for 30 min before the SEC-dRI study was conducted.
Cell Cultures.
Raji cells were purchased from the American Type Culture Collection (ATCC). B-Luciferase GFP Raji (Raji-Luc) cells were purchased from Biocytogen LLC (Worcester, MA). Karpas 299 (K299) cells were purchased from Sigma-Aldrich (St. Louis, MO). All lymphoblastic cell lines were cultured in an RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin in a 37 °C atmosphere supplemented with 5% CO2. The cell densities were determined with an Orflo Moxi Z Mini Automated Cell Counter (Orflo, Ketchum, ID). Phenol red-free culture media were used to culture the cancer cells used in all flow cytometry studies.
Quantification of CD20 Antigen Expressions.
CD20 antigen expressions in Raji and K299 cells were quantified using the commercial source anti-CD20 antibody FACS binding assay. Briefly, viable cancer cells were first washed with FACS buffer (1× PBS with0.5 w/v% BSA, 0.1 wt % sodium azide, and 2 mM EDTA) and blocked with 2% FBS before being stained with phycoerythrin (PE)-conjugated anti-CD20 antibody (clone: 2H7; ThermoFisher, catalog number: MA1–10137) according to the manufacturer’s instructions. The stained cells were analyzed on a CyAn ADP flow cytometer (Beckman Coulter) in the UNC Flow Cytometry Core Facility. A minimum of 20,000 cells were analyzed for each sample. Data analysis was performed with FlowJo software (version 10.0).
Direct FACS Binding Assay for Antibody Selection.
The direct FACS binding assay was employed to quantify the binding affinity and selectivity of different DBCO-functionalized α-CD20. In a typical binding study, cell suspension was first washed three times with FACS buffer (1× PBS with 0.5 w/v% BSA, 0.1 wt % sodium azide and 2 mM EDTA) before being blocked with FACS blocking buffer (FACS buffer with 2 v/v% FBS) at a cell density of 10 × 106 cells per mL at 4 °C for 30 min. The FBS-blocked cells were collected via centrifugation (3000 rpm, 5 min), and the cell pallet was resuspended in FACS buffer to achieve a cell density of 10 × 106 cells/mL. The calculated amount of antibody was added to the cell suspension and incubated at 4 °C for 60 min. Cells were washed three times with FACS buffer via centrifugation (3000 rpm, 5 min) to remove unbound antibodies. The stained cells were analyzed on a CyAn ADP flow cytometer (Beckman Coulter) in the UNC Flow Cytometry Core Facility. A minimum of 20,000 cells were analyzed for each sample. Data analysis was performed with FlowJo software (version 10.0). The microscopic dissociation constant (K d,micro) is defined as the concentration required to achieve half-saturated binding.
Pretargeted FACS Binding Assay.
The two-step pretargeting strategy was validated via flow cytometry. In a typical binding study, cell suspension was first washed three times with azide-free FACS buffer (1× PBS with 0.5 w/v% BSA, and 2 mM EDTA) before being blocked with FACS blocking buffer (FACS buffer with 2 v/v% FBS) at a cell density of 10 × 106 cells per mL at 4 °C for 30 min. The FBS-blocked cells were collected via centrifugation (3000 rpm, 5 min), and the cell pallet was resuspended in FACS buffer to achieve a cell density of 10 × 106 cells/mL. α-CD20(DBCO)10 (or preblocked α-CD20(DBCO)10 that pretreated with 0.1% NaN3) was added to the cell suspension to achieve an antibody concentration of 0.6 μg per million cell and incubated at 4 °C for 60 min. The antibody-treated cells were washed twice via centrifugation (3000 rpm, 5 min) to remove unbound antibodies. Desired amounts of azide-functionalized A488 or PAMAM(D-89Y)8(N)29(Rhod)8 were added to the α-CD20 treated cells and incubated at 37 °C for 30 min. The stained cells were then washed (3,000 rpm, 5 min) three times to remove unbound dye/dendrimers before being analyzed in a CyAn ADP flow cytometer (Beckman Coulter) in the UNC Flow Cytometry Core Facility. A minimum of 20,000 cells were analyzed for each sample. Data analysis was performed with FlowJo software (version 10.0). In a controlled study, comparable amounts of α-CD20(DBCO) 10 were first incubated with PAMAM(D-89Y)8(N)29(Rhod)8 at 37 °C for 30 min before the FBS-blocked cells were directly stained at 4 °C for 1 h. The premixture-stained cells were washed three times with FACS buffer before being analyzed in a CyAn ADP flow cytometer (Beckman Coulter) in the UNC Flow Cytometry Core Facility.
Cell Labeling and Detection by Confocal Microscopy.
Before the cell labeling, viable Raji and K299 cell suspensions were washed three times with ice cold PBS (1000 rpm, 5 min), and they were blocked with 2 v/v% FBS at 4 °C for 30 min. Washed cells (10 × 106 cells per mL) were then incubated with α-CD20(DBCO)10(A488)2 (0.6 μg per million cell), preblocked α-CD20(DBCO)10(A488)2 (0.6 μg per 1 × 106 cells), or PBS (as control) at 4 °C for 1 h. The cells were then washed three times with PBS before being either (i) fixed with 4% formaldehyde, (ii) further incubated with PAMAM-(D-89Y)8(N)29(Rhod)8 (0.12 μg per million cell), or (iii) incubated in a phenol red-free complete RPMI1640 culture medium at 37 °C for 1 or 7 h before fixing. After being incubated with PAMAM-(D-89Y)8(N)29(Rhod)8, cells were washed three times with PBS before being fixed with 10% neutral buffered formalin (NBF) or further incubated in a phenol red-free complete RPMI1640 culture medium at 37 °C for 6 h before fixing with 10% NBF. In a controlled study, α-CD20(DBCO)10(A488)2 was first incubated with PAMAM-(D-89Y)8(N)29(Rhod)8 at 37 °C for 30 min before the FBS-blocked cells were directly stained at 37 °C for 1 h. The stained cells were washed with PBS before fixing with 10% NBF or were resuspended in a phenol red-free complete RPMI1640 culture medium at 37 °C for another 6 h before fixing with 10% NBF. The nuclei of all fixed cells were stained with 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher) according to the manufacturer’s protocol. The stained cell suspensions were preserved with an equivalent volume of ProLong Gold (Invitrogen). Confocal fluorescence images were recorded using a Zeiss LSM 710 spectral confocal laser scanning microscope in the Microscopy Services Laboratory at the UNC School of Medicine. All confocal images were recorded in compensation mode to minimize fluorescence quenching due to FRET between A488 and rhodamine.
In Vitro Toxicity Studies.
Caspase 3 (Active) Red-DEVD-FMK Assay.
The early stage apoptosis of Raji and K299 cells after the different treatments were received was quantified with the caspase 3 (active) red-DEVD-FMK assay (Abcam, Cambridge). The in vitro study involved 11 control and treatment groups; they were: (1) nontreatment group, (2) nontreatment negative control group, (3) treatment with unmodified rituximab (0.6 μg/million cells), (4) treatment with α-CD20(DBCO)10 (0.6 μg/million cells), (5–7) treatment with 50–200 nM of PAMAM(D-89Y)8(N)29, (8–10) treatment with α-CD20(DBCO)10, followed by 50 to 200 nM of PAMAM(D-89Y)8(N)29, and (11) treatment with a premixture prepared from α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29, which contained 0.6 μg of α-CD20/million cells and 200 nM of dendrimers (preincubated at 37 °C for 30 min before treating the cells). In the pretargeting groups, cells were first treated with α-CD20(0.6 μg/million cell) and washed twice before being mixed with calculated amounts of dendrimers without further washing. In the direct treatment groups, cells were directly treated with desired amounts of antibodies, dendrimers, or a dendrimer-antibody premixture (without further washing except for the antibody treatment group). Treatment and control group cells were then plated at a density of 5000 cells/well in a black round-bottom 96-well plate and incubated at 37 °C for 20 h. Cells were then stained with the caspase 3 (active) red-DEVD-FMK assay according to the manufacturer’s protocol. The fluorescence intensities (and thus the relative caspase 3 activities) from the stained cells were recorded in a 96-well plate reader (Infinite 200 Pro, Tecan i-control) using an excitation filter at 539 ± 20 nm and an emission filter at 595 ± 20 nm.
Trypan Blue Exclusion Assay.
The proliferation of Raji and K299 cells after receiving different direct and pretargeted treatments was quantified with the trypan blue exclusion assay. Similarly to the caspase 3 assay study, the proliferation study involved 10 different groups: (1) nontreatment group, (2) treatment with unmodified rituximab (0.6 μg/million cells), (3) treatment with α-CD20(DBCO)10 (0.6 μg/million cells), (4–6) treatment with 50–200 nM of PAMAM-(D-89Y)8(N)29, (7–9) treatment with α-CD20(DBCO)10 followed by 50–200 nM of PAMAM(D-89Y)8(N)29, and (10) treatment with a premixture prepared from α-CD20(DBCO)10 and PAMAM-(D-89Y)8(N)29, which contained 0.6 μg of α-CD20/million cells and 200 nM of dendrimers. In the pretargeting groups, cells were first treated with α-CD20 (0.6 μg/million cell) and were washed twice before being mixed with calculated amounts of dendrimers without further washing. In the direct treatment groups, cells were directly treated with desired amounts of antibodies, dendrimers, or a dendrimer-antibody premixture (without further washing). In the direct treatment groups, cells were directly treated with desired amounts of antibodies, dendrimers, or a dendrimer-antibody premixture (without further washing except for the antibody treatment group). Treatment and control group cells were then plated at a density of 5000 cells/well in a black round-bottom 96-well plate and incubated at 37 °C for 4 days. The cells were then stained with an equal volume of a 0.4% trypan blue solution (Sigma). The number of viable cells (i.e., nonstained cells) and nonviable cells (i.e., stained in blue) were then counted under an optical microscope through a hemacytometer. For cell viability, % = (number of viable cells)/(total number of cells) × 100%.
Bioluminescence Assay.
The impact of β radiation on the proliferation of Raji-Luc cells was quantified with the luciferase reporter assay. Similarly to the trypan blue exclusion assay study, the bioluminescence assay study involved eight groups: (1) nontreatment group; (2) treatment with treatment with α-CD20(DBCO)10 (0.6 μg/million cells); (3) treatment with 100 nM of PAMAM(D-89Y)8(N)29; (4) treatment with α-CD20(DBCO)10 followed by 100 nM of PAMAM(D-89Y)8(N)29; (5) treatment with a α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29 premixture; (6) treatment with 100 nM of radioactive PAMAM(D-90Y)8(N)29; (7) treatment with α-CD20-(DBCO)10, followed by 100 nM of PAMAM(D-90Y)8(N)29; and (8) treatment with a radioactive α-CD20(DBCO)10 and PAMAM- (D-90Y)8(N)29 premixture. In the pretargeting groups, cells were first treated with α-CD20 (0.6 μg/million cell) and washed twice before mixed with calculated amounts of dendrimers without further washing. In the direct treatment groups, cells were directly treated with desired amounts of antibodies, dendrimers, or a dendrimer-antibody premixture (without further washing except for the antibody treatment group). In the direct treatment groups, cells were directly treated with desired amounts of antibodies, dendrimers, or a dendrimer-antibody premixture (without further washing). Treatment and control group cells were then plated at a density of 5000 cells/well in a black ultralow cluster 96-well plate (Corning, ME) and incubated at 37 °C for 4 days. Bioluminescence images were recorded before and 15 min after the addition of D-luciferin (30 μg/well) through an IVIS Kinetic Imaging System (Caliper Life Sciences, Hopkinton, MA) in the Small Animal Imaging Facility at UNC School of Medicine. All bioluminescence images were recorded with an exposure time of 60 s.
In Vivo Studies.
Animals were maintained in the Division of Comparative Medicine (an AAALAC-accredited experimental animal facility) under sterile environments at the University of North Carolina. All procedures involving experimental animals were performed in accordance with the protocols that the University of North Carolina Institutional Animal Care and Use Committee approved, and they conformed with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 86–23, revised 1985). All mice treated with radioactive 90Y-labeled dendrimers were isolated in a BSL-2 cubicle to prevent radio contamination.
In Vivo Toxicity Study.
The in vivo toxicities of DBCO-functionalized rituximab and nonradioactive PAMAM (PAMAM-(D-89Y)8(M)29 were evaluated in healthy tumor-free CD1 mice (10 weeks old, male; Charles River Laboratory, Durham, NC). The mice were divided into seven groups (five per group) for different systemic treatments. The treatments were: (1) PBS (nontreatment control);(2) unmodified rituximab (12 mg/kg); (3) α-CD20(DBCO)10 (12 mg/kg); (4) PAMAM(D-89Y)8(N)29 (40 mg/kg); (5) α-CD20-(DBCO)10 (12 mg/kg) followed by PAMAM(D-89Y)8(N)29 (40 mg/kg) 24 h later; (6) a premixture containing α-CD20(DBCO)10 and PAMAM(D-89Y)8(N)29 (40 mg/kg); and (7) preblocked α-CD20(DBCO)10 (12 mg/kg) followed by PAMAM(D-89Y)8(N)29 (40 mg/kg) 24 h later. All therapeutics were administrated via a tail-vein intravenous (i.v.) injection. 48 h after the final i.v. administration of the therapeutics, the mice were each anesthetized via a subcutaneous injection of 100 μL of a ketamine hydrochloride/xylazine hydro-chloride solution (Sigma; St Louis, MO). Circulating blood (0.6–0.8 mL) was collected directly from the heart; 100 μL of the blood sample was stored in an EDTA-coated tube at 4 °C before a blood toxicity study was conducted in the Animal Clinical Laboratory Core Facility at the UNC Medical School. The remaining blood was transferred to a microcentrifuge tube and stored at room temperature for 20 min before being centrifuged at 3000 g for 5 min to separate the red blood cells from the plasma. The isolated plasma was stored at 4 °C before being submitted to the Animal Clinical Laboratory Core Facility at the UNC School of Medicine for hepatotoxicity and nephrotoxicity studies.
Tumor Uptake and Biodistribution Study.
Time-dependent tumor uptake and the biodistribution of A488-labeled α-CD20 (α-CD20(DBCO)10(A488)2) and rhodamine-labeled PAMAM (PAMAM(D-89Y)8(M)29(Rhod)8) were determined via established fluorescence imaging method through an IVIS Kinetic Imaging System (Caliper Life Sciences, Hopkiton, MA) in the Small Animal Imaging Facility at UNC School of Medicine. A488 fluorescence data were collected with a 30 s exposure time, and a filter set was used with excitation at 465 ± 20 nm and emission at 520 ± 20 nm. Rhodamine fluorescence data were collected with 30 s exposure time and used a filter set with excitation at 570 ± 20 nm and emission at 620 ± 20 nm. All imaging parameters were kept constant for the whole imaging study. Region-of-interest values were recorded using Living Image software as the photon flux (also known as the radiance) in the total photon count per centimeter-squared per steradian (p s−1 cm−2 Sr1−). Fluorescent images of standard α-CD20(DBCO)10(A488)2 (0–22.5 μg/mL) and PAMAM(D-89Y)8(N)29(Rhod)8 (0–75.0 μg/mL) were recorded to verify the fluorescence imaging setting prior to the in vivo and ex vivo studies to confirm the radiances linear increases with concentrations of the standard samples. A further time-dependent quantitative fluorescence imaging study was performed by mixing PBS or 22.5 μg/mL (final concentration) of α-CD20(DBCO)10(A488)2 with 0 to 50 μg/mL (final concentration) of PAMAM-(D-89Y)8(N)29(Rhod)8 to verify the setting that could be used to monitor the FRET-based fluorescence quenching at λem = 520 ± 20 nm (λex = 465 ± 20 nm) due to SPAAC between the DBCO-functionalized α-CD20 and the azide-functionalized PAMAM.
Xenograft tumors were inoculated in the flanks of female athymic nude mice (Nu/Nu, 6–7 weeks old, female, 20–22 g; UNC Animal Services Core, Chapel Hill, NC) via the subcutaneous injection of 2 × 106 K299 cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI-1640/Matrigel solution in the left flank and 2 × 106 Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI-1640/Matrigel solution to the right flank. An in vivo imaging study was performed 10 days after tumor inoculation, when both xenograft tumors grew to about 100 mm3. Mice were randomized and separated into six groups (five mice per group) for different treatments. The treatments were as follows: (1) nontreatment control group, euthanized at day 12 post-inoculation; (2) single i.v. tail-vein administration of α-CD20- (DBCO)10(A488)2 (6 mg/kg) at day 10, euthanized at day 11 post-inoculation; (3) single i.v. tail-vein administration of α-CD20-(DBCO)10(A488)2 (6 mg/kg) at day 10, euthanized at day 12 post-inoculation; (4) i.v. tail-vein administration of α-CD20- (DBCO)10(A488)2 (6 mg/kg) at day 10, followed by another i.v. administration of PAMAM(D-89Y)8(N)29(Rhod)8 (20 mg/kg) at day 11 (24 h after the administration of an antibody), euthanized at day 12 post-inoculation; (5) single i.v. tail-vein administration of an antibody-dendrimer premixture containing 6 mg/kg of α-CD20-(DBCO)10(A488)2 and 20 mg/kg of PAMAM(D-89Y)8(N)29(Rhod)8 at day 11, euthanized at day 12; and (6) i.v. tail-vein administration of preblocked α-CD20(DBCO)10(A488)2 (6 mg/kg) at day 10, followed by another i.v. administration of PAMAM(D-89Y)8(N)29(Rhod)8 (20 mg/kg) at day 11 (24 h after the administration of an antibody), euthanized at day 12 post-inoculation. In group (5), the premixture was prepared by premixing desired amounts of α-CD20-(DBCO)10(A488)2 and PAMAM(D-89Y)8(N)29(Rhod)8 at 37 °C for 30 min before the administration.
For the mice in groups (1) and (4–6), time-dependent full body fluorescent images were recorded before as well as 1, 3, 5, 7, and 24 h after the i.v. administration of the rhodamine-labeled dendrimer/antibody-dendrimer premixture at day 11 post-inoculation. Rhodamine epifluorescence was observed through an IVIS Kinetic Imaging System (Caliper Life Sciences, Hopkiton, MA) in the Small Animal Imaging Facility at UNC School of Medicine. The excitation wavelength was 570 ± 20 nm, the emission wavelength was 620 ± 20 nm, and the exposure time was 30 s.
Twelve days post-inoculation (or 11 days post-inoculation for the mice in group (2)), mice were euthanized as described above, and xenograft tumors and key organs (liver, spleen, kidneys, two superficial cervical lymph nodes, lung, and heart) were harvested and weighed for an ex vivo imaging study through the IVIS Kinetic Imaging System. A488 fluorescence data were collected with a 30 s exposure time, and a filter set was used with excitation at 465 ± 20 nm and emission at 520 ± 20 nm. Rhodamine fluorescence data were collected with a 30 s exposure time, and a filter set was used with excitation at 570 ± 20 nm and emission at 620 ± 20 nm. Corrected A488 and rhodamine fluorescence data for the preserved organs in groups (2–6) were obtained by subtracting the averaged background A488 and rhodamine fluorescence from the nontreatment control group (1).
After the ex vivo imaging study, the xenograft tumors were fixed in 4% (v/v) neutral buffered formalin at 4 °C for 2 days, then 60% ethanol at 4 °C for another 2 days before being sectioned. Tumor sections were stained with DAPI to label the nucleus. The tumor sections were imaged on a Zeiss CLSM 710 spectral confocal laser scanning microscope in Microscopy Services Laboratory Core Facility at the UNC School of Medicine. The microscope was operated in compensation mode to minimize the fluorescence quenching due to FRET between A488 and rhodamine. Recorded confocal images were analyzed using the ImageJ Software package (NIH, USA).
In Vivo Anticancer Study In Raji B-Cell Non-Hodgkin’s Lymphoma Xenograft Tumor Model.
A xenograft tumor was established in the left flank of each of the male nude mice (Nu/Nu, 6–7 weeks old, 25–30 g; UNC Animal Services Core, Chapel Hill, NC) via a subcutaneous injection of 2 × 106 Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI-1640/Matrigel solution in the left flank. Twelve days after inoculation, tumor-bearing mice were randomized and separated into 11 groups (6 per group) for different treatments. Prior to treatment, all mice were ear tagged (for identification), and their initial tumor volumes and body weights were measured. The treatments were: (1) PBS (100 μL, nontreatment control group) at day 12 post-transplantation; (2) unmodified rituximab (7.5 mg/kg) at day 12 post-transplantation; (3) α-CD20(DBCO)10 (7.5 mg/kg) at day 12 post-transplantation; (4) PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 12 post-transplantation;(5) PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 12 post-trans-plantation; (6) α-CD20(DBCO)10 (7.5 mg/kg) at day 12 post-transplantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 13 post-transplantation; (7) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-89Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at day 13 post-transplantation; (8) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at day 12 post-transplantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 13 post-transplantation;(9) α-CD20(DBCO)10 (7.5 mg/kg) at day 12 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 13 post-trans-plantation; (10) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-90Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at day 13 post-transplantation; and (11) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at day 12 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 13 post-transplantation. The bodyweight and tumor volume were measured every 2–3 days with a digital scale and caliper, respectively. The tumor volume was calculated by measuring two perpendicular diameters with a caliper and using the formula V = 0.5 × a × b2, where a and b are the larger and smaller diameters, respectively.
Xenograft Tumor Histology.
A xenograft tumor was established in the left flank of each of the male nude mice (Nu/Nu, 6–7 weeks old, 20–22 g; UNC Animal Services Core, Chapel Hill, NC) via a subcutaneous injection of 2 × 106 Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI-1640/Matrigel solution in the left flank. Ten days after inoculation, tumor-bearing mice were randomized and separated into 11 groups for different treatments. The treatments were as follows: (1) PBS (100 μL, nontreatment control group) at day 10 post-transplantation; (2) unmodified rituximab (7.5 mg/kg) at day 10 post-transplantation; (3) α-CD20(DBCO)10 (7.5 mg/kg) at day 10 post-transplantation; (4) PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 10 post-transplantation; (5) PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 10 post-transplantation; (6) α-CD20(DBCO)10 (7.5 mg/kg) at day 10 post-transplantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 11 post-transplantation; (7) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-89Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at day 11 post-transplantation; (8) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at day 10 post-trans-plantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at day 11 post transplantation; (9) α-CD20(DBCO)10 (7.5 mg/kg) at day 10 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 11 post-transplantation; (10) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-90Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at day 11 post-transplantation; and (11) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at day 10 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at day 11 post-transplantation. Mice were euthanized with an overdose of carbon dioxide 48 h after the final treatment. The tumors were collected and fixed in 4% neutral-buffered formalin for 24 h at 4 °C and then stored in 70% ethanol at 4 °C for 3 weeks before being submitted to the Animal Histopathology Core Facility at UNC Medical School for sectioning. The xenograft tumors collected from the mice treated with radioactive PAMAM-(D-90Y)8(N)29 followed a similar protocol, except the weight and radioactivity of the xenograft tumors were recorded via a digital scale and Geiger counter, respectively. The radioactive tumors were first fixed at 4 °C for 24 h (in a lead-shielded container) and 70% ethanol at 4 °C for 3 weeks (in a lead-shield container). The fixed tumors were submitted to Animal Histopathology Core Facility at UNC Medical School for sectioning when the tumors were no longer radioactive. PCNA, caspase 3, and γ-H2AX immunohistochemistry stains were performed at the Translational Pathology Lab at the UNC Medical School. For quality control purposes, all staining was performed using a biological tissue automatic staining machine. All stained tumor sections were imaged on a Zeiss 710 Spectral CLSM confocal microscope in the Microscopy Services Laboratory Core Facility at the UNC School of Medicine.
In Vivo Anticancer Study In Disseminated Raji-Luc Non-Hodgkin Lymphoma Xenotransplant Model.
An in vivo anticancer efficacy study was performed with the disseminated Raji-Luc xenotransplant model. 80 athymic nude mice (5–6 weeks old, female, 20.7 ± 0.2 g, UNC Animal Services Core, Chapel Hill, NC) were randomized and divided into 11 groups (7–8 mice per group), ear punched (for identification), and weighed before the efficacy study took place. The disseminated xenotransplant model was established via a single-tail vein injection of Raji-Luc (2 × 106 cells per mouse suspended in 100 μL of PBS) in random order. The mice started treatments 5 days after the inoculation. The treatments were as follows: (1) PBS (100 μL, nontreatment control group) at days 5 and 12 post-transplantation; (2) unmodified rituximab (7.5 mg/kg) at days 5 and 12 post-transplantation; (3) α-CD20(DBCO)10 (7.5 mg/kg) at days 5 and 12 post-transplantation; (4) PAMAM(D-89Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation; (5) PAMAM-(D-90Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation; (6) α-CD20(DBCO)10 (7.5 mg/kg) at days 5 and 12 post-transplantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation; (7) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-89Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at days 6 and 13 post-transplantation; (8) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at days 5 and 12 post-transplantation, and PAMAM(D-89Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation; (9) α-CD20(DBCO)10 (7.5 mg/kg) at days 5 and 12 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation; (10) α-CD20(DBCO)10 (7.5 mg/kg) premixed with PAMAM(D-90Y)8(N)29 (25 mg/kg) and incubated at 37 °C for 30 min before i.v. at days 6 and 13 post-transplantation; and (11) preblocked α-CD20(DBCO)10 (7.5 mg/kg) at days 5 and 12 post-transplantation, and PAMAM(D-90Y)8(N)29 (25 mg/kg) at days 6 and 13 post-transplantation. Cerenkov bio-luminescence images were recorded at days 11 and 19 post-transplantation (before the administration of D-luciferin) to quantify the amount of radioactive 90Y-labeled dendrimers accumulated in the body. Time-dependent luciferase (bioluminescence) images were recorded before the xenotransplant model was established and at 5, 11, 19, 25, 32, and 46 days post-transplantation to quantify the luciferase expression from the Raji-Luc cells. Mice were intra-peritoneally administered with 3 mg of firefly D-luciferin 15 min before each bioluminescence imaging session. All bioluminescence images were recorded in an IVIS Kinetic Imaging System (Caliper Life Sciences, Hopkiton, MA) in the Small Animal Imaging Facility at UNC School of Medicine. The bodyweights and body movements of mice different controls and treatments groups were monitored every 2–3 days. The experiment end point was either the onset of hind-limb paralysis or the loss of more than 20% of the initial bodyweight.
STATISTICAL ANALYSIS
Quantitative data were expressed as mean ± SEM. The analysis of variance was completed using a one-way ANOVA in GraphPad Prism 6 software pack. The analysis of survival data was completed using a Gehan–Breslow–Wilcoxon test in GraphPad Prism 6 software pack; * p < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Microscopy Service Laboratory Core, Animal Study Core, Small Animal Imaging Facility, Animal Clinical Laboratory, Animal Histopathology Core Facility, Translation Pathology Lab, UNC Flow Cytometry Core Facility, UNC Macromolecular Interactions Facility and UNC Michael Hooker Proteomics Centre in the School of Medicine, and Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) at the University of North Carolina at Chapel Hill for their assistance with procedures in this manuscript. The UNC Flow Cytometry Core Facility, UNC Macromolecular Interactions Facility and UNC Michael Hooker Proteomics Center are supported in part by P30CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. Genentech, Inc (San Fransico, CA) is thanked for supplying rituximab and for permission to publish this work. This work was supported by the University Cancer Research Fund from the University of North Carolina and R01CA178748 (A.Z.W.) grant from the National Institutes of Health/National Cancer Institute. A.Z.W. was also supported by the National Institutes of Health Center for Nano-technology Excellence Grant U54-CA151652.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b08122.
Supporting figures and table (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2017. Ca-Cancer J. Clin 2017, 67, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Coiffier B; Lepage E; Briere J; Herbrecht R; Tilly H; Bouabdallah R; Morel P; Van Den Neste E; Salles G; Gaulard P; Reyes F; Lederlin P; Gisselbrecht C CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med 2002, 346, 235–42. [DOI] [PubMed] [Google Scholar]
- (3).Feugier P; Van Hoof A; Sebban C; Solal-Celigny P; Bouabdallah R; Ferme C; Christian B; Lepage E; Tilly H; Morschhauser F; Gaulard P; Salles G; Bosly A; Gisselbrecht C; Reyes F; Coiffier B Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J. Clin. Oncol 2005, 23, 4117–26. [DOI] [PubMed] [Google Scholar]
- (4).Molina A A decade of rituximab: improving survival outcomes in non-Hodgkin’s lymphoma. Annu. Rev. Med 2008, 59, 237–50. [DOI] [PubMed] [Google Scholar]
- (5).Boye J; Elter T; Engert A An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann. Oncol 2003, 14, 520–535. [DOI] [PubMed] [Google Scholar]
- (6).Smith MR Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene 2003, 22, 7359–68. [DOI] [PubMed] [Google Scholar]
- (7).Boross P; Leusen JH Mechanisms of action of CD20 antibodies. Am. J. Cancer Res 2012, 2, 676–90. [PMC free article] [PubMed] [Google Scholar]
- (8).Renaudineau Y; Devauchelle-Pensec V; Hanrotel C; Pers JO; Saraux A; Youinou P Monoclonal anti-CD20 antibodies: mechanisms of action and monitoring of biological effects. Jt., Bone, Spine 2009, 76, 458–63. [DOI] [PubMed] [Google Scholar]
- (9).Press OW Emerging immunotherapies for non-Hodgkin lymphomas: the tortoise approaches the finish line. Ann. Intern. Med 2000, 132, 916–8. [DOI] [PubMed] [Google Scholar]
- (10).DeNardo SJ; Kroger LA; DeNardo GL A new era for radiolabeled antibodies in cancer? Curr. Opin. Immunol 1999, 11, 563–9. [DOI] [PubMed] [Google Scholar]
- (11).Ma D; McDevitt MR; Barendswaard E; Lai L; Curcio MJ; Pellegrini V; Brechbiel MW; Scheinberg DA Radio-immunotherapy for model B cell malignancies using 90Y-labeled anti-CD19 and anti-CD20 monoclonal antibodies. Leukemia 2002, 16, 60–6. [DOI] [PubMed] [Google Scholar]
- (12).Knox SJ; Goris ML; Trisler K; Negrin R; Davis T; Liles TM; Grillo-Lopez A; Chinn P; Varns C; Ning SC; Fowler S; Deb N; Becker M; Marquez C; Levy R Yttrium-90-labeled anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma. Clin. Cancer Res 1996, 2, 457–70. [PubMed] [Google Scholar]
- (13).Dykes PW; Bradwell AR; Chapman CE; Vaughan AT Radioimmunotherapy of cancer: clinical studies and limiting factors. Cancer Treat. Rev 1987, 14, 87–106. [DOI] [PubMed] [Google Scholar]
- (14).Patra M; Zarschler K; Pietzsch HJ; Stephan H; Gasser G New insights into the pretargeting approach to image and treat tumours. Chem. Soc. Rev 2016, 45, 6415–6431. [DOI] [PubMed] [Google Scholar]
- (15).Goodwin DA; Meares CF Advances in pretargeting biotechnology. Biotechnol. Adv 2001, 19, 435–50. [DOI] [PubMed] [Google Scholar]
- (16).Goldenberg DM; Sharkey RM; Paganelli G; Barbet J; Chatal JF Antibody pretargeting advances cancer radioimmunode-tection and radioimmunotherapy. J. Clin. Oncol 2006, 24, 823–34. [DOI] [PubMed] [Google Scholar]
- (17).Park SI; Shenoi J; Pagel JM; Hamlin DK; Wilbur DS; Orgun N; Kenoyer AL; Frayo S; Axtman A; Back T; Lin Y; Fisher DR; Gopal AK; Green DJ; Press OW Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: a preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood 2010, 116, 4231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Park SI; Shenoi J; Frayo SM; Hamlin DK; Lin Y; Wilbur DS; Stayton PS; Orgun N; Hylarides M; Buchegger F; Kenoyer AL; Axtman A; Gopal AK; Green DJ; Pagel JM; Press OW Pretargeted radioimmunotherapy using genetically engineered antibody-streptavidin fusion proteins for treatment of non-hodgkin lymphoma. Clin. Cancer Res 2011, 17, 7373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sletten EM; Bertozzi CR Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed 2009, 48, 6974–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Majoros IJ; Williams CR; Baker JR Jr. Current dendrimer applications in cancer diagnosis and therapy. Curr. Top. Med. Chem 2008, 8, 1165–1179. [DOI] [PubMed] [Google Scholar]
- (21).Baker JR Jr. Dendrimer-based nanoparticles for cancer therapy. Hematology Am. Soc. Hematol Educ Program 2009, 2009, 708–19. [DOI] [PubMed] [Google Scholar]
- (22).Kobayashi H; Kawamoto S; Choyke PL; Sato N; Knopp MV; Star RA; Waldmann TA; Tagaya Y; Brechbiel MW Comparison of dendrimer-based macromolecular contrast agents for dynamic micro-magnetic resonance lymphangiography. Magn. Reson. Med 2003, 50, 758–66. [DOI] [PubMed] [Google Scholar]
- (23).Jiang X; Hu C; Arnovitz S; Bugno J; Yu M; Zuo Z; Chen P; Huang H; Ulrich B; Gurbuxani S; Weng H; Strong J; Wang Y; Li Y; Salat J; Li S; Elkahloun AG; Yang Y; Neilly MB; Larson RA; Le Beau MM; Herold T; Bohlander SK; Liu PP; Zhang J; Li Z; He C; Jin J; Hong S; Chen J miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat. Commun 2016, 7, 11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Murrey HE; Judkins JC; am Ende CW; Ballard TE; Fang Y; Riccardi K; Di L; Guilmette ER; Schwartz JW; Fox JM; Johnson DS Systematic Evaluation of Bioorthogonal Reactions in Live Cells with Clickable HaloTag Ligands: Implications for Intracellular Imaging. J. Am. Chem. Soc 2015, 137, 11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Karver MR; Weissleder R; Hilderbrand SA Bioorthogonal reaction pairs enable simultaneous, selective, multi-target imaging. Angew. Chem., Int. Ed 2012, 51, 920–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Reff ME; Carner K; Chambers KS; Chinn PC; Leonard JE; Raab R; Newman RA; Hanna N; Anderson DR Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–45. [PubMed] [Google Scholar]
- (27).Yamagishi K; Sawaki K; Murata A; Takeoka S A Cu-free clickable fluorescent probe for intracellular targeting of small biomolecules. Chem. Commun. (Cambridge, U. K.) 2015, 51, 7879–82. [DOI] [PubMed] [Google Scholar]
- (28).Zhang N; Khawli LA; Hu P; Epstein AL Generation of rituximab polymer may cause hyper-cross-linking-induced apoptosis in non-Hodgkin’s lymphomas. Clin. Cancer Res 2005, 11, 5971–5980. [DOI] [PubMed] [Google Scholar]
- (29).Chu TW; Yang J; Zhang R; Sima M; Kopecek J Cell surface self-assembly of hybrid nanoconjugates via oligonucleotide hybridization induces apoptosis. ACS Nano 2014, 8, 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Haun JB; Devaraj NK; Hilderbrand SA; Lee H; Weissleder R Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat. Nanotechnol 2010, 5, 660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Porter AG; Janicke RU Emerging roles of caspase-3 in apoptosis. Cell Death Differ 1999, 6, 99–104. [DOI] [PubMed] [Google Scholar]
- (32).Li L; Yang J; Wang J; Kopecek J Drug-Free Macromolecular Therapeutics Induce Apoptosis via Calcium Influx and Mitochondrial Signaling Pathway. Macromol. Biosci 2018, 18, 1700196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kelman Z PCNA: structure, functions and interactions. Oncogene 1997, 14, 629–40. [DOI] [PubMed] [Google Scholar]
- (34).Redon CE; Dickey JS; Bonner WM; Sedelnikova OA gamma-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv. Space Res 2009, 43, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ghetie MA; Richardson J; Tucker T; Jones D; Uhr JW; Vitetta ES Disseminated or localized growth of a human B-cell tumor (Daudi) in SCID mice. Int. J. Cancer 1990, 45, 481–5. [DOI] [PubMed] [Google Scholar]
- (36).Thorek D; Robertson R; Bacchus WA; Hahn J; Rothberg J; Beattie BJ; Grimm J Cerenkov imaging - a new modality for molecular imaging. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 163–73. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.