

SUMMARY
The cannabinoid receptor CB2 is predominately expressed in the immune system and selective modulation of CB2 without the psychoactivity of CB1 has therapeutic potential in inflammatory, fibrotic and neurodegenerative diseases. Here we report the crystal structure of human CB2 in complex with a rationally designed antagonist, AM10257, at 2.8 Å resolution. The CB2-AM10257 structure reveals a distinctly different binding pose compared to CB1. Yet, the extracellular portion of the antagonist-bound CB2 shares a high degree of conformational similarity with the agonist-bound CB1, which led to the discovery of AM10257’s unexpected opposing functional profile of CB2 antagonism vs CB1 agonism. Further structural analysis using mutagenesis studies and molecular docking revealed the molecular basis of their function and selectivity for CB2/CB1. Additional analyses of our designed antagonist/agonist pairs provide important insight into the activation mechanism of CB2. The present findings should facilitate rational drug design toward precise modulation of the endocannabinoid system.
Graphical Abstract
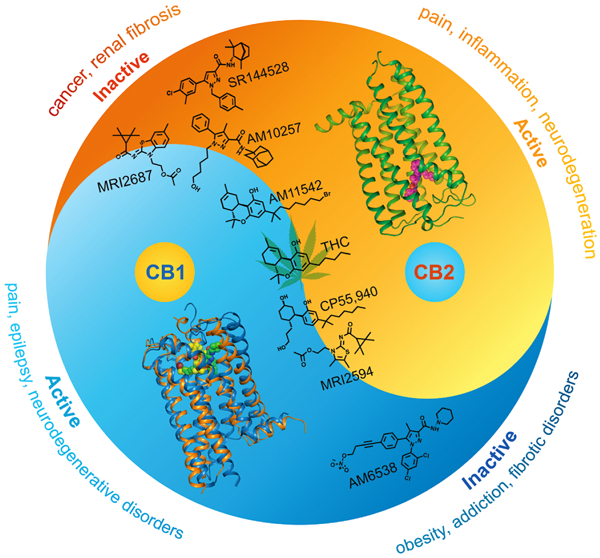
In Brief
The structure of the human cannabinoid receptor CB2 reveals how small molecules affect CB2 differently than CB1, and point to principles that could inform rational and selective drug design
INTRODUCTION
The cannabinoid receptors CB1 and CB2 serve as key components of the endocannabinoid system and are the principal targets of the widely consumed plant-derived phytocannabinoid ∆9-tetrahydrocannabinol (∆9-THC). CB1 is expressed throughout the body and is widely distributed in the central nervous system (CNS), while CB2 is mainly expressed in the immune system, and to a lesser extent in the CNS (Atwood and Mackie, 2010; Cabral et al., 2008; Herkenham et al., 1990; Stempel et al., 2016; Van Sickle et al., 2005). CB2 is emerging as an attractive therapeutic target for immunomodulation, the treatment of inflammatory and neuropathic pain, neuroinflammation and neurodegenerative disorders (Contino et al., 2017; Guindon and Hohmann, 2008; Lunn et al., 2008). Recent studies indicate that CB2 antagonists can ameliorate renal fibrosis (Zhou et al., 2018) and also delay tumor progression (Xiang et al., 2018) thus indicating their potential as compounds for treating fibrotic condition and cancer.
However, CB2 has a high degree of homology and shares 44% sequence identity with CB1 (Munro et al., 1993). Many cannabinergic compounds interact with both CB1 and CB2, making it difficult to delineate the individual signalling contributions required to modulate the two receptors. In this report, we have determined the crystal structure of CB2 in complex with a rationally designed CB2 antagonist. A comparative analysis of the antagonist-bound CB2 with our previously solved CB1 structures may clarify the determinants of ligand selectivity/function and will provide new insights into precise modulation of the endocannabinoid system for therapeutic applications.
RESULTS
Synthesis of CB2 Stabilizing Antagonist AM10257 for Structural Studies
To facilitate the crystallization, we designed a high-affinity CB2 antagonist, AM10257, which was obtained through systematic optimization of SR141716A (Rinaldi-Carmona et al., 1994), the first known CB1 antagonist (Figure 1A). Based on our early work, we had shown that substituting the phenyl ring at the N-1 position of SR141716A with an alkyl group imparted CB2 affinity. Further lead optimization of this template led to AM10257 in which the 5-hydroxypentyl chain at the N-1 position transformed the CB1 antagonist into a CB2 antagonist. Furthermore, the modified amide group with the bulkier 1-adamantyl group at C-3 further enhance the affinity for CB2 (Figure 1A), facilitated stabilization of the ligand-CB2 receptor complex, and promoted CB2 crystal formation (Figures S1 and S2).
Figure 1. Synthesis and Characterization of AM10257.

(A) Systematic lead optimization of AM10257, a CB2 antagonist, and radioligand binding affinity against [3H]CP55,940, Ki, inhibition constant. (B) AM10257 acts as a competitive antagonist/inverse agonist at CB2 against CP55,940, a potent agonist, in the forskolin-stimulated adenylyl cyclase assay as presented by the Schild plot where logKB = −8.30 ± 0.05; (EC50 for CP55,940 = 8.1 ± 0.6 nM). (C) AM10257 acts in a manner consistent with competitive antagonist/inverse agonist at CB2 in β-arrestin2 recruitment assay. CP55,940: EC50: 3.4 ± 0.6 nM, Emax (fold): 2.5 ± 0.2; AM10257: EC50: 0.53 ± 0.29 nM, Emax (fold): 0.70 ± 0.05. Data are presented as the mean ± s.e.m. of 3 experiments performed in duplicate.
See also Figures S1 and S2 and Table S1.
AM10257 shows a high affinity (Ki = 0.08 nM for CB2) as determined by radioligand competition assays against [3H]-CP55,940 (Kd = 0.86 nM) (Figure 1A). Importantly, the binding affinity of crystalized CB2 construct for AM10257 is still in sub-nanomolar range (Ki = 0.61 nM) (Figure S1F). In assays assessing the regulation of adenylyl cyclase and β-arrestin2 recruitment, AM10257 acted as an inverse agonist and competitive antagonist at CB2 (Figures 1B and 1C, Table S1).
Structural Determination of CB2-AM10257 Complex
To obtain crystals of CB2, we replaced residues Ser222-Ala235 in the third intracellular (ICL3) loop of the receptor with T4-lysozyme to generate a CB2-T4L fusion construct. Five mutations Gly782.48Leu, Thr1273.46Ala, Thr1534.45Leu, Arg2426.32Glu and Gly3048.48Glu were introduced (Figures S1 and S3 and METHODS) to further improve the protein homogeneity and thermostability. Additionally, the N terminal residues 1–20 and the C-terminal residues 326–360 were truncated to improve the crystal quality. Radioligand binding assays showed that these modifications had little effect on the binding affinity of CB2 toward AM10257, SR144528, a potent and selective CB2 antagonist (Rinaldi-Carmona et al., 1998) and CP55,940, a CB2/CB1 agonist (Figure S1F). Crystals of CB2-T4L in complex with AM10257 were grown in a lipidic cubic phase (LCP) cholesterol mixture and diffracted to 2.8 Å resolution (Table 1).
Table 1 |.
Crystallographic Data Collection and Structure Refinement Statistics. See also Figure S1.
| PDB code | CB2-AM0257 5ZTY |
|---|---|
| Data collection | |
| Number of crystals | 7 |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 34.29, 106.24, 183.40 |
| Number of reflections measured | 134347 |
| Number of unique reflections | 17142 |
| Resolution (Å) | 45.96–2.80 (2.90–2.80) * |
| Rmerge | 0.30 (0.97) |
| Mean I / sI | 14.70 (3.31) |
| CC1/2 | 0.99 (0.87) |
| Completeness (%) | 96.93 (92.81) |
| Redundancy | 7.80 (7.30) |
| Refinement | |
| Resolution (Å) | 45.96–2.80 |
| No. reflections | 16871 (856) |
| Rwork / Rfree (%) | 0.224/0.275 |
| No. atoms | |
| Protein | 3508 |
| Ligand | 31 |
| Lipid and other | 187 |
| Average B factors (Å) | |
| Wilson / Overall | 76.06 / 95.84 |
| Protein | 93.71 |
| Ligand/ion | 112.70 |
| Water | 58.72 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.00 |
| Ramachandran Plot Statistics (%) | |
| Favored regions | 97.08 |
| Allowed regions | 2.92 |
| Disallowed regions | 0.00 |
Values in parentheses are for highest-resolution shell.
Structural Features of CB2 in Complex with AM10257
The overall architecture of CB2 is comprised of a 7TM bundle (helices I-VII) and an intracellular amphipathic helix VIII (Figure 2A). Although the intracellular portion adopts an inactive conformation similar to antagonist-bound CB1, the extracellular portions of antagonist-bound CB2 and CB1 are significantly different, most notably within helices I and II (Figure 2B and Figures S5A and S5B). The non-truncated part of the N terminus in CB2, in contrast to the V-shaped loop in antagonist AM6538-bound (Hua et al., 2016) and taranabant-bound (Shao et al., 2016) CB1, forms a short helix over the orthosteric pocket with no direct involvement in the antagonist binding (Figure 2B and Figure S5B). Similar to CB1 (Hua et al., 2017; Hua et al., 2016; Shao et al., 2016) and the other two lipid binding receptors, sphingosine 1-phosphate receptor 1 (S1P1) (Hanson et al., 2012) and lysophosphatidic acid receptor 1 (LPA1) (Chrencik et al., 2015), CB2 exhibits a constrained conformation of extracellular loop 2 (ECL2), that is stabilized by a disulfide bond (Cys174 – Cys179) within ECL2 (Figure 2B and Figure S5C).
Figure 2. Overall structure of AM10257-CB2 Complex.

(A) Side view of the CB2-AM10257 complex. CB2 is shown in green with ligand AM10257 in magenta. (B) Overall structure comparison of antagonist-bound CB2 and CB1. CB2 color scheme as in (A). CB1 is shown in light blue with antagonist AM6538 (cyan sticks).
See also Figures S1 and S5.
AM10257 Interactions in CB2 ligand-Binding Pocket
In the CB2 structure, a strong electron density was observed in the orthosteric ligand-binding pocket (Figure S1D), enabling accurate placement of AM10257. The chemical scaffold of AM10257 consists of a core pyrazole ring substituted with three groups extending in different directions (‘three-arm pose’ Figure 3A). The interactions between AM10257 and CB2 are mainly hydrophobic and aromatic, consisting of residues from ECL2 as well as helices II, III, V, and VI (Figures 3A and 3B). The core pyrazole ring of AM10257 resides among helices II, III, and VII, forming π-π interactions with Phe183ECL2, and hydrophobic interactions with Phe872.57 and Val1133.32 (superscript denotes Ballesteros–Weinstein numbering (Juan A. Ballesteros, 1995)). The benzene ring (‘arm 1’) points downward and simultaneously engages in π-π interactions with Phe1173.36 and Trp2586.48 (Figures 3A and 3B). The 5-hydroxypentyl chain (‘arm 2’) extends between helices III and V, forming hydrophobic interactions with Thr1143.33, Phe183ECL2, Ile186ECL2, and Trp1945.53 while the terminal hydroxy group on the alkyl chain participates in a local hydrogen bond network formed by a water molecule and the carboxy oxygen of Ser1654.57 (Figure 3B). The adamantyl group (‘arm 3’) extends toward helices II and III establishing extensive hydrophobic interactions with Phe872.57, Phe912.61, Phe942.64, His952.65 and Phe183ECL2 (Figures 3A and 3B). The residues involved in AM10257 binding are nearly identical to those identified in the CB1 agonist-binding motif (Hua et al., 2017). Accordingly, mutations of Phe872.57, Phe912.61, Phe942.64 and His952.65 greatly reduce the potency of the CB2/CB1 agonist CP55,940 (Figure 3C).
Figure 3. Interactions between AM10257 and CB2.

(A) The ‘three-arm’ scaffold of AM10257. The benzene ring, 5-hydroxypentyl group, and adamantyl group in red, blue and green squares are termed arm 1, arm 2 and arm 3, respectively. The key residues, which form binding interactions with the arms, are also shown. (B) Key residues (green sticks) involved in AM10257 (magenta sticks) binding. The water is shown as a red sphere and the hydrogen bonds are shown as dashed lines. (C) Dose response studies of CP55,940 activity for each mutant compared to wild-type CB2. As determined in parallel studies: WT EC50: 1.6 ± 0.1 nM, Emax (fold): 2.2 ± 0.1; F87A EC50: 664.9 ± 57.4 nM, Emax (fold): 2.4 ± 0.1; F91A EC50: 56.6 ± 5.5 nM, Emax (fold): 2.4 ± 0.1; F94A EC50: 125.6 ± 11.6 nM, Emax (fold): 2.3 ± 0.1; H95A EC50: 349.9 ± 29.5, Emax (fold): 2.2 ± 0.1. Data are presented as mean ± s.e.m. of >3 experiments performed in duplicate.
See also Figures S3 and S5.
Structural Comparison with Antagonist-bound CB1 Receptor
Although both AM10257 and AM6538 encompass the core pyrazole ring and a ‘three-arm’ scaffold as in SR141716A, their binding poses in their respective receptors are quite different (Figure 4A). Instead of adopting the extended conformation of AM6538 in CB1 (Hua et al., 2016), AM10257 assumes a more constrained binding pose in CB2 (Figures 4C and 4D). In addition, the AM10257 binding position in CB2 is more akin to those observed within the two lipid receptors, S1P1 and LPA1 bound to antagonists ML056 (Hanson et al., 2012) and ONO9780307 (Chrencik et al., 2015), respectively (Figure S5C). Compared to AM6538 in CB1, AM10257 within CB2 shifts toward helices III and IV by 6.1 Å using the pyrazole ring as reference (Figures 4A and 4B). Synergistically, the extracellular parts of helices I and II experience a movement toward the ligand binding site, and residues Phe872.57 and Phe912.61 form hydrophobic interactions with ‘arm 3’ of AM10257. In contrast to the horizontally extended ‘arm 3’ in CB1, ‘arm 3’ of AM10257 assumes a near vertical pose in CB2 and the adamantyl group of AM10257 overlaps with the benzene ring of Phe102N-term of CB1 in the superimposed structures. As a result, the N terminus of CB2 does not insert into the ligand-binding pocket unlike the V-shaped loop in antagonist (AM6538 or taranabant) -bound CB1 (Hua et al., 2016; Shao et al., 2016). In comparison, the molecular docking pose of SR144528 resembles the binding pose of AM10257 (Figure S6A) where the main difference is the 4-methylbenzyl of SR144528 and the alkyl chain of AM10257 in ‘arm 2’, dock in similar positions, indicating that this is the common binding mode for CB2 antagonists.
Figure 4. Comparison of ligand-binding modes and binding pockets for cannabinoid receptors.

(A) Superposition of the CB2-AM10257 and CB1-AM6538 ligand binding pockets. The residues Phe2.57 and Phe2.61 are shown as spheres. AM10257 (magenta sticks)-bound CB2, and AM6538 (cyan sticks)-bound CB1 are shown in green and light blue cartoon, respectively. (B) Conformational difference of Phe3.36 and Trp6.48 in antagonist-bound CB2 and CB1. Color scheme as in (A). (C–E) Surface representations of ligand binding pocket for: CB2 with AM10257 (C), CB1 with AM6538 (D), and CB1 with AM11542 (E).
See also Figures S4 and S5.
Another key difference is the toggle switch residue Trp6.48 conformation in the antagonist-bound CB2 and CB1 structures (Figure 4B and Figure S4). ‘Arm 1’ of the ‘three-arm’ scaffold antagonist AM10257 (Figure 4B), confines the side chain of Trp2586.48, a residue conserved in 68% of class A GPCRs, to a relatively rare rotamer, which has only appeared in muscarinic acetylcholine receptors (Kruse et al., 2012; Kruse et al., 2013; Thal et al., 2016) and two neurotensin receptor (White et al., 2012) structures (Figure S5D). The confined Trp2586.48 conformation may restrict the outward movement of helix VI and stabilize the receptor in the inactive-state. Conversely, in the AM6538-bound CB1 structure, ‘arm 3’ of AM6538 extends itself to push helices I and II outwards and, as a result, ‘arm 1’ is positioned ~5 Å away from the toggle switch residue Trp3566.48. Here, Phe2003.36 functions as a latch to restrict the movement of Trp3566.48 and locks CB1 in the inactive state. These observations explain the high CB2/CB1 selectivity and unprecedented high affinity of these two antagonists for their respective receptors.
Structural Comparison with Agonist-bound CB1 Receptor
Surprisingly, our AM10257-bound CB2 structure closely resembles the agonist AM11542 bound CB1 structure (Hua et al., 2017) in the extracellular portion (Figure 5A) and the antagonist-binding motif in CB2 exhibits greater resemblance to the agonist-binding motif in CB1 (Figure 5B). The N termini adopt similar conformations, where each forms a short helix before Tyr24N-term of CB2 or Phe 108N-term of CB1 (Figure 5A). Additionally, the volume of the CB2 antagonist-binding pocket (447 Å3) is closer to that of the agonist AM11542-bound CB1 (384 Å3) (Hua et al., 2017) than to the antagonist AM6538-bound CB1 (822 Å3) (Hua et al., 2016) (Figures 4C, 4D and 4E). The parameters correlate well with the similar binding shapes of AM10257 and the CB1 agonist AM11542, as well as the near identical array of residues involved in AM10257 or AM11542 binding (Figure 5B). These observations inspired us to test the function of AM10257 within CB1 where we found it to behave as a CB1 partial agonist in the β-arrestin2 recruitment assay (Figure 5C).
Figure 5. Structural comparison of antagonist-bound CB2 and agonist-bound CB1.

(A) Overall structural comparison of AM10257 (magenta sticks)-bound CB2 and AM11542 (yellow sticks)-bound CB1. CB2 and CB1 receptors are shown in green and orange, respectively. (B) Binding pose comparison of AM10257 in CB2 and AM11542 in CB1. The residues involved in the ligand binding are shown as sticks. (C) AM10257 (Ki = 13 nM for CB1, as determined by radioligand competition assays against [3H]-CP55,940) and functions as a partial agonist of CB1 as determined by β-arrestin2 recruitment assay. CP55,940 EC50: 17.9 ± 1.4 nM, Emax (fold): 20.6 ± 0.9; AM10257 EC50: 51.5 ± 7.4 nM, Emax (fold): 2.4 ± 0.1. Data are presented as mean ± s.e.m. of >4 experiments performed in duplicate.
See also Figures S4 and S6.
Activation Mechanism of CB2
To delineate the activation mechanism of CB2, given the limited space for possible conformational changes of the ligand-binding pocket of CB2, we rationally designed two structurally related ligands, MRI2687 and MRI2594. These ligands only differ in ‘arm 1’, while featuring the extended central 6-methylbenzothiazole ring in MRI2687 and 4,5-dimethylthiazole ring in MRI2594 respectively (Figure 6A and Figure S2). Unexpectedly, MRI2687 behaves as an inverse agonist while MRI2594 acts as an agonist on CB2 (Figure 6C). Molecular docking showed these two ligands adopt similar binding poses within CB2 (Figure 6B), wherein the different central rings reside in the same position as ‘arm 1’ of AM10257. MRI2687 resembles AM10257 in the crystal structure, while ‘arm 1’ of MRI2687 forms π-π interactions with the side chain of Trp2586.48 and confines its conformation to the similar rotamer as in the CB2-AM10257 structure (Figure 6B). In contrast, since MRI2594 lacks a large substituent in ‘arm 1’, it does not extend sufficiently deep to constrain the conformation of Trp2586.48. This is consistent with our prediction that CP55,940 also has no moiety to constrain Trp2586.48 in CB2 (Figure S6B). Additionally, we performed molecular dynamic simulations (200ns) of ligands used in this study (Figures S6C–G) and found the rotamer of Trp2586.48 supports this conclusion.
Figure 6. Molecular docking and pharmacology of designed CB2 agonist/antagonist pair.

(A) Chemical structures of rationally designed agonist MRI2594 and antagonist/inverse agonist MRI2687. (B) The docking poses of MRI2594 (orange sticks) and MRI2687 (blue sticks) in AM10257-bound CB2 (AM10257 is removed). (C) MRI2594 performs as an agonist of CB2 and MRI2687 performs as an inverse agonist of CB2 as determined by β-arrestin2 recruitment assay. CP55,940 EC50: 2.3 ± 0.6 nM, Emax (fold): 2.3 ± 0.1; MRI2594 EC50: 0.17 ± 0.03 nM, Emax (fold): 2.0 ± 0.2; MRI2687 EC50: 0.24 ± 0.06 nM, Emax (fold): 0.75 ± 0.04; AM10257 EC50: 0.20 ± 0.04 nM, Emax (fold): 0.70 ± 0.05. (D) MRI2594 and MRI2687 perform as an agonist and a partial agonist of CB1 as determined by β-arrestin2 recruitment assay. CP55,940 EC50: 17.9 ± 1.4 nM, Emax (fold): 20.6 ± 0.9 (from Figure 5C); MRI2594 EC50: 310.0 ± 54.0 nM, Emax (fold): 28.0 ± 2.0; MRI2687 EC50: 114.0 ± 15.0 nM, Emax (fold): 6.0 ± 0.3. Data are presented as mean ± s.e.m. of > 4 experiments performed in duplicate.
See also Figures S2 and S6.
The above analysis indicated the critical role of the toggle switch residue Trp2586.48 during CB2 activation and essential function of ‘arm 1’ of the ligand for CB2 antagonism. Interestingly, our functional data showed that MRI2687 also acts as a partial agonist on CB1 (Figure 6D), further supporting the notion that CB2 antagonist/CB1 agonist profiles probably occur in certain compounds (Ogawa et al., 2015) representing a CB2/CB1 yin-yang functional relationship. The physiological implications of such opposing activation profiles between CB1 and CB2 are worth exploring further.
DISCUSSION
The endocannabinoid signaling system has emerged as an important target for therapeutic drug development, although the design of receptor-selective ligands has remained a challenge. The present findings represent an important step toward the rational design of drugs interacting with cannabinoid receptors, both in terms of their CB1/CB2 selectivity and their mode of action as agonists or antagonists/inverse agonists.
First, resolution of the crystal structure of the CB2 receptor in complex with our high affinity antagonist AM10257 has revealed that the CB2 antagonist-binding pocket is distinct, relatively smaller than the CB1 antagonist-binding pocket, and more similar with regard to size and the ligand-interacting residues, to the CB1 agonist-binding pocket. This provides a rational explanation for the empirical findings of a high degree of CB1/CB2 selectivity among cannabinoid receptor antagonists designed to date, and it also may account for the present finding that two structurally distinct CB2 antagonists also display CB1 partial agonist activity. Given their lack of CB1 antagonist activity, CB2 antagonists should not elicit the neuropsychiatric side effects similar to those that thwarted the clinical use of globally acting CB1 antagonists, such as rimonabant (Janero and Makriyannis, 2009).
A second important finding revealed a molecular mechanism critical for CB2 activation. This was achieved through molecular docking and functional studies using a rationally designed pair of structurally related ligands that unexpectedly acted as CB2 antagonist (MRI2687) or agonist (MRI2594) due to the length difference of their ‘arm 1’, which resulted in diverse interactions with the toggle switch residue Trp2586.48. Apart from providing strong support for the role of Trp2586.48 in the activation of CB2, the overlapping binding pose of these two compounds also highlights a close similarity of the interacting residues involved in the binding of CB2 agonists and antagonists. This conclusion is also supported by previous, homology-based studies of other CB2 agonist/inverse agonist pairs based on different chemical scaffolds (Han et al., 2015; Lucchesi et al., 2014). Because of the aforementioned similarity between the CB2 antagonist and CB1 agonist binding pockets, by inference, the CB1 and CB2 agonist binding pockets are also likely similar. Such structural observations may explain the low level of CB1/CB2 selectivity and the related psychotropic effects that are associated with CB1 activation for some of the classical cannabinoids and their synthetic analogs. Nevertheless, structure-based rational drug design could mitigate this problem by exploiting the subtle differences in interactions with critical residues within both receptors. While the agonist-bound structure of the CB2 remains to be determined, the knowledge obtained from the antagonist-bound CB2 crystal structure along with the findings relating to the opposing effects of CB2/CB1 should aid in the rational design of CB2 interactive ligands with improved functional selectivity.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhi-Jie Liu (liuzhj@shanghaitech.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Spodoptera frugiperda (Sf9) cells were used for CB2 expression and crystallization. Sf9 cells were grown in ESF 921 medium (Expression systems) at 27 °C and 125 rpm. Binding and functional experiments were performed with either CHO-K1 cell lines (ATCC, female) or U2OS cell lines (PathHunter® EA Parental Cell lines, female, DiscoveRx). CHO-K1 cells were maintained in DMEM/F12 media supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 5 mg/ml puromycin for stable cell line selection (Invitrogen, Waltham, MA). U2OS cells were selected by geneticin selection (500 µg/ml) in MEM media supplemented with 10% FBS, 1% penicillin/streptomycin and 250 µg/ml hygromycin.
METHOD DETAILS
Synthesis and Characterization of AM10257 (Scheme 1)
4-Methyl-5-phenyl-N-tricyclo[3.3.1.13,7]dec-1-yl-1H-pyrazole-3-carboxamide (2)
To a magnetically stirred solution of 4-methyl-5-phenyl-1H-pyrazole-3-carboxylic acid (CAS RN: 879770–33-9) (1, 2 g, 10 mmol) and 1-adamantylamine (1.5 g, 10 mmol) dissolved in 80 ml of dichloromethane was added Hünig’s base (1.75 ml, 10 mmol) followed by O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate (TBTU) (3.21 g, 10 mmol). The reaction mixture was stirred at room temperature for 30 min. The reaction contents were washed with 3 × 50 ml water and the organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated. The residue obtained was purified by flash column chromatography on silica gel to give amide 2 as a white solid (2.1 g, 65 %); m.p 272–275 0C; 1H NMR (500 MHz, CDCl3) δ ppm 7.46 (m, 4 H), 7.37 – 7.43 (m, 1 H), 6.70 (br. s., 1 H), 2.43 (s, 3 H), 2.13 (m, 6 H), 2.11 (m, 3 H), 1.62 – 1.81 (m, 6 H); 13C NMR (125 MHz, CDCl3) 162.28, 145.16, 142.79, 129.77, 129.02, 128.59, 127.68, 114.93, 51.83, 41.85, 36.46, 29.72, 29.57, 9.40; ES m/z 336.4508 ([M + H]+.
1-(5-Hydroxypentyl)-4-methyl-5-phenyl-N-tricyclo[3.3.1.13,7]dec-1-yl-1H-pyrazole-3-carboxamide (AM10257)
To a magnetically stirred solution of 1H-pyrazole amide (2, 500 mg, 1.5 mmol) in 20 ml of DMF was added NaH (60% dispersion in mineral oil, 35 mg, 1.5 mmol) at 0°C and the contents were stirred for 1 hr. Subsequently, a solution of 5-bromo-1-pentanol (250 mg, 1.5 mmol) in 5 ml of DMF was added and the resulting mixture was stirred at 0°C for another 1 hr. Water (50 ml) was added to the mixture, and the contents were extracted with 3 × 50 ml dichloromethane. The combined extracts were washed with 50 ml water, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by flash column chromatography on silica gel gave AM10257 as a white solid (391 mg, 62 %); m.p 125–127 °C; 1H NMR (500 MHz, CDCl3) δ ppm 7.38 – 7.53 (m, 3 H), 7.20 – 7.32 (m, 2 H), 6.73 (s, 1 H), 3.97 (t, J=7.32 Hz, 2 H), 3.57 (t, J=6.44 Hz, 2 H), 2.21 (s, 3 H), 2.23 – 2.20 (6H, m), 2.18 – 2.12 (3H, m), 1.64 – 1.82 (m, 8 H), 1.40 – 1.53 (m, 2 H), 1.18 – 1.36 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ ppm 162.7, 143.1, 142.9, 129.9, 128.7, 116.8, 62.5, 51.6, 49.6, 41.8, 36.5, 32.0, 29.9, 29.5, 22.6, 22.3; ES m/z 422.5763 ([M + H]+.
Synthesis and Characterization of MRI2687 and MRI2594 (Scheme 2/3)
General Procedure 1
Aminothiazole derivative (1 eq (equivalent, eq)) and 2-bromo ethanol (see Scheme, 1.5 eq) were heated in a sealed vessel at 90°C for 16 h. The resulting residue was triturated with the minimum amount of i-PrOH. The solid was filtered, dried under high vacuum, and used in the next step without further purification. 2,2,3,3-Tetramethylcyclo-propane acid (1 eq), Et3N (3 equiv.), and BOP (1.3 equiv.) were added to a suspension of the hydroxy alkyl 2-aminoimidazole (1 equiv.) in 7 ml of dichloromethane at room temperature, and the mixture was stirred at room temperature for 20 h. The reaction was quenched by the addition of 5 ml of water followed by extraction with dichloromethane (10 ml). The combined organic fractions were washed with 20 ml of brine, dried over Na2SO4, and concentrated by rotary evaporation. The obtained residue was purified by flash column chromatography (hexane:EtOAc 1:1) to give alcohol derivatives.
General Procedure 2.
The alcohol derivatives were taken in dichloromethane and cooled to 0°C. Et3N (2 eq) was added followed by the addition of acetyl chloride (1.2 eq). After stirring at room temperature for 4 h, the reaction was quenched with NaHCO3 and the organic layer was extracted with dichloromethane (10 ml). The organic fraction was washed with 5 ml of brine, dried over Na2SO4, and concentrated by rotary evaporation. The obtained residue was purified by flash column chromatography (hexane:EtOAc, 2:1) to give acetylated derivatives.
N-(3-(2-hydroxyethyl)-4,5-dimethylthiazol-2(3H)-ylidene)-2,2,3,3 tetramethylcyclopropane-1-carboxamide(Horti et al., 2010)(3)
2-(4,5-dimethyl-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)thiazol-3(2H)-yl)ethyl acetate (MRI2594)
Following General Procedure 2, 3 (200 mg, 0.68 mmol) gave MRI2594 (150 mg, 66 %) as a white powder. Mp 153–155 °C; 1HNMR (400 MHz, CDCl3): δ 4.36 (d, J = 5.1 Hz, 2H), 4.30 (d, J = 5.1 Hz, 2H), 2.16 (s, 3H), 2.13 (s, 3H), 2.02 (s, 3H), 1.50 (s, 1H), 1.30 (s, 6H), 1.18 (s, 6H). 13C NMR (101 MHz; CDCl3): δ 181.5, 170.6, 164.6, 127.2, 113.1, 61.7, 44.7, 42.3, 30.4, 24.0 (2C), 20.8 (2C), 16.9 (2C), 11.7, 11.2. HRMS (C17H27N2O3S) [M+H]+: found m/z, 339.1740; calcd. 339.1742
N-(3-(2-hydroxyethyl)-6-methylbenzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4)
Following General Procedure 2, 2amino-6-methylbenothiazole (1.0g, 6.0 mmol) gave compound 4 (600 mg, 30 %) as a white powder over two steps. Mp 158–160 °C; 1H NMR (400 MHz, CDCl3): δ 7.39 (s, 1H), 7.18 (q, J = 9.7 Hz, 2H), 4.88 (s, 1H), 4.49 (t, J = 4.3 Hz, 2H), 4.05 (t, J = 4.4 Hz, 2H), 2.40 (s, 3H), 1.55 (s, 1H), 1.31 (s, 6H), 1.20 (s, 6H). 13C NMR (101 MHz; CDCl3): δ 181.8, 166.4, 134.2, 133.8, 127.9, 126.7, 122.9, 110.6, 62.5, 48.6, 42.5, 31.9, 23.9(2C), 21.0(2C), 16.8(2C). (C18H25N2O2S) ES/[M+H]+: 333.1
2-(6-methyl-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI2687)
Following General Procedure 2, 4 (200 mg, 0.60 mmol) gave MRI2687 (170 mg, 76 %) as a white powder. Mp 125–127 °C; 1H NMR (400 MHz, CDCl3): δ 7.39 (s, 1H), 7.26 (s, 1H), 7.21 (s, 2H), 4.58 (t, J = 5.4 Hz, 2H), 4.48 (t, J = 5.4 Hz, 2H), 2.42 (s, 3H), 1.97 (s, 3H), 1.64 (s, 1H), 1.40 (s, 6H), 1.20 (d, J = 2.1 Hz, 6H). 13C NMR (101 MHz; CDCl3): δ 182.7, 170.9, 164.2, 134.5, 133.3, 127.6, 126.5, 122.8, 110.5, 61.1, 43.8, 42.7, 31.6 (2C), 24.0, (2C) 21.0, 20.8, 16.8 (2C). HRMS (C20H27N2O3S) [M+H]+: found m/z, 375.1736; calcd. 375.1742.
X-Ray data: The single crystal X-ray diffraction studies were carried out on a Bruker Kappa APEX-II CCD diffractometer equipped with Cu K radiation (λ = 1.5478). A 0.213 × 0.57 × 0.054 mm piece of a colourless rod was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using ϕ and ϖ scans. Crystal-to-detector distance was 40 mm using variable exposure time (5s-10s) depending on θ with a scan width of 2.0°. Data collection was 98.5% complete to 68.00° in θ. A total of 11,685 reflections were collected covering the indices, - 9<=h<=9, −11<=k<=11, −16<=l<=16. 3402 reflections were found to be symmetry independent with a Rint of 0.0270. Indexing and unit cell refinement indicated a primitive, triclinic lattice. The space group was found to be P-1. The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SHELXT) produced a complete phasing model consistent with the proposed structure. All nonhydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-2014). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-2014. Atomic coordinates for MRI2687 have been deposited with the Cambridge Crystallographic Data Centre (deposition number: 1856556). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223–336033 or e-mail: deposit@ccdc.cam.ac.uk.
Protein Engineering for Structural Studies
The codon-optimized human CB2 gene (Genewiz) was cloned into a modified pFastBac1 vector (Invitrogen) with the haemagglutinin (HA) signal sequence at the N terminus and a PreScission protease site followed by a 10 × His-tag and a Flag tag at the C terminus. To facilitate crystallization, T4-lysozyme was fused into the third intracellular loop (ICL3) of CB2 (Ser222-Ala235) with the truncation of the N terminal residues 1–20 and the C-terminal residues 326–360. The CB2-T4L gene was further modified by introducing five mutations Gly782.48Leu, Thr1273.46Ala, Thr1534.45Leu, Arg2426.32Glu and Gly3048.48Glu. The mutations were rationally designed using sequence-based and structure-based modules of the CompoMug V0.1 computational tool for prediction of stabilizing mutations (Popov et al., 2018).
Protein Expression and Purification
The modified CB2–T4L protein was expressed in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Sf9 cells were infected at a cell density of 2~2.5 × 106 cells per ml with high-titer viral stock MOI (multiplicity of infection) of 5.0. Cells were harvested by centrifugation for 48 hr post-infection and stored at −80°C for future use.
Frozen cell pellets were thawed and lysed by repeated washing and centrifugation in the hypotonic buffer of 10 mM HEPES (pH 7.5), 10 mM MgCl2, 20 mM KCl, and the high osmotic buffer of 10 mM HEPES (pH 7.5), 1.0 M NaCl, 10 mM MgCl2, 20 mM KCl, with EDTA-free complete protease inhibitor cocktail tablets (Roche). The washed membranes were suspended in hypotonic buffer with 30% glycerol, flash-frozen with liquid nitrogen and stored at −80°C. Purified membranes were thawed at room temperature and incubated with 20 µM AM10257 and inhibitor cocktail at 4°C for 3 hr. The membranes were further incubated with 1.0 mg/ml iodoacetamide (Sigma) for 1 hr and were solubilized in the buffer containing 50 mM HEPES (pH 7.5), 500 mM NaCl, 0.75% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) and 0.15% (w/v) cholesterol hemisucinate (CHS, Sigma-Aldrich) at 4°C for 2.5–3 hr. The supernatant was isolated by ultracentrifugation, and then incubated with TALON IMAC resin (Clontech) and 20 mM imidazole, at 4°C overnight. The resin was washed with 15 column volumes of washing buffer I containing 25 mM HEPES (pH 7.5), 500 mM NaCl, 10% (v/v) glycerol, 0.1% (w/v) LMNG, 0.02% (w/v) CHS, 30 mM imidazole and 20 µM AM10257, and 15 column volumes of washing buffer II containing 25 mM HEPES (pH 7.5), 500 mM NaCl, 10% (v/v) glycerol, 0.03% (w/v) LMNG, 0.006% (w/v) CHS, 50 mM imidazole and 20 µM AM10257. The protein was eluted using 3 column volumes of elution buffer containing 25 mM HEPES (pH 7.5), 500 mM NaCl, 10% (v/v) glycerol, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 250 mM imidazole and 25 µM AM10257. The purified receptor was then concentrated to 20 mg ml−1 using a 100kDa cut-off concentrator (Sartorius) for crystallization trials.
Crystallization in Lipidic Cubic Phase
Crystallization was performed using the lipidic cubic phase (LCP) method (Caffrey and Cherezov, 2009). The concentrated CB2 protein in complex with AM10257 was reconstituted into LCP by mixing with molten lipid (90% (w/v) monoolein and 10% (w/v) cholesterol) at volume ratio of 2:3 using a syringe mixer. The mesophase was dispensed onto 96-well glass sandwich plates in 50 nl drop and overlaid with 800 nl precipitant solution using an NT8-LCP crystallization robot (Formulatrix). Plates were incubated and imaged at 20°C using an automated incubator/imager (RockImager, Formulatrix). Crystals were obtained from precipitant conditions containing 100 mM sodium cacodylate trihydrate pH 6.2, 40% PEG 400, 400 mM lithium sulfate monohydrate, and reached a full size of 200 µm after 2 weeks. Crystals were harvested from the LCP matrix using MiTeGen micromounts and immediately flash frozen in liquid nitrogen.
Data Collection and Structure Determination
X-ray diffraction data were collected at the Spring-8 beam line 41XU and Diamond beam line I24, using a Pilatus3 6M detector (X-ray wavelength 1.0000 Å). A rastering and data-collection strategy was followed as previously described (Cherezov et al., 2009). Diffraction images were indexed, integrated and scaled using XDS (Kabsch, 2010a, b) and merged using XPREP. Initial phase was obtained by molecular replacement (MR) with Phaser (McCoy et al., 2007) using the T4L structure (PDB: 2LZM) as search model. The structure determination was completed iteratively by manually modelling the CB2 domain in the program COOT (Emsley et al., 2010) using both |2Fo|-|Fc| and |Fo|-|Fc | maps and refined with Phenix (Adams et al., 2010) and Buster (Smart et al., 2012).
Radioligand Binding Assay
Stably transfected HEK293F cell lines expressing the hCB2 WT receptors and the Sf9 membrane for crystallized CB2 receptors were used for saturation and competition binding assays with [3H]-CP55,940 (specific activity: 81.1 Ci/mmol, NDSP, NIDA). Binding assays were performed in a 96 well plate format wherein hCB2 membrane pellets were resuspended in TME buffer (25 mM Tris-HCl, 5 mM MgCl2, 1mM EDTA, pH7.4) with 0.1% BSA (TME-BSA) and aliquots of resuspended membranes (containing 25 µg or 8 µg protein for WT hCB2 or the crystallized CB2 receptors, respectively) were added to each assay well. [3H]-CP55,940 was diluted in TME-BSA at concentrations ranging from 0.75 – 23.5 nM (WT hCB2) or 0.25 to 254 nM for crystallized CB2. Nonspecific binding was evaluated in the presence of 5 µM unlabelled CP55,940. The assays were performed at 37°C for 1 hr prior to collection of membranes by rapid filtration, washing and scintillation facilitated detection of tritium retained on the membranes according to standard procedures (Hua et al., 2016; Mercier et al., 2010; Pei et al., 2008). Bound radioactivity was quantified using a Packard TopCount Scintillation Counter (Perkin Elmer). Nonlinear regression analysis was used to calculate Bmax (pmol/mg protein) and Kd; values were determined by fitting the saturation binding data to a one site binding equation using GraphPad Prism (GraphPad Software, San Diego, CA), n=3.
CB2 Cell Line Generation for Functional Studies
CHO cell lines expressing human CB2 receptors were generated and maintained as described previously (Hua et al., 2016). Briefly, the 3xHA (haemagglutinin)-N-terminus tagged human CB2 (3xHA-hCB2) cDNA purchased from cDNA.org was subcloned into a MSCV retroviral vector (pMSCV-puro, Clontech). Point mutations were introduced by using Q5 site-Directed Mutagenesis kit (New England Biolabs) (Gly782.48Leu, Thr1273.46Ala, Thr1534.45Leu, Arg2426.32Glu, Gly3048.48Glu, Phe872.57Ala, Phe912.61 Ala, Phe942.64 Ala and His952.65 Ala). Retroviruses carrying wild-type and mutant hCB2 cDNAs were packaged by using the Phoenix package system, and the produced retroviruses were applied to CHO-K1 cells for gene transduction. Cells were maintained in DMEM/F12 media supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 5 μg/ml puromycin for stable cell line selection (Invitrogen, Waltham, MA).
For generation of the U2OS-hCB2 DiscoveRx cell line, the 3xHA-hCB2 cDNA was first subcloned into the pCMV-ProLink™ 1 Vector. Then, the plasmid was transfected into PathHunter® EA Parental Cell Lines by electroporation. Transfected cells were subjected to flow cytometry to select the cell population with high expression levels of 3xHA-hCB2 after geneticin selection (500 µg/ml) in MEM media supplemented with 10% FBS, 1% penicillin/streptomycin and 250 µg/ml hygromycin.
Cell-surface expression of the HA-tagged hCB2 receptors in CHO-K1 cells or PathHunter® EA Parental U2OS cells was visually confirmed by confocal microscopy. Briefly, cells were stained by anti-HA-Alexa 488 antibody (1:100, Thermo Fisher Scientific Inc.) for 10 min at 37°C after serum starvation in DMEM/F12 or MEM media for 60 min at 37°C. Cells were then fixed by 4% paraformaldehyde and subjected to nucleus staining by Hoechst (1:1000) for 10 min at room temperature.
Cyclic AMP (cAMP) Accumulation Assay
Inhibition of forskolin-stimulated cAMP was determined using CISBIO® cAMP Homogeneous Time-Resolved Fluorescence resonance energy transfer (FRET) (HTRF) HiRange assay according to the manufacturer’s instructions (Cisbio Assays, Bedford, MA). 5000 CHO-hCB2 cells were plated in low-volume 384-well plates with Opti-MEM media supplemented with 2% BSA after suspended in 5 mM EDTA/PBS and washed twice with PBS. Cells were then cotreated at room temperature for 30 min with 25 µM RO-20–1724 (phosphodiesterase inhibitor), and 20 µM forskolin (Sigma-Aldrich); test compounds were dissolved in DMSO and diluted to final solvent concentrations of 1% at concentrations ranging from 0.03 – 10,000 nM for 10 min at room temperature. Cells were then incubated with cAMP-d2 antibody in assay diluent and cryptate solution in lysis buffer for 60 min at room temperature (Cisbio Assays). Fluorescence was measured at 620/665 nm using a Perkin-Elmer EnVision plate reader (Waltham, MA). FRET was calculated as fluorescence at 665/620 nm. Vehicle (1% DMSO) treated cells in the presence of forskolin served as the vehicle-treated control and data are normalized as fold over vehicle or as the percent of CP55,940 maximal stimulation (all studies were performed with CP55,940 curves run in parallel to assure cellular performance). For antagonist competition studies, both agonist and antagonist were added simultaneously (Hua et al., 2017; Hua et al., 2016).
β-arrestin2 Recruitment Assay
For β-arrestin2 enzyme fragment complementation, the PathHunter® β-arrestin assay was performed following the manufacturer’s protocol (DiscoveRx). Briefly, U2OS-βarrestin2-EFC-hCB2 cells were plated at 5000 cells per well in Bio-one CELLSTAR 384 well white plates (Greiner) in AssayComplete™ Cell Plating 2 Reagent (DiscoveRx) overnight at 37°C. The next day, cells were treated with compounds at the indicated doses (prepared as described for cAMP assay) at concentrations ranging from 0.03 – 10,000 nM for 90 min at 37°C followed by 1 hr incubation of PathHunter® detection reagent at room temperature protected from light. Luminescence levels were determined by using a Synergy HT luminometer (BioTek, Winooski, VT) (Hua et al., 2016). All data points are normalized to vehicle only controls (1% DMSO) included in the assay; all studies were performed with CP55,940 curves run in parallel to assure cellular performance.
Quantitative Flow Cytometry
Parental CHO-K1 cells or CHO-K1 cells expressing WT or mutant hCB2 receptors were serum starved with DMEM/F12 media (Invitrogen) for 30 min at 37°C. The cells were then suspended with 5 mM EDTA/DPBS, centrifuged at 2,000 rpm for 2 min and then resuspended with serum free DMEM/F12 media for 30 min immunostaining with anti-HA-Alexa 488 antibody (1:250, Thermo Fisher Scientific Inc.) at 37°C followed by two washes with 5 ml of DPBS. The cells were then resuspended with 500 µl of ice-cold flow buffer containing 1 mM EDTA, 1% FBS and 10 ng/ml DAPI nuclear stain in DPBS. The receptor expression on the cell surface was evaluated by quantitative flow cytometry by using the Beckman Coulter’s Gallios flow cytometer. 50,000 events were recorded for each cell line, and the data were analysed using the FlowJo software. The data were presented as the percentage of positive-fluorescent cells recorded from the 50,000 events (CHO-WT hCB2 = 93.6%, parental CHO = 0.01%) and the percentage relative to the CHO-WT hCB2 cell line (100%).
Molecular Docking of Cannabinoid Receptor Ligands
Prediction of ligand binding to CB2 was done with Scrodinger Suite 2015–4. Processing of the protein structures was performed with the ‘Protein Preparation Wizard’. Converting of ligands from 2D to 3D structures was performed using ‘LigPrep’. Molecular docking was performed with Glide 6.9 (Friesner et al., 2004; Friesner et al., 2006; Halgren et al., 2004) for molecular docking.
Molecular Dynamics Simulations of CB2 in Complex with Ligands
Molecular dynamics simulation was performed using GROMACS 5.1.2 (Abraham et al., 2015) with force field CHARMM36 (Best et al., 2012). The topology files of ligands were generated using the CGenFF tool (Vanommeslaeghe et al., 2010; Yu et al., 2012) and converted to GROMACS format with the cgenff_charmm2gmx script (mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/cgenff_charmm2gmx.py). CB2 in complex with AM10257 (crystal structure) in the pocket was embedded into a pre-equilibrated POPC (1-palmytoil-2-oleoyl-sn-glycerol-3-phosphatidylcholine) lipid bilayer with TIP3 water molecules using the ‘membed’ tool in GROMACS program. Sodium ions were added to 0.15 M in water, and chloride ions were added to neutralize the system. Molecular Dynamics simulations were performed in the NPT ensemble, at temperature of 310 K and pressure of 1 atm using semi-isotropic coupling. Firstly, each system was balanced in position-restrained MD for 15 ns (total energy was stable). Then 200 ns molecular dynamics simulations with no position restraints were performed to each system for two independent runs, and these trajectories are used for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was performed in Prism 7 (GraphPad, La Jolla, CA) to produce sigmoidal concentration-response curves by using the standard three-parameter equation with the exception of the Schild analysis in Figure 1B; wherein data were fit to a four parameter nonlinear regression allowing for a variable slope for subsequent Schild analysis. The values of EC50 and Emax (fold over basal) for all compounds were obtained for individual experiments performed in duplicate and then averaged and presented as the mean ± S.E.M. (N ≥ 3). Statistical analyses comparing the data obtained for individual experimental EC50 and Emax between WT and mutant hCB2 cell lines were performed using one-way ANOVA followed by Dunnett’s post-hoc test in Prism 7.
Supplementary Material
Related to Figures 1 and 2. (A) Analytical size exclusion chromatography profile. (B) Crystal image. (C) The overall structure of CB2-AM10257 complex with receptor in green and T4L fusion protein in pink. The antagonist AM10257 is shown as magenta sticks and the five single mutations are shown as yellow sticks. (D) The 2|Fo|-|Fc| and |Fo|-|Fc| maps of AM10257 are contoured at 1.6 σ and 2.5 σ, respectively, at 2.8 Å. (E) Crystal packing of CB2-AM10257. Color scheme as in (B). (F) Radioligand binding affinity of AM10257, SR144528, CP55,940 and MRI2687 on the crystallized CB2 construct against [3H]CP55,940, Ki, inhibitory constant.
{kind=link}
Related to Figures 1 and 6. Reagents and conditions: Scheme 1: (a) TBTU, Hünig’s base, DCM, 1-adamantylamine. (b) NaH, DMF, 5-bromo-1-pentanol; Scheme 2: (a) Et3N, DCM, Acetyl Chloride; Scheme 3: (a) 2-bromoethanol 90 °C. (b) BOP, ET3N, DCM, 2,2,3,3-tetramethylcyclopropane carboxylic acid. (c) ET3N, DCM, Acetyl Chloride.
{kind=link}
Related to Figures 2 and 3. (A) Expression of wild-type and mutant CB2 receptors in transient CHO cells. Live-cell fluorescence microscopy verified all CB2 mutants to be properly exported to the cell membrane like the wild-type receptor. (B) The summary of the flow cytometry analysis of mutant receptor expression relative to WT receptor expression as determined by impermeabilized cell N-terminal HA-tag staining. (C-D) Assessment of the effect of the individual point mutations, made to stabilize the receptor in absence of the T4L insert, on receptor activity. CP55,940, WT EC50: 1.8 ± 0.2 nM, Emax (fold): 2.0 ± 0.1; G78L EC50: 1.2 ± 0.2 nM, Emax (fold): 2.5 ± 0.1; T127A EC50: 5.4 ± 2.3 nM, Emax (fold): 1.31 ± 0.02; T153L EC50: 2.1 ± 0.4 nM, Emax (fold): 2.5 ± 0.1; R242E EC50: 2.2 ± 0.1 nM, Emax (fold): 2.2 ± 0.1; G304E EC50: 4.0 ± 0.1 nM, Emax (fold): 2.1 ± 0.1. AM10257 +100 nM CP, WT IC50: 35.9 ± 3.2 nM; G78L IC50: 59.7 ± 8.1 nM; T127A IC50: 40.1 ± 4.5 nM; T153L IC50: 46.2 ± 4.6 nM; R242E IC50: 46.7± 11.3 nM; G304E IC50: 32.1 ± 6.2 nM. Data are presented as the mean ± s.e.m. of 3 experiments performed in triplicate.
{kind=link}
Related to Figure 4. (A) The conformations of Phe3.36 and Trp6.48 in CB2-AM10257, CB1-AM6538 and CB1-AM11542 structures with the residues shown in green, blue, and orange sticks, respectively. (B) Key dihedrals (N-CA-CB-CG of Phe3.36 and CA-CB-CG-CD1 of Trp6.48) in 200 ns MD simulations of CB2-AM10257 comparing to crystal structures.
{kind=link}
Related to Figures 2 and 4. (A) Overall structure comparison of AM10257 (magenta sticks)-bound CB2 (green cartoon) and taranabant (orange sticks)-bound CB1 (yellow cartoon). (B) The extracellular part comparison of AM10257-bound CB2 and taranabant-bound CB1. The color scheme as in (A). (C) Binding pose comparison of AM10257 (magenta sticks), AM6538 (cyan sticks), ML056 (orange sticks) and ONO9780307 (yellow sticks) in their receptors, which are shown in green, light blue, grey and light grey cartoons, respectively. (D) In total, 43 receptors of 217 available structures were downloaded from the Protein Data Bank (PDB). The corresponding dihedral angles’ value was calculated with a unified definition of chi1 (χ1, N-CA-CB-CG) and chi2 (χ2, CA-CB-CG-CD1). The data shown in this figure only include the receptors in which the 6.48 residues are Trp. Note that some structures of the M2 and NTS1 receptors are in the upper focused area (χ2 ~ 100°) such as 4MQS and 4GRV, while M1, M3 and M4 are all in the lower focused area (χ2 ~ 40°).
{kind=link}
Related to Figures 4, 5 and 6. (A–B) The docking poses of SR144528 (A) and CP55,940 (B) are shown. (C–G) The r.m.s.d. values of ligand heavy atoms show that the docked poses are stable during the 200ns molecular dynamics simulations: AM10257 (C), SR144528 (D), CP55,940 (E), MRI2687 (F) and MRI2594 (G).
{kind=link}
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA Epitope Tag Antibody, Alexa Fluor® 488 conjugate (16B12) | Thermo Fisher Scientific Inc. | Cat#A-21287; RRID: AB_2535829 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EDTA-free complete protease inhibitor cocktail tablets | Roche | Cat#5056489001 |
| Iodoacetamide | Sigma | Cat#I1149 |
| Lauryl Maltose Neopentyl Glycerol | Anatrace | Cat#4216588 |
| Cholesterol hemisucinate (CHS) | Sigma | Cat#C6512 |
| N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) | Invitrogen | Cat#D10251 |
| TALON IMAC resin | Clontech | Cat#635507 |
| 1-Oleoyl-rac-glycerol (monoolein) | Sigma | Cat#M7765 |
| Magnesium Chloride | Fisher Scientific | Cat#BP214–500 |
| Sucrose | Fisher Scientific | Cat#S5–3 |
| Protein Assay Dye | Biorad | Cat#5000006 |
| Trizma base | Sigma | Cat#1503–1KG |
| Microscint-20 | PerkinElmer | Cat#6013621 |
| Cholesterol | Sigma | Cat#C8667 |
| CP55,940 | Tocris | Cat#0949 |
| AM10257 | This paper | N/A |
| MRI2594 | This paper | N/A |
| MRI2687 | This paper | N/A |
| [3H]CP 55,940 | NIDA Drug Supply Program | Cat#NOCD-092 |
| SR144528 | NIDA Drug Supply Program | Cat#NOCD-085 |
| CP 55,940 | NIDA Drug Supply Program | Cat#NOCD-091 |
| 4-methyl-5-phenyl-1H-pyrazole-3-carboxylic acid | ChemBridge | Cat#4036397 |
| 1-adamantylamine | Sigma-Aldrich | Cat#138576 |
| O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate | Sigma-Aldrich | Cat#12806 |
| Hünig’s base | Sigma-Aldrich | Cat#D125806 |
| Sodium Hydride | Sigma-Aldrich | Cat#452912 |
| 5-Bromo-1-pentanol | Sigma-Aldrich | Cat#670855 |
| N,N-Dimethylformamide | Sigma-Aldrich | Cat#227056 |
| dichloromethane | Sigma-Aldrich | Cat#D65100 |
| sodium sulfate | Sigma-Aldrich | Cat#239313 |
| silica gel columns | Biotage | Cat#FSUL-0442 |
| DMEM/F-12 (1:1) cell culture media | Invitrogen | Cat#11330–057 |
| Opti-MEM cell culture media | Invitrogen | Cat#11058–021 |
| FreeStyle™ 293 Expression Medium | Life Technologies | Cat#12338–026 |
| DMEM | Invitrogen | Cat#11965–118 |
| MEM | Invitrogen | Cat#11095–098 |
| Penicillin/Streptomycin | Invitrogen | Cat#15140–122 |
| Puromycin | Invitrogen | Cat#A11138–03 |
| Hygromycin B | Life Technologies | Cat#10687–010 |
| Geneticin | Life Technologies | Cat#10131–035 |
| Bovine Serum Albumin, Fraction V, Cold-ethanol Precipitated | Fisher Scientific | Cat#BP1605100 |
| Trypsin 0.5% EDTA | Invitrogen | Cat#25300–120 |
| Dulbecco’s phosphate-buffered saline (DPBS) | Invitrogen | Cat#14190250 |
| 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) | Sigma | Cat#D8417 |
| Fetal Bovine Serum (FBS) | Life Technologies | Cat#16140089 |
| Ethylenediamine Tetraacetic Acid (EDTA) | Fisher Scientific | Cat#S311–500 |
| Collagen I | Fisher | Cat#CB40231 |
| Lipofectamine® 2000 | Thermo Scientific | Cat#11668027 |
| Critical Commercial Assays | ||
| HTRF HiRange cAMP Assay Kit | CISBIO | Cat# 62AM6PEC |
| PathHunter Detection Kit | DiscoveRx | Cat#93–0001 |
| Q5 site directed mutagenesis kit | NEB | Cat#E0554S |
| In-Fusion® HD EcoDry™ Cloning System | Clontech | Cat#639684 |
| Deposited Data | ||
| CB2_AM10257 complex structure | This paper | PDB: 5ZTY |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda (Sf9) | A gift from Dr. Beili Wu (SIMM, CAS) | N/A |
| Phoenix-AMPHO | Allele Biotechnology | Cat#ABP-RCV-10001 |
| CHO-K1 | ATCC | Cat#CCL-61 |
| PathHunter® U2OS EA β-Arrestin Parental Cell Line | DiscoveRx | Cat#93–0166 |
| CHO-hCB1R PathHunter DiscoveRx | DiscoveRx | Cat#93–0959C2 |
| HA11_hCB2_pcDNA3.0 plasmid | This paper | N/A |
| HEK293 human cell line | ATCC | Cat#CRL-1573 |
| Oligonucleotides | ||
| Primers for site-direct mutagenesis | This paper, see Table S2 | N/A |
| Recombinant DNA | ||
| pMSCVpuro vector | Clontech | Cat#634401 |
| pcDNA 3.1 (+) vector | Thermo Scientific | Cat#V79020 |
| 3HA-human CB2 receptor cDNA | Missouri S&T cDNA Resource Center | Cat#CNR020TN00 |
| pCMV-ProLink™ 1 Vector | DiscoveRx | Cat#93–0167 |
| Software and Algorithms | ||
| Schrödinger Suite 2015–4 | Schrödinger | www.schrodinger.com |
| GROMACS 5.1.2 | Abraham et al., 2015 | www.gromacs.org |
| SwissParam | Zoete et al., 2011 | www.swissparam.ch |
| XDS | Kabsch, 2010 | Xds.mpimf-heidelberg.mpg.de |
| SCALA | Collaborative Computational Project, 1994 | www.ccp4.ac.uk/html/scala.html |
| Phaser | McCoy et al., 2007 | www.phenix-online.org |
| Phenix | Adams et al., 2010 | www.phenix-online.org |
| Buster | Smart et al., 2012 | www.globalphasing.com/buster |
| COOT | Emsley et al., 2010 | www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Prism v.7.0 | GraphPad Software Inc. | N/A |
| FlowJo® v.10.5.0 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| FluoView™ FV1000 | Olympus - Life Science Solutions | https://www.olympus-lifescience.com/en/ |
| Other | ||
| Solid white 384-well assay plates | VWR | Cat#82051–278 (CS) |
| Low-volume (20 µL) 384-well assay plates | VWR | Cat#784080 |
| Phoenix package system | Stanford University | N/A |
| Glass bottom dishes-uncoated (confocal plates) | MatTek | Cat#P35G-10–14-C |
| 100kDa cutoff concentrators | Sartorius | Cat#VS0642 |
| PD Minitrap G-25 coulmn | GE Healthcare | Cat#28–9180-07 |
| 96-well glass sandwich plates for LCP crystallization | NOVA | Cat#NOA90020 |
| 96-well white microplate with bonded GF/B filter | PerkinElmer | Cat#6005177 |
| 500 cm2 Square Cell Culture Dish | Corning | Cat#431110 |
Highlights:
Crystal structure of human CB2 in complex with antagonist AM10257 is determined
A high degree of conformational similarity with the agonist-bound CB1 is uncovered
The yin-yang relationship of CB2/CB1 will facilitate the design of selective drugs
ACKNOWLEDGMENTS
This work was supported by the National Nature Science Foundation of China grants 31870744 (T.H.) and 31330019 (Z.J.L.), the Ministry of Science and Technology of China grant 2015CB910104 (Z.J.L.), Key R&D Program of China grant 2016YCF0905902 (S.Z.), National Institutes of Health grants R01DA045020 (A.M., L.M.B., R.C.S), R01DA041435 (R.C.S., A.M), P01DA009158 (A.M., L.M.B.), Intramural funds of the National Institute on Alcohol Abuse and Alcoholism, NIH (G.K., R.C., M.R.I. and N.J.C) and NSF grants. P.P. acknowledges the Russian Science Foundation Research Grant (RSF No.18-74-00117). We thank the Shanghai Municipal Government and ShanghaiTech University for financial support. The synchrotron radiation experiments were performed at the BL41XU of Spring-8 with approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2017A2708 and 2017B2707), at X06SA beamline at the Swiss Light Source of the Paul Scherrer Institute, and at I24 beamline of Diamond light source. We thank the Cloning, Cell Expression and Protein Purification Core Facilities of iHuman Institute for their support. We thank Julie Liu for graphical abstract design.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
All authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the coordinates and structure factors of CB2-AM10257 is PDB: 5ZTY.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, and Mackie K (2010). CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol 160, 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, and Mackerell AD Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, and Marciano-Cabral F (2008). CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153, 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, and Cherezov V (2009). Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4, 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, and Stevens RC (2009). Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 microm size X-ray synchrotron beam. J R Soc Interface 6 Suppl 5, S587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrencik JE, Roth CB, Terakado M, Kurata H, Omi R, Kihara Y, Warshaviak D, Nakade S, Asmar-Rovira G, Mileni M, et al. (2015). Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 161, 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contino M, Capparelli E, Colabufo NA, and Bush AI (2017). Editorial: The CB2 Cannabinoid System: A New Strategy in Neurodegenerative Disorder and Neuroinflammation. Front Neurosci 11, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, and Mainz DT (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Guindon J, and Hohmann AG (2008). Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. (2012). Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, and Rice KC (1990). Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87, 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horti AG, Gao Y, Ravert HT, Finley P, Valentine H, Wong DF, Endres CJ, Savonenko AV, and Dannals RF (2010). Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg Med Chem 18, 5202–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho JH, et al. (2017). Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature [DOI] [PMC free article] [PubMed] [Retracted]
- Hua T, Vemuri K, Pu M, Qu L, Han Gye W., Wu Y, Zhao S, Shui W, Li S, Korde A, et al. (2016). Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 167, 750–762.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janero DR, and Makriyannis A (2009). Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs 14, 43–65. [DOI] [PubMed] [Google Scholar]
- Ballesteros Juan A., H.W. (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences 25, 1995, 366–428. [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, et al. (2012). Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn CA, Reich EP, Fine JS, Lavey B, Kozlowski JA, Hipkin RW, Lundell DJ, and Bober L (2008). Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br J Pharmacol 153, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier RW, Pei Y, Pandarinathan L, Janero DR, Zhang J, and Makriyannis A (2010). hCB2 ligand-interaction landscape: cysteine residues critical to biarylpyrazole antagonist binding motif and receptor modulation. Chem Biol 17, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, and Abu-Shaar M (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365, 61–65. [DOI] [PubMed] [Google Scholar]
- Ogawa G, Tius MA, Zhou H, Nikas SP, Halikhedkar A, Mallipeddi S, and Makriyannis A (2015). 3’-functionalized adamantyl cannabinoid receptor probes. J Med Chem 58, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, and Makriyannis A (2008). Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol 15, 1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov P, Peng Y, Shen L, Stevens RC, Cherezov V, Liu ZJ, and Katritch V (2018). Computational design of thermostabilizing point mutations for G protein-coupled receptors. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D, et al. (1994). SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350, 240–244. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, et al. (1998). SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther 284, 644–650. [PubMed] [Google Scholar]
- Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, and Rosenbaum DM (2016). High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature [DOI] [PMC free article] [PubMed]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, and Bricogne G (2012). Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempel AV, Stumpf A, Zhang HY, Ozdogan T, Pannasch U, Theis AK, Otte DM, Wojtalla A, Racz I, Ponomarenko A, et al. (2016). Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 90, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Sun B, Feng D, Nawaratne V, Leach K, Felder CC, Bures MG, Evans DA, Weis WI, Bachhawat P, et al. (2016). Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, et al. (2005). Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 310, 329–332. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, et al. (2012). Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Shi R, Kang X, Zhang X, Chen P, Zhang L, Hou A, Wang R, Zhao Y, Zhao K, et al. (2018). Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat Commun 9, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, He X, Vanommeslaeghe K, and MacKerell AD Jr. (2012). Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhou S, Yang P, Tian Y, Feng Z, Xie XQ, and Liu Y (2018). Targeted inhibition of the type 2 cannabinoid receptor is a novel approach to reduce renal fibrosis. Kidney Int 94, 756–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figures 1 and 2. (A) Analytical size exclusion chromatography profile. (B) Crystal image. (C) The overall structure of CB2-AM10257 complex with receptor in green and T4L fusion protein in pink. The antagonist AM10257 is shown as magenta sticks and the five single mutations are shown as yellow sticks. (D) The 2|Fo|-|Fc| and |Fo|-|Fc| maps of AM10257 are contoured at 1.6 σ and 2.5 σ, respectively, at 2.8 Å. (E) Crystal packing of CB2-AM10257. Color scheme as in (B). (F) Radioligand binding affinity of AM10257, SR144528, CP55,940 and MRI2687 on the crystallized CB2 construct against [3H]CP55,940, Ki, inhibitory constant.
Related to Figures 1 and 6. Reagents and conditions: Scheme 1: (a) TBTU, Hünig’s base, DCM, 1-adamantylamine. (b) NaH, DMF, 5-bromo-1-pentanol; Scheme 2: (a) Et3N, DCM, Acetyl Chloride; Scheme 3: (a) 2-bromoethanol 90 °C. (b) BOP, ET3N, DCM, 2,2,3,3-tetramethylcyclopropane carboxylic acid. (c) ET3N, DCM, Acetyl Chloride.
Related to Figures 2 and 3. (A) Expression of wild-type and mutant CB2 receptors in transient CHO cells. Live-cell fluorescence microscopy verified all CB2 mutants to be properly exported to the cell membrane like the wild-type receptor. (B) The summary of the flow cytometry analysis of mutant receptor expression relative to WT receptor expression as determined by impermeabilized cell N-terminal HA-tag staining. (C-D) Assessment of the effect of the individual point mutations, made to stabilize the receptor in absence of the T4L insert, on receptor activity. CP55,940, WT EC50: 1.8 ± 0.2 nM, Emax (fold): 2.0 ± 0.1; G78L EC50: 1.2 ± 0.2 nM, Emax (fold): 2.5 ± 0.1; T127A EC50: 5.4 ± 2.3 nM, Emax (fold): 1.31 ± 0.02; T153L EC50: 2.1 ± 0.4 nM, Emax (fold): 2.5 ± 0.1; R242E EC50: 2.2 ± 0.1 nM, Emax (fold): 2.2 ± 0.1; G304E EC50: 4.0 ± 0.1 nM, Emax (fold): 2.1 ± 0.1. AM10257 +100 nM CP, WT IC50: 35.9 ± 3.2 nM; G78L IC50: 59.7 ± 8.1 nM; T127A IC50: 40.1 ± 4.5 nM; T153L IC50: 46.2 ± 4.6 nM; R242E IC50: 46.7± 11.3 nM; G304E IC50: 32.1 ± 6.2 nM. Data are presented as the mean ± s.e.m. of 3 experiments performed in triplicate.
Related to Figure 4. (A) The conformations of Phe3.36 and Trp6.48 in CB2-AM10257, CB1-AM6538 and CB1-AM11542 structures with the residues shown in green, blue, and orange sticks, respectively. (B) Key dihedrals (N-CA-CB-CG of Phe3.36 and CA-CB-CG-CD1 of Trp6.48) in 200 ns MD simulations of CB2-AM10257 comparing to crystal structures.
Related to Figures 2 and 4. (A) Overall structure comparison of AM10257 (magenta sticks)-bound CB2 (green cartoon) and taranabant (orange sticks)-bound CB1 (yellow cartoon). (B) The extracellular part comparison of AM10257-bound CB2 and taranabant-bound CB1. The color scheme as in (A). (C) Binding pose comparison of AM10257 (magenta sticks), AM6538 (cyan sticks), ML056 (orange sticks) and ONO9780307 (yellow sticks) in their receptors, which are shown in green, light blue, grey and light grey cartoons, respectively. (D) In total, 43 receptors of 217 available structures were downloaded from the Protein Data Bank (PDB). The corresponding dihedral angles’ value was calculated with a unified definition of chi1 (χ1, N-CA-CB-CG) and chi2 (χ2, CA-CB-CG-CD1). The data shown in this figure only include the receptors in which the 6.48 residues are Trp. Note that some structures of the M2 and NTS1 receptors are in the upper focused area (χ2 ~ 100°) such as 4MQS and 4GRV, while M1, M3 and M4 are all in the lower focused area (χ2 ~ 40°).
Related to Figures 4, 5 and 6. (A–B) The docking poses of SR144528 (A) and CP55,940 (B) are shown. (C–G) The r.m.s.d. values of ligand heavy atoms show that the docked poses are stable during the 200ns molecular dynamics simulations: AM10257 (C), SR144528 (D), CP55,940 (E), MRI2687 (F) and MRI2594 (G).