

Abstract
Cellular senescence is associated with the induction of a proinflammatory phenotype. Notably, senescent endothelial cells are detected at the sites of atherosclerotic lesions, suggesting the involvement of senescent endothelial cells in atherogenesis. Moreover, bacterial infection has been speculated to contribute to the pathogenesis of atherosclerosis. The present study investigated the effects of Gram-negative bacterial lipopolysaccharide (LPS) and LL-37 (a human antimicrobial peptide of the cathelicidin family), on senescent endothelial cells, using serially passaged human endothelial cells. The results indicated that senescent endothelial cells exhibited the basal proinflammatory phenotype, as evidenced by higher intercellular adhesion molecule-1 (ICAM-1) expression and NF-κB p65 phosphorylation, compared with non-senescent cells. Additionally, exposure to LPS and LL-37 further enhanced the expression of ICAM-1 in senescent endothelial cells, compared with non-senescent cells. Of note, the NF-κB p65 pathway was more activated in senescent endothelial cells stimulated with LPS and LL-37. Furthermore, the expression levels of the receptors for LPS and LL-37 [toll-like receptor 4 (TLR4) and purinergic receptor P2X 7 (P2X7), respectively] were upregulated in senescent endothelial cells. These observations indicated that LPS and LL-37 enhanced the ICAM-1 expression and NF-κB p65 activation in senescent endothelial cells, potentially via the upregulated TLR4 and P2X7. Thus, senescent endothelial cells may contribute to the pathogenesis of atherosclerosis via the basal proinflammatory phenotype and the enhanced inflammatory responses against atherogenic factors, including LPS and LL-37.
Keywords: cellular senescence, endothelial cells, senescence- associated secretory phenotype, lipopolysaccharide, LL-37, NF-κB, atherosclerosis, antimicrobial peptide
Introduction
Cellular senescence is defined as an irreversible growth arrest of cells after repeated population doublings (1). Upon senescence, cells have several characteristic features, such as larger and flattened cell morphology, protein expression representing cell cycle arrest, and increased activity of senescence-associated β-galactosidase (SA-β-Gal) (2). Although cellular senescence was originally recognized as an antitumor mechanism to inhibit the proliferation of DNA-damaged cells (3), recent studies have established the proinflammatory characteristics of senescent cells, termed senescence-associated secretory phenotype (SASP) (2). Thus, it has been proposed that senescent cells contribute to the pathogenesis of age-associated inflammatory diseases (4). For instance, atherosclerosis is a representative chronic inflammatory disease of the arteries, which is closely related to aging. Notably, senescent endothelial cells are detected at the sites of atherosclerotic lesions, suggesting the involvement of senescent endothelial cells in atherogenesis (5).
In addition to classical atherogenic conditions, such as dyslipidemia and hypertension, persistent bacterial infection can contribute to the pathogenesis of atherosclerosis (6). Lipopolysaccharide (LPS), an outer membrane component of Gram-negative bacteria, is a potential pathogenic factor for atherosclerosis (7), although the mechanism by which LPS accelerates the formation of atherosclerotic lesions is unclear. LPS acts on immune cells as well as vascular cells, and induces proinflammatory or procoagulant responses via the activation of NF-κB signaling. While the expression of LPS receptors [CD14 and toll-like receptor 4 (TLR4)] is considerably low in endothelial cells compared with myeloid cells, TLR4 is upregulated in endothelial cells localized in atherosclerotic lesions (8), suggesting an augmentation of LPS-induced inflammatory responses in endothelial cells via upregulated TLR4.
LL-37 is a human antimicrobial peptide of the cathelicidin family, consisting of 37 amino acids, and predominantly produced by neutrophils and epithelial cells following cleavage from the precursor of human cationic antibacterial protein of 18 kDa (9). In addition to its broad spectrum of bactericidal activity (10), LL-37 binds with host cell surface molecules, such as chemokine receptors or growth factor receptors, and induces multidirectional immunomodulatory responses in leukocytes and epithelial cells (11). LL-37 also acts on endothelial cells and induces angiogenesis via formyl peptide receptor 2 (FPR2; also known as FPRL1 and ALX) (12), or increases endothelial cell stiffness via purinergic receptor P2X 7 (P2X7) (13). Recent studies strongly suggest the involvement of LL-37 in the pathogenesis of atherosclerosis; LL-37 was found to be highly accumulated on endothelial cells of human atherosclerotic lesions (14), and knockout (KO) of cathelin-related antimicrobial peptide (CRAMP; the mouse homolog of LL-37) reduced atherosclerosis in a hyperlipidemic ApoE-deficient mice (15). In addition, extrinsic LL-37 promoted monocyte adhesion to endothelial cells in CRAMP KO mice (16), and CRAMP activated T cells in atherosclerotic ApoE-deficient mice (17). These observations suggested that LL-37 may function as a possible pathogenic factor for atherosclerosis by acting on endothelial cells and immune cells.
The NF-κB molecule p65 subunit (RelA) has a pivotal role in the SASP induction (18). The present study aimed to evaluate the response of senescent endothelial cells to LPS and LL-37 by assessing putative atherogenic molecules. Therefore, the present study focused on NF-κB p65 activation by LPS and LL-37. The results indicated that both LPS and LL-37 potently induced intercellular adhesion molecule-1 (ICAM-1) expression and NF-κB p65 activation in senescent cells compared with non-senescent cells. Furthermore, the receptors for LPS and LL-37 (TLR4 and P2X7, respectively) were upregulated in senescent endothelial cells. These observations suggested that LPS and LL-37 enhanced ICAM-1 expression and p65 activation in senescent endothelial cells, potentially via TLR4 and P2X7.
Materials and methods
Reagents
LPS from Escherichia coli serotype O111:B4, was purchased from Sigma-Aldrich (Mer ck KGaA). The 37-mer peptide LL-37 of the human cathelicidin family (L1L GDF FRK SKE KIG KEF KRI VQR IKD FLR MLV PRTES37) was synthesized with the solid phase method on a peptide synthesizer (model PSSM-8; Shimadzu Corporation) by F-moc chemistry and purified as described previously (19). The FPR2 antagonist WRW4 peptide (sequence: WRWWWW) was from Alomone Labs; the P2X7 inhibitor KN-62 was from Sigma-Aldrich (Merck KGaA).
Antibodies
Anti-CD14-phycoerythrin (PE; MY4; cat. no. CO6603262; Beckman Coulter, Inc.), anti-TLR4-PE (HTA125; cat. no. 12-9917-42; eBioscience; Thermo Fisher Scientific, Inc.), anti-FPR2-PE (GM1D6; cat. no. sc-57141; Santa Cruz Biotechnology, Inc.) and isotype control immunoglobulin G (IgG)-PE (eBM2a; cat. no. 12-4724-42; eBioscience; Thermo Fisher Scientific, Inc.) were used for flow cytometry. Polyclonal anti-P2X7 (cat. no. APR-008; Alomone Labs), normal rabbit IgG (cat. no. PM035; Medical and Biological Laboratories, Ltd.) and PE-conjugated donkey anti-rabbit IgG (cat. no. 406421; BioLegend, Inc.) were also used for flow cytometry. Anti-ICAM-1 (H-108; cat. no. sc-7891; Santa Cruz Biotechnology, Inc.), anti-phosphorylated (p-) NF-κB p65 Ser 536 (93H1; cat. no. 3033; Cell Signaling Technology, Inc.), anti-NF-κB p65 (D14E12; cat. no. 8242; Cell Signaling Technology, Inc.), anti-cyclin-dependent kinase inhibitor 1A (CDKN1A; also known as p21 Waf1/Cip1; 12D1; cat. no. 2947; Cell Signaling Technology, Inc.), horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (cat. no. 12-348; Chemicon International; Merck KGaA) and anti-GAPDH-HRP (5A12; cat. no. 015-25473; Wako Pure Chemical Industries, Ltd.) were used for western blot analysis.
Cell culture
Human umbilical vein endothelial cells (HUVECs; tested negative for mycoplasma, bacteria, yeast, fungi, HIV-1, hepatitis B and hepatitis C) were purchased from Lonza Group, Ltd. and cultured in endothelial cell growth medium 2 (EGM-2), containing 2% FBS, endothelial cell growth supplements and antibiotics (EGM-2 Bullet kit; Lonza Group, Ltd.) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were maintained in 10 cm-diameter tissue culture-treated dishes and passaged every 3 or 4 days. HUVECs with population doubling level <4 (PDL; the total number of times the cells in the population have doubled in vitro since their primary isolation) were used as non-senescent cells; whereas senescent HUVECs were prepared by serial passage of the cells (PDL, >32) and exhibited senescent characteristics, as shown in Fig. 1.
Figure 1.
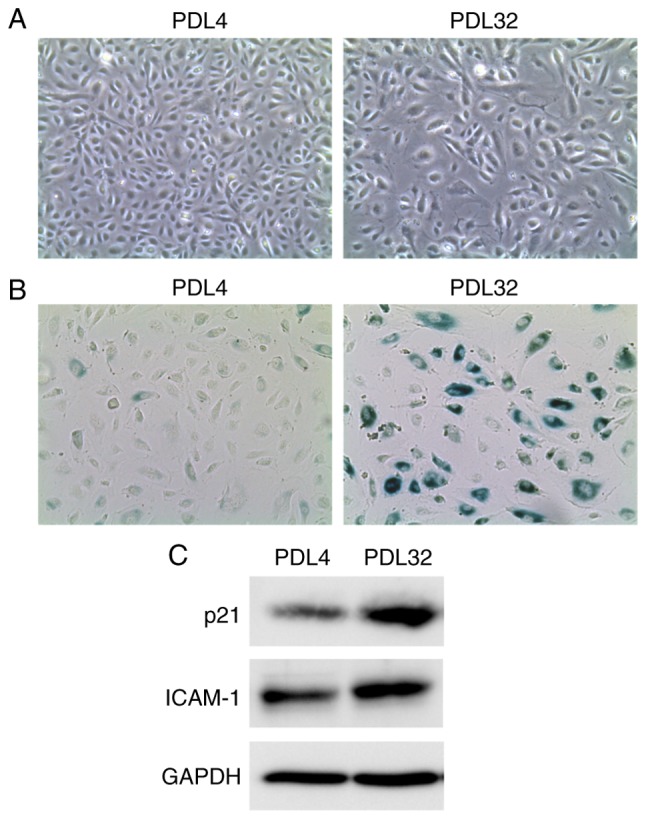
Features of senescent endothelial cells prepared by serial passage of HUVECs. HUVECs were cultured in endothelial cell growth medium-2 in 10 cm-diameter tissue culture dishes and passaged every 3 or 4 days. (A) Phase contrast images of PDL4 (left) and PDL32 cells (right). Original magnification, ×50. (B) Images of senescence-associated β-galactosidase staining in PDL4 (left) and PDL32 cells (right). Original magnification, ×100. (C) The protein expression levels of p21 Waf1/Cip1 and ICAM-1 were analyzed in PDL4 and PDL32 cells by western blotting. GAPDH was evaluated as an internal control. Images are representative of three independent experiments. HUVECs, human umbilical vein endothelial cells; PDL, population doubling level; p21 Waf1/Cip1, cyclin-dependent kinase inhibitor 1A; ICAM-1, inter-cellular adhesion molecule-1.
SA-β-Gal assay
SA-β-Gal staining was performed using Senescence Detection kit (BioVision, Inc.) according to the manufacturer's protocol. Images were captured using a Leica DM IL LED light microscope and LAS EZ software (Leica Microsystems GmbH).
Expression analysis by flow cytometry
Senescent or non-senescent HUVECs were detached with 0.05% trypsin/EDTA, washed with 1% FBS/PBS, suspended in undiluted Clear Back (a human Fc receptor blocking reagent; Medical and Biological Laboratories, Ltd.) for 15 min on ice, incubated with anti-CD14-PE, anti-TLR4-PE, anti-FRP2-PE or isotype IgG-PE (5 µg/ml final) for 30 min on ice, and then analyzed by flow cytometry (FACSCalibur; Becton, Dickinson and Company) using CellQuest Pro software (version 6.0; Becton, Dickinson and Company). For the P2X7 expression analysis, cells were detached with Cell Dissociation Solution (Sigma-Aldrich; Merck KGaA), washed, blocked with Clear Back as aforementioned, and incubated with anti-P2X7 or normal rabbit IgG (5 µg/ml final) for 30 min on ice, followed by anti-rabbit IgG-PE (5 µg/ml final) for 30 min on ice.
Western blot analysis
Senescent or non-senescent HUVECs in 6-well plates were incubated with LPS (10 or 100 ng/ml) or LL-37 (2, 5 or 10 µg/ml) for the indicated periods. For the experiments using receptor antagonists, cells were pre-incubated with WRW4 peptide (0.1 or 1 µM), KN-62 (0.1 or 1 µM) or DMSO as a solvent control for 30 min, and then incubated with LL-37 (5 µg/ml) for 24 h. Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40 and 0.1% SDS; Wako Pure Chemical Industries, Ltd.) with complete protease inhibitor cocktails and PhosSTOP phosphatase inhibitor cocktails (Roche Diagnostics). Cell lysates (5 µg protein/lane, determined by bicinchoninic acid assay) were subjected to 8% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Immobilon-P; EMD Millipore) using a semi-dry transfer system (Trans-Blot; Bio-Rad Laboratories, Inc.). The membranes were blocked with undiluted BlockAce (DS Pharma Biomedical Co., Ltd.) for 1 h at room temperature (RT), probed with anti-ICAM-1 (1:1,000), anti-p-NF-κB p65 (Ser536; 1:3,000), anti-total NF-κB p65 (1:3,000), or anti-p21 Waf1/Cip1 (1:1,000) primary antibodies overnight at 4°C, followed by HRP-conjugated goat anti-rabbit IgG (1:5,000) for 2 h at RT. Signals were developed with SuperSignal West Pico/Dura Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific, Inc.) and detected using FUSION FX luminescent image analyzer (Vilber Lourmat) and FUSION-Capt Advance software (version 17.03b; Vilber Lourmat). GAPDH was used as an internal control.
NF-κB p65 localization analysis with immunofluorescence
HUVECs were seeded into Lab Tek II CC2 chamber slides (Nunc; Thermo Fisher Scientific, Inc.) and cultured overnight at 37°C and 5% CO2. Cells were then incubated with LL-37 (10 µg/ml) at 37°C for 4 h, washed, fixed with 2% paraformaldehyde for 10 min at RT, permeabilized with 0.2% Triton X-100, blocked with undiluted BlockAce for 1 h at RT, and incubated with the primary antibody against NF-κB p65 (D14E12; 1:500) overnight at 4°C. After washing, cells were further incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (cat. no. A27034; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.) overnight at 4°C, followed by mounting with an aqueous media (Vectashield Hardset with DAPI; Vector laboratories, Inc.). Images were captured using a BZ-X710 fluorescence microscope (Keyence Corporation).
Statistical analysis
Data are presented as means ± SD. Statistical significance was determined by unpaired t-test or two-way ANOVA followed by Dunnett's multiple comparisons test (GraphPad Prism 8; GraphPad Software, Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
Preparation of senescent endothelial cells
To prepare senescent endothelial cells, HUVECs were serially passaged. A previous report revealed that HUVECs of PDL30 became non-proliferative with larger cell size, and the numbers of β-Gal-positive cells were significantly higher compared with cells of PDL9 (20), indicating that HUVECs at PDL >30 acquire a senescence phenotype. Similarly, in the present study, the cell proliferation rate appeared decreased at PDL ~30, and the cells no longer proliferated at PDL40 (data not shown). During serial passage, a morphological change of the cells was also observed from spindle shape (PDL4 cells) to enlarged and irregular shape (PDL32 cells; Fig. 1A). In addition, a marked increase in SA-β-Gal-positive cell ratio was found in PDL32 cells compared with PDL4 cells (Fig. 1B). Furthermore, western blot analysis revealed the upregulation of p21 Waf1/Cip1 (an inhibitory protein of cyclin-dependent kinase) in the PDL32 cells (Fig. 1C), indicating cell cycle arrest. Of note, it has been established that cell senescence is accompanied by the induction of the SASP inflammatory phenotype, as characterized by the upregulation of proinflammatory cytokines, adhesion molecules, growth factors or proteases (2). In the PDL32 cells generated in the present study, ICAM-1 protein expression levels were markedly increased in the PDL32 cells compared with the PDL4 cells (Fig. 1C), confirming the SASP induction. These observations indicated that senescence was successfully induced in the endothelial cells. Thus, in the present study and for subsequent experiments, cells of PDL >32 were used as senescent, while cells of PDL <4 were used as non-senescent.
LPS-induced ICAM-1 expression in senescent endothelial cells
Persistent bacterial infection or increased serum LPS are associated with the pathogenesis of atherosclerosis (6,7). Importantly, aging is a major risk factor for the development of atherosclerosis (4). Therefore, it was hypothesized that senescent endothelial cells may be more sensitive to LPS. Senescent HUVECs were incubated with LPS (10 or 100 ng/ml), and the ICAM-1 protein expression levels were compared with that of non-senescent cells by western blot analysis. As shown in Fig. 2, LPS stimulation (100 ng/ml) significantly induced ICAM-1 expression in both non-senescent cells and senescent cells. Of note, ICAM-1 protein expression levels in LPS-stimulated senescent cells (100 ng/ml) were significantly higher compared with the levels in LPS-stimulated non-senescent cells (Fig. 2).
Figure 2.
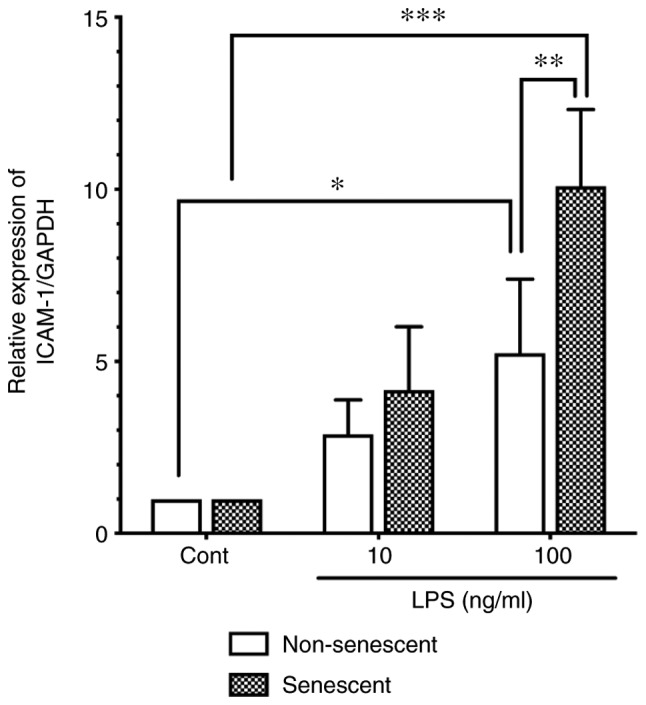
Effect of LPS on ICAM-1 expression in senescent and non-senescent endothelial cells. Senescent or non-senescent HUVECs were incubated with LPS (10 or 100 ng/ml) for 24 h. ICAM-1 protein expression levels were analyzed by western blotting, and the signals were quantified using an image analyzer. Relative expression of ICAM-1/GAPDH was expressed as a ratio to control untreated cells. Data are presented as the mean ± SD of four independent experiments. *P<0.05, **P<0.01 and ***P<0.001, with comparisons indicated by lines. LPS, lipopolysaccharide; ICAM-1, intercellular adhesion molecule-1; HUVECs, human umbilical vein endothelial cells; Cont, control.
LPS-induced NF-κB p65 activation in senescent endothelial cells
NF-κB p65 has a pivotal role in the inflammatory responses, especially in myeloid cells, and is also responsible for endothelial inflammatory responses, such as LPS-induced ICAM-1 expression (21). To evaluate the role of NF-κB p65 in senescent endothelial cells, first the basal expression and phos-phorylation levels were investigated in senescent HUVECs. Notably, the total protein expression levels of p65 were increased in senescent cells compared with non-senescent cells (Fig. 3A). In addition, the phosphorylated p65 (at Ser536) levels were increased in senescent cells compared with non-senescent cells (Fig. 3B). Thus, senescent endothelial cells express intrinsically increased levels of phosphorylated (activated) NF-κB p65 compared with non-senescent endothelial cells.
Figure 3.

Basal and LPS-stimulated levels of NF-κB p65 in senescent and non-senescent endothelial cells. (A) The expression levels of total p65 and (B) p-p65 were analyzed in senescent and non-senescent HUVECs by western blotting. Relative expression was normalized to GAPDH and presented as relative to non-senescent cells. Data are presented as the mean ± SD of six independent experiments. (C) Senescent or non-senescent HUVECs were incubated with LPS (10 or 100 ng/ml) for 1 h. The protein expression levels of total and p-p65 were analyzed by western blotting. (D) Relative expression of p-p65/total p65 was expressed as a ratio to control untreated cells. Data are presented as the mean ± SD of four independent experiments. *P<0.05 and **P<0.01, with comparisons indicated by lines. LPS, lipopolysaccharide; p-, phosphorylated; HUVECs, human umbilical vein endothelial cells; Cont, control.
Next, in order to elucidate the mechanism for the enhanced level of LPS-induced ICAM-1 expression in senescent endothelial cells (Fig. 2), the phosphorylation levels of p65 were evaluated following LPS stimulation. LPS stimulation (100 ng/ml) induced the phosphorylation of p65 (as evidenced by an increase in the p-p65/total p65 ratio) in both non-senescent and senescent cells (Fig. 3C and D); notably, p65 phosphorylation was significantly more pronounced in senescent cells, compared with non-senescent cells (Fig. 3C and D). These observations suggested that NF-κB p65 activation is enhanced by LPS stimulation, which may be associated with the enhanced expression of ICAM-1 in senescent endothelial cells.
Expression of LPS receptors in senescent endothelial cells
It has been demonstrated that aging is associated with changes of TLR expression levels in mice (22) and humans (23). Since the LPS-induced ICAM-1 expression and p65 phosphorylation were enhanced in senescent cells (Figs. 2 and 3), it was next speculated that an increase of LPS receptor expression may be present in senescent cells. Thus, the cell surface expression levels of the LPS receptors CD14 and TLR4 were analyzed in non-senescent and senescent cells by flow cytometry. The results indicated that TLR4 was upregulated in senescent endothelial cells compared with non-senescent cells (Fig. 4A). By contrast, CD14 was downregulated in senescent endothelial cells compared with non-senescent cells (Fig. 4B).
Figure 4.
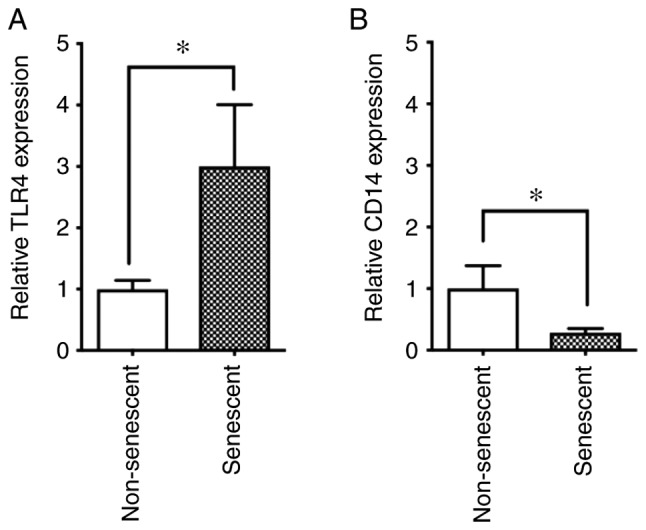
Expression levels of TLR4 and CD14 in senescent and non-senescent endothelial cells. Cell surface expression levels of the LPS receptors TLR4 and CD14 were analyzed with phycoerythrin-conjugated specific antibodies in senescent and non-senescent HUVECs by flow cytometry. (A) Relative expression of TLR4 and (B) CD14 in senescent cells was expressed as a ratio to non-senescent cells. Data are presented as the mean ± SD of at least three independent experiments. *P<0.05, with comparisons indicated by lines. TLR, toll-like receptor; LPS, lipopolysaccharide; HUVECs, human umbilical vein endothelial cells.
LL-37-induced ICAM-1 expression in senescent endothelial cells
Human antimicrobial peptide LL-37 has been speculated to be involved in the pathogenesis of atherosclerosis (14,15). Moreover, in vitro studies have demonstrated the direct action of LL-37 on immune or vascular cells; LL-37 induces integrin activation in monocytes (16), and induces ICAM-1 and monocyte chemoattractant protein-1 expression in endothelial cells (14). These observations suggest that LL-37 promotes monocyte/macrophage-endothelial cell interaction in atherosclerosis. Thus, to assess the effect of LL-37 on senescent endothelial cells, senescent HUVECs were incubated with LL-37 (2, 5 or 10 µg/ml), and the ICAM-1 protein expression levels were compared with those of non-senescent cells by western blot analysis. LL-37 stimulation induced ICAM-1 expression in senescent HUVECs (Fig. 5). Notably, LL-37 stimulation (5 and 10 µg/ml) more potently induced ICAM-1 expression in senescent cells compared with non-senescent cells (Fig. 5).
Figure 5.
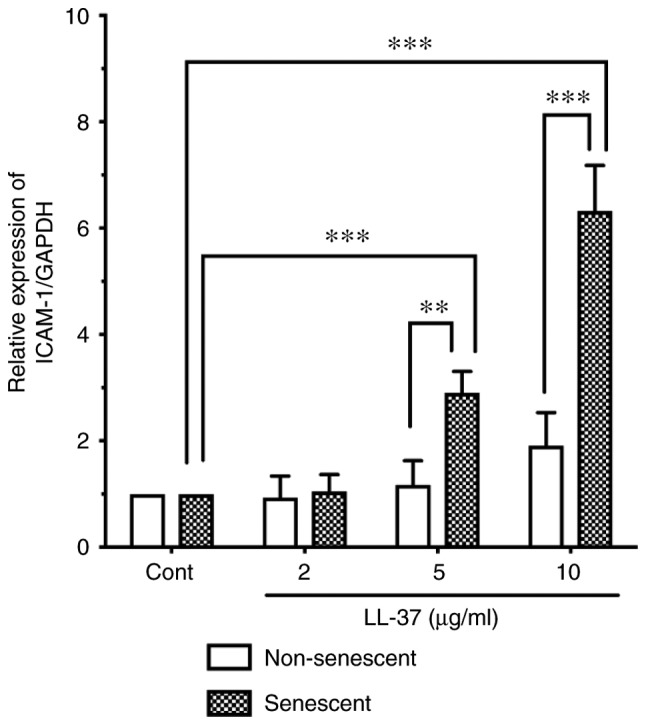
Effect of LL-37 on ICAM-1 expression in senescent and non-senescent endothelial cells. Senescent or non-senescent HUVECs were incubated with LL-37 (2, 5 or 10 µg/ml) for 24 h. ICAM-1 expression was analyzed by western blotting, and the signals were quantified using an image analyzer. Relative expression of ICAM-1/GAPDH was expressed as a ratio to control untreated cells. Data are presented as the mean ± SD of at least three independent experiments. **P<0.01 and ***P<0.001, with comparisons indicated by lines. ICAM-1, intercellular adhesion molecule-1; HUVECs, human umbilical vein endothelial cells; Cont, control.
LL-37-induced NF-κB p65 activation in senescent endothelial cells
The signaling molecules involved in the LL-37-induced ICAM-1 expression of endothelial cells has not been elucidated. Koczulla et al (12) reported that NF-κB p65 was involved in the LL-37-induced endothelial cell proliferation, by demonstrating nuclear translocation of p65 following LL-37 stimulation (12). Thus, the present study investigated the localization of p65 in HUVECs following LL-37 stimulation by immunofluorescence. The results revealed that LL-37 stimulation (10 µg/ml) induced the translocation of p65 from cytoplasm to nucleus (Fig. 6A), confirming the activation of NF-κB signaling by LL-37 in endothelial cells.
Figure 6.
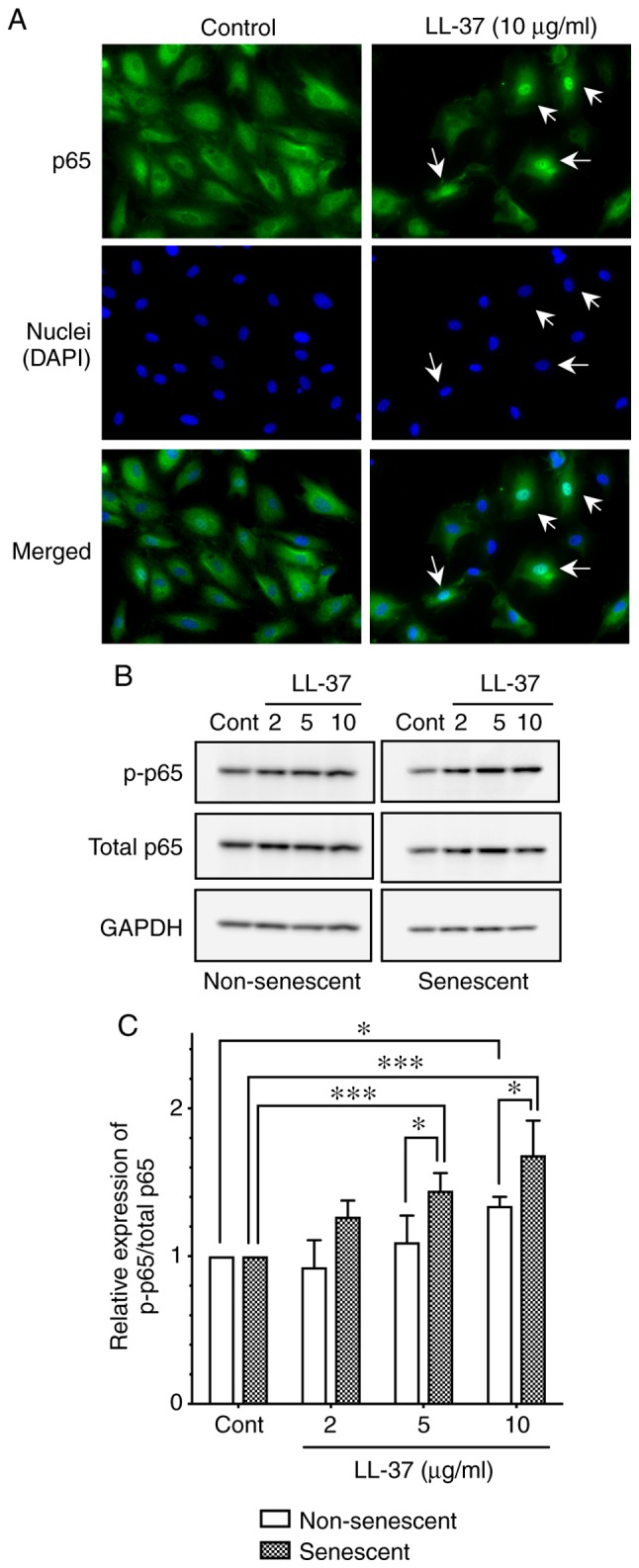
Effect of LL-37 on NF-κB p65 activation in senescent and non-senescent endothelial cells. (A) Non-senescent HUVECs were seeded into chamber slides and incubated without (Control) or with LL-37 (10 µg/ml) for 4 h. Cells were then analyzed for p65 expression (green) by immunofluorescence. Nuclei were counterstained with DAPI (blue). Images are representative of three independent experiments. Arrows indicate the cells with nuclear localization of p65. Original magnification, ×400. (B) Senescent or non-senescent HUVECs were incubated with LL-37 (2, 5 or 10 µg/ml) for 4 h. Total and p-p65 protein expression levels were analyzed by western blotting. (C) Relative expression of p-p65/total p65 was calculated as a ratio to untreated control cells, in senescent and non-senescent cells. Data are presented as the mean ± SD of at least four independent experiments. *P<0.05 and ***P<0.001, with comparisons indicated by lines. HUVECs, human umbilical vein endothelial cells; p-, phosphorylated; Cont, control.
Next, to elucidate the mechanism underlying the enhanced levels of LL-37-induced ICAM-1 expression in senescent endothelial cells (Fig. 5), the present study evaluated the activation of p65 following LL-37 stimulation by western blot analysis. LL-37 stimulation (10 µg/ml) induced the phosphorylation of p65 (as evidenced by the increased ratio of phosphorylated p65/total p65) in both non-senescent and senescent cells (Fig. 6B and C); notably, p65 phosphorylation was significantly enhanced in senescent cells, compared with non-senescent cells (Fig. 6B and C). These observations suggested that NF-κB p65 was more potently activated (phosphorylated) by LL-37 in senescent HUVECs, which may contribute to the enhanced expression of ICAM-1 in senescent endothelial cells.
Expression of LL-37 receptors in senescent endothelial cells
Several cell surface receptors are involved in the LL-37-induced host cell activation. FRP2 is known as an LL-37 receptor in endothelial cells; LL-37 induces cell proliferation of HUVECs via FRP2 (12), or acts on FRP2 in endothelial cells to induce vascular smooth muscle relaxation in veins (24). However, the LL-37 receptor responsible for ICAM-1 induction is unknown. To identify the LL-37 receptor involved in ICAM-1 induction, the present study first assessed the effect of an FPR2 antagonist on the LL-37-induced ICAM-1 expression. As shown in Fig. 7A, the FPR2 antagonist WRW4 peptide (1 µM) attenuated the LL-37-induced ICAM-1 expression, suggesting that LL-37 induced ICAM-1 expression via FPR2. Additionally, P2X7 is also reported as a receptor for LL-37 in immune cells (25) and endothelial cells (13). In the present study, the P2X7 antagonist KN-62 (1 µM) attenuated the LL-37-induced ICAM-1 expression (Fig. 7A), confirming P2X7 as an LL-37 receptor involved in ICAM-1 induction. Of note, the combination of WRW4 and KN-62 (0.1 µM each) suppressed the LL-37-induced ICAM-1 expression, although WRW4 or KN-62 at the same concentration alone did not affect ICAM-1 expression (Fig. 7A), suggesting that FRP2 and P2X7 may cooperatively function as receptors for the LL-37-induced ICAM-1 expression. These observations indicated that LL-37 induced ICAM-1 expression in endothelial cells via both FRP2 and P2X7.
Figure 7.
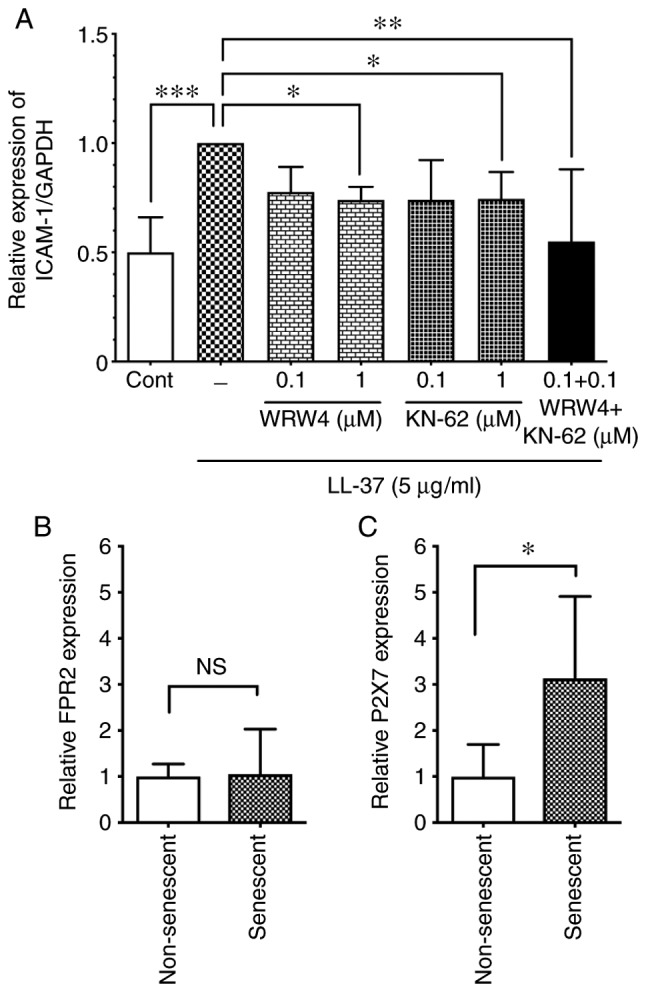
Role of FPR2 and P2X7 receptors in senescent and non-senescent endothelial cells. (A) Non-senescent HUVECs were preincubated with WRW4 (0.1 or 1 µM) or KN-62 (0.1 or 1 µM) for 30 min, and then incubated with LL-37 (5 µg/ml) for 24 h. Alternatively, non-senescent HUVECs were preincubated with a combination of WRW4 (0.1 µM) and KN-62 (0.1 µM) for 30 min, and then incubated with LL-37 (5 µg/ml) for 24 h. ICAM-1 protein expression levels were analyzed by western blotting. Relative expression of ICAM-1/GAPDH was calculated as a ratio to LL-37-stimulated cells without antagonists. Data are presented as the mean ± SD of at least three independent experiments. (B) Cell surface expression levels of LL-37 receptors FPR2 and (C) P2X7 were analyzed in senescent and non-senescent HUVECs by flow cytometry. Relative expression in senescent cells was calculated as a ratio to non-senescent cells. Data are presented as the mean ± SD of at least four independent experiments. *P<0.05, **P<0.01 and ***P<0.001, with comparisons indicated by lines. FPR2, formyl peptide receptor 2; P2X7, purinergic receptor P2X 7; HUVECs, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molecule-1; Cont, control; NS, not significant.
Since LL-37 induced ICAM-1 expression and p65 phos-phorylation more potently in senescent cells compared with non-senescent cells (Figs. 5 and 6), it was speculated that the LL-37 receptors may be upregulated in senescent cells. Thus, the cell surface expression levels of FRP2 and P2X7 were analyzed in senescent cells by flow cytometry. The results demonstrated that P2X7 was upregulated in senescent cells compared with non-senescent cells (Fig. 7C), whereas no difference was observed in the FRP2 levels between senescent and non-senescent cells (Fig. 7B).
Discussion
Cellular senescence is associated with the induction of the proinflammatory phenotype, termed SASP (2). ICAM-1 is recognized as a representative SASP marker in endothelial cells, since overexpression of ICAM-1 is commonly observed in several senescent endothelial cell models, including stress stimulation (26), serial passage (20) and oncogene transfer (5). In the present study, the basal expression levels of ICAM-1 were compared between senescent and non-senescent HUVECs, and, as expected, ICAM-1 was demonstrated to be upregulated in senescent cells (Fig. 1C). Furthermore, p65 activation (phosphorylation) was compared between senescent and non-senescent endothelial cells, since NF-κB p65 has a pivotal role in basal SASP induction (27). The present results demonstrated that p65 phosphorylation, as well as the total amount of p65, was upregulated in senescent endothelial cells. Thus, the present study revealed that senescent endothelial cells exhibited intrinsic proinflammatory features, as indicated by higher ICAM-1 expression and p65 activation.
Localization of senescent endothelial cells at the sites of atherosclerotic lesions suggests the involvement of senescent endothelial cells in atherogenesis (5). The present study compared the response to LPS, a potential atherogenic factor, between senescent and non-senescent HUVECs, and revealed that both ICAM-1 expression and p65 phosphorylation were increased in senescent endothelial cells compared with non-senescent endothelial cells. This observation suggested that the NF-κB p65 pathway was more potently activated by LPS in senescent endothelial cells, which may result in the enhanced expression of ICAM-1. Expression of ICAM-1 (an adhesion molecule) is important for the interaction of monocyte/macrophage to endothelial cells, the initial step of vascular inflammation. Thus, the enhanced ICAM-1 expression is expected to promote monocyte/macrophage adhesion to senescent endothelial cells and migration of these cells into the atherosclerotic lesion.
Previously, the effect of LPS on senescent cells was evaluated using fibroblasts in vitro; LPS from Campylobacter rectus, which is associated with adult periodontitis, induces higher production of interleukin (IL)-6 and plasminogen activator in senescent gingival fibroblasts prepared by serial passage (28,29). These results are consistent with the present finding that the LPS response is more enhanced in senescent cells compared with non-senescent cells.
The important role of TLR4-mediated signaling has been demonstrated in atherogenesis, based on the finding that the KO of TLR4 or myeloid differentiation factor 88 (MyD88) reduces the aortic plaque area in ApoE-deficient atherosclerotic mouse (30). To elucidate the mechanism for enhanced p65 activation and ICAM-1 expression in senescent endothelial cells, the present study compared the expression levels of LPS receptors TLR4 and CD14 between non-senescent and senescent cells by flow cytometry. Of note, TLR4 was upregulated in senescent endothelial cells, whereas CD14 was downregulated in senescent endothelial cells. Therefore, the LPS-induced enhanced p65 phosphorylation and ICAM-1 expression observed in senescent endothelial cells may be mediated via the upregulated TLR4. However, the involvement of CD14 in the LPS response in senescent cells cannot be excluded, because endothelial cells have also been reported to use a soluble form of CD14 for the transfer of LPS to TLR4 (31).
The present study revealed the upregulation of TLR4 in serial passage-induced senescent endothelial cells. Notably, high glucose and oxidative stress are recognized as inducers of cellular senescence (32), and TLR4 is upregulated by high glucose in human retinal vascular endothelial cells isolated from patients with diabetic retinopathy (32), and by oxidative stress in rat and human cerebral endothelial cells (33). Therefore, it can be speculated that TLR4 is upregulated in senescent endothelial cells under various conditions including high glucose and oxidative stress.
It is proposed that bacterial infection with Chlamydia pneumoniae or Porphyromonas gingivaris is associated with the pathogenesis of atherosclerosis (34). We have previously indicated that Chlamydiaceae LPS has low affinities for LPS recognition molecules (such as CD14 and LPS-binding protein) and exhibits weak biological activities against monocytes, thereby possibly contributing to the persistent inflammatory response during infection (35). Furthermore, P. gingivaris LPS, at a higher concentration (1 µg/ml), was demonstrated to induce proinflammatory responses including ICAM-1 expression in HUVECs (36). By contrast, gut-derived bacterial LPS (such as E. coli LPS) possesses a potent biological activity and can be detected in the atherosclerotic plaque of carotid arteries (37); in addition, the serum levels of E. coli LPS is significantly higher in the atherosclerosis patients (37). Thus, the present study used E. coli LPS for evaluating the LPS response of senescent cells, and revealed that E. coli LPS potently induced ICAM-1 expression and p65 phosphorylation in senescent endothelial cells.
LL-37 was originally identified as a human antimicrobial peptide of the cathelicidin family; it is released by neutrophils and epithelial cells upon infection and participates in bacterial killing (9,10). However accumulating evidence has revealed that LL-37 acts on host cells (immune cells, epithelial cells and endothelial cells) and exhibits immunomodulatory action (11). Previous studies suggested the involvement of LL-37 in the pathogenesis of atherosclerosis (14-17). The current study compared the response of senescent and non-senescent HUVECs to LL-37, and revealed that ICAM-1 expression and p65 activation were induced more by LL-37 in senescent endothelial cells. This observation suggested that the NF-κB pathway was more potently activated by LL-37 in senescent endothelial cells to induce the enhanced expression of ICAM-1.
In the present study, the ICAM-1 induction by LL-37 in senescent endothelial cells was greater compared with non-senescent cells; LL-37 stimulation (10 µg/ml) increased ICAM-1 expression by 6.3 times in senescent cells but only 1.9 times in non-senescent cells (Fig. 5). By contrast, the induction of p65 phosphorylation by LL-37 was not apparent between senescent and non-senescent cells (1.7 times in senescent cells and 1.3 times in non-senescent; Fig. 6C). These observations suggested that signaling molecules other than NF-κB p65 may be involved in the LL-37-induced ICAM-1 expression. It has been reported that LL-37 can activate the ERK1/2, p38 MAPK and PI3K pathways, as well as the NF-κB pathway, in several types of cells (38).
FPR2 and P2X7, the receptors for LL-37, have been identified to be present in endothelial cells (12,13). To clarify the involvement of FPR2 and P2X7 in the ICAM-1 induction by LL-37, the present study utilized FPR2 and P2X7 antagonists (WRW4 peptide and KN-62, respectively), and revealed that the two antagonists inhibited the LL-37-induced ICAM-1 expression in non-senescent endothelial cells (Fig. 7A), indicating that LL-37 induced ICAM-1 expression via FPR2 and P2X7 in non-senescent endothelial cells.
In order to further clarify the contribution of the two LL-37 receptors to the enhanced ICAM-1 expression in senescent endothelial cells, the expression levels of FPR2 and P2X7 were compared between senescent and non-senescent cells, by flow cytometry (Fig. 7) and western blot analysis (data not shown). The results demonstrated that both FPR2 and P2X7 were expressed in non-senescent and senescent cells, and that P2X7 expression, but not FPR2 expression, was upregulated in senescent endothelial cells compared with non-senescent endothelial cells. These observations suggested that upregulated P2X7 may have a more important role in the enhanced response to LL-37 in senescent endothelial cells. In this regard, it is interesting to note that endothelial P2X7 is increased in atherosclerotic lesions of mouse aorta (39) and is required for inflammatory signaling in endothelial cells exposed to low shear stress mimicking atherogenic conditions (39). These observations suggest that LL-37 localized on endothelial cells of human atherosclerotic lesion (14) may contribute to the enhanced inflammatory response during atherogenesis possibly via the upregulated P2X7.
The effect of LPS or LL-37 on a different type of senescent endothelial cells (such as aortic endothelial cells) has not been investigated in the present study. However, the expression of endothelial TLR4 is augmented in human atherosclerotic aorta lesions, where senescent endothelial cells have been detected (8). Additionally, endothelial P2X7 is increased in atherosclerotic lesions of mouse aorta (39). Based on these observations, it can be speculated that enhanced inflammatory responses could be induced by LPS or LL-37 in different types of senescent endothelial cells (such as HUVECs and aortic endothelial cells).
In addition to its broad spectrum of bactericidal function, LL-37 directly binds to LPS released from Gram-negative bacteria, and neutralizes its biological activity (40). Due to the LPS-neutralizing activity, LL-37 suppresses the LPS-induced TNF-α production by monocytes/macrophages (19), IL-1β release and pyroptosis of monocytes/macrophages (41) and apoptosis of endothelial cells (42). Thus, it would be of interest to examine the effect of LL-37 on the LPS-induced expression of ICAM-1 in non-senescent and senescent endothelial cells. The present study evaluated the effect of simultaneous stimulation with LPS and LL-37, and the results indicated that in non-senescent endothelial cells, LL-37 stimulation (5 µg/ml) did not induce the ICAM-1 expression (Fig. 5) and almost completely suppressed the LPS (100 ng/ml)-induced expression of ICAM-1 due to its LPS-neutralizing activity (data not shown). By contrast, in senescent endothelial cells, LL-37 stimulation (5 µg/ml) itself significantly upregulated ICAM-1 expression (Fig. 5), but suppressed the LPS-induced expression of ICAM-1; however, the suppression was partial, and ICAM-1 expression was retained (data not shown). Thus, LL-37 could be an atherogenic molecule for senescent endothelial cells even in the presence of LPS.
In conclusion, the present study revealed that senescent endothelial cells exhibited basal proinflammatory phenotype, as evidenced by higher ICAM-1 expression and NF-κB p65 phosphorylation. In addition, ICAM-1 expression was potently enhanced in senescent endothelial cells upon exposure to LPS and LL-37. Furthermore, NF-κB p65 signaling was more activated by LPS and LL-37 in senescent endothelial cells compared with non-senescent cells, possibly via the upregulation of their respective receptors TLR4 and P2X7. Altogether, the present results indicated that senescent endothelial cells may contribute to the pathogenesis of atherosclerosis via the basal proinflammatory phenotype and the enhanced inflammatory response against atherogenic factors, including LPS and LL-37.
Acknowledgments
We thank members of the Laboratory of Proteomics and Biomolecular Science (Dr Yoshiki Miura), the Division of Molecular and Biochemical Research (Professor Hiroshi Koide), and the Division of Cell Biology (Drs Akemi Koyanagi and Tamami Sakanishi), Research Support Center, Juntendo University Graduate School of Medicine for synthesizing LL-37 peptide or technical assistance. We are also grateful to Dr Akimasa Someya (Department of Host Defense and Biochemical Research, Juntendo University Graduate School of Medicine) for helpful discussion.
Funding
This study was supported by the Strategic Research Foundation Grant-aided Project for Private Universities, 2014-2018 (grant no. S1411007), and the MEXT KAKENHI (grant no. JP16K08789) from the Ministry of Education, Culture, Sport, Science, and Technology, Japan.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
KS designed the study. KS and MO performed the experiments and analyzed data. KS and IN wrote and edited the manuscript. All the authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 2.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campisi J, d'Adda di Fagagna F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 4.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.CIR.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 6.Aimetti M, Romano F, Nessi F. Microbiologic analysis of periodontal pockets and carotid atheromatous plaques in advanced chronic periodontitis patients. J Periodontol. 2007;78:1718–1723. doi: 10.1902/jop.2007.060473. [DOI] [PubMed] [Google Scholar]
- 7.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007;27:1433–1439. doi: 10.1161/ATVBAHA.106.138743. [DOI] [PubMed] [Google Scholar]
- 8.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation. 2002;105:1158–1161. doi: 10.1161/circ.105.10.1158. [DOI] [PubMed] [Google Scholar]
- 9.Nagaoka I, Hirata M, Sugimoto K, Tsutsumi-Ishii Y, Someya A, Saionji K, Igari J. Evaluation of the expression of human CAP18 gene during neutrophil maturation in the bone marrow. J Leukoc Biol. 1998;64:845–852. doi: 10.1002/jlb.64.6.845. [DOI] [PubMed] [Google Scholar]
- 10.Travis SM, Anderson NN, Forsyth WR, Espiritu C, Conway BD, Greenberg EP, McCray PB, Jr, Lehrer RI, Welsh MJ, Tack BF. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect Immun. 2000;68:2748–2755. doi: 10.1128/IAI.68.5.2748-2755.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi KY, Chow LNY, Mookherjee N. Cationic host defence peptides: Multifaceted role in immune modulation and inflammation. J Innate Immun. 2012;4:361–370. doi: 10.1159/000336630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrücker K, Unterberger P, Zaiou M, Lebherz C, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665–1672. doi: 10.1172/JCI17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byfield FJ, Wen Q, Leszczynska K, Kulakowska A, Namiot Z, Janmey PA, Bucki R. Cathelicidin LL-37 peptide regulates endothelial cell stiffness and endothelial barrier permeability. Am J Physiol Cell Physiol. 2011;300:C105–C112. doi: 10.1152/ajpcell.00158.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edfeldt K, Agerberth B, Rottenberg ME, Gudmundsson GH, Wang XB, Mandal K, Xu Q, Yan ZQ. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1551–1557. doi: 10.1161/01.ATV.0000223901.08459.57. [DOI] [PubMed] [Google Scholar]
- 15.Doering Y, Drechsler M, Wantha S, Kemmerich K, Lievens D, Vijayan S, Gallo RL, Weber C, Soehnlein O. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ Res. 2012;110:1052–1056. doi: 10.1161/CIRCRESAHA.112.265868. [DOI] [PubMed] [Google Scholar]
- 16.Wantha S, Alard JE, Megens RT, van der Döes AM, Döring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL, et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. 2013;112:792–801. doi: 10.1161/CIRCRESAHA.112.300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mihailovic PM, Lio WM, Yano J, Zhao X, Zhou J, Chyu KY, Shah PK, Cercek B, Dimayuga PC. The cathelicidin protein CRAMP is a potential atherosclerosis self-antigen in ApoE(−/−) mice. PLoS One. 2017;12:e0187432. doi: 10.1371/journal.pone.0187432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaoka I, Hirota S, Niyonsaba F, Hirata M, Adachi Y, Tamura H, Heumann D. Cathelicidin family of antibacterial peptides CAP18 and CAP11 inhibit the expression of TNF-alpha by blocking the binding of LPS to CD14(+) cells. J Immunol. 2001;167:3329–3338. doi: 10.4049/jimmunol.167.6.3329. [DOI] [PubMed] [Google Scholar]
- 20.Yanaka M, Honma T, Sato K, Shinohara N, Ito J, Tanaka Y, Tsuduki T, Ikeda I. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci Biotechnol Biochem. 2011;75:1098–1103. doi: 10.1271/bbb.100909. [DOI] [PubMed] [Google Scholar]
- 21.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- 22.Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: Impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–4701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 23.Qian F, Wang X, Zhang L, Chen S, Piecychna M, Allore H, Bockenstedt L, Malawista S, Bucala R, Shaw AC, et al. Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly. Aging Cell. 2012;11:104–110. doi: 10.1111/j.1474-9726.2011.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berkestedt I, Nelson A, Bodelsson M. Endogenous antimicrobial peptide LL-37 induces human vasodilatation. Br J Anaesth. 2008;100:803–809. doi: 10.1093/bja/aen074. [DOI] [PubMed] [Google Scholar]
- 25.Nagaoka I, Tamura H, Hirata M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J Immunol. 2006;176:3044–3052. doi: 10.4049/jimmunol.176.5.3044. [DOI] [PubMed] [Google Scholar]
- 26.Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, Stuppner H, Lipovac M, Uhrin P, Breuss JM. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep. 2017;7:39501. doi: 10.1038/srep39501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-κB in aging and disease. Aging Dis. 2011;2:449–465. [PMC free article] [PubMed] [Google Scholar]
- 28.Ogura N, Matsuda U, Tanaka F, Shibata Y, Takiguchi H, Abiko Y. In vitro senescence enhances IL-6 production in human gingival fibroblasts induced by lipopolysaccharide from Campylobacter rectus. Mech Ageing Dev. 1996;87:47–59. doi: 10.1016/0047-6374(96)01701-0. [DOI] [PubMed] [Google Scholar]
- 29.Mochizuki K, Yamaguchi M, Abiko Y. Enhancement of LPS-stimulated plasminogen activator production in aged gingival fibroblasts. J Periodontal Res. 1999;34:251–260. doi: 10.1111/j.1600-0765.1999.tb02251.x. [DOI] [PubMed] [Google Scholar]
- 30.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lloyd-Jones KL, Kelly MM, Kubes P. Varying importance of soluble and membrane CD14 in endothelial detection of lipopoly-saccharide. J Immunol. 2008;181:1446–1453. doi: 10.4049/jimmunol.181.2.1446. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Wang J, Fang J, Zhou H, Liu X, Su SB. High glucose induces and activates Toll-like receptor 4 in endothelial cells of diabetic retinopathy. Diabetol Metab Syndr. 2015;7:89. doi: 10.1186/s13098-015-0086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Haskó J, Krizbai IA. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Borel N, Pospischil A, Dowling RD, Dumrese C, Gaydos CA, Bunk S, Hermann C, Ramirez JA, Summersgill JT. Antigens of persistent Chlamydia pneumoniae within coronary atheroma from patients undergoing heart transplantation. J Clin Pathol. 2012;65:171–177. doi: 10.1136/jclinpath-2011-200270. [DOI] [PubMed] [Google Scholar]
- 35.Tsutsumi-Ishii Y, Shimada K, Daida H, Toman R, Nagaoka I. Low potency of Chlamydophila LPS to activate human mononuclear cells due to its reduced affinities for CD14 and LPS-binding protein. Int Immunol. 2008;20:199–208. doi: 10.1093/intimm/dxm129. [DOI] [PubMed] [Google Scholar]
- 36.An N, Andrukhov O, Tang Y, Falkensammer F, Bantleon HP, Ouyang X, Rausch-Fan X. Effect of nicotine and Porphyromonas gingivalis lipopolysaccharide on endothelial cells in vitro. PLoS One. 2014;9:e96942. doi: 10.1371/journal.pone.0096942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carnevale R, Nocella C, Petrozza V, Cammisotto V, Pacini L, Sorrentino V, Martinelli O, Irace L, Sciarretta S, Frati G, et al. Localization of lipopolysaccharide from Escherichia coli into human atherosclerotic plaque. Sci Rep. 2018;8:3598. doi: 10.1038/s41598-018-22076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agier J, Efenberger M, Brzezińska-Blaszczyk E. Cathelicidin impact on inflammatory cells. Cent Eur J Immunol. 2015;40:225–235. doi: 10.5114/ceji.2015.51359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green JP, Souilhol C, Xanthis I, Martinez-Campesino L, Bowden NP, Evans PC, Wilson HL. Atheroprone flow activates inflammation via endothelial ATP-dependent P2X7-p38 signalling. Cardiovasc Res. 2018;114:324–335. doi: 10.1093/cvr/cvx213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC. Human CAP18: A novel antimicrobial lipopolysaccha-ride-binding protein. Infect Immun. 1995;63:1291–1297. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, Nagaoka I. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS One. 2014;9:e85765. doi: 10.1371/journal.pone.0085765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki K, Murakami T, Kuwahara-Arai K, Tamura H, Hiramatsu K, Nagaoka I. Human anti-microbial cathelicidin peptide LL-37 suppresses the LPS-induced apoptosis of endothelial cells. Int Immunol. 2011;23:185–193. doi: 10.1093/intimm/dxq471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.