

Abstract
Virotherapy using oncolytic viruses is an upcoming therapy strategy for cancer treatment. A variety of preclinical and clinical trials have indicated that adenoviruses may be used as potent agents in the treatment of a variety of cancers, and also for the treatment of brain tumors. In these studies, it has also been shown that oncovirotherapy is safe in terms of toxicity and side effects. In addition, previous studies have presented evidence for a significant role of oncovirotherapy in the activation of anti-tumor immune responses. With regard to oncolytic adenoviruses, we have demonstrated previously that the multifunctional protein Y-box binding protein-1 (YB-1) is a potent factor that was used to develop an YB-1-dependent oncolytic adenovirus (XVir-N-31). XVir-N-31 provides the opportunity for tumor-selective replication and exhibited marked oncolytic properties in a mouse glioma tumor model using therapy-resistant brain tumor initiating cells (BTICs). In a number of, but not all, patients with glioma, YB-1 is primarily located in the nucleus; this promotes XVir-N-31-replication and subsequently tumor cell lysis. However, in certain BTICs, only a small amount of YB-1 has been identified to be nuclear, and therefore virus replication is suboptimal. YB-1 in BTICs was demonstrated to be translocated into the nucleus following irradiation, which was accompanied by an enhancement in XVir-N-31 production. R28 glioma spheres implanted in living organotypic human brain slices exhibited a significantly delayed growth rate when pre-irradiated prior to XVir-N-31-infection as compared with single treatment methods. Consistent with the in vitro data, R28 glioma-bearing mice exhibited a prolonged mean and median survival following single tumor irradiation prior to intratumoral XVir-N-31 injection, compared with the single treatment methods. In conclusion, the present study demonstrated that in an experimental glioma model, tumor irradiation strengthened the effect of an XVir-N-31-based oncovirotherapy.
Keywords: glioma, oncolytic adenovirus, irradiation
Introduction
Glioblastoma (GBM) is the most malignant type of brain tumor in humans, and ~3,000 incident cases are diagnosed in Germany every year. Even with the highest level of care and use of novel therapeutic strategies, including tumor treating fields, the median survival of GBM patients is <20 months (1), and survival times have only marginally improved during the previous decade. Therefore, the development of novel therapeutic strategies is necessary. The high levels of malignancy of GBM is a result of its characteristic behaviors, including immunosuppression, and high levels of resistance towards irradiation, most chemotherapeutics and hypoxia. In addition, the tumor exhibits extensive neo-angiogenesis and grows invasively into the healthy brain tissue, making complete surgical resection impossible. Brain tumor initiating cells (BTICs), also termed glioma stem cell like cells, are an extremely resistant subpopulation of GBM cells that are considered to be responsible for tumor initiation and recurrence (2). It has also been demonstrated that temozolomide (TMZ), the standard chemotherapeutic drug used to treat GBM, leads to a dedifferentiation of GBM cells towards a more stem cell like phenotype (3). Involved in this processes are signaling cascades including the PI3K/AKT and RAS/mitogen active protein kinase pathway, which are constitutively activated in GBM cells (4,5). A downstream substrate of these pathways is Y-box binding protein-1 (YB-1), a protein that exhibits both transcriptional and translational activity (6,7). In a number of tumor types, YB-1 expression is correlated with malignancy and progression (8-10). Cellular stress, for example by DNA damage induced by irradiation or chemotherapy, induces several post-translational YB-1 modifications including phos-phorylation, acetylation or SUMOylation. These modifications are suggested to promote its nuclear translocation (11-14). Subsequently, nuclear YB-1 activates the expression of multi-drug resistance protein/P-glycoproteins, classical mediators of resistance towards tumor treatment (15).
YB-1 is involved in the replication of therapeutic adenoviruses. The YB-1-dependent oncolytic adenovirus XVir-N-31 developed by this study group, also termed Ad-Delo3-RGD, replicates in tumor cells harboring nuclear YB-1 (16,17). In mice bearing R28-cell-derived glioma, a single intratumoral injection of XVir-N-31 prolonged survival rates (18). In ~60% of BTICs, YB-1 is upregulated and located primarily in the nucleus, which correlates with good virus replication and virus-mediated lysis of these cells. However, in ~40% of BTICs, YB-1 is only slightly expressed and located primarily in the cytoplasm, which mitigates virus replication 100-1,000-fold (18). We demonstrated previously that irradiation of established cancer cell lines leads to the translocation of YB-1 into the nucleus (6,19). Whether this also occurs in BTICs has not yet been identified. The present study evaluated whether a therapeutically relevant irradiation dose induced the translocation of YB-1 into the nucleus, promoted XVir-N-31 replication and subsequently oncolysis of BTICs in vitro and in a BTIC-derived mouse glioma model, with aim of developing a clinically relevant therapeutic strategy that combines irradiation and XVir-N-31-based oncovirotherapy for patients with recurrent glioma.
Materials and methods
Cell culture
BTIC R11, R28 and R49 cell lines, kindly provided by Dr C. Beier (University Hospital Regensburg, Center for Brain Tumors, Regensburg, Germany), were obtained from patients with primary GBM, as described previously (20), and were maintained as tumor spheres in stem cell-permissive Dulbecco's modified Eagle's medium/F12 medium (Merck KGaA) supplemented with human recombinant epidermal growth factor (EGF; BD Biosciences), human recombinant basic fibroblast growth factor (bFGF; R&D Systems Europe, Ltd.), human leukemia inhibitory factor (EMD Millipore; 20 ng/ml each) and 2% B27 supplement (Thermo Fisher Scientific, Inc.) for preservation of the original molecular characteristics of the tumors and to minimize differentiation. R28mCherry BTIC were generated by transduction of R28 cells with Lenti-mCherry (BioCat GmbH) followed by selection with puromycin. In brief, 4×105 cells were infected with pre-made lentiviral particles encoding the fluorescent protein mCherry, at multiplicity of infection (MOI) 5. At 24 h post-infection, the medium was changed to fresh growth medium. At 48 h post-infection, puromycin (2 µg/ml; Merck KGaA) was added. Selected surviving cells were monitored for mCherry fluorescence at ×50 magnification using a Zeiss Axio Imager Z1 fluorescence microscope (Carl Zeiss AG). For adenoviral infection, BTIC spheres were separated into single cells by trypsinization. Irradiation of BTICs and R28-tumor-sphere containing brain slices was performed in a Gammacell GC40 device [Nordion (Canada) Inc.].
Brain tissue slices and BTIC implantation. Written informed consent was obtained from all patients, allowing spare tissue from resective surgery to be included in the present study. Ethical approval was obtained from the Ethics Commission of Tübingen (approval no. 338/2016A). Tissues from healthy cortex areas were obtained during surgery of patients with epilepsy. Tissue preparation was performed according to previously published protocols (21). The tissue was carefully micro-dissected and resected with only minimal use of bipolar forceps to ensure tissue integrity, transferred into ice-cold artificial cerebrospinal fluid (aCSF: 110 mM choline chloride, 26 mM NaHCO3, 10 mM D-glucose, 11.6 mM Na-ascorbate, 7 mM MgCl2, 3.1 mM Na-pyruvate, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2) equilibrated with carbogene (95% O2, 5% CO2) and immediately transported to the laboratory. Tissue was kept submerged in cool and carbogenated aCSF at all times. Following removal of the pia, tissue chunks were trimmed perpendicular to the cortical surface and 250-350 µm thick slices were prepared using a Microm HM 650V vibratome (Thermo Fisher Scientific, Inc.). Following slicing of the cortical tissue, slices were cut into several evenly sized pieces (~5.0×2.0 mm). Subsequently, the slices were transferred onto culture membranes of uncoated 30 mm Millicell-CM tissue culture inserts with 0.4 µm pores (Merck KGaA) and maintained in 6-well culture dishes (BD Biosciences). For long-term culture the plates were stored in at 37°C, 5% CO2 and 100% humidity in human CSF isolated from non-tumor patients, as described previously (22). Neurons were stained for 1 h at 37°C with NeuO (0.25 µM; STEMCELL Technologies, Inc.). Then, the NeuO-containing CSF was removed and fresh CSF was added. To calculate the total amount of cells in a tissue slice, nuclei were stained with DAPI (Abcam), according to the manufacturer's protocol. Z-stacks were obtained microscopically using a Zeiss Axio Imager Z1 fluorescence microscope (Carl Zeiss AG) at ×50 magnification. Nuclei were counted using the ImageJ software (version win63, Fiji) (23). For BTIC implantation into the tissue slices, R28mCherry spheres measuring 20-50 cells in size were used. Implantation was performed manually under microscopic observation using a 10 µl pipette tip. Slices and spheres were grown in human CSF supplemented with recombinant human bFGF and EGF (20 ng/ml each) for up to 23 days. The CSF was changed every 3rd day. Images were captured using Zeiss AxioVision (version SE64 Rel. 4.8.) and ZEN2 software (version 1.0; Carl Zeiss AG) on a Zeiss Axio Imager Z1 fluorescence microscope (Carl Zeiss AG) at ×50 or ×100 magnification. Quantification of tumor growth was performed using Image J software (version win64, Fiji) (23). If the tumor area reached the borders of the photographically detectable area, the highest measurable value was used for quantification.
Virus preparation and infection
XVir-N-31 was prepared, purified and titrated as previously described (18). The transduction efficacy of R11, R28 and R49 BTICs was determined using a green fluorescent protein (GFP)-expressing adenovirus (Ad-RGD-GFP) that possessed the same RGD-motif-modified capsid as XVir-N-31. Infections were performed by adding the appropriate amount of virus into the growth medium. At 48 h post-infection, the cells were separated into single cells by trypzination, and analyzed for GFP expression by flow cytometry. Analyses were performed on a Cyan ADP flow cytometer (Beckman Coulter, Inc.) using the Summit software (version 4.3; Beckman Coulter, Inc.).
The MOI of BTICs using XVir-N-31 was optimized for each in vitro experiment. Infections were performed, as described above. After 72 h, cells and medium were collated and freeze-thaw lysates were prepared. Viral titers in these lysates were determined in 293 cells (Microbix Biosystems, Inc.) using the Adeno-Rapid-X-Titration kit (Takara Bio Europe SAS).
Western blot analysis
Total cell lysates were prepared as described previously (18). Nuclear and cytoplasmic protein lysates were prepared according to the protocol described by Toulany et al (6). Western blot analysis was performed as previously described (18). The following antibodies were used: Anti-YB-1 (1:1,000, cat. no. SC-101198, Santa Cruz Biotechnology, Inc.; or cat. no. 9744, Cell Signaling Technology, Inc.), anti-Histone H3 (1:10,000; cat. no. 39163; Active Motif), anti-calreticulin (1:5,000; cat. no. C7492; Sigma-Aldrich; Merck KGaA) and GAPDH (1:1,000; cat. no. MAB374; Merck KGaA). Immunoreactive specific proteins were detected using the CLARITY Western ECL substrate on a ChemiDoc MP imaging device using the ImageLab 5.1 software (all from Bio-Rad Laboratories, Inc.). For quantification of YB-1 expression, Image J (version win64; Fiji) (23) was used.
Intracranial tumor model
A total of 1×105 R28 cells were implanted under peritoneally applied anesthesia (0.05 mg/kg fentanyl, 5 mg/ml midazolam and 0.5 mg/kg medetomin) (24) in the right striatum of 5-6 week old female Rj:NMRI Foxn1nu/Foxn1nu nude mice (Janvier Labs) using a mouse stereotactic device with automated infusion pump (Stoelting Co) and a 10 µl Hamilton syringe (Type 701SN; Medchrom Medizinischer Chromatographiebedarf GmbH). Carprofen (5 mg/kg) was injected subcutaneously as an analgesic. Following surgery, anesthesia was antagonized by a subcutaneous injection of naloxon (1.2 mg/kg), flumazenil (0.5 mg/kg) and atipamezol (2.5 mg/kg). Mice were housed in IVC cages under standard pathogen-free conditions in the animal facility of the Hertie Institute (Tübingen, Germany). Animal health and behavior was monitored 3 times per week. At day 11 after tumor cell implantation, the 30 tumor cell-injected mice were randomly split into four groups (n=5-7 animals per group). A total of two groups were tumor-irradiated with 3 Gy (6 MV photons) using a LINAC linear accelerator as described previously (25) and received an intratumoral injection of either PBS (sham) or 1.5×108 infectious units (IFU) XVir-N-31 the next day. One group received only XVir-N-31 and one group received an intracranial PBS injection (sham). The mice were euthanized by lethal CO2 inhalation (10-30 volume percentage discharge of CO2/per minute in their homing cage) at the onset of tumor-related symptoms. Human endpoints were defined according to the score sheet of regional council approval N12/14 and include the early onset of hemi- or paraplegia, paroxism, gait disorder, or loss of body weight of >19%. Euthanasia was confirmed by the lack of respiration and faded eye color. After 150 days the experiment was terminated by euthanasia of the surviving animals. All animal research was performed in accordance with the German Animal Welfare Act and its guidelines for the care and use of laboratory animals (https://www.gesetze-im-internet.de/tierschg/BJNR012770972.html) and was approved by the regional council of Tübingen (Regierungspräsidium Tübingen, approval no. N12/14).
Statistical analysis
For the in vitro assays, results were calculated as means ± standard deviation (SD) and compared using the one-way analysis of variance, followed by Bonferroni post-hoc correction for multiple testing using GraphPad Prism software (version 7.04; GraphPad Software, Inc.). If not otherwise indicated, the data were from at least 3 repeat experiments. For the in vivo assays, survival and mean or median survival times were analysed using the Kaplan-Meier method and compared using a log-rank test with JMP v13 software (SAS Institute Inc.), followed by Bonferroni correction for multiple testing. P<0.05 was considered to indicate a statistically significant difference.
Results
Irradiation induces YB-1 expression, its nuclear translocation and promotes XVir-N-31 production in BTICs
For established glioma and breast cancer cell lines, it has been demonstrated that irradiation leads to the translocation of YB-1 into the nucleus (6,19). The present study examined whether this was also true in TMZ-resistant BTICs (18). Using 3 different BTIC lines, we identified an upregulation of total YB-1 in the cells 8 h after irradiation, however to a different extent (Fig. 1). Using nuclear and cytoplasmic cell extracts, an additional, higher molecular weight YB-1 band (YB-160 kD) was detected in the nuclear fraction of irradiated R28 and R49 cells, but not in non-irradiated control cells. In the R11 cells, this band was present in the nuclear extracts of both irradiated and non-irradiated cells. In R49 cells, an additional YB-1 protein variant (YB-165 kD) was detected at a high level at 24 h after irradiation (Fig. 2A). As it has been established that under stress stimuli, like irradiation, the YB-1 protein will be post-translationally modified, all YB-1 bands were used to quantify the amount of nuclear YB-1 (Fig. 2B). Nuclear YB-1 levels were enhanced in both irradiated R28 and R49 cells. However, doses of only 2 Gy were sufficient to relocate YB-1 into the nucleus in R28 cells whilst doses of 6 Gy were required for nuclear translocation of YB-1 in R49 cells (Fig. 2 and data not shown). Notably, in R49 cells, YB-165 kD, a highly post-translationally modified YB-1, was prominent at high levels and was not observed in R11 nor in R28 cells. No relevant elevated nuclear YB-1 levels and no YB-165 kD bands were observed in R11 cells, which harbor a high basal level of nuclear YB-1 in contrast to R28 and R49 cells (Fig. 2). R11 and R28 cells were also irradiated using a dose of 6 Gy. However, this irradiation dose did not increase the level of nuclear YB-1 in R11 nor in R28 cells, and even induced cell death in R28 cells (data not shown).
Figure 1.
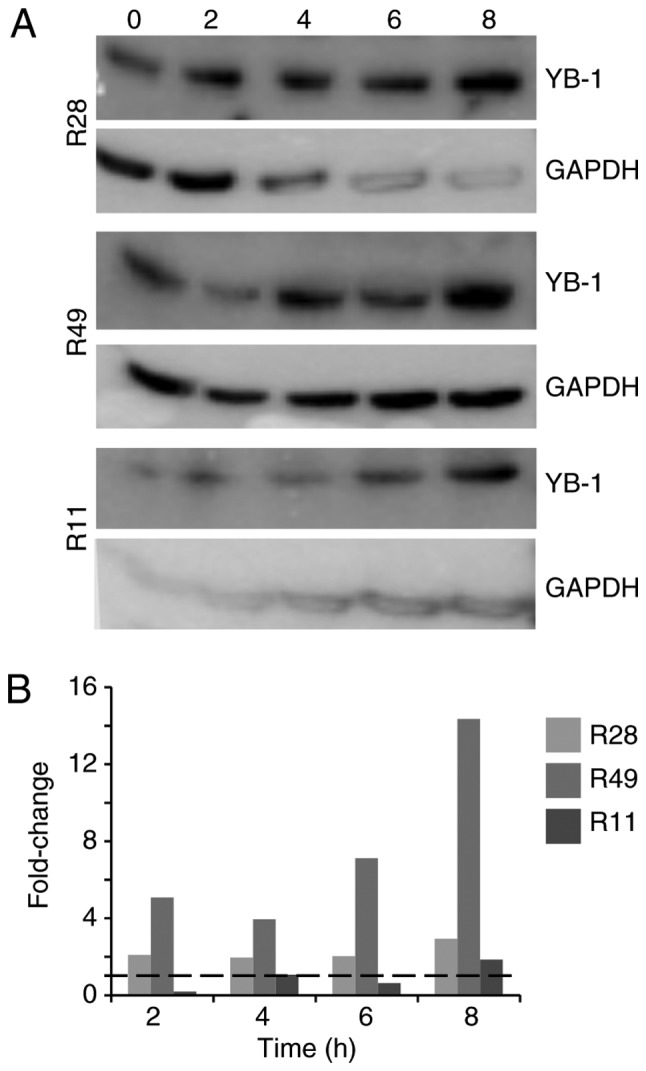
Irradiation induces YB-1 expression in brain tumor initiating cells. (A) R28, R11 and R49 cells were irradiated with 2 Gy. Cell lysates were prepared at the indicated time periods (2-8 h) following irradiation. YB-1 expression was determined by western blot analysis. GAPDH was used as a control. (B) Upregulation of total YB-1 following irradiation at the indicated time points. YB-1, Y-box binding protein-1.
Figure 2.
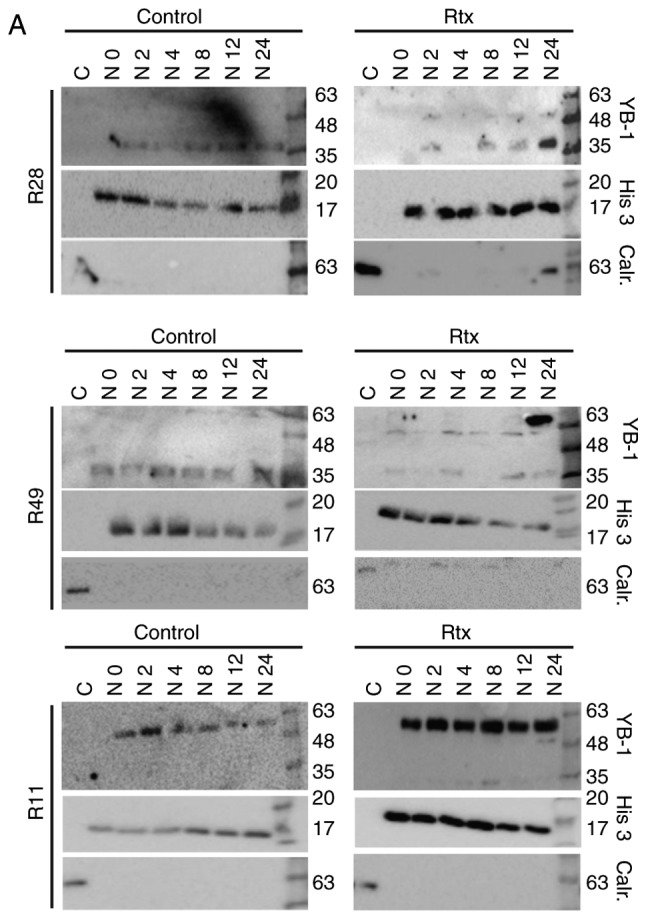

Irradiation induces YB-1 nuclear translocation in brain tumor initiating cells. (A) The cells were irradiated with either 2 Gy (R11 and R28 cells) or 6 Gy (R49 cells). At the indicted time periods nuclear extracts were generated. As a control, a cytoplasmic protein extract was prepared. YB-1 expression was detected by western blot analysis. To control extract purity, detection of the nuclear protein histone H3 and the cytoplasmic protein calreticulin was performed. (B-D) Upregulation of the different isoforms of nuclear YB-1 in non-irradiated and irradiated (B) R28, (C) R49 and (D) R11 cells at the indicated time points. YB-1, Y-box binding protein-1; Rtx, irradiation; C, cytoplasmic extract; N, nuclear extract.
To determine the effect of nuclear YB-1 on XVir-N-31 production, the adenoviral transduction efficacy of glioma stem cells was first examined, which was 6.55±0.1% at 1 MOI and 16.0±1.1% at 3 MOI for R11 cells, 2.5±0.1% at 1 MOI and 6.05±0.1% at 3 MOI for R28 cells and 2.3±0.1% at 1 MOI and 5.35±0.9% for R49 cells (Fig. 3A). To examine whether irradiation enhanced XVir-N-31 production, the cells were irradiated 24 h prior to XVir-N-31 infection. For infection, only the aforementioned low infection rates were used, as it has been established that XVir-N-31 replicates efficiently in glioma cells at a dose of 5 MOI (19). Although the levels were not significant due to a high variability in virus production in the independent experiments, the nuclear translocation of YB-1 in R28 and R49 cells, and in particular the level of nuclear YB-165 kD, correlated well with the enhanced production of XVir-N-31 in these cells. Whilst combined irradiation and infection of R28 and R49 cells using 1 MOI of XVir-N-31 exhibited almost no enhancement in virus production, the increase of XVir-N-31 production in irradiated R49 cells infected with 3 MOI was 1.6-130-fold, depending on the irradiation dose, and was superior to that observed in R28 cells (1.2-7-fold; Fig. 3B; Table I). In R28 cells, a dose of 6 Gy of irradiation led to cell death (data not shown), paralleled by a decrease in virus production (Fig. 3). As expected, in irradiated R11 BTICs that exhibited high basal nuclear YB-1 expression but no relevant irradiated-mediated nuclear transfer of YB-1, no significant enhancement of XVir-N-31 production was detected.
Figure 3.

Irradiation of brain tumor initiating cells prior to XVir-N-31 infection potentiates the production of XVir-N-31. (A) Transduction efficacy of the adenovirus in glioma stem cells, as determined by flow cytometry analysis at 48 h post-infection, using either 1 or 3 MOI (n=2). Error bars represent standard deviation. (B) R11, R28 and R49 cells were irradiated with 2, 4 or 6 Gy and were infected with either 1 or 3 MOI of XVir-N-31 24 h later. After 72 h, cells and medium were collated and total infection units were determined (n=3). Data are presented as mean ± standard error of the mean, and analyzed by one-way analysis of variance. MOI, multiplicity of infection.
Table I.
XVir-N-31 production in irradiated brain tumor initiating cells.
| 2 | 4 | 6 | 2 | 4 | 6 | Gy | |
|---|---|---|---|---|---|---|---|
| Cell lines | 1 | 1 | 1 | 3 | 3 | 3 | MOI |
| R28 | 0.4-1.4 | 0.8-1.4 | 0.5-0.9 | 1.2-5.0 | 1.6-7.0 | 0.5-0.9 | - |
| R49 | 0.5-2.1 | 0.4-1.2 | 0.4-31.8 | 1.0-7.5 | 1.6-91.4 | 4.9-130.4 | - |
| R11 | 1.0-1.3 | 0.6-1.8 | 1.1-2.8 | 1.4-2.0 | 0.8-1.5 | 0.7-3.0 | - |
Data are presented as fold induction compared with non-irradiated cells. MOI, multiplicity of infection.
Irradiation in combination with XVir-N-31 infection significantly decreases the growth of R28mCherry spheres implanted in human cortical brain slices
To analyze the effect of a combined irradiation and XVir-N-31-based oncovirotherapy in therapy-resistant BTICs, an organotypic in vitro glioma model was established, in which R28mCherry spheres were implanted in human cortical brain tissue slices. The slices were irradiated with either 3 Gy, infected with 5×107 IFU of XVir-N-31 or were irradiated and infected 24 h later. Tumor sphere growth was quantified over a period of 14 days. A longer period of quantification was not possible, as most tumor sphere sizes expanded the microscopic observation area after this time point. As indicated in Figs. 4 and 5, all (6/6) untreated control R28mCherry tumor spheres grew quickly, whilst irradiation or infection alone resulted in a decrease in sphere growth rate. By single infection with XVir-N-31, the growth of 3/6 spheres was almost completely inhibited, whilst 3/6 spheres continued to grow, but to a lesser extent compared with the controls. Nevertheless, in this group a number of tumor cells exhibited the typical characteristic of an oncolytic adenovirus infection, including decreased cell size and a rounded cell shape, indicating that these cells were dying (Fig. 6). When irradiation was used as a single treatment, all spheres (6/6) were delayed in growth. However, surviving tumor cells grew in a highly invasive manner into the brain tissue (Fig. 6). By contrast, combined therapy was very efficient and significantly delayed tumor sphere growth; 1/6 spheres was nearly completely eradicated upon treatment, 2/6 spheres exhibited a highly delayed growth and 3/6 grew slowly. The morphology of these slow-growing spheres was therefore assessed. As indicated in Fig. 6, in these spheres many cells exhibited the characteristics of oncolysis, however, a population of non-infected viable tumor cells that continued to proliferate was visible.
Figure 4.

Irradiation prior to XVir-N-31 infection delayed the growth of R28 tumor spheroids in human cortical brain slice cultures. R28mCherry spheroids were implanted in human brain slices stained with NeuO. The cultures were left either untreated (Co), irradiated with 3 Gy, infected with 5×107 IFU XVir-N-31, or were irradiated with 3 Gy and infected using 5×107 IFU XVir-N-31 24 h later. Images were captured every second day starting at the day of XVir-N-31 infection. Green staining represents NeuO staining in the brain slices. Red staining represents R28mCherry-expressing cells. Representative 1/6 spheres from 2 independent experiments is presented (scale bar=200 µm). IFU, infectious units; Co, control; Combi, 3 Gy tumor irradiation + an intratumoral XVir-N-31 injection (1.5×108 IFU) 24 h later.
Figure 5.

Irradiation prior to XVir-N-31 infection significantly decreases the growth of R28mCherry spheres in human cortical brain slice cultures. Quantification of R28mCherry spheroid growth in human brain slice cultures. R28mCherry spheroids were implanted as described. Images were captured every second day and tumor areas were quantified (n=6 spheres). Data from 2 independent experiments are presented. Data were analyzed using one-way analysis of variance. *P<0.05 and **P<0.01. Co, control; Combi, 3 Gy tumor irradiation + an intratumoral XVir-N-31 injection (1.5×108 infectious units) 24 h later.
Figure 6.

Morphological characteristics of R28mCherry spheroids. High magnification of treated R28 spheroids growing in human brain slice cultures at day 17 after treatment. (scale bar =500 µm). Co, control; Combi, 3 Gy tumor irradiation + an intratumoral XVir-N-31 injection (1.5×108 infectious units) 24 h later.
Irradiation in combination with XVir-N-31 infection prolongs the median survival of R28 tumor-bearing nude mice
The promising in vitro results prompted the analysis of the therapeutic effect of a combined XVir-N-31-based oncoviro-irradiation therapy in mice. For this purpose, R28 BTICs were intracranially implanted into the striatum of nude mice. The mice were treated as follows: i) Sham (no irradiation + intratumoral injection of PBS); ii) irradiation (3 Gy tumor irradiation) + intratumoral PBS injection 24 h later; iii) XVir-N-31 (no irradiation + intratumoral injection of 1.5×108 IFU XVir-N-31). This virus amount represents one-half of the recently determined effective therapeutic dose (18); iv) Combi [3 Gy tumor irradiation + an intratumoral XVir-N-31 injection (1.5×108 IFU) 24 h later; Fig. 7A]. A suboptimal dose of XVir-N-31 was selected for the XVir-N-31 and Combi groups to identify any possible augmentation of the oncolytic effect of XVir-N-31 by tumor irradiation prior to infection. Large tumors were observed visually in the sham treated mice (data not shown), and these animals succumbed at around day 85. The mean survival of the single treatment groups was increased compared with that of the controls, but was almost the same for both mono-therapies (95 days for irradiation and 96 days for XVir-N-31 infection groups, respectively). The median survival of the virus-treatment group was improved (96 days) compared with that of the irradiated group (88 days), but did not reach significance. As expected, based on previous data, oncovirotherapy using a suboptimal amount of infectious XVir-N-31 particles (1.5×108 IFU) did not reach the median survival of ~120 days, as was observed previously when using the double amount of XVir-N-31 (3×108 IFU) (18). Nevertheless, there was a trend towards a prolonged survival in the group of mice subjected to tumor irradiation followed by oncovirotherapy, although this did not reach significance. The median survival in this group was 101 days, and the mean survival 108 days. One mouse within this group did not exhibit any tumor-associated symptoms, even at a period of 150 days after tumor implantation. This mouse was sacrificed, but no visible tumors were detected (data not shown).
Figure 7.
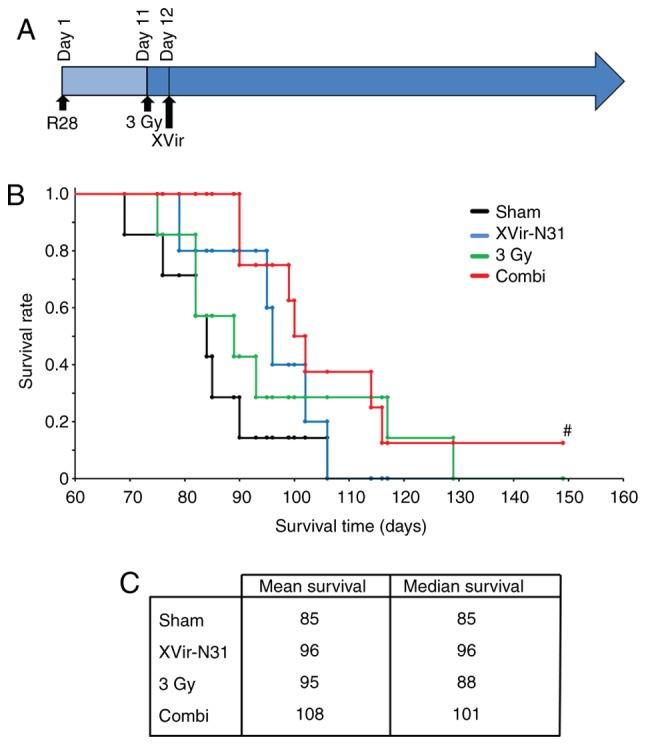
Irradiation prior to XVir-N-31 infection prolongs the survival of R28 tumor bearing mice. The mice (n=5-8 per group) were treated as described. (A) Treatment schema. (B) Kaplan-Meier survival analysis. (C) Mean and median survival times of the treatment groups. Combi, 3 Gy tumor irradiation + an intratumoral XVir-N-31 injection (1.5×108 infectious units) 24 h later. #No tumor visible.
Discussion
The results of the present study demonstrated that irradiation of BTICs enhanced the lytic activity of the YB-1-dependent oncolytic adenovirus XVir-N-31. BTICs are highly therapy-resistant cells that exhibit stem cell characteristics, and have been proposed as the primordial cells responsible for disease initiation and its recurrence (2). Due to the fact that not many therapeutic options are available to efficiently treat recurrent glioma, the results of the present study may have substantial consequences on the development of future combined treatment strategies. We previously determined the effects of XVir-N-31 in BTICs and demonstrated that XVir-N-31 replicated in BTICs. In a BTIC-derived mouse glioma model, treatment of mice with a single intratu-moral injection of XVir-N-31 significantly prolonged their survival (18). XVir-N-31 replication is dependent on the presence of nuclear YB-1 (19). YB-1 is primarily expressed in all high grade glioma, conveys resistance towards TMZ, the standard chemotherapeutic drug to treat GBM, and serves as a prognostic biomarker for tumor progression (18,26,27). However, in some BTICs only a small amount of YB-1 was identified to be nuclear (18), therefore virus replication is suboptimal. In previous years, several studies have demonstrated that YB-1, in response to genotoxic stress, becomes post-translationally modified and is transferred from the cytoplasm into the nucleus. This in turn enhances the replication and oncolytic effect of XVir-N-31 in established tumor cell lines and experimental glioma models (6,19,28).
Since oncovirotherapy is still not a standardized therapy option to treat glioma, it is expected to be primarily used as second line therapy for recurrent glioma. As it has been established that the irradiation of established glioma cell lines induced the translocation of YB-1 into the nucleus and supported XVir-N-31 replication, we hypothesized that irradiation prior to oncovirotherapy may augment the therapeutic effect of a XVir-N-31-based oncovirotherapy in patients with recurrent glioma. To examine this hypothesis, the effect of irradiation on the subcellular localization of YB-1 in BTICs, the primary target cells in and drivers of recurrent glioma, was investigated. Elevated nuclear YB-1 was detected in R28 and R49 BTICs, however, in R11 cells no irradiation-mediated nuclear translocation of YB-1 was observed. In the present study, in contrast to R28 and R49 BTICs, R11 cells were observed to possess a high basal level of nuclear YB-1. This observation is not concordant with previous data that revealed the presence of both cytoplasmic and nuclear YB-1 in R11 cells (18), but this may be explained by the use of different YB-1 antibodies between these studies. Nevertheless, the high basal level of nuclear YB-1 in R11 BTICs may explain why these cells possess a higher basal XVir-N-31 replication rate compared with the R28 and R49 cells, why there is no additional increase in nuclear YB-1 levels detected following irradiation of these cells, and why irradiation did not enhance XVir-N-31 production.
In the present study, R49 cells required a higher irradiation dose (6 Gy) to initiate the transfer of YB-1 into the nucleus. In concordance with the observed elevated nuclear level of YB-1 in R49 and R28 BTICs, irradiation prior to infection facilitated XVir-N-31-production in these cells up to 130-fold. However, induction of virus production was variable. As YB-1 serves as a stress protein, its posttranslational modification and nuclear translocation, and consequently its activity as a transcription factor that is necessary for XVir-N-31 replication, is very sensitive to external conditions. Additionally, virus production itself is a complex process including transcription of viral proteins, DNA replication, and the self-assembly of viral proteins and the genome to create infectious viral particles. This may explain the variability of virus production observed in the irradiated R28 and R49 cells.
Notably, in the R49 cells, which exhibited superior XVir-N-31 production following irradiation compared with the R28 cells, the presence of nuclear YB-165 kD, which was suggested to represent a highly post-translationally modified version of YB-1, was detected. We hypothesized that not only the nuclear translocation of YB-1, but also its post-translational modification, is important for its function as a transcription factor necessary for XVir-N-31 replication. Whether this modification includes only phosphorylation or also acetylation or SUMOylation (11-14), or even presently unknown modifications, requires additional studies. In summary, the data from the present study demonstrated a direct association between nuclear (modified) YB-1 and XVir-N-31-replication and its production in established tumor cell lines, which was in concordance with our previous observations (19).
Subsequently, the therapeutic effect of a combined XVir-N31-based oncoviro-radiotherapy was assessed in human cortical brain slice cultured harboring R28mCherry glioma spheroids as well as in R28 glioma bearing mice. Infection of slice cultures was performed with a suboptimal dose of XVir-N-31 (5×107 IFU) to determine the synergistic effects of irradiation and virus infection. The specific time point of virus infection (24 h after irradiation) was selected as elevated levels of nuclear (post-translationally modified) YB-1 were detectable after this period. Due to the ability of XVir-N-31 to also infect neurons, glial cells and astrocytes, the virus dose selected only equated to ~0.1 IFU/cell. Therefore, the therapeutic effect of the XVir-N-31-based monotherapy was only moderate. However, a delay in R28mCherry tumor spheroid growth was detectable even at this concentration. A total of 3/6 analyzed spheroids exhibited a marked delay in growth, and the others a moderate delay. The variance in growth rates may not only be caused by virus infection, but also by the variable size of the implanted spheroids. When examined more closely, an oncolytic cytopathic effect and dying BTICs were observed in the XVir-N-31-infected tissues, as indicated by a rounded cell shape and the occurrence of cell debris in the spheroids (Fig. 6). Irradiation also delayed the growth of BTICs, however no dead cells were observed and the surviving cells massively invaded the surrounding brain tissue, an effect that has been described in glioma cells in the past (29). By contrast, irradiation in combination with XVir-N-31-infection significantly decreased R28mCherry tumor spheroid growth. Whilst 1 spheroid did not grow at all, 2 exhibited a massive and 3 a considerable decrease in growth. This effect appeared to be comparable to, or even exceed, the effect of irradiation alone. The structure of spheroids with considerable growth reduction were then microscopically examined and a number of dead cells and cell debris were detected. In addition, viable cells that had started to divide again were observed in these samples, but at a later time point following treatment (days 17-23). The outgrowth of tumor cells following combined therapy may be an effect provoked by the low amount of infectious XVir-N-31 used to treat the spheroids, and the fact that certain cells may not have been infected at all. If these cells invade the surrounding brain tissue at a faster rate compared with virus replication in the adjacent infected tumor cells occurs, the cancer cells will be protected from virus infection due to a protective wall of non-neoplastic brain cells that forms a barrier for virus distribution in which the virus is unable to replicate.
The therapeutic effects of a XVir-N-31-based oncovirotherapy were previously determined in vivo in a mouse glioma model, in which it was demonstrated that ~3×108 IFU XVir-N-31 were required to significantly decrease tumor growth, leading to a significant survival prolongation (18). To confirm the therapeutic effect of a combined oncoviro-radiotherapy, as was observed in R28mCherry BTIC-derived glioma containing organotypic brain cultures in vitro, and to demonstrate the support of tumor irradiation on oncolysis-mediated tumor cell death and therefore on tumor growth and survival, only a suboptimal amount of XVir-N-31 (1.5×108 IFU) was intra-tumorally injected in the disease model in the present study. This virus dose was in itself not able to significantly prolong survival. Additionally, radiotherapy was administered prior to infection by a single 3 Gy tumor irradiation, a dose that is used during fractional irradiation of patients with glioma (30). Although non-significant, a prolongation of the mean survival in both monotherapy groups for 10-11 days was observed. The survival rates in the mice that received the oncoviro-irradiation therapy instead increased by 23 days. The median survival was determined in this group, and the combined treatment exhibited superior effects compared with the monotherapies. The effects achieved by the combination of tumor-irradiation and XVir-N-31-based oncovirotherapy were not statistically significant, however suboptimal total doses of irradiation and of XVir-N-31 were used in the therapeutic glioma mouse model. Therefore, we hypothesize that optimizing the therapeutic schema, for example repeated irradiation and injection of a therapeutic relevant dose of XVir-N-31, may result in improved effects of this combination on tumor growth and survival.
Although the interaction between irradiation and oncolytic adenoviruses is complex, given the diversity of cellular responses to irradiation and the diverse oncovirus-mediated antitumoral immune effects, the irradiation-mediated post-translational modifications of YB-1 and its nuclear transport, due to its central role in XVir-N-31-replication, may explain the superior effects of a combined oncoviro-radiotherapy in the glioma mouse model established in the present study using therapy-resistant BTICs. The results from the present study suggest that irradiation of patients with recurrent glioma prior to XVir-N-31 based oncovirotherapy should be performed to optimize the effect of the oncolytic virotherapy in these patients. Finally, as it is well known that both virus infection and tumor-irradiation cause a broad-range immune cell activity against tumors (31,32), the combined radio-oncovirotherapy treatment may in turn support the therapeutic outcome of patients with recurrent GBM.
Acknowledgments
Not applicable.
Abbreviations
- BTIC
brain tumor initiating cell
- EGF
epithelial growth factor
- bFGF
basic fibroblast growth factor
- Gy
Gray
- IFU
infectious unit
- MOI
multiplicity of infection
- TMZ
temozolomide
- YB-1
Y-box binding protein-1
Funding
The present study was supported by grants awarded to UN by the Else-Übelmesser-Foundation.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contribution
RC, NS, HK, SS, SH; TW and UN contributed to the data collection, analysis and interpretation. PH and UN contributed to the study design. UN wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
For patient data and material, written informed consent was obtained from all patients, allowing spare tissue from resective surgery to be included in the present study. Ethical approval was obtained from the Ethics Commission of Tübingen (approval no. 338/2016A). Animal experiments were performed according to the German Animal Welfare Act and its guidelines for the care and use of laboratory animals (approval N12/14 of the regional council Tübingen).
Patient consent for publication
Not applicable.
Competing interests
Per S. Holm is CEO and co-founder of XVir Therapeutics GmbH, 80335 Munich, Germany. All other authors declare that they have no competing interests.
References
- 1.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318:2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother. 2015;15:741–752. doi: 10.1586/14737175.2015.1051968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina JR, Hayashi Y, Stephens C, Georgescu MM. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12:453–463. doi: 10.1593/neo.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, Kitanaka C. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28:1930–1939. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- 6.Toulany M, Schickfluss TA, Eicheler W, Kehlbach R, Schittek B, Rodemann HP. Impact of oncogenic K-RAS on YB-1 phosphorylation induced by ionizing radiation. Breast Cancer Res. 2011;13:R28. doi: 10.1186/bcr2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinnberg T, Sauer B, Holm P, Spangler B, Kuphal S, Bosserhoff A, Schittek B. MAPK and PI3K/AKT mediated YB-1 activation promotes melanoma cell proliferation which is counteracted by an autoregulatory loop. Exp Dermatol. 2012;21:265–270. doi: 10.1111/j.1600-0625.2012.01448.x. [DOI] [PubMed] [Google Scholar]
- 8.Lee Wu J, Yokom C, Jiang D, Cheang H, Yorida MC, Turbin E, Berquin D, Mertens IM, Iftner PRT, et al. Disruption of the Y-box binding protein-1 results in suppression of the epidermal growth factor receptor and HER-2. Cancer Res. 2006;66:4872–4879. doi: 10.1158/0008-5472.CAN-05-3561. [DOI] [PubMed] [Google Scholar]
- 9.Kosnopfel C, Sinnberg T, Schittek B. Y-box binding protein 1-a prognostic marker and target in tumour therapy. Eur J Cell Biol. 2014;93:61–70. doi: 10.1016/j.ejcb.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Schittek B, Psenner K, Sauer B, Meier F, Iftner T, Garbe C. The increased expression of Y box-binding protein 1 in melanoma stimulates proliferation and tumor invasion, antagonizes apoptosis and enhances chemoresistance. Int J Cancer. 2007;120:2110–2118. doi: 10.1002/ijc.22512. [DOI] [PubMed] [Google Scholar]
- 11.Alemasova EE, Pestryakov PE, Sukhanova MV, Kretov DA, Moor NA, Curmi PA, Ovchinnikov LP, Lavrik OI. Poly(ADP-ribosyl)ation as a new posttranslational modification of YB-1. Biochimie. 2015;119:36–44. doi: 10.1016/j.biochi.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Tiwari A, Rebholz S, Maier E, Dehghan Harati M, Zips D, Sers C, Rodemann HP, Toulany M. Stress-induced phosphorylation of nuclear YB-1 depends on nuclear trafficking of p90 ribosomal S6 kinase. Int J Mol Sci. 2018;19:pii: E2441. doi: 10.3390/ijms19082441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewert L, Fischer A, Brandt S, Scurt FG, Philipsen L, Müller AJ, Girndt M, Zenclussen AC, Lindquist JA, Gorny X, Mertens PR. Cold shock Y-box binding protein-1 acetylation status in monocytes is associated with systemic inflammation and vascular damage. Atherosclerosis. 2018;278:156–165. doi: 10.1016/j.atherosclerosis.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Pagano C, di Martino O, Ruggiero G, Maria Guarino A, Mueller N, Siauciunaite R, Reischl M, Simon Foulkes N, Vallone D, Calabrò V. The tumor-associated YB-1 protein: New player in the circadian control of cell proliferation. Oncotarget. 2017;8:6193–6205. doi: 10.18632/oncotarget.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lage H, Surowiak P, Holm PS. YB-1 as a potential target in cancer therapy. Pathologe. 2008;2(Suppl 2):S187–S190. doi: 10.1007/s00292-008-1030-2. In German. [DOI] [PubMed] [Google Scholar]
- 16.Holm PS, Bergmann S, Jurchott K, Lage H, Brand K, Ladhoff A, Mantwill K, Curiel DT, Dobbelstein M, Dietel M, et al. YB-1 relocates to the nucleus in adenovirus-infected cells and facilitates viral replication by inducing E2 gene expression through the E2 late promoter. J Biol Chem. 2002;277:10427–10434. doi: 10.1074/jbc.M106955200. [DOI] [PubMed] [Google Scholar]
- 17.Holm PS, Lage H, Bergmann S, Jürchott K, Glockzin G, Bernshausen A, Mantwill K, Ladhoff A, Wichert A, Mymryk JS, et al. Multidrug-resistant cancer cells facilitate E1-independent adenoviral replication: Impact for cancer gene therapy. Cancer Res. 2004;64:322–328. doi: 10.1158/0008-5472.CAN-0482-2. [DOI] [PubMed] [Google Scholar]
- 18.Mantwill K, Naumann U, Seznec J, Girbinger V, Lage H, Surowiak P, Beier D, Mittelbronn M, Schlegel J, Holm PS. YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J Transl Med. 2013;11:216. doi: 10.1186/1479-5876-11-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bieler A, Mantwill K, Holzmüller R, Jürchott K, Kaszubiak A, Stärk S, Glockzin G, Lage H, Grosu AL, Gansbacher B, Holm PS. Impact of radiation therapy on the oncolytic adenovirus dl520: Implications on the treatment of glioblastoma. Radiother Oncol. 2008;86:419–427. doi: 10.1016/j.radonc.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 21.Verhoog MB, Goriounova NA, Obermayer J, Stroeder J, Hjorth JJ, Testa-Silva G, Baayen JC, de Kock CP, Meredith RM, Mansvelder HD. Mechanisms underlying the rules for associative plasticity at adult human neocortical synapses. J Neurosci. 2013;33:17197–17208. doi: 10.1523/JNEUROSCI.3158-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz N, Hedrich UBS, Schwarz H, P A H, Dammeier N, Auffenberg E, Bedogni F, Honegger JB, Lerche H, Wuttke TV, Koch H. Human Cerebrospinal fluid promotes long-term neuronal viability and network function in human neocortical organotypic brain slice cultures. Sci Rep. 2017;7:12249. doi: 10.1038/s41598-017-12527-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erhardt W, Henke J, Haberstroh J, editors. Anästhesie und Analgesie beim klein-und heimtier. Schattauer Verlag; 2004. [Google Scholar]
- 25.Mutschelknaus L, Azimzadeh O, Heider T, Winkler K, Vetter M, Kell R, Tapio S, Merl-Pham J, Huber SM, Edalat L, et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci Rep. 2017;7:12423. doi: 10.1038/s41598-017-12403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng J, Dong W, Zhang J, Li G, Gong H. YB-1, a new biomarker of glioma progression, is associated with the prognosis of glioma patients. Acta Biochim Biophys Sin (Shanghai) 2016;48:318–325. doi: 10.1093/abbs/gmw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong H, Zhao K, Zhang J, Zhu J, Xiao J. YB-1 modulates the drug resistance of glioma cells by activation of MDM2/p53 pathway. Drug Des Devel Ther. 2019;13:317–326. doi: 10.2147/DDDT.S185514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, Stratford AL, Astanehe A, Dunn SE. YB-1 is a transcription/translation factor that orchestrates the oncogenome by hardwiring signal transduction to gene expression. transl Oncogenomics. 2007;2:49–65. [PMC free article] [PubMed] [Google Scholar]
- 29.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61:2744–2750. [PubMed] [Google Scholar]
- 30.Khan L, Soliman H, Sahgal A, Perry J, Xu W, Tsao MN. External beam radiation dose escalation for high grade glioma. Cochrane Database Syst Rev. 2016;19:CD011475. doi: 10.1002/14651858.CD011475.pub2. [DOI] [PubMed] [Google Scholar]
- 31.Woller N, Gürlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23:1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahebjam S, Sharabi A, Lim M, Kesarwani P, Chinnaiyan P. Immunotherapy and radiation in glioblastoma. J Neurooncol. 2017;134:531–539. doi: 10.1007/s11060-017-2413-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.