

Abstract
Neuropathic pain (NP) is a complex, chronic pain condition caused by injury or dysfunction affecting the somatosensory nervous system. This study aimed to identify crucial genes and miRNAs involved in NP. Microarray data (access number GSE91396) were downloaded from the Gene Expression Omnibus (GEO). Murine RNA-seq samples from three brain regions [nucleus accumbens, (NAc); medial prefrontal cortex, (mPFC) and periaqueductal gray, (PAG)] were compared between the spared nerve injury (SNI) model and a sham surgery. After data normalization, differentially expressed RNAs were screened using the limma package and functional enrichment analysis was performed with Database for Annotation, Visualization and Integrated Discovery. The microRNA (miRNA/miR)-mRNA regulatory network and miRNA-target gene-pathway regulatory network were constructed using Cytoscape software. A total of 2,776 differentially expressed RNAs (219 miRNAs and 2,557 mRNAs) were identified in the SNI model compared with the sham surgery group. A total of two important modules (red and turquoise module) were found to be related to NP using weighed gene co-expression network analysis (WGCNA) for the 2,325 common differentially expressed RNAs in three brain regions. The differentially expressed genes (DEGs) in the miRNA-mRNA regulatory network were significantly enriched in 21 Gene Ontology terms and five pathways. A total of four important DEGs (CXCR2, IL12B, TNFSF8 and GRK1) and five miRNAs (miR-208a-5p, miR-7688-3p, miR-344f-3p, miR-135b-3p and miR-135a-2-3p) were revealed according to the miRNA-target gene-pathway regulatory network to be related to NP. Four important DEGs (CXCR2, IL12B, TNFSF8 and GRK1) and five miRNAs (miR-208a-5p, miR-7688-3p, miR-344f-3p, miR-135b-3p and miR-135a-2-3p) were differentially expressed in SNI, indicating their plausible roles in NP pathogenesis.
Keywords: neuropathic pain, differentially expressed RNA, regulatory network
Introduction
Neuropathic pain (NP) is a severe chronic condition caused by injury or dysfunction affecting the somatosensory nervous system (1). A total of >100 million Americans are thought to be affected by chronic pain (2). NP is characterized by a wide range of sensory, cognitive and affective symptoms, such as tactile allodynia (burning pain resulting from noxious stimuli), hyperalgesia, and spontaneous pain (3). Most NP patients may suffer from depression, anxiety disorders, or other negative moods and various therapies offer only partial relief to 40-60% of NP patients (4). Despite great progress in understanding NP's prognosis, a number of patients respond poorly to current therapies. Therefore, it is necessary and urgent to explore NP's molecular mechanisms, especially the association between NP and anxiety, depression, and other mood disorders.
Chronic pain is associated with adaptations in several brain networks involved in mood, motivation and reward. Evidence indicates that the nucleus accumbens (NAc) is necessary for expressing NP-like behavior (5), whereas the medial prefrontal cortex (mPFC) is linked to a wide variety of cognitive functions critical for social behavior, including personal traits (6). NP can lead to mPFC remodeling, which associates it with emotional regulation of chronic pain (7). Periaqueductal gray (PAG) is the primary center for descending pain modulation (8) that receive schronic pain and temperature signals from the spinomesencephalic tract, hinting at its key role in NP progression (9). Microarray profiling was previously used to explore the altered gene expression of the murine nervous system following nerve injury (10,11). This led to the identification of IL-6, c-Jun and Plau as crucial genes (12) and DNA binding, cell cycle, and forkhead box protein as a major signaling pathway involved in NP (13). Despite these investigations profiling post nerve injury gene expression using microarray analysis, NP's pathological mechanisms remain poorly understood.
Recently, Descalzi et al (11) used RNA-sequencing technology to explore NP; they found NP can affect the expression of multiple genes in three distinct brain regions. However, they did not explore differentially expressed mRNAs (DEMs) and microRNAs (miRNAs/miR), especially the interactions between them. Based on the microarray data deposited by Descalzi et al, DEMs and differentially expressed miRNAs were screened from three distinct brain regions (NAc, mPFC, and PAG) of the spared nerve injury (SNI) and sham surgery murine model.
miRNA is a subset of non-coding, small RNAs, about 22nt long, that can combine with the 3′ untranslated region of messenger RNA (mRNA) to regulate post-transcription gene expression (14). miRNA binds to mRNA, forming a complex regulatory network that plays a vital role in a number of biological processes, such as cellular proliferation, apoptosis, differentiation and metabolism (15). Biological networks have provided a systems biology approach using data from DNA microarray, RNA-seq, miRNA and signaling pathways (16). Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were used to analyze biological networks to reveal potential synergism between gene and the organism (17). To achieve this, a miRNA-mRNA regulatory network was constructed by integrating DEMs, related miRNAs and major signaling pathways.
The present study results might provide further understanding of molecular mechanisms involved in NP progression and how differentially expressed mRNAs and miRNAs may serve as potential targets for NP therapy.
Materials and methods
Microarray database
The microarray data under access number GSE91396 (11) were downloaded from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/) (18). In GSE91396, RNA-seq samples were derived from three brain regions (NAc, mPFC and PAG) of animals two and a half months after sham surgery or SNI on the sciatic nerve. Bilateral punches were collected from the NAc, mPFC and PAG regions from 12 adult male C57BL/6 mice in the sham or SNI surgery group. The punches were pooled from two mice per sample. Thus, 36 samples in GSE91396 were processed, including 18 (6 mPFC, 6 NAc and 6 PAG) from sham surgery and 18 (6 mPFC, 6NAc and 6 PAG) from the SNI surgery group. The SNI model of neuropathic pain was performed under Avertine general anesthesia according to the study of Descalzi et al (11). Briefly, skin and muscle incisions were made on the left hind leg at mid-thigh level, revealing the sciatic nerve and its three branches. The common peroneal and sural nerves were carefully ligated with 6.0 silk sutures, transected, and 1-2 mm sections of each of these nerves were removed. The tibial nerve was left intact. Skin was then sutured with silk 4.0 sutures. Sham surgery mice underwent the same procedure, but all nerves were left intact.
Gene expression profiles in TXT format were downloaded from the NCBI database. The R3.4.1 preprocessCore (19) version 1.40.0 (bioconductor.org/packages/2.4/bioc/html/preprocessCore.html) was used to process the fragments per kilobase per million mapped reads (FPKM) values. The limma package (20) version 3.32.5 (bioconductor.org/packages/release/bioc/html/limma.html) was used to identify differentially expressed RNAs (mRNAs and miRNAs) in the three brain regions that are significantly different (P<0.05) and |log2 fold change (FC)| >0.585 were considered as thresholds. The numbers of miRNAs and genes were calculated. Subsequently, pheatmap (21) version 1.0.8 package (cran.r-project.org/web/packages/pheatmap/index.html) in R3.4.1 software were used for unsupervised hierarchical clustering analysis based on a correlation algorithm. Finally, the Database for Annotation, Visualization and Integrated Discovery (DAVID) (22,23) version 6.8 (david.ncifcrf. gov/) was used to perform GO (Biology Process, Cellular Component, and Molecular Function) and KEGG pathway enrichment analysis for the identified differentially expressed genes (DEGs).
Analysis of the common differentially expressed RNAs for all the three brain regions
Venn diagram (24) (cran.r-project. org/web/packages/VennDiagram/index.html) version 1.6.17 in R3.4.1 software was used to visualize differentially expressed RNAs. The common differentially expressed RNAs from all the three brain regions were selected for further analysis.
The weighed gene co-expression network analysis (WGCNA) algorithm provided topology properties for co-expression network analysis (25). This allows construction of scale-free networks, defines the co-expression matrix and adjacency function, calculates different node coefficients, and identifies functional modules associated with disease from high through put data (26). The WGCNA package (27) version 1.61 in R3.4.1 software (cran.r-project.org/web/packages/WGCNA/) was used to analyze the correlation with disease status for common differentially expressed RNAs. Genes in major modules were screened by GO functional and KEGG pathway enrichment analysis; P<0.05 was considered to indicate a statistically significant difference.
Construction and analysis of miRNA-mRNA regulatory network
To construct an miRNA-mRNA regulatory network and explore the correlations between miRNA and mRNA for mining for the potential roles of these RNAs in NP, the TargetScan Release7.1 (28) (www.targetscan.org/vert_71/) database was used to predict the target mRNAs of miRNAs. The miRNA-mRNA interactions involving common DEMs were selected. According to co-expression WGCNA results, the negative miRNA-mRNA interactions were used to construct the miRNA-mRNA regulatory network that was visualized using Cytoscape3.5.1 (29) (www.cytoscape.org/). GO annotation and KEGG pathway analysis was performed for target genes in the regulatory network.
miRNA-mRNA-pathway network construction
The Comparative Toxicogenomics Database (30) (CTD; ctd.mdibl. org/), a public website and research tool launched by Mount Desert Island Biological Laboratory, elucidates relationships between genesproteins, diseases, phenotypes, GO annotations, pathways, and interaction modules (31). CTD's primary objective is to advance understanding of how gene-environment interactions affect human health (32). 'Neuropathic pain' was used as the keyword to search the KEGG pathways related to NP disease in CTD. Pathways thus retrieved were compared with enriched pathways for target genes in the regulatory network. The overlapping pathways and related genes were hypothesized to play important roles in NP pathogenesis.
Results
Data preprocessing and screening of differentially expressed RNA
The transcriptional profiles downloaded from the GEO database were preprocessed and, after data normalization, 21,193 RNAs were identified, including 1,123 miRNAs and 20,070 mRNAs (Fig. 1A and B). RNAs with a zero median value were excluded, yielding 383 miRNAs and 17,654 mRNAs. A total of 2,776 differentially expressed RNAs were identified (219 miRNAs and 2,557 mRNAs) in SNI samples compared with sham surgery samples for further analysis (Table I). The clustering heatmaps showed significant differences in expression levels of differentially expressed RNAs between SNI and sham surgery groups pertaining to the three distinct brain regions (NAc, mPFC and PAG; Fig. 1C-E). These results show that clustering heatmaps of differentially expressed RNAs, identified from microarray datasets, may accurately distinguish samples from SNI and sham surgeries.
Figure 1.


Data normalization and cluster analysis for differentially expressed RNAs. (A) Data before normalization. (B) Data after normalization. Volcano and clustering heatmaps for the differential expressed RNAs in three distinct regions of brain samples (C) NAc and (D) mPFC. Volcano and clustering heatmaps for the differential expressed RNAs in three distinct regions of brain samples (E) PAG. The horizontal axis in Volcano represents log2FC; vertical axis refers to −1*log10 (P-value). Red triangles and blue triangles represent the significantly upregulated and downregulated RNAs respectively in SNI samples. Black dots represent the RNAs that are not differentially expressed. Crosswise red dashed lines represent threshold of P=0.05 and vertical red dashed lines refer to the threshold of |log2FC|=0.585. FPKM, fragments per kilobase per million mapped reads; NAc, nucleus accumbens; mPFC, medial prefrontal cortex; PAG, periaqueductal gray; FC, fold-change; SNI, spared nerve injury.
Table I.
Differentially expressed mRNAs and miRNAs in three distinct brain regions (NAc, mPFC, PAG) related to neuropathic pain.
| Type | NAc
|
mPFC
|
PAG
|
|||
|---|---|---|---|---|---|---|
| Down | Up | Down | Up | Down | Up | |
| miRNA | 50 | 50 | 49 | 56 | 7 | 7 |
| mRNA | 489 | 491 | 437 | 422 | 95 | 623 |
| Total | 539 | 541 | 486 | 478 | 102 | 630 |
| 1,080 | 964 | 732 | ||||
miRNA, microRNA; NAc, nucleus accumbens; PAG, periaqueductal grey; medial prefrontal cortex.
GO functional and KEGG pathway enrichment analysis for the DEGs
GO functional and KEGG pathway enrichment analyses were performed for the DEGs identified above (Fig. 2). The DEGs in NAc were mainly involved in DNA binding (GO:0003677, n=99), extracellular region part (GO:0044421, n=82), transcription regulator activity (GO:0030528, n=75), transcription factor activity (GO:0003700, n=64) and extracellular space (GO:0005615, n=57; Fig. 2A). The DEGs in mPFC were mainly involved in immune response (GO:0006955, n=41), transcription factor activity (GO:0003700, n=41), extracellular region part (GO:0044421, n=35), sequence-specific DNA binding (GO:0043565, n=32) and positive regulation of the macromolecule metabolic process (GO:0010604, n=32; Fig. 2B). The DEGs in PAG were mainly involved in DNA binding (GO:0003677, n=83), transcription regulator activity (GO:0030528, n=59), regulation of transcription from RNA polymerase II promoter (GO:0006357, n=34), protein kinase activity (GO:0004672, n=31) and structural molecule activity (GO:0005198, n=30; Fig. 2C).
Figure 2.


GO functional and KEGG pathway enrichment analysis for the differentially expressed genes related to neuropathic pain. The pie chart slice sizes correspond to the number of annotated differentially expressed genes. GO functional enrichment analysis results for the differentially expressed genes in three distinct regions of brain (A) NAc, (B) mPFC and periaqueductal gray, (C) PAG. The yellow, pink and green colors represent Biological Process, Molecular Function and Cellular Component, respectively. (D) KEGG pathway enrichment analysis results for the differentially expressed genes in three distinct regions of brain (NAc, mPFC and PAG). The pink, yellow and green represent NAc, mPFC and PAG groups, respectively, and black dots represent -log2 (P-value). GO, gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; NAc, nucleus accumbens; mPFC, medial prefrontal cortex; PAG, periaqueductal gray.
In addition, nine, six and four significant pathways were identified for the DEGs identified in NAc, mPFC, and PAG, respectively (Fig. 2D). There were five overlapping pathways for NAc and mPFC, including cytokine-cytokine receptor interaction (mmu04060), chemokine signaling pathway (mmu04062), neuroactive ligand-receptor interaction (mmu04080), JAK-signal transducer and activator of transcription signaling pathway (mmu04630), and metabolism of xenobiotics by cytochrome P450 (mmu00980). The DEGs in PAG were mainly enriched in vascular endothelial growth factor (mmu04370) and Notch (mmu04330) signaling pathways.
Analysis of critical gene modules related to NP
A total of 2,325 common differentially expressed RNAs were identified in all the three brain regions (NAc, mPFC and PAG), including 173 miRNAs and 2,152 mRNAs. These common differentially expressed RNAs were analyzed using the WGCNA algorithm to identify disease-related RNAs. The topological matrix's scale-free distribution was calculated based on the GSE91396 dataset. Once the square value of the correlation coefficient reached 0.9 for the first time, the corresponding power value (power=7) was selected to calculate community dissimilarity of RNAs. After constructing the clustering dendrogram, the minimum gene number was set as 50 and the cut Height= 0.99. This process resulted in the identification of six modules-module-red, module-turquoise, module-blue, module-green, module-brown and module-yellow (Fig. 3).
Figure 3.

Differentially expressed RNAs-identification and critical functional modules analysis. (A) Venn diagram of differentially expressed RNAs in three brain regions (NAc, mPFC and PAG). (B) The adjacency function definition for the RNAs: Left chart represents the power selection diagram of the adjacency matrix weight parameter. The horizontal axis represents the weight parameters power, while the vertical axis represents the square values of correlation coefficient between log (k) and log [p (k)]. The red line represents the standard as the square value reached 0.9. The right chart represents the mean connectivity of RNAs under a different adjacency matrix weight parameter. The red line shows that the average connection degree of the node is under differential values of power parameter. (C) Identification of major functional modules. The left chart refers to a cluster dendrogram based on the dynamic tree. Each dendrogram color represents a unique module. The right chart represents the heatmap for the correlation between each module and clinical factors. The number in the grid represents the correlation coefficient while the number in parentheses represents P-value. NAc, nucleus accumbens; mPFC, medial prefrontal cortex; PAG, periaqueductal gray.
Correlations between the RNA module and disease characteristics (different regions and disease states) were calculated and are shown in Table II. As shown, genes in module-red (correlation=0.33, P=4.0×10-66) and module-turquoise (correlation=0.82, P<0.001) were positively correlated with disease characteristics. Furthermore, the correlation values were significantly increased compared with the module-gray (correlation with disease=−0.10, P=2.0×10−07). Thus, the differentially expressed RNAs in the red and turquoise modules were used for further analysis.
Table II.
Differentially expressed RNAs in gene modules related to neuropathic pain.
| Module color | Correlation with disease | P-value | #RNA | #miRNA | #mRNA |
|---|---|---|---|---|---|
| Blue | −0.32 | 2.0×10−63 | 412 | 11 | 401 |
| Brown | −0.79 | <0.0001 | 209 | 11 | 198 |
| Green | −0.05 | 6.0×10−3 | 87 | 0 | 87 |
| Grey | −0.10 | 2.0×10−7 | 665 | 118 | 547 |
| Red | 0.33 | 4.0×10−66 | 71 | 2 | 69 |
| Turquoise | 0.82 | <0.0001 | 781 | 31 | 750 |
| Yellow | −0.31 | 7.0×10−59 | 100 | 0 | 100 |
Correlations between the RNA module and disease were calculated using weighed gene co-expression network analysis package version 1.61 in R3.4.1 software (cran.r-project.org/web/packages/WGCNA/).
Pathway enrichment analyses by GO functional and KEGG for DEGs in red and turquoise modules (Table III) showed 18 GO terms and seven pathways (Fig. 4). The DEGs in the two modules were mainly enriched in immune response (GO:0006955), defense response (GO:0006952) and response to wounding (GO:0009611). The major pathway categories for DEGs in the two modules were cytokine-cytokine receptor interaction (mmu04060) and neuroactive ligand-receptor interaction (mmu04080).
Table III.
GO terms and KEGG pathways for the differential expressed mRNAs in module-red and module-turquoise.
| A, Biology process
| ||
|---|---|---|
| Term | Count | P-value |
| GO:0006955 immune response | 33 | 6.36×10−5 |
| GO:0007218 neuropeptide signaling pathway | 11 | 2.01×10−4 |
| GO:0009611 response to wounding | 25 | 4.02×10−4 |
| GO:0006952 defense response | 29 | 7.03×10−4 |
| GO:0006814 sodium ion transport | 13 | 7.38×10−4 |
| GO:0042742 defense response to bacterium | 12 | 7.42×10−4 |
| GO:0050954 sensory perception of mechanical stimulus | 11 | 7.70×10−4 |
| GO:0007605 sensory perception of sound | 10 | 1.55×10−3 |
| GO:0009617 response to bacterium | 14 | 1.78×10−3 |
| GO:0006954 inflammatory response | 17 | 2.72×10−3 |
| GO:0006811 ion transport | 38 | 3.05×10−3 |
| GO:0007601 visual perception | 10 | 5.42×10−3 |
| GO:0050953 sensory perception of light stimulus | 10 | 5.79×10−3 |
| GO:0002684 positive regulation of immune system process | 15 | 7.24×10−3 |
| GO:0007155 cell adhesion | 30 | 9.03×10−3 |
| GO:0022610 biological adhesion | 30 | 9.42×10−3 |
| GO:0007423 sensory organ development | 17 | 9.56×10−3 |
| GO:0030182 neuron differentiation | 23 | 1.06×10−2 |
| B, KEGG pathway
| ||
|---|---|---|
| Term | Count | P-value |
| mmu04060: Cytokine-cytokine receptor interaction | 22 | 5.36×10−6 |
| mmu04080: Neuroactive ligand-receptor interaction | 18 | 1.20×10−3 |
| mmu00980: Metabolism of xenobiotics by cytochrome P450 | 8 | 2.68×10−3 |
| mmu00982: Drug metabolism | 8 | 5.50×10−3 |
| mmu04512: ECM-receptor interaction | 8 | 9.50×10−3 |
| mmu00830: Retinol metabolism | 7 | 1.30×10−2 |
| mmu04350: TGF-β signaling pathway | 7 | 3.85×10−2 |
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; ECM, extracellular matrix; TGF, transforming growth factor.
Figure 4.

Functional and pathway enrichment analysis for differentially expressed mRNAs in turquoise and red modules. (A) GO functional and (B) KEGG pathway analyses of the differentially expressed mRNAs in turquoise and red modules. The horizontal axis refers to the number of mRNAs in functional analysis, while the vertical axis represents GO terms and KEGG pathway categories. The colors range from red to green, representing the change in P-values from low to high. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
The mRNA-miRNA regulatory network construction
Target gene prediction of 33 miRNAs in red (2 miRNAs) and turquoise (31 miRNAs) modules using TargetScan Release 7.1 database resulted in overlapping target genes, which were matched with 819 mRNAs in red (69 mRNAs) and turquoise (750 mRNAs) modules, involving 558 pairs of negative miRNA-mRNA regulatory interactions. The correlation coefficients >0.6 for these relationships, based on the WGCNA algorithm, yielded 59 miRNA-mRNA interactions and 172 mRNA-mRNA interactions that facilitated construction of a miRNA-mRNA regulatory network (Fig. 5), which consisted of 58 nodes, including 10 miRNAs and 48 mRNAs.
Figure 5.

Construction and analyses of miRNA-mRNA regulatory network. (A) The miRNA-mRNA regulatory network: The squares and circles represent miRNAs and mRNAs respectively, and the node colors from green to red indicate change from downregulated to upregulated in gene expression. The red 'T' shape represents a negative connection between miRNA and mRNA. The gray solid line refers to a positive connection. (B) GO analysis for the differential expressed mRNAs in miRNA-mRNA regulatory network. The horizontal axis refers to the number of mRNAs in functional analysis, while the vertical axis represents GO terms. The colors range from red to green, representing the change in P-values from low to high. (C) The pie chart of KEGG pathway analysis results: Each part represents one pathway category, while the number refers to the number of mRNAs. miRNA/miR, microRNA; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
The GO functional and KEGG pathway enrichment analysis showed that DEGs in the miRNA-mRNA regulatory network were enriched in 21 GO terms and five pathways. The GO terms included regulating T cell proliferation (GO:0042129) and T cell differentiation (GO:0030217). The major pathway was cytokine-cytokine receptor interaction (mmu04060; Table IV).
Table IV.
GO terms and KEGG pathways for the critical genes in mRNA-microRNA regulatory network.
| A, Biology process
| |||
|---|---|---|---|
| Term | Count | P-value | Genes |
| GO:0042129 regulation of T cell proliferation | 3 | 0.011 | FOXJ1, IL12B, SPN |
| GO:0042981 regulation of apoptosis | 6 | 0.016 | COL18A1, PLEKHF1, GFRAL, MGMT, |
| COL2A1, SPN | |||
| GO:0043067 regulation of programmed cell death | 6 | 0.017 | COL18A1, PLEKHF1, GFRAL, MGMT, COL2A1, SPN |
| GO:0010941 regulation of cell death | 6 | 0.017 | COL18A1, PLEKHF1, GFRAL, MGMT, COL2A1, SPN |
| GO:0030217 T cell differentiation | 3 | 0.018 | CD3D, IL12B, SPN |
| GO:0032944 regulation of mononuclear cell proliferation | 3 | 0.020 | FOXJ1, IL12B, SPN |
| GO:0050670 regulation of lymphocyte proliferation | 3 | 0.020 | FOXJ1, IL12B, SPN |
| GO:0070663 regulation of leukocyte proliferation | 3 | 0.021 | FOXJ1, IL12B, SPN |
| GO:0007601 visual perception | 3 | 0.031 | OPN5, PDE6G, CNGB3 |
| GO:0050953 sensory perception of light stimulus | 3 | 0.031 | OPN5, PDE6G, CNGB3 |
| GO:0050863 regulation of T cell activation | 3 | 0.035 | FOXJ1, IL12B, SPN |
| GO:0002820 negative regulation of adaptive immune response | 2 | 0.035 | FOXJ1, SPN |
| GO:0002704 negative regulation of leukocyte mediated immunity | 2 | 0.037 | FOXJ1, SPN |
| GO:0002707 negative regulation of lymphocyte mediated immunity | 2 | 0.037 | FOXJ1, SPN |
| GO:0030098 lymphocyte differentiation | 3 | 0.038 | CD3D, IL12B, SPN |
| GO:0042110 T cell activation | 3 | 0.040 | CD3D, IL12B, SPN |
| GO:0045061 thymic T cell selection | 2 | 0.040 | CD3D, SPN |
| GO:0042325 regulation of phosphorylation | 4 | 0.044 | DOK7, RAPGEF3, PDE6G, GRK1 |
| GO:0002520 immune system development | 4 | 0.046 | CD3D, FOXJ1, IL12B, SPN |
| GO:0051174 regulation of phosphorus metabolic process | 4 | 0.048 | DOK7, RAPGEF3, PDE6G, GRK1 |
| GO:0019220-regulation of phosphate metabolic process | 4 | 0.048 | DOK7, RAPGEF3, PDE6G, GRK1 |
| B, KEGG pathway
| |||
|---|---|---|---|
| Term | Count | P-value | Genes |
| mmu04060: Cytokine-cytokine receptor interactiona | 3 | 0.010 | CXCR2, IL12B, TNFSF8 |
| mmu04512: ECM-receptor interaction | 2 | 0.017 | ITGB6, COL2A1 |
| mmu04062: Chemokine signaling pathwaya | 2 | 0.034 | CXCR2, GRK1 |
| mmu04510: Focal adhesion | 2 | 0.037 | ITGB6, COL2A1 |
| mmu04144: Endocytosisa | 2 | 0.037 | CXCR2, GRK1 |
Overlapping pathways identified by comparing pathways selected from Comparative Toxicogenomics Database with enriched KEGG pathways. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of genes and genomes.
Crucial miRNAs, mRNAs and pathways related to NP
Keyword ('neuropathic pain') search of the CTD resulted in 88 pathways. Three pathways overlapped when the 88 pathways were compared to previously-identified enriched pathways for the DEGs in the mRNA-miRNA regulatory network. These pathways, cytokine-cytokine receptor interaction, chemokine signaling pathway and endocytosis were involved in four DEGs, including interleukin (IL)-8 receptor β (CXCR2), subunit β of interleukin (IL)-12 (IL12B), CD153 (also known as TNFSF8), and rhodopsin kinase (GRK1). The CXCR2 can be targeted by miR-7688-3p, IL12B by miR-208a-5p, GRK1 by miR-135b-3p and TNFSF8 by miR-344f-3p, miR-135b-3p, and miR-135a-2-3p, respectively (Fig. 6).
Figure 6.
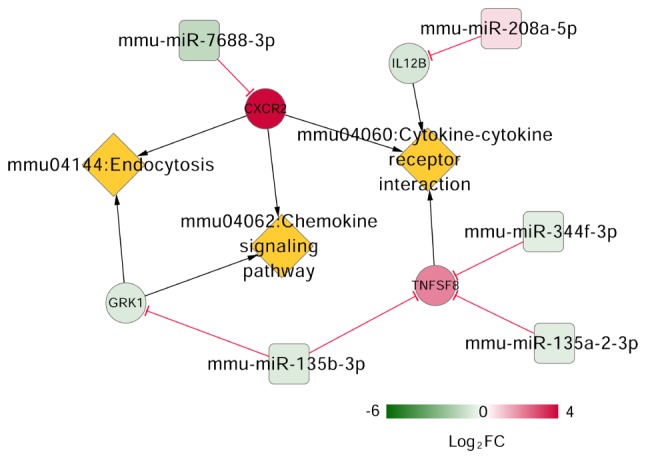
mRNA-miRNA-pathway regulatory network. The square, circle and rhombus shapes represent miRNA, mRNA, and pathway respectively. The variation of node colors from green to red indicates change from down-regulated to up-regulated in gene expression. The red 'T' shape represents a negative connection between miRNA and mRNA. The black arrow represents a connection between mRNA and the participating pathway. miRNA/miR, microRNA.
Discussion
In this study, 2,776 differentially expressed RNAs (219 miRNAs and 2,557 mRNAs) were identified in SNI compared with the sham surgery samples. In the three brain regions (NAc, mPFC and PAG), there were 2,325 common differentially expressed RNAs (173 miRNAs and 2,152 mRNAs). Two important modules (red and turquoise module) were identified as related to NP using WGCNA for the common differentially expressed RNAs. The miRNA-mRNA regulatory network was constructed based on the differentially expressed RNAs in the red and turquoise modules. The DEGs in miRNA-mRNA regulatory network were enriched in 21 GO terms and 5 pathways. Three pathways in the miRNA-target gene-pathway regulatory network, including cytokine-cytokine receptor interaction, chemokine signaling pathway and endocytosis, comprised four important DEGs (CXCR2, IL12B, TNFSF8, and GRK1) that were related to NP.
Cytokines are a group of small proteins (5-20 kDa) produced by a range of cells, including immune cells-macrophages, B lymphocytes, T lymphocytes, mast cells, as well as endothelial cells and various stromal cells (33). Cytokines have important roles in the immune system (34). Cytokines, along with chemokines, interferons (IFN), ILs, colony stimulation factor and tumor necrosis factor (TNF) are involved in responsiveness to trauma, pain, and infection (35). In the present study, three DEGs (CXCR2, IL12B and TNFSF8) participated in cytokine-cytokine receptor interaction. IL12B located on chromosome 5q31-33 encodes the p40 subunit of IL-12, an immunomodulatory cytokine (36). IL12B polymorphisms are associated with asthma and psoriasis (37). A recent study showed that significant decreases in systemic concentrations of chemokines, IL-12 and IFNγ, were observed in nerve-injured Foxp3+ regulatory T cell-depleted transgenic mice; decrease in IL-12 promoted pain hypersensitivity in this model (38). The immune system, particularly T cells, plays a key role in mediating NP. IL-12p70, also referred to as IL-12, is a pro-inflammatory cytokine secreted by activated hematopoietic phagocytic cells; Chen et al (39) investigated pain response following systemic administration of IL-12p70 and IL-12p40 homodimer and found that IL-12p40 exhibited significant anti-nociceptive effects in a rat model of chronic NP. The present study found that IL12B was dysregulated in brain tissues of NP mice, which is consistent with previous studies (37-39). This dysregulation may be effectuated by miRNA, one of which, miR-208a-5p, was found in the miRNA-target gene-pathway regulatory network, to target IL12B. Findings in the literature show miR-208a to regulate cardiac hypertrophy and conduction in mice (40), whereas the expression level of miR-208b progressively declined after spinal cord injury in humans (41). The results of the present study suggest miR-208a-5p may play a vital role in NP pathogenesis by regulating IL12B expression levels.
TNFSF8, a ligand of cluster of differentiation (CD)30 (or CD153), exhibits polymorphisms that showed significant associations with spondylarthritis in a French cohort (42). In addition, TNFSF8 is a susceptibility gene in excessive inflammatory responses (43). Since inflammation is a key pathophysiological process in NP (44), TNFSF8 might be a major regulator in NP inflammatory responses. The miRNA-target gene-pathway regulatory network revealed differentially expressed TNFSF8 could be targeted by three miRNAs (miR-344f-3p, miR-135b-3p and miR-135a-2-3p). It has been reported that miR-135a modulates inflammatory molecules, IL-6, IL-1β and TNF-α, which enhances inflammatory responses of vascular smooth muscle cells involved in vascular disease complications (45,46). Another study showed that overexpressing the miR-344b-1-3p inhibitor in alveolar macrophages significantly increased the expression of TNF-α, IL-1β and macrophage inflammatory protein (MIP)-2 (47). Together, the results of the present study indicate that differentially expressed IL12B targeted by miR-208a-5p and TNFSF8 targeted by miR-344f-3p, miR-135b-3p and miR-135a-2-3p, might play critical roles in NP through cytokine-cytokine receptor interaction.
CXCR2 and GRK1 are chemokines that were identified in this study to be involved in NP progression. CXCR2 (or IL 8 receptor β) is a chemokine receptor whose interaction with MIP elicits chronic neuroinflammation through neutrophil accumulation and hyperacetylation of histone H3 leading to NP (48).
CXCL1/CXCR2 signaling plays a vital role in pathological pain, including peripheral and central sensitization (49). CXCL1 sensitizes primary peripheral neurons by directly acting on CXCR2 (50). In the central nervous system, CXCL1/CXCR2 signaling increases N-methyl-D-aspartate receptor currents in neurons and promotes expression of genes related to neuroplasticity, contributing to prolonged chronic pain (51).
The enzyme phosphorylating rhodopsin receptor, rhodopsin kinase (GRK1), was identified in the late 1970s as controlling vision (52). Nerve injury, high norepinephrine concentration and abnormal β2-adrenoreceptor functions indicate high sympathetic nerve activity and dysfunction (53). Phosphorylation by GRK1/2, under high norepinephrine concentrations and high sympathetic nerve activity, induced β2-adrenoreceptor internalization by recruiting β-arrestin-1 to the receptor, leading adrenoreceptors to either recycle to the membrane or traffic to lysosomes for degradation (54). Taken together, it was speculated that CXCR2 targeted by miR-7688-3p and GRK1 targeted by miR-135b-3p might be involved in NP progression through chemokine signaling pathway and endocytosis.
The present study has limitations. The small sample size available for analysis may have obscured rare interactions. Additional validation of the roles of DEGs and miRNAs in NP progression is warranted.
In conclusion, 2,776 differentially expressed RNAs (219 miRNAs and 2,557 mRNAs) were identified in the NP SNI model. Differentially expressed IL12B, targeted by miR-208a-5p, as well as TNFSF8 dysregulated by miR-344f-3p, miR-135b-3p and miR-135a-2-3p, might play critical roles in NP through cytokine-cytokine receptor interaction. CXCR2 targeted by miR-7688-3p and GRK1 targeted by miR-135b-3p might be involved in NP progression through chemokine signaling pathway and endocytosis. The present study has provided new insights into the regulatory mechanisms of NP, which merits their potential as candidates for therapies for this debilitating condition.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable requests.
Authors' contributions
SXL and CGZ made substantial intellectual contributions to the study design. HL, HQW, SXL and HJZ searched and downloaded microarray data from the Gene Expression Omnibus database. HL, HQW, SXL and HJZ made substantial contributions to the analysis and interpretation of microarray dataset. SXL and CGZ were involved in revising the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 2.Institute of Medicine (US) Committee on Advancing Pain Research Care, and Education . Relieving pain in America: A blueprint for transforming prevention, care, education, and research. Institute of Medicine (US); 2011. [PubMed] [Google Scholar]
- 3.Baron R. Mechanisms of disease: Neuropathic pain-a clinical perspective. Nat Clin Pract Neurol. 2006;2:95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]
- 4.Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, Kalso EA, Loeser JD, Miaskowski C, Nurmikko TJ, et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain. 2007;132:237–251. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 5.Chang PC, Pollema-Mays SL, Centeno MV, Procissi D, Contini M, Baria AT, Martina M, Apkarian AV. Role of nucleus accumbens in neuropathic pain: Linked multi-scale evidence in the rat transitioning to neuropathic pain. Pain. 2014;155:1128–1139. doi: 10.1016/j.pain.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weaver KE, Richardson AG. Medial prefrontal cortex, secondary hyperalgesia, and the default mode network. J Neurosci. 2009;29:11424–11425. doi: 10.1523/JNEUROSCI.3263-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seifert F, Bschorer K, De Col R, Filitz J, Peltz E, Koppert W, Maihöfner C. Medial prefrontal cortex activity is predictive for hyperalgesia and pharmacological antihyperalgesia. J Neurosci. 2009;29:6167–6175. doi: 10.1523/JNEUROSCI.4654-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh MY, Abosch A, Kim SH, Lang AE, Lozano AM. Long-term hardware-related complications of deep brain stimulation. Neurosurgery. 2002;50:1268–1274. doi: 10.1097/00006123-200206000-00017. [DOI] [PubMed] [Google Scholar]
- 9.Du L, Wang SJ, Cui J, He WJ, Ruan HZ. The role of HCN channels within the periaqueductal gray in neuropathic pain. Brain Res. 2013;1500:36–44. doi: 10.1016/j.brainres.2013.01.035. [DOI] [PubMed] [Google Scholar]
- 10.Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–8708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Descalzi G, Mitsi V, Purushothaman I, Gaspari S, Avrampou K, Loh YE, Shen L, Zachariou V. Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci Signal. 2017;10 doi: 10.1126/scisignal.aaj1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Xia T, Xu F, Ma Z, Gu X. Identification of the key genes associated with neuropathic pain. Mol Med Rep. 2018;17:6371–6378. doi: 10.3892/mmr.2018.8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CJ, Liu DZ, Yao WF, Gu Y, Huang F, Hei ZQ, Li X. Identification of key genes and pathways associated with neuropathic pain in uninjured dorsal root ganglion by using bioinformatic analysis. J Pain Res. 2017;10:2665–2674. doi: 10.2147/JPR.S143431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habibi I, Emamian ES, Abdi A. Quantitative analysis of intracellular communication and signaling errors in signaling networks. BMC Syst Biol. 2014;8:89. doi: 10.1186/s12918-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013;39:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan M, Song M, Bai R, Cheng S, Yan W. Identification of potential therapeutic targets for colorectal cancer by bioinformatics analysis. Oncol Lett. 2016;12:5092–5098. doi: 10.3892/ol.2016.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diao C, Xi Y, Xiao T. Identification and analysis of key genes in osteosarcoma using bioinformatics. Oncol Lett. 2018;15:2789–2794. doi: 10.3892/ol.2017.7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Boutros PC. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011;39:3864–3878. doi: 10.1093/nar/gkq1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, Peterson KJ. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet. 2015;49:213–242. doi: 10.1146/annurev-genet-120213-092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis AP, King BL, Mockus S, Murphy CG, Saraceni-Richards C, Rosenstein M, Wiegers T, Mattingly CJ. The comparative toxicogenomics database: Update 2011. Nucleic Acids Res. 2011;39:D1067–D1072. doi: 10.1093/nar/gkq813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattingly CJ, Rosenstein MC, Davis AP, Colby GT, Forrest JN, Jr, Boyer JL. The comparative toxicogenomics database: A cross-species resource for building chemical-gene interaction networks. Toxicol Sci. 2006;92:587–595. doi: 10.1093/toxsci/kfl008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattingly CJ, Colby GT, Rosenstein MC, Forrest JN, Jr, Boyer JL. Promoting comparative molecular studies in environmental health research: An overview of the comparative toxicogenomics database (CTD) Pharmacogenomics J. 2004;4:5–8. doi: 10.1038/sj.tpj.6500225. [DOI] [PubMed] [Google Scholar]
- 33.Lackie J. A Dictionary of Biomedicine. 1st ed. Oxford University Press Inc; New York, NY: 2010. [Google Scholar]
- 34.Rittner HL, Brack A, Stein C. Pain and the immune system. Br J Anaesth. 2008;101:40–44. doi: 10.1093/bja/aen078. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA. Pro- and anti-inflammatory cytokine gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain. Exp Neurol. 2001;169:386–391. doi: 10.1006/exnr.2001.7677. [DOI] [PubMed] [Google Scholar]
- 36.Huang D, Cancilla MR, Morahan G. Complete primary structure, chromosomal localisation, and definition of polymorphisms of the gene encoding the human interleukin-12 p40 subunit. Genes Immun. 2000;1:515–520. doi: 10.1038/sj.gene.6363720. [DOI] [PubMed] [Google Scholar]
- 37.Randolph AG, Lange C, Silverman EK, Lazarus R, Silverman ES, Raby B, Brown A, Ozonoff A, Richter B, Weiss ST. The IL12B gene is associated with asthma. Am J Hum Genet. 2004;75:709–715. doi: 10.1086/424886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lees JG, Duffy SS, Perera CJ, Moalem-Taylor G. Depletion of Foxp3+ regulatory T cells increases severity of mechanical allodynia and significantly alters systemic cytokine levels following peripheral nerve injury. Cytokine. 2015;71:207–214. doi: 10.1016/j.cyto.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 39.Chen IF, Khan J, Noma N, Hadlaq E, Teich S, Benoliel R, Eliav E. Anti-nociceptive effect of IL-12 p40 in a rat model of neuropathic pain. Cytokine. 2013;62:401–406. doi: 10.1016/j.cyto.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 40.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boon H, Sjögren RJ, Massart J, Egan B, Kostovski E, Iversen PO, Hjeltnes N, Chibalin AV, Widegren U, Zierath JR. MicroRNA-208b progressively declines after spinal cord injury in humans and is inversely related to myostatin expression. Physiol Rep. 2015;3 doi: 10.14814/phy2.12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zinovieva E, Kadi A, Letourneur F, Cagnard N, Izac B, Vigier A, Said-Nahal R, Elewaut D, de Vlam K, Pimentel-Santos F, et al. Systematic candidate gene investigations in the SPA2 locus (9q32) show an association between TNFSF8 and susceptibility to spondylarthritis. Arthritis Rheum. 2011;63:1853–1859. doi: 10.1002/art.30377. [DOI] [PubMed] [Google Scholar]
- 43.Fava VM, Cobat A, Van Thuc N, Latini AC, Stefani MM, Belone AF, Ba NN, Orlova M, Manry J, Mira MT, et al. Association of TNFSF8 regulatory variants with excessive inflammatory responses but not leprosy per se. J Infect Dis. 2015;211:968–977. doi: 10.1093/infdis/jiu566. [DOI] [PubMed] [Google Scholar]
- 44.Shi J, Jiang K, Li Z. MiR-145 ameliorates neuropathic pain via inhibiting inflammatory responses and mTOR signaling pathway by targeting Akt3 in a rat model. Neurosci Res. 2018;134:10–17. doi: 10.1016/j.neures.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Lu X, Yin D, Zhou B, Li T. MiR-135a promotes inflammatory responses of vascular smooth muscle cells from db/db mice via downregulation of FOXO1. Int Heart J. 2018;59:170–179. doi: 10.1536/ihj.17-040. [DOI] [PubMed] [Google Scholar]
- 46.Du XJ, Lu JM. MiR-135a represses oxidative stress and vascular inflammatory events via targeting toll-like receptor 4 in atherogenesis. J Cell Biochem. 2018;119:6154–6161. doi: 10.1002/jcb.26819. [DOI] [PubMed] [Google Scholar]
- 47.Xu H, Wu Y, Li L, Yuan W, Zhang D, Yan Q, Guo Z, Huang W. MiR-344b-1-3p targets TLR2 and negatively regulates TLR2 signaling pathway. Int J Chron Obstruct Pulmon Dis. 2017;12:627–638. doi: 10.2147/COPD.S120415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiguchi N, Kobayashi Y, Maeda T, Fukazawa Y, Tohya K, Kimura M, Kishioka S. Epigenetic augmentation of the macrophage inflammatory protein 2/C-X-C chemokine receptor type 2 axis through histone H3 acetylation in injured peripheral nerves elicits neuropathic pain. J Pharmacol Exp Ther. 2012;340:577–587. doi: 10.1124/jpet.111.187724. [DOI] [PubMed] [Google Scholar]
- 49.Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol Dis. 2017;105:109–116. doi: 10.1016/j.nbd.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Manjavachi MN, Costa R, Quintão NL, Calixto JB. The role of keratinocyte-derived chemokine (KC) on hyperalgesia caused by peripheral nerve injury in mice. Neuropharmacology. 2014;79:17–27. doi: 10.1016/j.neuropharm.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 51.Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137:2193–2209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem. 2011;286:1420–1428. doi: 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elenkov IJ, Iezzoni DG, Daly A, Harris AG, Chrousos GP. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation. 2005;12:255–269. doi: 10.1159/000087104. [DOI] [PubMed] [Google Scholar]
- 54.Bellinger DL, Lorton D. Sympathetic nerve hyperactivity in the spleen: Causal for nonpathogenic-driven chronic immune-mediated inflammatory diseases (IMIDs)? Int J Mol Sci. 2018;19 doi: 10.3390/ijms19041188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable requests.