

Abstract
The retina is sensitive to injury resulting from oxidative stress (OS) due to its high oxygen consumption. Patients with retinitis pigmentosa suffer from excessive OS. N-acetylcysteine (NAC) is used as a mucolytic agent for the clinical treatment of disorders, such as chronic bronchitis and other pulmonary diseases. The aim of the present study was to investigate the role of hexokinase 2 (HKII) in retinal OS injury. Amyloid β (Aβ)1-40 was used to establish a cellular model of OS. Cell viability was measured with a Cell Counting Kit-8 assay, and the apoptosis, reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) of cells were analyzed via flow cytometry with corresponding kits. The mRNA and protein levels were detected by reverse transcription-quantitative PCR and western blot analyses, respectively. It was observed that Aβ1-40 reduced the expression of HKII in the mitochondria of retinal pigment epithelial ARPE cells and impaired mitochondrial antioxidant functions. Additionally, knockdown of HKII promoted apoptosis, and increased ROS levels and the MMP. NAC attenuated the inhibition of mitochondrial functions induced by Aβ1-40. The knockdown of HKII was revealed to decrease the levels of Bcl-2, manganese superoxide dismutase (SOD) and copper-zinc-SOD, and increase the levels of cleaved caspase-3, Bax and cytochrome c. The present findings suggested that the dissociation of HKII induced by OS induces apoptosis and mitochondrial damage. This study provided improved understanding of the mechanisms underlying the effects of OS on retinal epithelial cells.
Keywords: N-acetylcysteine, hexokinase II, oxidative stress, ARPE-19 cells
Introduction
Oxidative stress (OS), which was initially conceptualized in 1985 (1), is regarded as an important phenomenon in redox biology and medicine. OS was initially defined as an imbalance between oxidants and antioxidants, which leads to a disruption of redox signaling and/or molecular damage (2). Reactive oxygen species (ROS) were considered to be damaging agents in living organisms; however, they were subsequently also determined to serve positive roles in living organisms (3). Thus, the definition of OS was revised to refer to an imbalance between ROS generation and elimination (4). OS can be classified as basal, low intensity, intermediate intensity OS or high intensity OS (4).
The eyes are susceptible to OS injury due to the production of ROS, as eyes are exposed to various adverse environments that are able to shift the cell redox status towards oxidizing conditions, including ionizing radiation, light exposure, ultraviolet rays, chemical pollutants and pathogenic microbes (5). Additionally, the retina is vulnerable to OS due to its high oxygen consumption (6,7). OS can induce peroxidation of nucleic acids, bases, lipids, proteins and carbohydrates, resulting in a number of eye conditions [dry eye syndrome, diabetic retinopathy, autoimmune and inflammatory uveitis, corneal and conjunctive diseases, cataracts, glaucoma, age-related macular degeneration (AMD) and retinitis pigmentosa (RP)], as well as chronic inflammation (5,8). Amyloid β (Aβ), which promotes the progression of AMD (9), can induce OS; Aβ has been used in animal or cell models of OS and AMD (10-12). In the present study, ROS were investigated in ARPE-19 cells treated with Aβ1-40.
Hexokinases (HKs) catalyze the first step of glucose metabolism; glucose, which is transported through glucose transporters in the mitochondrial membrane, is phosphorylated by HKs, producing glucose-6-phosphate (G6P) (13-16); G6P also provides feedback regulation of HK activity. HKs serve important roles in the regulation of metabolic process, as G6P is a precursor of ATP, glycogenesis, and pentose phosphate and hexosamine biosynthetic pathways (13,14,16,17). HK has four isomers (I, II, III and IV) in mammalian cells; hexokinase II (HKII) serves important roles in insulin-sensitive tissues, such as skeletal muscle, heart and adipose tissue (18).
HKII has been reported to induce important effects on mitochondrial function in myocardial cells (19). HKII, which binds to mitochondria, can suppress the mitochondrial translo-cation of Bax and the release of cytochrome c (Cyt c) (20,21), therefore preventing cell apoptosis (22). It has been reported that ischemia or glucose deprivation in adult hearts or isolated cardiomyocytes can result in the dissociation of HKII from mitochondria, thereby releasing mitochondrial Cyt c to induce apoptosis (23,24).
N-acetylcysteine (NAC), an antioxidant, is used as a mucolytic agent for treating various disorders, including paracetamol intoxication, doxorubicin cardiotoxicity and ischemia-reperfusion cardiac injury in clinical settings (25,26). The effects of HKII on Aβ1-40-induced OS injury were studied in retinal pigment epithelial (RPE) cells, using NAC as a control.
Materials and methods
Cell culture, oxidative stress model and morphological observation
The human RPE cell line (ARPE-19/HPV-16) was purchased from the American Type Culture Collection. The cell line was cultured at 37°C with 5% CO2 in an incubator (Thermo Fisher Scientific, Inc.) with DMEM/F-12 medium (cat. no. 11330057; Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% 10,000 U/ml penicillin/10,000 µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc,.). The cells were subcultured every 3 days in a 35-mm culture flask (Corning Inc.). After the cells (2×104 cells/well) were seeded in 96-well plates (Corning Inc.), and different concentrations of Aβ1-40 (0, 0.01, 0.1, 0.5, 1, 5 and 10 µmol/ml; Sigma-Aldrich; Merck KGaA) mixed with culture medium were added to the cells in order to establish OS, the cells were incubated at 37°C for 24 h. For subsequent experiments, 0.5 µmol/ml Aβ1-40 was used. Following culture for 24 h, the morphology of cells in the control and 0.5 µmol/ml Aβ1-40 groups was observed under an inverted phase contrast microscope (magnification, ×200; Olympus Corporation).
Cell viability
Cell viability was measured via a Cell Counting Kit-8 (CCK-8) assay. ARPE-19 cells (2×104 cells/well) were seeded in a 96-well plates (Corning Inc.) and cultured in an incubator for 24 h at 37°C. Cells were cultured with various concentrations (0.1, 1 or 10 µmol/ml) of NAC (Shanghai Aladdin Bio-Chem Technology Co., Ltd.); for subsequent experiments, 1 µmol/ml NAC was used. A CCK-8 kit (Sigma-Aldrich; Merck KGaA) was diluted with serum-free DMEM (1:9). The culture medium was then replaced, and the cells were washed three times with PBS (Gibco; Thermo Fisher Scientific, Inc.). CCK-8 working solution (10 µl) was added to each well, and plates were incubated in an incubator for a further 2 h. The optical density at 490 nm was then detected using a microplate reader (Thermo Fisher Scientific, Inc.).
Cell transfection
Small interfering RNA specific for HKII (siHKII; 5′-GACCCTCTACAAGCTACAT-3′) and negative control (NC) siRNA (5′-GGTAAGCAAGGGAGATCAA-3′) were synthesized by Orbigen, Inc. (Allele Biotechnology). Serum-free DMEM was diluted (1:1) with Lipofectamine® (Invitrogen; Thermo Fisher Scientific, Inc.). Then, 50 nmol/l siHKII or NC mixed with the Lipofectamine solution was added to the cells for 1-2 h, following which the solution was replaced with normal culture medium. Subsequent experiments were performed 72 h following transfection.
Apoptosis and ROS analysis
After the cells had been treated with Aβ1-40 (0.5 µmol/ml), NAC (1 µmol/ml), Aβ1-40 (0.5 µmol/ml) + NAC (1 µmol/ml) or Aβ1-40 (0.5 µmol/ml) + NAC (1 µmol/ml) + siHKII (50 nM) for 24 h, respectively, they were collected by trypsin (Gibco; Thermo Fisher Scientific, Inc.) via centrifugation at 800 × g for 5 min at 4°C. An Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (BD Biosciences) and a fluorometric intracellular ROS kit (cat. no. MAK143; Sigma-Aldrich; Merck KGaA) were applied to detect the apoptosis and ROS contents of ARPE cells using a flow cytometer (FACSCalibur; BD Biosciences). The cells were incubated with Annexin V-FITC and PI in the dark for 20 min at room temperature; the same temperature and duration were used for ROS assays. The fluorescence intensity was analyzed using CellQuest software (version 3.3; BD Biosciences). Experiments were conducted according to the manufacturers' protocols. The apoptosis rate was calculated as the percentage of early + late apoptotic cells.
Mitochondrial membrane potential (MMP) analysis
The JC-1 fluorescent probe can be used to detect changes in the MMP; the color of fluorescence is altered when the MMP changes (27). Cells were seeded in a 6-well plate at 1×105 cells/well for 24 h, and the cells were treated as aforementioned for 24 h; untreated cells were regarded as a control group. The cells were collected with 0.25% trypsin for 5 min at 37°C and via centrifugation at 800 × g for 5 min at 4°C, and 105 cells were resuspended in 0.5 ml DMEM. A JC-1 MMP assay kit (Beijing Leagene Biotech Co., Ltd.) was used to analyze the MMP. The experimental procedure was performed according to the manufacturer's protocols. The fluorescence was detected using a flow cytometer (FACSCalibur) and CellQuest version 3.3 software at wavelengths of 530 and 590 nm.
Western blotting
The cells were seeded in 90-mm petri dishes (Corning Inc.) at 106 cells/dish, and were treated as aforementioned for 24 h. The medium was discarded, and PBS was used to wash the cells three times. Total protein was extracted by a cell scraper (Thermo Fisher Scientific, Inc.) with 300 µl cell lysis buffer (cat. no. RABLYSIS1; Sigma-Aldrich; Merck KGaA) on ice, and the cells were centrifuged at 4°C and 12,000 × g for 15 min. Mitochondria in ARPE-19 cells were extracted with a mitochondria isolation kit (Sigma-Aldrich; Merck KGaA). Cell lysis buffer (Sigma-Aldrich; Merck KGaA) was used to extract protein from mitochondria via centrifugation at 12,000 × g for 15 min at 4°C. A BCA assay kit (Sigma-Aldrich; Merck KGaA) was used for determining the amount of protein. Then, 40 µg protein was separated via 12% SDS-PAGE and transferred to PVDF membranes (Sigma-Aldrich; Merck KGaA). Protein membranes were blocked with 3% bovine serum albumin (Sigma-Aldrich; Merck KGaA) for 2 h at room temperature. Primary antibodies were diluted in TBS with 0.1% Tween-20 (TBST). Cleaved caspase-3 (1:1,000; cat. no. 9661; Cell Signaling Technology, Inc.), Bcl-2 (1:1,000; cat. no. 4223; Cell Signaling Technology, Inc.), Bax (1:1,000; cat. no. 5023; Cell Signaling Technology, Inc.), manganese superoxide dismutase (MnSOD; 1:1,000; cat. no. 13141; Cell Signaling Technology, Inc.), copper/zinc superoxide dismutase (CuZnSOD; 1:1,000; cat. no. 2770; Cell Signaling Technology, Inc.), Cyt c (1:1,000; cat. no. 11940; Cell Signaling Technology, Inc.), HKII (1:1,000; cat. no. 2867; Cell Signaling Technology, Inc.), GAPDH (1:1,000; cat. no. 5174; Cell Signaling Technology, Inc.) and mitochondrial marker Cyt c oxidase subunit IV (1:1,000; cat. no. 4850; Cell Signaling Technology, Inc.) antibodies were used to incubate the protein membranes for 12 h at 4°C. Following three washes with TBST, horseradish peroxidase-conjugated anti-rabbit secondary antibodies (1:10,000; cat. no. 7074; Cell Signaling Technology, Inc.) were added to the membranes, and the protein membranes were incubated for 1.5 h at room temperature. Bands were visualized using ECL reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The protein bands were analyzed using ImageJ v1.45s (National Institutes of Health).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocols. cDNA was synthesized from mRNA by using a PrimeScript First Strand cDNA synthesis kit (Takara Bio, Inc.); the RT reaction was performed at 45°C for 20 min and 95°C for 5 min. qPCR was performed using an SYBR Premix Ex Taq kit (Takara Biotechnology Co., Ltd.) under the following conditions: 94°C for 75 sec, then 50 cycles of 55°C for 45 sec and 72°C for 10 min. The primers (Sangon Biotech Co., Ltd.) used for qPCR were as follows: HKII, forward 5′-AGA CTG TCC TTT CCA CAT GG-3′, reverse 5′-TTC CAG GTG CAT TCG ACA AG-3′; GAPDH, forward 5′-ACG GAT TTG GTC GTA TTG GG-3′, reverse 5′-CGC TCC TGG AAG ATG GTG AT-3′. The 2−∆∆Cq method was employed to analyze the relative levels of gene expression (28).
Statistical analysis
All values were presented as the mean ± SD. All experiments were repeated three times. For comparison, one-way ANOVA followed by a Tukey's post hoc test was performed with GraphPad Prism 5.0 software (GraphPad Software, Inc.). The untreated experimental groups were regarded as the control group. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of Aβ1-40 on ARPE-19 cells
ARPE-19 cells were treated with different concentrations of Aβ1-40 for 24 h (0, 0.01, 0.1, 0.5, 1, 5, 10 µmol/ml). Aβ1-40 reduced ARPE-19 cell viability; the extent of inhibition increased with increasing concentrations of Aβ1-40 (Fig. 1) increased. Then, 0.5 µmol/ml Aβ1-40 was selected to treat ARPE-19 cell; the extent of inhibition increased with prolonged duration of treatment (Fig. 2A). As observed via microscopy, the cell morphology was altered by Aβ1-40 treatment; cells shrunk, and cell fragments were detected in the 0.5 µmol/ml Aβ1-40 group (Fig. 2B). In addition, the number of cells notably decreased. Additionally, 0.5 µmol/ml Aβ1-40 significantly increased the apoptosis and ROS content of ARPE-19 cells (Fig. 2C-E). Notably, the expression of HKII in ARPE-19 cells was not significantly altered in the 0.5 µmol/ml Aβ1-40 group compared with the control group; however, HKII expression in ARPE-19 cell mitochondria was significantly decreased following Aβ1-40 treatment (Fig. 2F-H).
Figure 1.
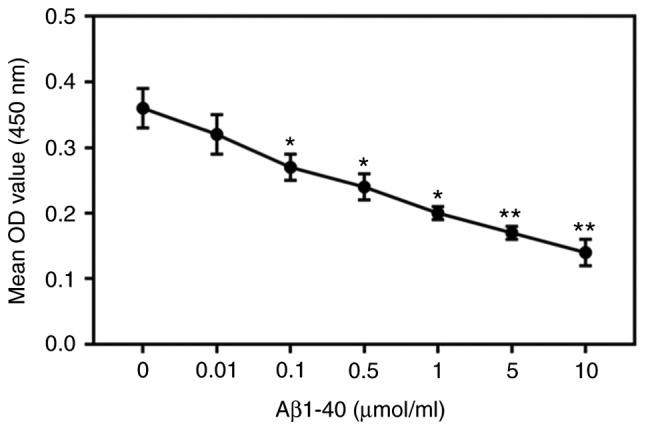
Aβ1-40 inhibits ARPE-19 retinal pigment epithelial cell viability. ARPE-19 cells were treated with various concentrations of Aβ1-40 (0, 0.01, 0.1, 0.5, 1, 5 and 10 µmol/ml) for 24 h. The viability of cells was assessed using a Cell Counting Kit-8 assay. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. control. Aβ1-40, amyloid β1-40; OD; optical density.
Figure 2.

Effects of Aβ1-40 on ARPE-19 retinal pigment epithelial cells. ARPE-19 cells were treated with 0.5 µmol/ml Aβ1-40; untreated cells were used as a control group. (A) Aβ1-40 (0.5 µmol/ml) were added to cells for 12, 24, 48 and 72 h; cell viability was determined using a Cell Counting Kit-8 assay. (B) Images of ARPE-19 cells following treatment with 0.5 µmol/ml Aβ1-40 for 24 h. Scale bar, 100 µm. After 0.5 µmol/ml Aβ1-40 was added to cells for 24 h, (C and D) apoptosis and (E) ROS levels were determined via flow cytometry. Effects of Aβ1-40 (0.5 µmol/ml) on (F) HKII mRNA levels as determined by a reverse transcription-quantitative PCR assay, and (G) cellular and (H) mitochondrial HKII protein levels as determined by western blotting. Data are presented as the mean ± standard deviation. **P<0.01 vs. control. Aβ1-40, amyloid β1-40; COX IV, cytochrome c oxidase subunit IV; HKII, hexokinase II; OD, optical density; PI, propidium iodide; ROS, reactive oxygen species.
Effects of siHKII on ARPE-19 cells
siHKII significantly suppressed HKII expression in ARPE-19 cells, and significantly decreased the viability and increased the apoptosis of cells (Fig. 3A-D). Additionally, the ROS content of ARPE-19 cells was also significantly increased (Fig. 3E). The percentage of early and late apoptotic cells was notably increased in the siHKII group compared with the two control groups (Fig. 3F). These findings indicated that inhibition of HKII damaged RPE cells.
Figure 3.

Effects of siHKII on ARPE-19 retinal pigment epithelial cells. Cells were transfected with siHKII for 1-2 h, and analyzed 24 h later. (A) HKII mRNA levels were determined via reverse transcription-quantitative PCR analysis. (B) Protein levels of HKII were determined via western blot analysis. (C) Cell viability was determined using a Cell Counting Kit-8 assay. (D-F) Apoptosis and ROS levels were determined via flow cytometry. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. control; $P<0.05, $$P<0.01 vs. NC. Con, control; HKII, hexokinase II; NC, negative control; PI, propidium iodide; ROS, reactive oxygen species; si, small interfering (RNA).
Knockdown of HKII alleviates the protective role of NAC in Aβ1-40-induced OS injury
High concentrations of NAC damaged ARPE-19 cells (Fig. 4); therefore, 1 µmol/ml NAC was selected for subsequent experiments. NAC attenuated the effects of Aβ1-40 on ARPE-19 cell viability; this was reversed by knockdown of HKII (Fig. 5). NAC reduced the rate of apop-tosis and ROS content in ARPE-19 cells treated with Aβ1-40 (Fig. 6A-C). Conversely, siHKII induced ARPE-19 cell apoptosis in the Aβ1-40 + NAC group. Furthermore, Aβ1-40 treatment significantly decreased the MMP in ARPE-19 cells (Fig. 6D); NAC attenuated the effects of Aβ1-40 treatment, but siHKII reversed the effects of NAC, reducing the MMP in Aβ1-40 + NAC-treated cells.
Figure 4.
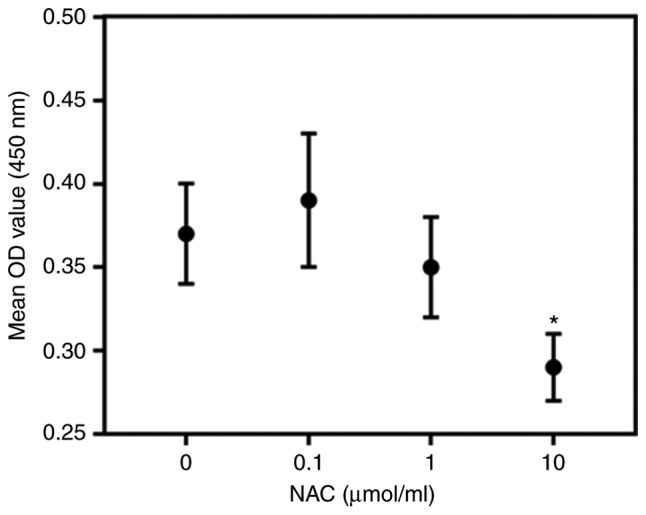
Effects of NAC on ARPE-19 retinal pigment epithelial cell viability ARPE-19 cells were treated with various concentrations of NAC (0, 0.1, 1 or 10 µmol/ml) for 24 h. The viability of cells was assessed using a Cell Counting Kit-8 assay. Data are presented as the mean ± standard deviation. *P<0.05 vs. control. NAC, N-acetylcysteine; OD, optical density.
Figure 5.
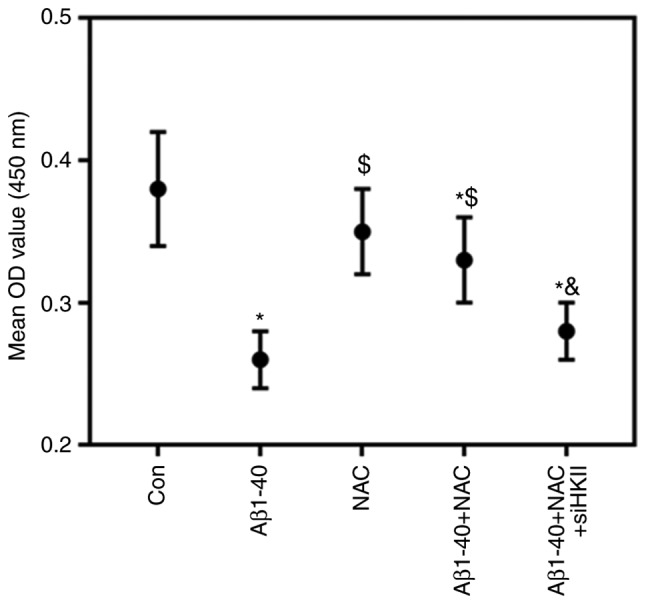
NAC improves, and HKII knockdown reduces Aβ1-40-treated ARPE-19 retinal pigment epithelial cell viability. ARPE-19 cells transfected with siHKII or negative control siRNA were treated for 24 h with Aβ1-40 (0.5 µmol/ml) and/or NAC (1 µmol/ml). The viability of cells was assessed using a Cell Counting Kit-8 assay. Data are presented as the mean ± standard deviation. *P<0.05 vs. Con; $P<0.05 vs. Aβ1-40; &P<0.05 vs. Aβ1-40 + NAC. Aβ1-40, amyloid β1-40; Con, control; HKII, hexokinase II; NAC, N-acetylcysteine; OD, optical density; si(RNA), small interfering (RNA).
Figure 6.

Effects of Aβ1-40, NAC and siHKII treatment on ARPE-19 retinal pigment epithelial cells. ARPE-19 cells transfected with siHKII or negative control siRNA were treated for 24 h with Aβ1-40 (0.5 µmol/ml) and/or NAC (1 µmol/ml). (A and B) Apoptosis and (C) ROS levels were determined via flow cytometry. (D) Analysis of the MMP in treated ARPE-19 cells. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. Con; $P<0.05, $$P<0.01 vs. Aβ1-40; #P<0.05, ##P<0.01 vs. NAC; &P<0.05 vs. Aβ1-40 + NAC. Aβ1-40, amyloid β1-40; Con, control; HKII, hexokinase II; MMP, mitochondrial membrane potential; NAC, N-acetylcysteine; PI, propidium iodide; ROS, reactive oxygen species; si(RNA), small interfering (RNA).
Effects of Aβ1-40, NAC and siHKII treatments on the levels of apoptosis-associated and ROS-associated proteins in ARPE-19 cells
As presented in Fig. 7, it was demonstrated that Aβ1-40 treatment significantly downregulated Bcl-2, MnSOD and CuZnSOD protein expression levels, and significantly upregulated cleaved caspase-3, Bax and Cyt c protein expression. NAC significantly attenuated the effects of Aβ1-40 on the expression of these proteins in ARPE-19 cells; however, siHKII significantly decreased Bcl-2, MnSOD and CuZnSOD expression, and increased cleaved caspase-3, Bax and Cyt c levels in ARPE-19 cells treated with Aβ1-40 and NAC.
Figure 7.

Effects of Aβ1-40, NAC and siHKII treatment on the levels of apoptosis- and reactive oxygen species-associated proteins in ARPE-19 retinal pigment epithelial cells. ARPE-19 cells transfected with siHKII or negative control siRNA were treated for 24 h with Aβ1-40 (0.5 µmol/ml) and/or NAC (1 µmol/ml). (A) Western blotting was performed to determine the protein expression levels of (B) caspase-3, (C) Bcl-2, (D) Bax, (E) MnSOD, (F) CuZnSOD and (G) Cyt c. Expression was quantified using ImageJ. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. Con; $P<0.05, $$P<0.01 vs. Aβ1-40; #P<0.05, ##P<0.01 vs. NAC; &P<0.05, &&P<0.01 vs. Aβ1-40 + NAC. Aβ1-40, amyloid β1-40; Con, control; Cyt c, cytochrome c; CuZnSod, copper-zinc-SOD; HKII, hexokinase II; MnSOD, manganese-SOD; NAC, N-acetylcysteine; si(RNA), small interfering (RNA); SOD, superoxide dismutase.
Effects of Aβ1-40, NAC and siHKII treatments on the expression of HKII in ARPE-19 cells and mitochondria
NAC treatment markedly increased HKII mRNA levels in ARPE-19 cells; however, siHKII resulted in a significant reduction in HKII mRNA expression compared with all other groups (Fig. 8A). Furthermore, siHKII also signifi-cantly reduced HKII protein levels in ARPE-19 cells treated with Aβ1-40 and NAC; HKII protein levels were not significantly altered by Aβ1-40 or NAC (Fig. 8B). Conversely, Aβ1-40 significantly downregulated HKII protein levels in mitochondria, whereas NAC attenuated the effects of Aβ1-40 on mitochondrial HKII protein levels (Fig. 8C). siHKII significantly reduced HKII protein levels in the mitochondria of ARPE-19 cells treated with Aβ1-40 and NAC (Fig. 8C), indicating that the changes in HKII induced by siHKII mainly occurred in the mitochondria.
Figure 8.

Effects of Aβ1-40, NAC and siHKII treatment on the expression of HKII in ARPE-19 retinal pigment epithelial cells and mitochondria. ARPE-19 cells transfected with siHKII or negative control siRNA were treated for 24 h with Aβ1-40 (0.5 µmol/ml) and/or NAC (1 µmol/ml). (A) HKII mRNA levels in ARPE-19 cells, as determined via reverse transcription-quantitative PCR analysis. (B) Whole cell and (C) mitochondrial levels of HKII in ARPE-19 cells, as determined by western blotting. The stains were analyzed using Image J system. Expression was quantified using ImageJ. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. Con; $P<0.05, $$P<0.01 vs. Aβ1-40; #P<0.05, ##P<0.01 vs. NAC; &P<0.05, &&P<0.01 vs. Aβ1-40 + NAC. Aβ1-40, amyloid β1-40; Con, control; COX IV, cytochrome c oxidase subunit IV; HKII, hexokinase II; NAC, N-acetylcysteine; si(RNA), small interfering (RNA).
Discussion
Retinal degeneration, including AMD and RP, is one of the most common neurodegenerative diseases globally (29). AMD and RP are characterized by gradual faded central vision due to photoreceptor cell degeneration; RPE cells, which are a layer of cells in the retina, support the photoreceptors function, thus serving an important role in maintaining retinal homeostasis (30,31). Patients with RP exhibit ocular OS and damage from the absence of the ability to overcome systemic OS; an imbalance between the generation and elimination of ROS leads to OS, oxidation of macromolecules and eventually retinal disease (32,33). ARPE-19 cell viability was inhibited, and apoptosis and ROS contents were increased following induction of OS via Aβ1-40 treatment.
OS, inflammation and endoplasmic reticulum stress are involved in glutamate excitotoxicity, contributing to mitochondrial dysfunction (34). The mechanism via which dissociation of HKII induces mitochondrial dysfunction and apoptosis in the retina remains unclear. It was previously reported that when HKII dissociates from the mitochondrial membrane, Bax translocates into the mitochondria and binds to unoccupied voltage-dependent anion channels (VDACs) to form a large pore, which exhibits 4- and 10-fold higher conductance levels than VDACs and Bax channels, respectively; however, the large pore lacks the ion selectivity of individual channels (21,35). Furthermore, the VDAC-Bax pore can result in the release of Cyt c to the cytosol (36). Additionally, others have also suggested that disaggregation of the VDAC-HKII interaction could open the Bax-independent mitochondrial permeability transition pore (PTP) (37,38), a multiprotein complex including cyclophilin D in the matrix, adenine nucleotide translocator in the inner membrane, and VDACs in the outer membrane (39,40). The opening of the PTP can lead to rapid MMP depolarization and matrix swelling, therefore resulting in the unfolding of cristae and breaches in the outer mitochondrial membrane, rendering it permeable to proteins (41). In the present study, the results showed that Aβ1-40 treatment decreased the MMP, which is consistent with the research of Moreira et al (42). It has been suggested that MMP depolarization is associated with apoptosis. Conversely, it has been reported that Aβ can potentiate Ca2+-induced PTP formation in liver mitochondria (42). In the present study, Aβ1-40 may reduce the transmembrane potential by reducing the concentration of Ca2+, thereby causing changes in mitochondrial membrane permeability, leading to the release of proapoptotic substances and activation of the caspase family, and promoting cell apoptosis. Mitochondria are not only the primary cellular energy source under aerobic conditions, but also an important an important component in apoptotic cell death (43). A previous study reported that the MMP regulates matrix configuration and Cyt c release during apoptosis (44). Additionally, outer mitochondrial membrane permeabilization is a crucial signal for apoptosis, resulting in the liberation of proapoptotic molecules such as cytochrome c and procaspase activation (45). Regardless of the mechanism, the findings from the present study suggested that the dissociation of HKII from the mitochondrial membrane inhibits proliferation, induces apoptosis, increases the ROS levels and decreases the MMP in RPE cells.
NAC, an antioxidant, has been reported to reduce retinal superoxide radicals and promote cone cell survival in mouse models (46). The present study further revealed that NAC improved the viability and reduced the apoptosis of HPE cells under OS induced by Aβ1-40 treatment. Previous studies reported that increased expression of HKII provided protection (47,48), and that decreased expression of HKII promoted cell apoptosis (21). For example, overexpression of HKII provided protection against peroxide in cardiomyocytes (19,37). The present study demonstrated that the inhibition of HKII mRNA expression decreased ARPE-19 cell viability, and promoted cell apoptosis and ROS, indicating that decreased expression of HKII promoted cell damage. However, a potential association between NAC and HKII, and the mechanisms via which altered HKII expression induces cell damage remain unclear.
Akt, a member of the AGC kinase group, is important in various cell functions, including proliferation, apoptosis and metabolism (49). Previous studies reported that Akt was upregulated in tumors or following insulin treatment; HKII levels were also increased under these conditions (19,50), suggesting a potential association between the Akt pathway and HKII. A number of studies have demonstrated that the Akt pathway was associated with apoptosis pathways, and that apoptosis was induced by inhibiting the Akt pathway (51,52). The present study suggested that NAC reduced Aβ1-40-induced damage by upregulating HKII levels in the mitochondria, and that the downregulation of HKII promoted apoptosis pathways, including upregulated cleaved caspase-3, Bax and Cyt c, and decreased Bcl-2 expression, all of which are involved in the mitochondrial apoptosis pathway (53-55).
A previous study observed that activation of Akt increased HK activity in mitochondria, and that mitochondrial HK was required for the antiapoptotic properties of Akt signaling (56). The function of living cells and mechanisms of energy can be assessed via the MMP (57); a study indicated that antiproliferative and proapoptotic effects occurred following loss of the MMP (58). Downregulation of mitochondrial HKII, either by Aβ1-40-induced OS or siHKII-mediated knockdown, resulted in a lower MMP, increased apoptosis and downregulation of SOD. NAC treatment attenuated the proapoptotic and antiproliferative effects of Aβ1-40-induced OS in ARPE-19 cells, which was accompanied with restoration of the normal MMP; however, knockdown HKII reversed these effects and again decreased the MMP. CuZnSOD and MnSOD eliminate ROS and maintain redox balance in the immune system (59). NAC reduced ROS levels in Aβ1-40-induced ARPE-19 cells; HKII knockdown enhanced ROS levels, potentially via the down-regulation of CuZnSOD and MnSOD.
The aims of the present study were to investigate the roles of HKII in OS-induced injury in RPE cells. The present findings suggested that NAC reduced OS-associated damage, and increased the viability of RPE cells subjected to Aβ1-40-induced OS. It was further suggested that the effects of NAC on OS involved upregulation of mitochondrial HKII levels, as HKII serves roles in regulating mitochondrial apoptotic pathways. It was also observed that decreased expression of HKII promoted the expression of proapoptotic proteins associated with the mitochondrial apoptosis pathway, and reduced the levels of MnSOD and CuZnSOD in RPE cells under OS. However, there are certain limitations to the present study; for example, the levels of VDACs, and the expression and activation Akt were not detected. These and other limitations will be resolved in future investigations.
Acknowledgments
Not applicable.
Funding
This study was supported by the Beijing Shijitan Hospital Fund (grant no. 2016-c08), the Beijing Talents Training Fund (grant no. 2010D003034000006) and the Beijing High-level Talents Training Fund for Health System (grant no. 2014-3-049).
Availability of data and materials
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
LC made substantial contributions to the conception and design of the study. LX, BX and JX were involved in data acquisition, analysis and interpretation. LC drafted the article and critically revised it for important intellectual content. All authors approved the final version of the manuscript to be published.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wei EP, Christman CW, Kontos HA, Povlishock JT. Effects of oxygen radicals on cerebral arterioles. Am J Physiol. 1985;248:H157–H162. doi: 10.1152/ajpheart.1985.248.2.H157. [DOI] [PubMed] [Google Scholar]
- 2.Sies H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell EG, Cotter TG. New insight into the role of reactive oxygen species (ROS) in cellular signal-transduction processes. Int Rev Cell Mol Biol. 2015;319:221–254. doi: 10.1016/bs.ircmb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact. 2014;224:164–175. doi: 10.1016/j.cbi.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Kruk J, Kubasik-Kladna K, Aboul-Enein HY. The role oxidative stress in the pathogenesis of eye diseases: Current status and a dual role of physical activity. Mini Rev Med Chem. 2015;16:241–257. doi: 10.2174/1389557516666151120114605. [DOI] [PubMed] [Google Scholar]
- 6.Ames A., III Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: A commentary based on studies on retina. Can J Physiol Pharmacol. 1992;(70 Suppl):S158–S164. doi: 10.1139/y92-257. [DOI] [PubMed] [Google Scholar]
- 7.Guo X, Dason ES, Zanon-Moreno V, Jiang Q, Nahirnyj A, Chan D, Flanagan JG, Sivak JM. PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am J Pathol. 2014;184:1017–1029. doi: 10.1016/j.ajpath.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Moreno ML, Mérida S, Bosch-Morell F, Miranda M, Villar VM. Autophagy dysfunction and oxidative stress, two related mechanisms implicated in retinitis pigmentosa. Front Physiol. 2018;9:1008. doi: 10.3389/fphys.2018.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Zhu X, Zhang K, Yao Y, Zhuang M, Tan C, Zhou F, Zhu L. Puerarin inhibits amyloid β-induced NLRP3 inflammasome activation in retinal pigment epithelial cells via suppressing ROS-dependent oxidative and endoplasmic reticulum stresses. Exp Cell Res. 2017;357:335–340. doi: 10.1016/j.yexcr.2017.05.030. [DOI] [PubMed] [Google Scholar]
- 10.Briyal S, Shepard C, Gulati A. Endothelin receptor type B agonist, IRL-1620, prevents beta amyloid (Aβ) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol Biochem Behav. 2014;120:65–72. doi: 10.1016/j.pbb.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Yang R, Wang Q, Min L, Sui R, Li J, Liu X. Monosialoanglioside improves memory deficits and relieves oxidative stress in the hippocampus of rat model of Alzheimer's disease. Neurol Sci. 2013;34:1447–1451. doi: 10.1007/s10072-012-1263-y. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida T, Ohno-Matsui K, Ichinose S, Sato T, Iwata N, Saido TC, Hisatomi T, Mochizuki M, Morita I. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest. 2005;115:2793–2800. doi: 10.1172/JCI24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- 14.Wilson JE. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 15.Ardehali H, Printz RL, Whitesell RR, May JM, Granner DK. Functional interaction between the N- and C-terminal halves of human hexokinase II. J Biol Chem. 1999;274:15986–15989. doi: 10.1074/jbc.274.23.15986. [DOI] [PubMed] [Google Scholar]
- 16.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 17.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the 'Warburg Effect', i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 18.Heikkinen S, Suppola S, Malkki M, Deeb SS, Jänne J, Laakso M. Mouse hexokinase II gene: Structure, cDNA, promoter analysis, and expression pattern. Mamm Genome. 2000;11:91–96. doi: 10.1007/s003350010019. [DOI] [PubMed] [Google Scholar]
- 19.Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013;288:23798–23806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 22.Das S, Steenbergen C, Murphy E. Does the voltage dependent anion channel modulate cardiac ischemia-reperfusion injury? Biochim Biophys Acta. 2012;1818:1451–1456. doi: 10.1016/j.bbamem.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2012;2:e005645. doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calmettes G, John SA, Weiss JN, Ribalet B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J Gen Physiol. 2013;142:425–436. doi: 10.1085/jgp.201310968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141:150–159. doi: 10.1016/j.pharmthera.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Garner DL, Thomas CA. Organelle-specific probe JC-1 identifies membrane potential differences in the mitochondrial function of bovine sperm. Mol Reprod Dev. 1999;53:222–229. doi: 10.1002/(SICI)1098-2795(199906)53:2<222::AID-MRD11>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Wert KJ, Lin JH, Tsang SH. General pathophysiology in retinal degeneration. Dev Ophthalmol. 2014;53:33–43. doi: 10.1159/000357294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geerlings MJ, de Jong EK, den Hollander AI. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol Immunol. 2017;84:65–76. doi: 10.1016/j.molimm.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petit L, Punzo C. mTORC1 sustains vision in retinitis pigmentosa. Oncotarget. 2015;6:16786–16787. doi: 10.18632/oncotarget.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campochiaro PA, Strauss RW, Lu L, Hafiz G, Wolfson Y, Shah SM, Sophie R, Mir TA, Scholl HP. Is there excess oxidative stress and damage in eyes of patients with retinitis pigmentosa? Antioxid Redox Signal. 2015;23:643–648. doi: 10.1089/ars.2015.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitra RN, Conley SM, Naash MI. Therapeutic approach of nanotechnology for oxidative stress induced ocular neurodegenerative diseases. Adv Exp Med Biol. 2016;854:463–469. doi: 10.1007/978-3-319-17121-0_62. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Li J, Li S, Li Y, Wang X, Liu B, Fu Q, Ma S. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol Appl Pharmacol. 2015;286:53–63. doi: 10.1016/j.taap.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu S, Ide T, Yanagida T, Tsujimoto Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J Biol Chem. 2000;275:12321–12325. doi: 10.1074/jbc.275.16.12321. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee J, Ghosh S. Phosphorylation of rat brain mitochondrial voltage-dependent anion as a potential tool to control leakage of cytochrome c. J Neurochem. 2006;98:670–676. doi: 10.1111/j.1471-4159.2006.03853.x. [DOI] [PubMed] [Google Scholar]
- 37.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: Another view. Biochimie. 2002;84:153–166. doi: 10.1016/S0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 41.Woldetsadik AD, Vogel MC, Rabeh WM, Magzoub M. Hexokinase II-derived cell-penetrating peptide targets mitochondria and triggers apoptosis in cancer cells. FASEB J. 2017;31:2168–2184. doi: 10.1096/fj.201601173R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreira PI, Santos MS, Moreno A, Rego AC, Oliveira C. Effect of amyloid beta-peptide on permeability transition pore: A comparative study. J Neurosci Res. 2002;69:257–267. doi: 10.1002/jnr.10282. [DOI] [PubMed] [Google Scholar]
- 43.Gottlieb RA. Mitochondria: Execution central. FEBS Lett. 2000;482:6–12. doi: 10.1016/S0014-5793(00)02010-X. [DOI] [PubMed] [Google Scholar]
- 44.Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- 45.Birkinshaw RW, Czabotar PE. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol. 2017;72:152–162. doi: 10.1016/j.semcdb.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Lee SY, Usui S, Zafar AB, Oveson BC, Jo YJ, Lu L, Masoudi S, Campochiaro PA. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J Cell Physiol. 2011;226:1843–1849. doi: 10.1002/jcp.22508. [DOI] [PubMed] [Google Scholar]
- 47.McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. 2013;2:e000355. doi: 10.1161/JAHA.113.000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corona JC, Gimenez-Cassina A, Lim F, Diaz-Nido J. Hexokinase II gene transfer protects against neurodegeneration in the rotenone and MPTP mouse models of Parkinson's disease. J Neurosci Res. 2010;88:1943–1950. doi: 10.1002/jnr.22357. [DOI] [PubMed] [Google Scholar]
- 49.Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. 2015;74:38–48. doi: 10.1016/j.vph.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuo B, Li Y, Li Z, Qin H, Sun Q, Zhang F, Shen Y, Shi Y, Wang R. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem Biophys Res Commun. 2015;464:401–406. doi: 10.1016/j.bbrc.2015.06.092. [DOI] [PubMed] [Google Scholar]
- 51.Hyun S, Kim MS, Song YS, Bak Y, Ham SY, Lee DH, Hong J, Yoon DY. Peroxisome proliferator-activated receptor-gamma agonist 4-O-methylhonokiol induces apoptosis by triggering the intrinsic apoptosis pathway and inhibiting the PI3K/Akt survival pathway in SiHa human cervical cancer cells. J Microbiol Biotechnol. 2015;25:334–342. doi: 10.4014/jmb.1411.11073. [DOI] [PubMed] [Google Scholar]
- 52.Sun D, Sawada A, Nakashima M, Kobayashi T, Ogawa O, Matsui Y. MK2206 potentiates cisplatin-induced cytotoxicity and apoptosis through an interaction of inactivated Akt signaling pathway. Urol Oncol. 2015;33:111.e17–e26. doi: 10.1016/j.urolonc.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 53.Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol. 2003;199:221–228. doi: 10.1002/path.1289. [DOI] [PubMed] [Google Scholar]
- 54.Liu YQ, Liu YF, Ma XM, Xiao YD, Wang YB, Zhang MZ, Cheng AX, Wang TT, Li JL, Zhao PX, et al. Hydrogen-rich saline attenuates skin ischemia/reperfusion induced apoptosis via regulating Bax/Bcl-2 ratio and ASK-1/JNK pathway. J Plast Reconstr Aesthet Surg. 2015;68:e147–e156. doi: 10.1016/j.bjps.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Liang S, Sun K, Wang Y, Dong S, Wang C, Liu L, Wu Y. Role of Cyt-C/caspases-9,3, Bax/Bcl-2 and the FAS death receptor pathway in apoptosis induced by zinc oxide nanoparticles in human aortic endothelial cells and the protective effect by alpha-lipoic acid. Chem Biol Interact. 2016;258:40–51. doi: 10.1016/j.cbi.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 56.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lobanova EG, Kondrat'eva EV. Measurement of mitochondrial membrane potential in leukocyte suspension by fluorescent spectroscopy. Bull Exp Biol Med. 2014;157:288–290. doi: 10.1007/s10517-014-2547-4. [DOI] [PubMed] [Google Scholar]
- 58.Tian Q, Zang YH. Antiproliferative and apoptotic effects of the ethanolic herbal extract of Achillea falcata in human cervical cancer cells are mediated via cell cycle arrest and mitochondrial membrane potential loss. J BUON. 2015;20:1487–1496. [PubMed] [Google Scholar]
- 59.Lu X, Wang C, Liu B. The role of Cu/Zn-SOD and Mn-SOD in the immune response to oxidative stress and pathogen challenge in the clam Meretrix meretrix. Fish Shellfish Immunol. 2015;42:58–65. doi: 10.1016/j.fsi.2014.10.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.