

Abstract
PARP inhibitors (PARPi) are potentially effective therapeutic agents capable of inducing synthetic lethality in tumors with deficiencies in homologous recombination (HR)-mediated DNA repair such as those carrying BRCA1 mutations. However, BRCA mutations are rare, the majority of tumors are proficient in HR repair, and thus most tumors are resistant to PARPi. Previously, we observed that ionizing radiation (IR) initiates cytoplasmic translocation of BRCA1 leading to suppression of HR-mediated DNA repair and induction of synthetic PARPi lethality in wild-type BRCA1 and HR-proficient tumor cells. The tumor suppressor p53 was identified as a key factor that regulates DNA damage-induced BRCA1 cytoplasmic sequestration following IR. However, the role of p53 in IR-induced PARPi sensitization remains unclear. This study elucidates the role of p53 in IR-induced PARPi cytotoxicity in HR-proficient cancer cells and suggests p53 status may help define a patient population that might benefit from this treatment strategy. Sensitization to PARPi following IR was determined in vitro and in vivo utilizing human breast and glioma tumor cells carrying wild-type BRCA1 and p53, and in associated cells in which p53 function was modified by knockdown or mutation. In breast and glioma cells with proficient HR-repair, IR-induced BRCA1 cytoplasmic sequestration, HR-repair inhibition, and subsequent PARPi sensitization in vitro and in vivo was dependent upon functional p53.
Keywords: PARP, BRCA1, p53, GBM, ABT-888
Introduction
PARP inhibitors (PARPi) demonstrate synthetic lethality in cells with impaired homologous recombination (HR)-mediated DNA repair function, particularly BRCA1/2-associated tumors (1, 2). Unfortunately, BRCA1/2-associated HR-repair deficiencies occur in only a small percentage of tumors greatly limiting the clinical efficacy of PARPi based therapies (3, 4). Methods to expand the population of cancer patients that may benefit from PARPi beyond those with BRCA-associated tumors, including the combination of PARPi with other cytotoxic agents, have thus far failed to be efficacious in the clinic. Despite these setbacks, the development of novel paradigms to elicit tumor-specific PARPi synthetic lethality in HR-competent cancers remains an intriguing treatment strategy and recently reported studies in experimental models of HR-proficient cancers have demonstrated an increase in treatment efficacy when PARPi are used in combination with other cytotoxic agents such as ionizing radiation (IR) (5, 6). Existing combinatorial strategies utilize PARPi as an inducer of single strand DNA breaks, but fail to tap into the synthetic lethality seen in HR-deficient tumors. Recently, we developed a novel combinatorial paradigm in which IR is combined with PARPi (7, 8). Our strategy utilizes IR not just as a cytotoxic agent but more importantly as a means to export BRCA1 from the nucleus to inhibit HR repair and create synthetic PARPi lethality.
The essential roles of BRCA1 in DNA damage response and HR-mediated repair following DNA double strand breaks (DSBs) have been well characterized (9–11). While BRCA1 deficiencies are commonly associated with inherited breast and ovarian cancers, BRCA1 status is prognostic of outcome and treatment response in a variety of cancers including glioblastomas (12) and prostate tumors (13). Cells with HR-repair deficiencies rely heavily on alternative mechanisms to repair damaged DNA such as single strand break (SSB) base excision repair. PARP1 is the rate-limiting enzyme in SSB base excision repair, and thus, inhibition of PARP in HR-deficient cancer cells leaves these tumors greatly compromised in their ability to repair damaged DNA accounting for the increased cytotoxicity of PARPi in these tumors (1). While dysfunction of BRCA1 activity due to mutation occurs only in a small subset of cancers, BRCA1 function may be positively or negatively regulated by a variety of mechanisms including subcellular localization in both normal and cancer cells (14–16). Nuclear localization is required for BRCA1 to participate in HR-mediated DNA repair and initiate cell-cycle checkpoints following DNA damage; cytosolic BRCA1, on the other hand, not only is unable to contribute to DNA repair, but is also involved in the induction of apoptosis through a p53-independent mechanism (17–19). Therefore, the nuclear/cytoplasmic localization of BRCA1 is a critical factor in determining whether a cell effectively repairs damaged DNA or dies following genotoxic stress. We have previously demonstrated that nuclear export of BRCA1 suppresses its nuclear function resulting in inhibition of HR-mediated DNA damage repair and, subsequently, increased cytotoxicity to DNA-damaging agents (8, 15, 19, 20). Furthermore, our group and others have demonstrated that IR can induce the export of BRCA1 from the nucleus to the cytoplasm resulting in decreased HR-repair and increased cytotoxicity of PARPi in breast tumor models (8, 15, 19).
Like BRCA1 and PARP1, the tumor suppressor p53 is a critical guardian of genome stability (21), and thus, it is not surprising that cross-talk exists between these important factors to coordinate the decision of DNA-repair or cell death following genotoxic stress. Recent work (22) has demonstrated that in glioblastoma cell lines the therapeutic efficacy of PARPi plus IR combination therapy is dependent upon p53 status due to the requirement of PARP1 enzymatic activity for p53 nuclear accumulation and transcriptional activity following DNA damage. Work by our laboratory has established an important role for p53 in the subcellular distribution of BRCA1. We have previously reported that p53 dysfunction leads to nuclear accumulation and reduced cytoplasmic translocation of BRCA1 following exposure to IR (15, 20). These data demonstrate that p53-mediated shuttling of BRCA1 is an important mechanism regulating the choice of cell death or DNA repair following genomic damage. We hypothesized that p53 status would be an essential determinant of PARPi sensitivity in our treatment strategy of IR preceding PARPi and, given the high frequency of p53 mutation in solid tumors, sought to elucidate the role of p53 status on the effectiveness of our treatment paradigm to more precisely define those tumors that are likely to benefit.
The results presented herein demonstrate that BRCA1 sequestration to the cytoplasm following exposure to IR can be used to induce HR-mediated DNA repair deficiency and increase PARPi cytotoxicity in multiple tumor models. Importantly, we find that p53 status is a significant determining factor of tumor cell sensitization to PARPi following IR. Together our results demonstrate that IR can suppress HR-mediated DNA repair in a p53 dependent-manner in variety of cancer types to confer PARPi synthetic lethality, which, if translated to the clinic, may significantly increase the population of cancer patients who benefit from PARPi based therapies.
Materials and Methods
Cell Culture
Stable MCF7/E6 and their associated control cell line were established from cells purchased from ATCC as described previously (15). The U87 human glioblastoma cell line was purchased from ATCC. SF767 shCtrl and SF767 shp53 human glioma cell lines were a gift from Dr. Jay Fitzgerald Dorsey (Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA) and have been previously described (23). Cell lines were cultured at low passage number and authenticated by morphology and growth characteristics. Cells were tested for Mycoplasma by PCR (ATCC) every three months. All cell lines were maintained in DMEM supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin/streptomycin. Transient p53 knockdown was accomplished using p53-targeting siRNAs (L-003329–00, Dharmacon) with siRNA A4 (Dharmacon) serving as a non-targeting control and Lipofectamine2000 transfection reagent (Invitrogen) as recommended by the manufacturer. Cells were irradiated as indicated using a RS-2000 biological irradiator (Rad Source). Cells were treated with ABT-888 (Enzo) as described in the text.
Immunohistochemistry
Immunohistochemical staining using antibodies to BRCA1, gamma-H2AX, and Rad51 was performed as previously described (7, 24).
Clonogenic survival
Clonogenic survival assays were performed as previously described (25). Briefly, cells were treated as described in the text and then maintained in culture for approximately three weeks until colonies were of sufficient size to be stained and counted. Colonies were then fixed in a 1:7 mixture of acetic acid and methanol prior to staining with 0.5% crystal violet. Colonies of greater than 50 cells were counted and the survival fraction was calculated as previously described (25).
In vivo tumor growth
All animal procedures were approved by the Institutional Animal Care and Use Committee at The Ohio State University. 1×107 MCF7 or MCF7/E6 cells, or 5×106 U87, SF767 shCtrl or SF767 shp53 cells were injected into the left flank of 6 week old female athymic Foxn1 (nu/nu) mice (Harlan Sprague Dawley Inc. or Jackson Laboratories). For experiments utilizing MCF7 and MCF7/E6 cells, mice were implanted with 17β-estradiol pellets (0.72mg, 60 day release; Innovative Research of America) 6 days prior to injection of the cells. Mice were randomly assigned to an experimental treatment arm once a tumor volume of ~100 mm3 was achieved. As indicated, 25 mg/kg ABT-888 (Enzo) suspended in H20 was delivered by oral gavage daily for five days. Mice receiving radiation treatment were anesthetized and ionizing radiation was delivered to the tumor as a single dose of 3 Gy (MCF7 control and E6 tumors) or 4 Gy (SF767 shCtrl and shp53 tumors). Mice receiving a combination of radiation and ABT-888 treatment were irradiated as described and then treated with ABT-888 by oral gavage daily for 5 consecutive days starting 24h after radiation.
Western blot analysis
Whole cell lysates were prepared as previously described (15) and subjected to SDS-PAGE analysis. Anti-BRCA1 antibody (Ab-1; Calbiochem) was used at a 1:100 dilution while antibodies to β-actin, α-tubulin and lamin a/c were purchased from Cell Signaling Technology and used at working dilutions of 1:1000. Protein bands were visualized and photographed by means of a Versadoc Imaging System (BioRad) using HRP-conjugated anti-rabbit and anti-mouse (Cell Signaling Technologies) secondary antibodies at working dilutions of 1:5000 and Immobilon western ECL reagent (Miilipore). Quantitation of western blots was performed using Image-J software (NIH). Isolation of nuclear and cytoplasmic protein fractions was accomplished using a Cell Fractionation Kit (Cell Signaling Technologies) according to the manufacturer’s recommendations.
Cell cycle analysis
Unsynchronized, subconfluent MCF7 control or E6 cells were irradiated with 4 Gy or mock treated. 24 h later cells were fixed in 70% ethanol. Cells were then rehydrated in PBS prior to incubation for 30 m in a solution containing 0.1 % Triton X-100, 0.2 mg/mL DNase-free RNase A and 0.02 mg/mL propidium iodide prior to sorting using an LSR II (BD Biosciences) and analysis using BD FACSDiva software (BD Biosciences).
Assay for HR-mediated repair of DNA double strand breaks
MCF7 cells with a stably integrated DR-GFP chromosomal reporter system were utilized for this assay. This reporter system contains two differently mutated GFP genes oriented as direct repeats. The upstream repeat contains a single I-SceI site while the downstream copy is a 5’-3’ truncated GFP fragment. Expression of I-SceI in these cells generates a DSB that, when repaired by HR, results in expression of GFP. Reporter cells were seeded overnight then transfected with control siRNA, p53-targeting siRNA, empty vector, or vector encoding p53R273H respectively. 16 h later cells were treated with 4 Gy IR or mock treated. After an additional 4 h, cells were transfected a control vector or vector encoding I-SceI. 48 h later cells were collect and analyzed for GFP expression by flow cytometry.
Statistical analyses
Statistical analysis between groups was performed by ANOVA while direct comparisons between two groups were done by a two-tailed t-test. P < 0.05 was considered statistically significant.
Results
p53 function is required for IR-induced cytoplasmic translocation of BRCA1 and sensitization of breast cancer cells to PARPi in vitro and in vivo
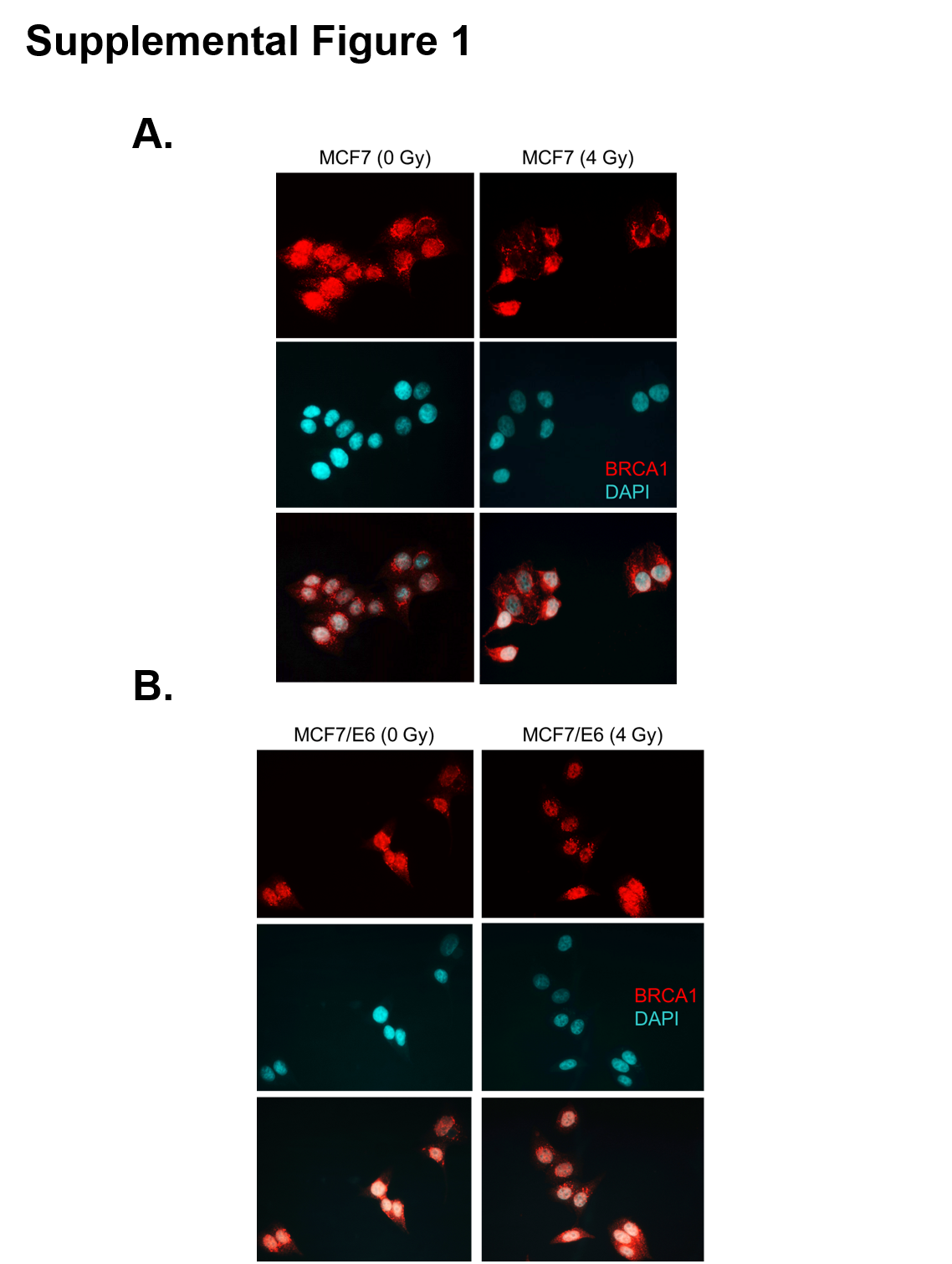
We previously demonstrated that p53 function is required for cytoplasmic translocation of BRCA1 following IR (15, 20). In addition, IR-induced cytoplasmic sequestration of BRCA1 results in inhibition of HR-mediated DNA repair and increased sensitivity to PARPi (7). It remains to be elucidated if functional p53 is required to render HR-proficient cancer cells sensitive to PARPi subsequent to IR. To begin to address this clinically relevant question we inhibited p53 function in MCF7 breast cancer cells by stable expression of the HPV16-E6 viral protein which catalyzes the degradation of p53 (MCF7/E6) (26). In agreement with our previously published findings (7), treatment of control MCF7 cells with 4 Gy ionizing radiation resulted in a significant shift in BRCA1 localization from the nucleus to the cytoplasm 24h post-IR as determined by immunofluorescence (Figure 1A, Supplemental Figure 1A). In contrast, no significant cytoplasmic translocation of BRCA1 was observed following radiation in MCF7/E6 cells (Figure 1B, Supplemental Figure 1B).
Figure 1. p53 function is required for IR-induced cytoplasmic translocation of BRCA1 and sensitization of breast cancer cells to PARPi in vitro.

A and B, the percentage of MCF7 control (A) and MCF7/E6 (B) cells that demonstrated nuclear (N), nuclear and cytoplasmic (NC), or cytoplasmic (C) immunohistochemical staining of BRCA1 was analyzed 24 h after treatment with 0 or 4 Gy IR. Results are the average of three independent experiments. C and D, the cell cycle distribution of unsynchronized MCF7 control (C) and E6 (D) cells 24h after mock treatment or treatment with 4 Gy IR. E and F, the surviving fraction of MCF7 (E) or MCF7/E6 (F) was determined by clonogenic survival assay. Cells were first treated with 0 or 4 Gy IR and then treated 24h later with 0, 5, 10, 15, 20, or 25 µM ABT-888. Results are the average of three independent experiments.
Subcellular localization of BRCA1 is influenced by the cell cycle (15). During S phase BRCA1 is primarily confined to the nucleus and is exported to the cytoplasm during G2 resulting in an almost exclusively cytoplasmic distribution in M phase. As cells leave G1 and re-enter S phase, BRCA1 is trafficked back to the nucleus. Given that p53 is a critical mediator of G1 and G2 arrest following DNA damage (27), we next asked whether the differences in BRCA1 localization following DNA damage observed between MCF7 control and E6 cells could be explained by changes in the cell cycle distribution of these cells following radiation. As expected, radiation elicited a G1 arrest in MCF7 cells as indicated by increased cells in G1 and significantly fewer cells in S phase after radiation (Figure 1C). In contrast, no G1 arrest was detected following radiation of MCF/E6 cells (Figure 1D). The lack of G1 arrest in MCF7/E6 cells following radiation may account for some of the reduction in BRCA1 nuclear export seen in these cells following radiation. However, the difference in the percentage of the population in S phase between MCF7 control and E6 cells after radiation was only 7.39% and does not fully account for the almost 25% difference in cells with nuclear BRCA1 after radiation seen between these two cell lines (Figure 1 A and B). This is in agreement with our previous data that demonstrated that, while BRCA1 distribution is dependent upon cell cycle, DNA damage-induced BRCA1 nuclear export is independent of cell cycle and occurs during all phases of the cell cycle (15).
To determine the role of p53 in IR-induced sensitization of MCF7 cells to PARPi, we performed in vitro clonogenic survival assays utilizing MCF7 control and E6-expressing cells treated first with 0 or 4 Gy IR then treated 24h later with 0, 5, 10, 15, 20, or 25 μM of the PARP inhibitor ABT-888. In agreement with previously published findings (8), MCF7 cells pre-treated with 4 Gy IR demonstrated significantly increased sensitivity to ABT-888 relative to cells that received no radiation (Figure 1E). However, in MCF7/E6 cells, pretreatment with radiation did not significantly sensitize these cells to ABT-888 relative to un-irradiated MCF7/E6 cells (Figure 1F). These results suggest that p53 function is required for IR-induced sensitization of MCF7 cells to PARPi in vitro.
To analyze effect of p53 on the sensitization of MCF7 control and E6 cells to PARPi subsequent to radiation in vivo, MCF7 and MCF7/E6 tumors were established in nude mice and the animals were randomly divided into one of four treatment arms: control animals receiving no treatment, animals receiving a single dose of 3 Gy radiation, animals receiving 25 mg/kg ABT-888 daily for 5 days, and animals receiving a single dose of 3 Gy radiation followed by daily treatments of 25 mg/kg ABT-888 for 5 days commencing 24h after radiation treatment. In animals bearing MCF7 control tumors, the combination treatment of radiation plus PARPi resulted in a significant reduction in tumor volume relative to mice in either mono-therapy arm (p < 0.05, Figure 2A). Treatment with radiation alone or with ABT-888 alone did not result in a significant reduction in tumor volume relative to untreated controls (p < 0.05, Figure 2A). In mice bearing MCF7/E6 tumors, radiation plus PARPi combination therapy did not result in a significant change in tumor volume relative to untreated animals or animals treated with either of the mono-therapy arms suggesting that p53 function is essential for in vivo sensitization of MCF7 tumors to PARPi following IR (Figure 2B). To determine if BRCA1 translocation to the cytosol is indeed depend on p53 in tumors in vivo following IR, we harvested representative tumors 24h post-irradiation and assessed BRCA1 subcellular localization by immunofluorescence. In agreement with our in vitro results, in control MCF7 tumors, treatment with IR resulted in a significant shift in localization of BRCA1 from the nucleus to the cytoplasm 24h after treatment (Figure 2C, Supplemental Figure 2A). In contrast, cytoplasmic sequestration of BRCA1 following IR treatment was not observed in MCF7/E6 tumors (Figure 2D, Supplemental Figure 2B) confirming that p53 function is essential for translocation of BRCA1 to the cytosol in response to IR providing a mechanism for subsequent sensitization of MCF7 tumors to PARPi after IR treatment in vivo.
Figure 2. p53 function is required for IR-induced cytoplasmic translocation of BRCA1 and sensitization of breast cancer cells to PARPi in vivo.

A and B, relative tumor volumes of mice bearing MCF7 (A) or MCF7/E6 (B) tumors treated with no treatment (Control, n = 3), 25mg/kg of ABT-888 for 5 consecutive days (ABT-888, n = 3), 3 Gy ionizing radiation (IR, n = 3), or the combination of 3 Gy IR followed by 25mg/kg ABT-888 for 5 days (ABT-888 + IR, n = 3). C and D, the percentage of cells demonstrating nuclear and nuclear plus cytoplasmic (N + NC), or primarily cytoplasmic (C) staining of BRCA1 in MCF7 control (C) and MCF7/E6 (D) tumors (n = 5 per group) harvested 24 h after treatment with 3 Gy IR. *, p < 0.05.
Reduced HR-mediated DNA repair following radiation requires p53 function
Decreased HR-function is a prerequisite for synthetic lethality to PARP1 inhibitors. We hypothesized that p53-depenent nuclear export of BRCA1 following radiation inhibited HR-function in these cells to increase their sensitivity to PARPi. To test this hypothesis, the efficiency of HR-mediated DSB repair was analyzed in MCF7 cells carrying a stably chromosomal integrated, HR-specific reporter system which contains in tandem repeat two differently mutated GFP genes. Expression of I-SceI in these cells generates a DSB within the reporter that, when repaired only by HR, will result in expression of GFP which can be detected and quantified by flowcytometry. Knockdown of p53 by siRNA (siP53), which causes BRCA1 nuclear accumulation and resistance to IR-induced BRCA1 nuclear export (15, 20), resulted in a significant increase in HR repair in MCF-siP53 cells with or without 4 Gy radiation (Figure 3A, Supplemental Figure 3A). Importantly, MCF7-siP53 cells had approximately 2-fold higher HR efficiency than control MCF7-siCtrl cells regardless of IR treatment. This demonstrated that p53 knockdown promotes HR function even after radiation, most likely due to the accumulation of nuclear BRCA1 (15, 20). Given that p53 is typically mutated, rather than deleted, in cancer; as an additional test, we expressed a tumor-associated, transcriptional activity-deficient mutant form of p53 (R273H) which we have previously demonstrated act in a dominant-negative fashion to inhibit nuclear export of BRCA1 (20). Expression of R273H significantly increased HR repair relative to vector-control cells in both cells with or without 4 Gy radiation (Figure 3B, Supplemental Figure 3B) and R237H expressing cells that were treated 4 Gy radiation had equivalent HR activity to unirradiated vector-only cells again demonstrating that p53’s function in BRCA1 nuclear export following DNA damage is important for reduced HR function and increased sensitivity to PARPi.
Figure 3. Reduced HR-mediated DNA repair following radiation requires p53 function.

A and B, HR repair was analyzed in HR-reporter MCF7 cells treated with control siRNA (siCtrl) or siRNA targeting p53 (siP53) (A) or in MCF7 cells transfected by empty vector or vector encoding R273H mutant p53 (B). C and D, the surviving fraction of MCF7 siCtrl and siP53 cells (C) or MCF7 empty vector and R273 H mutant p53 expressing cells (D) was determined by clonogenic survival assay. Cells were first treated with 0 or 4 Gy IR and then treated 24h later with 0, 5, 7.5, or 10 µM ABT-888. Results are the average of three independent experiments.
To determine if these observed changes in p53-mediated HR activity correlated with sensitivity to PARPi we performed in vitro clonogenic survival assays utilizing MCF7 treated with control siRNA or siRNA targeting p53. These cells were first treated with 0 or 4 Gy IR then treated 24h later with 0, 5, 7.5, or 10 μM of ABT-888 (Figure 3C). In agreement with our earlier data, knockdown of p53 partially offset the sensitization to ABT-888 achieved by pretreatment with radiation in these cells. In complementary experiments, expression of dominant-negative R273H mutant-p53 also partially, but significantly (P < 0.05) nullified sensitization to ABT-888 following pretreatment with 4 Gy radiation (Figure 3D).
BRCA1 controls HR-mediated DNA repair and susceptibility to PARPi in a cell line model of malignant glioma
Our studies to date have examined the sensitization of HR-proficient breast cancer cells to the cytotoxic effects of PARPi following IR-mediated cytosolic sequestration of BRCA1. However, this strategy should be applicable to a wide-variety of tumors. Glioblastomas (GBM) are highly aggressive primary brain tumors with poor prognoses and few treatment options. BRCA mutations are rarely reported with malignant gliomas and, while PARPi are currently being evaluated in novel combination therapies for the treatment of GBM and other brain malignancies (28–30), strategies utilizing PARPi have not yet proven effective for treatment of these tumors. To evaluate the role of BRCA1 in HR-mediated repair in malignant glioma cells we analyzed the expression and function of BRCA1 in the GBM cell line U87. As demonstrated in Figure 4A, U87 cells express BRCA1 at a level similar to MCF7 breast cancer cells and western blot analysis of protein lysates from both MCF-7 and U87 show a shift in the molecular weight of BRCA1 following treatment with 4 Gy IR indicative of increased phosphorylation of functional BRCA1 following IR which has been reported previously (15, 31). Next we examined if knockdown of BRCA1 decreased HR-mediated repair in U87 cells. Similar to our findings in the MCF7 breast cancer cell line, silencing of BRCA1 in U87 cells (Figure 4B) decreased Rad51 foci (Figure 4C) and increased persistent γ-H2AX foci (Figure 4D) both indicators of impaired HR-mediated repair of radiation-induced double strand breaks. To determine if deficient HR-mediated DNA repair following silencing of BRCA1 rendered GBM cells more sensitive to PARPi, we compared the surviving fractions of U87 cells treated with either control siRNA or siBRCA1 then treated with 0, 5, 10, or 15 μM ABT-888 (Figure 4E). Knockdown of BRCA1 significantly increased the sensitivity of U87 cells to ABT-888 confirming that U87 cells can be sensitized to the cytotoxic effects of PARPi after inhibition of HR-mediated DNA repair.
Figure 4. BRCA1 controls HR-mediated DNA repair and susceptibility to PARPi in a cell line model of glioblastoma.

A, western blot showing hyper-phosphorylation of BRCA1 in MCF7 breast cancer and U87 GBM cells in response to 4 Gy IR. B, western blot demonstrating BRCA1 expression in U87 cells treated with control or BRCA1-targeting siRNA. C, representative photos illustrating fluorescent immunohistochemical staining of nuclear Rad51 foci (green). Cell nuclei are stained blue with DAPI. Bar graph summarizes the percentage of U87 cells treated with control siRNA (siCtrl, white bars) or siRNA to BRCA1 (siBRCA1, black bars) with Rad51 foci 0 or 4h after treatment with 4 Gy IR. Results are the average of three experiments. D, representative photos illustrating fluorescent immunohistochemical staining of nuclear γ-H2AX foci (green). Cell nuclei are stained blue with DAPI. A bar graph summarizes the number of foci in U87 control siRNA (white bars) or siBRCA1 treated cells (black bars) at 0, 0.5, or 4h after treatment with 4Gy IR. Results are the summary of 3 experiments. E, graph summarizing the surviving fraction of U87 cells treated initially with control or BRCA1 siRNA and then treated 48h later with the indicated concentration of ABT-888. Results summarize duplicate experiments. *, p < 0.05.
IR sensitizes GBM cancer cells to PARPi through cytoplasmic translocation of BRCA1
Having established that loss of BRCA1 sensitizes GBM cells to PARPi we next sought to determine if GBM cells could be sensitized to PARPi following IR-induced cytoplasmic sequestration of BRCA1. To determine if radiation initiates translocation of BRCA1 from the nucleus to the cytoplasm in GBM cells we treated U87 cells with 0 or 4 Gy IR and examined the subcellular localization of BRCA1 by western blot of nuclear and cytoplasmic protein fractions (Figure 5A). Treatment with IR caused a significant translocation of BRCA1 from the nucleus to the cytoplasm 24h post IR treatment. Cytoplasmic translocation of BRCA1 in U87 was confirmed by examining the subcellular localization of immunofluorescent stained BRCA1 following IR treatment (Figure 5B). To determine if IR-mediated cytoplasmic translocation of BRCA1 decreased DNA-repair efficiency and increased PARPi cytotoxicity in U87 GBM cells we assessed persistent DNA damage by the number of γ-H2AX foci in cells pre-treated with 0 or 4 Gy IR and then treated with 10 µM ABT-888 or vehicle 24h post-radiation. Pretreatment with IR significantly increased γ-H2AX foci 24h after ABT-888 treatment relative to cells treated with ABT-888 but no radiation (Figure 5C). To determine more specifically if HR-mediated DNA repair is compromised in U87 cells following IR-induced BRCA1 cytoplasmic sequestration we examined the number of Rad51 foci in cells treated initially with 0 or 4 Gy IR and then treated with 10 µM ABT-888 or vehicle 24h post-radiation. Rad51 foci were tallied 24h after treatment with ABT-888 or vehicle. In cells that were not treated with radiation, ABT-888 significantly increased Rad51 foci indicative of active DNA repair in response to PARPi mediated damage in these cells (Figure 5D). In contrast, in cells pre-treated with radiation, no increase in Rad51 foci is seen following PARPi treatment indicating decreased HR-repair capability. These results suggest that, in U87 GBM cells, pre-treatment with IR results in increased persistent DNA damage after PARPi due to decreased HR-mediated DNA repair capacity. We next analyzed the change in PARPi toxicity in U87 cells following IR treatment in vitro utilizing colony formation assays. U87 cells were first treated with 0 or 4Gy IR and then 24h later treated with 0, 5, 10 or 15 μM PARPi. Pre-treatment of U87 cells with IR significantly reduced the surviving fraction of cells at each concentration of ABT-888 tested (Figure 5E). To test sensitization of U87 cells to PARPi following IR treatment in vivo we first established U87 tumors in the flanks of Nu/Nu mice. Once the tumors reached an average volume of ~100 mm3 the mice were divided into four treatment arms: control animals receiving no treatment, mice receiving a single dose of 4 Gy IR, mice receiving 25 mg/kg ABT-888 daily for 5 consecutive days, or mice receiving a single dose of 4 Gy IR and 25 mg/kg ABT-888 daily for 5 consecutive days starting 24h after radiation treatment. While a single dose of radiation or five daily treatments of ABT-888 alone failed to significantly alter the growth of U87 tumors relative to control tumors, the combination of ABT-888 and radiation significantly reduced the growth of these tumors relative to control tumors or tumors from either single treatment arm (Figure 5F). These results demonstrate that pre-treatment with a sub-optimal dose of IR can sensitize GBM cells to PARPi cytotoxicity in vivo.
Figure 5. IR sensitizes glioblastoma cancer cells to PARPi through cytoplasmic translocation of BRCA1.

A, BRCA1 cytoplasmic translocation is measured by western blot of BRCA1 in the nuclear and cytoplasmic protein fractions of U87 cells treated with 0 or 4 Gy IR. α-tubulin was used as a cytoplasmic protein loading control while lamin A/C served as a loading control for the nuclear fractions. Bar graph summarizes the intensity of the BRCA1 band in the respective fractions from three independent experiments. B, representative images illustrate cytoplasmic (C), nuclear (N), or nuclear and cytoplasmic (NC) fluorescent immunohistochemical staining of BRCA1 (red). Nuclei are stained blue with DAPI. Bar graph illustrates the percentage of cells demonstrating nuclear (N), nuclear and cytoplasmic (NC), or cytoplasmic (C) staining of BRCA1 after treatment with 0 (grey bars) or 4Gy (black/white checkered bars). The result is the average of three experiments. C, representative photos illustrating fluorescent immunohistochemical staining of nuclear-γ-H2AX foci (green). Cell nuclei are stained blue with DAPI. Cells were pre-treated with 0 or 4 Gy IR, then treated 24h later with 0 or 10 µM ABT-888. After an additional 24h, cells were fixed, stained and assayed for γ-H2AX foci. Results are the summary of three experiments. D, representative photos illustrate fluorescent immunohistochemical staining of Rad51 foci (green). U87 cells were pre-treated with 0 or 4 Gy IR, then treated 24h later with 0 or 10 µM ABT-888 after an additional 24h cells were fixed and stained and assayed for Rad51 foci. Bar graph summarizes the percentage of U87 control (white bars) or ABT-888 treated cells (10 µM, black bars) with Rad51 foci after treatment with no radiation or 4h after treatment with 4Gy IR from three independent experiments. E, U87 cells were first treated with 0 (square, solid line) or 4Gy IR (circles, dashed line), 24h later the cells were treated with the indicated concentration of ABT-888 and the surviving cells were assayed by their ability to form colonies. Results summarize three independent experiments. F, U87 tumors were established in nu/nu mice and then subjected to no treatment (control, n = 6), treatment with a single dose of 4Gy IR (IR, n = 4), treatment with 25mg/kg ABT-888 daily for 5 days (ABT-888, n = 5), or a combination of 4 Gy IR followed by 5 daily treatments of 25mg/kg ABT-888 beginning 24h post-radiation (ABT-888 + IR, n = 6). *, p < 0.05.
p53 is essential for IR-induced sensitization of malignant glioma cells to PARPi cytotoxicity
We demonstrated earlier that p53 function is required for BRCA1 cytosolic sequestration and increased PARPi cytotoxicity in MCF7 breast cancer cells. We thus hypothesized that functional p53 is also required for sensitization of malignant glioma cells to PARPi cytotoxicity following IR. Given the frequency of p53 mutations in cancer, it is critical to evaluate p53 as a universal determinate of HR-deficiency following IR in order to precisely define those tumors may most benefit from PARPi following IR. Rather than utilize HPV16-E6 to degrade p53 protein, we utilized siRNA against p53 to transiently manipulate p53 expression in U87 cells for this set of experiments. U87 cells were first treated with siRNA to p53 or a non-targeting control siRNA. Cells were then irritated with 0 or 4 Gy IR 24h later, and after an additional 24h were exposed to 0, 5, 10, 15, or 20 µM ABT-888. The surviving cells were then assessed by their ability to form colonies. In U87 cells treated with non-targeting control siRNA, exposure to 4 Gy IR prior to PARPi significantly sensitized these cells to the cytotoxic effects of PARPi at all doses tested (Figure 6A). In contrast, in U87 cells treated with siRNA to p53, exposure to 4 Gy IR did not significantly sensitize the cells to subsequent treatment with PARPi at any dose of ABT-888 tested (Figure 6B). We confirmed these findings utilizing a second malignant glioma cell line, SF767 which like the U87 line is wild-type for BRCA1 and p53. In these cells p53 expression was stably manipulated by shRNA. In agreement with our findings in U87 cells, SF767 cells expressing a control non-targeting shRNA were significantly sensitized to PARPi following a single dose of 4 Gy IR particularly at the lower doses of ABT-888 tested (Figure 6C). In SF767 cells treated with shRNA to p53, much like our observations in U87 cells treated with siRNA to p53, exposure to 4 Gy IR did not sensitize the cells to subsequent treatment with PARPi at any dose of ABT-888 tested (Figure 6D). These results demonstrate, in two independent glioma cell lines, that p53 expression is required for increased cytotoxicity to PARPi following IR in vitro. To confirm these results in vivo, SF767 shCtrl or shp53 tumors were established in Nu/Nu mice and grown to a volume of ~100 mm3. Tumor bearing mice were then divided into four treatment arms: control mice receiving no treatment, mice receiving a single dose of 4 Gy IR, mice receiving 25 mg/kg ABT-888 daily for 5 consecutive days, or mice receiving a single dose of 4 Gy IR in combination with 25 mg/kg ABT-888 daily for 5 consecutive days starting 24h after radiation treatment. In mice bearing SF767 shCtrl tumors, neither treatment with a single suboptimal dose of IR nor treatment with PARPi alone reduced the relative tumor volume compared to untreated tumors (Figure 6E). However, in SF767 shCtrl tumor bearing mice the combination treatment of IR followed by ABT-888 did significantly reduce the relative tumor volume compared to either control tumors or tumors from either single treatment arm (Figure 6E). In contrast, in mice with SF767 shp53 tumors, IR plus PARPi combination therapy did not result in a significant change in tumor volume relative to control animals or animals in either of the mono-therapy arms (Figure 6F). These results illustrate that p53 is essential for sensitization of cancer cells to PARPi cytotoxicity following IR.
Figure 6. p53 is essential for IR-induced sensitization of malignant glioma cells to PARPi cytotoxicity.

A and B, U87 cells were treated with control siRNA (A) or siRNA targeting p53 (B), 24h later cells were treated with 0 (black circle, solid line) or 4 Gy IR (open circle, dashed line), after an additional 24h cells were plated in media containing the indicated concentration of ABT-888 and assessed for their ability to form colonies. Graphs summarize the results of three independent experiments. C and D, SF767 cells stably expressing control shRNA (C) or shRNA to p53 (D) were treated with 0 (black circle, solid line) or 4 Gy IR (open circle, dashed line), after an additional 24h cells were plated in media containing the indicated concentration of ABT-888 and assessed for their ability to form colonies. Graphs summarize the results of three independent experiments. E and F, SF767 shCtrl (E) or SF767 shp53 (F) tumors were established in nu/nu mice and then subjected to one of the following treatments: no treatment (control, green circle, n = 3), a single treatment of 4Gy IR (IR only, red diamond, n = 3), 5 daily treatments of 25mg/kg ABT-888 (ABT-888, yellow squares, n = 3), or the combination of 4Gy IR followed by 5 daily treatments of ABT-888 (ABT-888 + IR, blue triangles, n = 3). *, p < 0.05.
Discussion
PARPi are an effective, well-tolerated therapy for treatment of cancers with familial BRCA1/2 mutations (2). However, the clinical efficacy of PARPi has been greatly limited by the relative rarity of BRCA1/2 mutations (3, 4). We previously demonstrated a treatment paradigm utilizing IR to trigger cytoplasmic translocation of BRCA1 and inhibition of HR, which subsequently renders breast cancer cells susceptible to PARPi toxicity (8). Here we demonstrate that this treatment scheme may be applicable to a wide range of HR-proficient tumors including malignant gliomas. Importantly, given the fact that p53 is frequently mutated and dysfunctional in solid tumors, we establish that p53 function is essential for IR-induced inhibition of BRCA1 nuclear function in HR repair and subsequent sensitization of cancer cells to the synthetic lethality of PARPi. These results provide a treatment strategy that potentially increases the population of cancer patients for whom PARPi therapy may be efficacious and provides p53 as a putative biomarker to more precisely define the benefiting population.
As a single agent therapy, PARPi efficacy is currently limited to tumors with BRCA1/2 mutations or tumors that exhibit “BRCAness” i.e. phenotypes commonly associated with BRCA1/2-mutant tumors (32, 33). PARPi combinatorial therapies have been developed with the goals of increased cytotoxicity in susceptible tumors and to expand the efficacy of PARPi beyond BRCA1/2-associated cancers (34, 35). In pursuit of these goals, PARPi have been combined with a variety of DNA damaging agents including temozolomide, cisplatin, carboplatin, and topotecan in efforts to determine which combinations best improve tumor cytotoxicity (36–41). While there is evidence, including the results of two recent clinical trials (42,43) that these combinations may provide benefit to patients, particularly those with HR-deficient tumors, identifying the patients that would most benefit from addition of PARPi remains elusive (44). A common limitation of the aforementioned combinations is the utilization of PARPi primary as a sensitizing agent with the hope that decreased single strand break repair will increase the cytotoxicity of DNA damaging agents. The unfortunate short fall of this strategy is that cancer cells with intact HR-repair can efficiently repair the ensuing double strand breaks to circumvent cell death. Recognizing that these strategies fail to tap into the synthetic lethality of PARPi seen in HR-deficient tumors we developed a treatment paradigm to create an artificial HR-deficiency and render increased sensitivity to PARPi. Future studies are warranted to determine if other DNA damage agents such as temozolomide or cisplatin can be combined with our treatment paradigm to further increase the cytotoxic response without unwanted off-target toxicity.
Recent work has expanded the potential use of PARPi through identification of additional mutations in DNA repair pathways such as PTEN, RAD51, and ATM mutations which, like BRCA1/2 mutations, might predict PARPi sensitivity (45–49). The success of this approach was recently highlighted by the finding that mutations in any of 12 DNA repair genes predict response of metastatic castrate-resistant prostate cancer to olaparib (50). While these important studies have succeeded in identifying additional patient populations that are intrinsically sensitive to PARPi toxicity, the benefiting population is still limited to patients harboring a defined set of mutations which compromise HR-mediated DNA repair much like those with BRCA1/2 mutations. Our treatment strategy creates an artificial HR-deficiency, and thus, is not constrained to use in tumors with endogenous DNA-repair defects which should significantly increase the percentage of patients that may benefit. Additional studies are warranted to determine if response to our treatment paradigm is enhanced in tumors that harbor mutations in other DNA-repair genes.
Our previous studies revealed that functional p53 is essential for translocation of BRCA1 to the cytoplasm following IR. Here we expand upon this finding to demonstrate that functional p53 is a critical determinant of sensitization to PARPi following IR. We employ several independent and complementary methods including siRNA, shRNA and E6 mediated p53 knockdown to demonstrate that p53 is required for PARPi sensitization after IR. While p53 is typically mutated rather than lost outright in cancer, we have previously shown that p53 mediates BRCA1 nuclear export via protein–protein binding, rather than by modulation of its transcription. We have also shown that R273H a common p53 dominant-negative mutation demonstrates impaired binding to BRCA1 and is defective in regulating IR-induced BRCA1 cytoplasmic translocation (15, 20). While some p53 mutations, such as H179Q, retain their ability to bind with and export BRCA1 to the cytoplasm (15, 20) the ability of any specific p53 mutant to retain its BRCA1 shuttling function will need to be determined experimentally. It is also possible that p53 mutations that demonstrate impaired p53 binding may play an important role in resistance to PARPi in cancers with wtBRCA1. Future studies will address whether these p53 mutants are enriched in the tumors that grow following our treatment strategy. p53 dysfunction is typically accompanied by increased p53 protein stability and nuclear accumulation and we have demonstrated that p53 nuclear accumulation correlates with BRCA1 nuclear accumulation (15, 20). Future studies will determine if elevated p53 nuclear accumulation can be used as a biomarker screen to identify tumors that are unlikely to benefit from PARPi following IR due to p53 dysfunction.
PARPi were first introduced to the clinic in 2003 and have since been developed as both a single agent therapy and as combination therapy in conjunction with other cytotoxic agents (2, 51). PARPi directly inhibit the catalytic function of PARP1/2 and for some PARPi, such as veliparib/ABT-888, this is the major mode of activity (52). However, other PARPi such as olaparib and niraparib are also capable of trapping PARP upon DNA interfering with DNA replication leading to a second mechanism of cytotoxicity (52). Veliparib was employed in our studies as it has proven to be efficacious in the treatment of BRCA1/2 associated tumors, can cross the blood-brain barrier making it suitable for the treatment of GBM and brain metastases, and it exhibits little PARP trapping ability simplifying the mechanism of action (52, 53). The results presented here should be representative of those that can be achieved with other PARPi that directly inhibit PARP1/2 catalytic function. Additional studies are needed to determine if PARPi with greater PARP-trapping ability are more efficacious in our treatment strategy or result in increased unwanted toxicity in vivo.
Combined, our data demonstrate that IR-induced cytoplasmic sequestration of BRCA1 can be used to sensitize a variety of cancer models to PARPi in vitro and in vivo and reveal that functional p53 is necessary for this sensitization. These results have the potential to significantly increase the potential population of patients that may benefit from PARPi. Future studies will focus on the molecular mechanisms of p53 regulation of BRCA1 nuclear/cytoplasmic shuttling and the translation of these findings to the clinical setting and determine the spectrum of p53 mutations that are defective in BRCA1 shuttling may potentially be used as biomarkers to precisely identify the patient population that is most likely to benefit from this treatment paradigm.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Implications:
Implications: p53 status determines PARP inhibitor sensitization by ionizing radiation in multiple BRCA1 and HR-proficient tumor types and may predict which patients are most likely to benefit from combination therapy.
Acknowledgments
Financial Support: This work was supported by research grants R01 CA188500 (F. Xia and A. Chakravarti) and R01 CA163838 (F. Xia) from the National Institutes of Health and 81XWH-08–1-0571 (F. Xia) from the Department of Defense.
Footnotes
Conflicts of Interest: No author claims a conflict of interest.
References
- 1.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434: 917–21. [DOI] [PubMed] [Google Scholar]
- 2.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol 2014;25: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer 2000;83: 1301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malone KE, Daling JR, Neal C, et al. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer 2000;88: 1393–402. [DOI] [PubMed] [Google Scholar]
- 5.Russo AL, Kwon HC, Burgan WE, et al. In vitro and in vivo radiosensitization of glioblastoma cells by the poly (ADP-ribose) polymerase inhibitor E7016. Clin Cancer Res 2009;15: 607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen RD, Gajjar MK, Jensen KE, Hamerlik P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol oncol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang ES, Xia F. BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J 2010;277: 3079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang ES, Nowsheen S, Rahman MA, Cook RS, Xia F. Targeting BRCA1 localization to augment breast tumor sensitivity to poly(ADP-Ribose) polymerase inhibition. Cancer Res 2012;72: 5547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson LH. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 2012;751: 158–246. [DOI] [PubMed] [Google Scholar]
- 10.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003;22: 5784–91. [DOI] [PubMed] [Google Scholar]
- 11.Xia F, Powell SN. The molecular basis of radiosensitivity and chemosensitivity in the treatment of breast cancer. Semin Radiat Oncol 2002;12: 296–304. [DOI] [PubMed] [Google Scholar]
- 12.Chai KM, Wang CY, Liaw HJ, Fang KM, Yang CS, Tzeng SF. Downregulation of BRCA1-BRCA2-containing complex subunit 3 sensitizes glioma cells to temozolomide. Oncotarget 2014;5: 10901–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 2013;31: 1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada S, Ouchi T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affect its subcellular localization. J Biol Chem 2003;278: 2015–20. [DOI] [PubMed] [Google Scholar]
- 15.Feng Z, Kachnic L, Zhang J, Powell SN, Xia F. DNA damage induces p53-dependent BRCA1 nuclear export. J Biol Chem 2004;279: 28574–84. [DOI] [PubMed] [Google Scholar]
- 16.Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem 2002;277: 21315–24. [DOI] [PubMed] [Google Scholar]
- 17.Shao N, Chai YL, Shyam E, Reddy P, Rao VN. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 1996;13: 1–7. [PubMed] [Google Scholar]
- 18.Fabbro M, Schuechner S, Au WW, Henderson BR. BARD1 regulates BRCA1 apoptotic function by a mechanism involving nuclear retention. Exp Cell Res 2004;298: 661–73. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA damage-induced cytotoxicity is dissociated from BRCA1’s DNA repair function but is dependent on its cytosolic accumulation. Cancer Res 2010;70: 6258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang J, Yang ES, Jiang G, et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res 2011;71: 5546–57. [DOI] [PubMed] [Google Scholar]
- 21.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8: 275–83. [DOI] [PubMed] [Google Scholar]
- 22.Sabbatino F, Fusciello C, Somma D, et al. Effect of p53 activity on the sensitivity of human glioblastoma cells to PARP-1 inhibitor in combination with topoisomerase I inhibitor or radiation. Cytometry A 2014;85: 953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorsey JF, Mintz A, Tian X, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and paclitaxel have cooperative in vivo effects against glioblastoma multiforme cells. Mol Cancer Ther 2009;8: 3285–95. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Wang H, Yang ES, Arteaga CL, Xia F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res 2008;68: 9141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1: 2315–9. [DOI] [PubMed] [Google Scholar]
- 26.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990;63: 1129–36. [DOI] [PubMed] [Google Scholar]
- 27.Masutani M, Nozaki T, Wakabayashi K, Sugimura T. Role of polu(ADP-ribose) polymerase in cell-cycle checkpoint mechanisms following gamma-irradiation. Biochimie 1995;77: 462–5. [DOI] [PubMed] [Google Scholar]
- 28.Robins HI, Zhang P, Gilbert MR, et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: an NRG oncology RTOG group study. J Neurooncol 2016;126: 309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su JM, Thompson P, Adesina A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro Oncol 2014;16: 1661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith MA, Hampton OA, Reynolds CP, et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer 2015;62: 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scully R, Chen J, Ochs RL, et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997;90: 425–35. [DOI] [PubMed] [Google Scholar]
- 32.Sessa C Update on PARP1 inhibitors in ovarian cancer. Ann Oncol 2011;22 Suppl 8: viii72–viii6. [DOI] [PubMed] [Google Scholar]
- 33.De Summa S, Pinto R, Sambiasi D, et al. BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol 2013;24 Suppl 8: viii13–viii21. [DOI] [PubMed] [Google Scholar]
- 34.Palma JP, Rodriguez LE, Bontcheva-Diaz VD, et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res 2008;28: 2625–35. [PubMed] [Google Scholar]
- 35.Lajud SA, Nagda DA, Yamashita T, et al. Dual disruption of DNA repair and telomere maintenance for the treatment of head and neck cancer. Clin Cancer Res 2014;20: 6465–78. [DOI] [PubMed] [Google Scholar]
- 36.Gupta SK, Kizilbash SH, Carlson BL, et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J Natl Cancer Inst 2015;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussain M, Carducci MA, Slovin S, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs 2014;32: 904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awada A, Campone M, Varga A, et al. An open-label, dose-escalation study to evaluate the safety and pharmacokinetics of CEP-9722 (a PARP-1 and PARP-2 inhibitor) in combination with gemcitabine and cisplatin in patients with advanced solid tumors. Anticancer Drugs 2016. [DOI] [PubMed] [Google Scholar]
- 39.Rodler ET, Kurland BF, Griffin M, et al. Phase I Study of Veliparib (ABT-888) Combined with cisplatin and vinorelbine in advanced triple-negative breast cancer and/or BRCA mutation-associated breast cancer. Clin Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karginova O, Siegel MB, Van Swearingen AE, et al. Efficacy of carboplatin alone and in combination with ABT888 in intracranial murine models of BRCA-mutated and BRCA-wild-type triple-negative breast cancer. Mol Cancer Ther 2015;14: 920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kummar S, Chen A, Ji J, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res 2011;71: 5626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramalingam SS, Blais N. Mazieres J, et al. Randomized, placebo-controlled, phase II study of veliparib in combination with carboplatin and paclitaxel for advanced/metastatic non-small cell lung cancer. Clin Cancer Res 2017;23:1937–44. [DOI] [PubMed] [Google Scholar]
- 43.Rugo HS, Olopade OI, DeMichele A, et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N Engl J Med 2016;375:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonnenblick A, de Azambuja E, Azim HA Jr., Piccart M. An update on PARP inhibitors--moving to the adjuvant setting. Nat Rev Clin Oncol 2015;12: 27–41. [DOI] [PubMed] [Google Scholar]
- 45.Majuelos-Melguizo J, Rodriguez MI, Lopez-Jimenez L, et al. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 2015;6: 4790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin F, de Gooijer MC, Roig EM, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res 2014;20: 2703–13. [DOI] [PubMed] [Google Scholar]
- 47.Somyajit K, Mishra A, Jameei A, Nagaraju G. Enhanced non-homologous end joining contributes toward synthetic lethality of pathological RAD51C mutants with poly (ADP-ribose) polymerase. Carcinogenesis 2015;36: 13–24. [DOI] [PubMed] [Google Scholar]
- 48.Gilardini Montani MS, Prodosmo A, Stagni V, et al. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res 2013;32: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williamson CT, Kubota E, Hamill JD, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med 2012;4: 515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and Olaparib in metastatic prostate cancer. N Engl J Med 2015;373: 1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front Oncol 2014;4: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012;72: 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 2007;13: 2728–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.