

Abstract
Local axonal translation of specific mRNA species plays an important role in axon maintenance, plasticity during development and recovery from injury. Recently, disrupted axonal mRNA transport and translation have been linked to neurodegenerative disorders. To identify mRNA species that are actively transported to axons and play an important role in axonal physiology, we mapped the axonal transcriptome of human induced pluripotent stem cell (iPSC)-derived motor neurons using permeable inserts to obtain large amounts of enriched axonal material for RNA isolation and sequencing. Motor neurons from healthy subjects were used to determine differences in gene expression profiles between neuronal somatodendritic and axonal compartments. Our results demonstrate that several transcripts were enriched in either the axon or neuronal bodies. Gene ontology analysis demonstrated enrichment in the axonal compartment for transcripts associated with mitochondrial electron transport, microtubule-based axonal transport and ER-associated protein catabolism. These results suggest that local translation of mRNAs is required to meet the high-energy demand of axons and to support microtubule-based axonal transport. Interestingly, several transcripts related to human genetic disorders associated with axonal degeneration (inherited axonopathies) were identified among the mRNA species enriched in motor axons.
Keywords: Human motor neurons, Axonal transcriptome, Induced pluripotent stem cells, RNA sequencing
1. Introduction
Local translation of proteins from messenger RNAs that are selectively transported from the neuronal cell body to axons and the synaptic terminal (Wang et al., 2010) allows for local modulation of protein synthesis in response to synaptic activation (Sutton and Schuman, 2006). The local translation of proteins in axons seems to also have a role in regulating axon outgrowth and regeneration (Zheng et al., 2001; Taylor et al., 2009), as well as in synapse formation and remodelling (Sutton and Schuman, 2006).
Disrupted axonal RNA metabolism has recently emerged as a new pathomechanism of distinct neuromuscular disorders, including Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis (Fallini et al., 2016; Rotem et al., 2017). Moreover, given the significant disruption in axonal transport observed in several types of axonal Charcot-Marie-Tooth disease (CMT), including those associated with mutations in MFN2 (Baloh et al., 2007; Misko et al., 2010; Misko et al., 2012), NEFL (Saporta et al., 2015; Gentil et al., 2012) and HSPB1 (D’Ydewalle et al., 2011), it is likely that the transport of mRNAs to the distal axons is also impaired in these inherited axonopathies. As a first step towards the goal of identifying mRNA species that may be dysregulated in human axonopathies, we mapped the axonal transcriptome of several healthy induced pluripotent stem cell (iPSC)-derived motor neuron lines. Interestingly, multiple transcripts known to be associated with inherited axonopathies, including CMT and Hereditary Spastic Paraplegia (HSP), were found to be enriched in the axons of human motor neurons, suggesting that their local expression is essential for axonal maintenance. The transcriptome map assembled by this study will be a useful tool to investigate axonal mRNA dysregulation in human neurodegenerative diseases that specifically affect spinal motor neuron axons.
2. Results
2.1. Production of an enriched population of mature motor neurons
Human iPSCs were differentiated into spinal motor neurons using a modified dual SMAD inhibition protocol (Supplementary Fig. 1A). This protocol produces around 21–25% mature motor neurons (ISL1/HB9 positive cells) (Supplementary Fig. 1B), in a heterogeneous cell population. This heterogeneity is a limiting factor, as unbalanced cell populations may create artificial differences between study groups. In order to enrich cell cultures with mature motor neurons, we used L1CAM (CD171) magnetic bead sorting at day 24 of differentiation (Saporta, 2015) (Fig. 1A; Supplementary Fig. 1). A mixed culture of neural progenitor cells (PAX6) and neurons (SMI32) is present in unsorted cell fraction (Supplementary Fig. 1C and D). However, after cell sorting, progenitor cells are removed, and an enriched population of SMI32-positive motor neurons are observed (Supplementary Fig. 1C and D). We also demonstrate by qPCR from two independent spinal motor neuron differentiations, that neuronal markers such as NFM, L1CAM, Synapsin and β-tubulin III are overexpressed in the positive cell fraction (Supplementary Fig. 2A-2D). At the same time, neuroprogenitor markers such as OTX2 and PAX6 (Supplementary Fig. 2E and F) are downregulated compared to unsorted cells. We also show that all mature motor neuron markers (HB9, ISL1 and CHAT) are present and overexpressed in the positive cell fraction compared to the unsorted cell fraction (Supplementary Fig. 2G-2I). After cell sorting (Fig. 1A), the resultant neuronal cultures were enriched for over 59.74% ± 2.86 s.e.m. HB9/SMI32-double positive spinal cord motor neurons (Fig. 1B), and 65.38% ± 2.57 s.e.m. positive for CHAT (Fig. 1C), in differentiations from 3 different cell lines. While these results and our previously published work (Saporta et al., 2015) support the efficient elimination of neural stem cells and iPSCs from the sorted culture, SMI32-negative cells can still be observed after magnetic sorting. GFAP staining have been consistently negative in multiple cell lines and differentiation and sorting rounds, suggesting that glial cells are not found in significant numbers. It is possible that these SMI32-negative cells are either less mature neurons or motor neurons that were outside the focal plane of the images, which are challenging to obtain due to the configuration of the filter inserts.
Fig. 1.
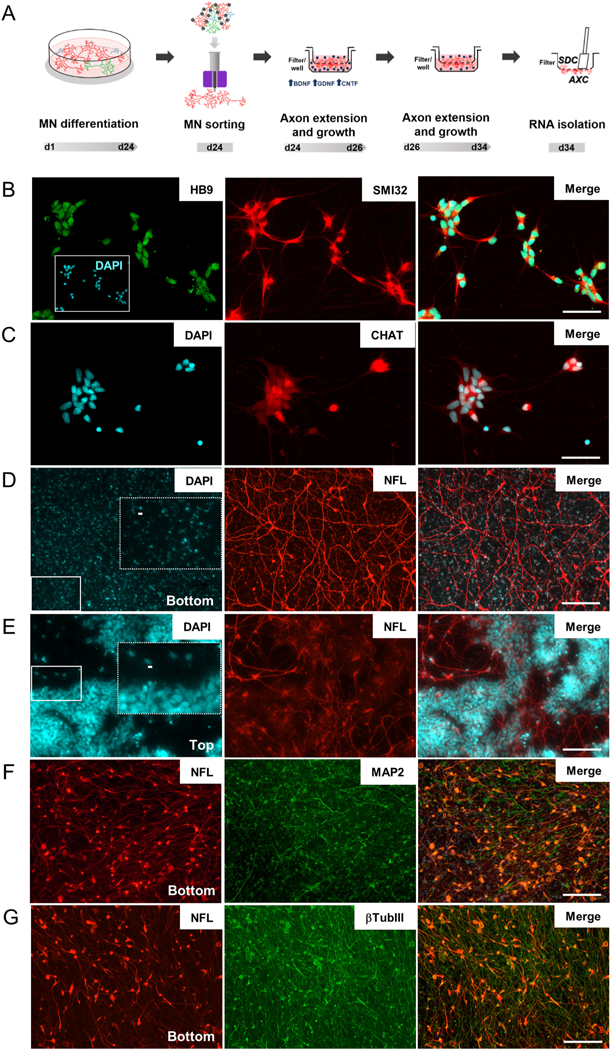
Isolation of axons from iPSC-derived motor neurons. (A) Timeline showing motor neuron differentiation protocol and separation of cell compartments for RNA isolation. Representative images of sorted motor neurons stained for (B) HB9 and SMI32 and (C) CHAT. (D & E) Permeable inserts bearing a 1.0-μm porous membrane were used to obtain large amounts of (D) enriched axonal material from iPSC-derived motor neurons. No neuronal nuclei were identified on DAPI staining at filter-bottom compartment, as opposed to the (E) numerous nuclei observed in the filter top. Dashed rectangles in D and E are zoomed images from fields indicated by solid rectangles showing (D) blue auto fluorescence of filter pores in bottom compartment and (E) nuclei staining in top compartments. Scale bar 5 μm. (F & G) Representative images of bottom compartments showing that neuronal projections (β-TubIII) are mainly composed of axons (NFL) and few dendrites (MAP2). HB9: Homeobox HB9; SMI32: neurofilament heavy chain; CHAT: choline acetyltransferase; NFL: neurofilament light chain; MAP2: microtubule associated protein 2; βTubIII: Tubulin, beta 3 class III. Scale bar 100 μm.
2.2. Isolation of axons from iPSC-derived human motor neurons
Sorted iPSC-derived motor neurons were cultured on inserts containing a 1 μm pore filter (Fig. 1A) in order to isolate the axonal compartment from the somatodendritic compartment (AXC and SDC, respectively) (Unsain et al., 2014; Torre and Steward, 1996; Poon et al., 2006). Motor neuron axons crossed the filter pores populating the bottom part of the membrane (Fig. 1D), while neuronal bodies (and nuclei) were retained in the top part (Fig. 1E). Of note, only a very small fraction of neurites observed in the axonal compartment were MAP2-positive dendrites (Fig. 1F) and known dendritically localized mRNAs were not enriched in the AXC compared to other mRNAs. For example, MAP2, CAMK2A, and DLG4 [PSD95] demonstrated 0.7, 0.11 and 0.1 log2 fold change, respectively (Supplementary Table 1), confirming that this strategy successfully isolated a large number of NFL-and βTubIII-positive axons (Fig. 1G). Axonal RNA was extracted from the bottom membrane, followed by extraction of somatodendritic RNA from the top membrane (Fig. 1A).
2.3. Differential gene expression between axons and neuronal cell bodies from human motor neurons
Whole transcriptome sequencing was performed on ribosomal-depleted RNA from the axonal and somatodendritic compartments of the motor neuron cultures (n = 3 for each compartment). Sequencing was performed in two separate batches. Batch 1 included axonal and somatodendritic RNA from sample 1, while batch 2 included axonal and somatodendritic RNA from samples 2 and 3. Regularized log-transformation was applied to the count data using the DESeq2 R module (Love et al., 2014). Correlation analysis was performed on genes with an average transformed count > 1 (n = 26,902, Fig. 2A); however, of the 26,902 transcripts identified, only 14,129 transcripts had enough data for statistical analysis of differential expression by DESeq2 (Supplementary Table 1). The correlation between technical replicates demonstrates minimal variation across different cell lines and for each compartment. The degree of variation is highest between replicate 1 with replicates 2 and 3, reflecting the batch structure of sequencing runs (Fig. 2A).
Fig. 2.
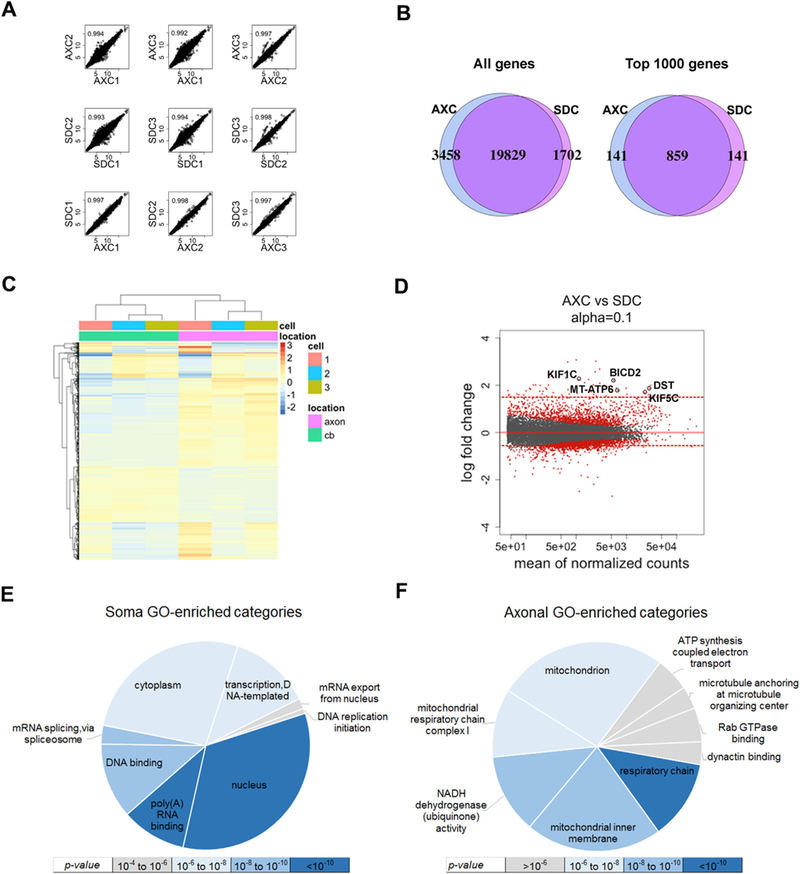
Transcriptome profiling of iPSC-derived motor neuron axonal and somatodendritic compartments (AXC and SDC). (A) Correlation analysis of regularized-log-transformed (rld) read counts for biological replicates (rows 1 and 2) and compartments derived (row 3) from each individual line (n = 3). The unit axes are rld read counts and the plots contain the Pearson correlation coefficient. (B) Venn diagrams displaying the total detectable genes (read count ≥ 1) in all3 biological replicates of each compartment (left), and the 1000 genes with the highest average read count of each compartment (right). (C) Unsupervised clustering of the top 1000 most significantly differentially localized genes, annotated with cell line number and subcellular compartment. The scale bar units are the row-average-normalized rld read counts. (D) MA-plot showing the log2 fold change between compartments (y-axis) versus the gene abundance (x-axis), highlighting significant genes in red (q ≤ 0.1). Red dotted lines represent thresholds fold change used for differentially localized transcripts (≥ 1.5 or fold change ≤ 0.5). Neurological disorders-and axonal transport-related genes are labeled (KIF1C, KIF5C, DST, BICD2 and MT-ATP6). (E & F) Gene Ontology enrichment analysis of differentially localized transcript sets colored by significance: (E) upregulated in soma (log fold-change ≤ 0.5 and q-value ≤ 0.1) and (F) upregulated in axon (log fold-change ≥ 1.5 and q-value ≤ 0.1). BICD2: bicaudal D homolog 2; DST: dystonin; KIF5C: kinesin family member 5C; MT-ATP6: mitochondrially encoded ATP synthase 6; KIF1C: kinesin family member 1C.
Sequence read alignment produced 24,989 detectable genes at which each technical replicate contained at least one non-transformed aligned read (Fig. 2B). Although more than nineteen thousand genes were detected in both compartments, 3458 and 1702 genes are exclusively detected in axonal and somatodendritic compartments, respectively (Fig. 2B, left). A comparison of genes with the highest averaged expression in each compartment (n = 1000) revealed a significant overlap of 859 genes (Fig. 2B, right), consistent with what has been shown in a similar study using mouse neuronal cultures (Briese et al., 2015). Nonetheless, almost 150 transcripts were enriched in either the axonal or the somatodendritic compartment.
To explore the transcriptomic similarities between AXC and SDC, the top 1000 most significantly differentially localized genes (q-value ≤ 0.004, n = 1000) underwent unsupervised hierarchical clustering (Fig. 2C) and revealed that genes from three different lines are grouped by location, despite batch differences. Using thresholds fold change ≥ 1.5 or fold change ≤ 0.5 and q-value ≤ 0.1, we identified 2297 differentially localized transcripts (Fig. 2D). 57 genes were identified as having a > 1.5-fold increase in their levels in the axonal compartment (FC ≥ 1.5).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially localized transcript sets (log fold-change ≥ 1.5 or ≤ 0.5 and q-value ≤ 0.1) on SDC revealed enrichment for pathways associated with nucleus and cytoplasm metabolism (Fig. 2E). Conversely, in the axonal compartment pathways related to microtubule-based axonal transport and mitochondrial metabolism, including oxidative phosphorylation, respiratory chain complex, ATP synthesis and NADH dehydrogenase activity (Fig. 2F), were enriched (Supplementary Table 2).
2.4. Enrichment analysis reveals several transcripts associated with neurological disorders enriched in axons of human motor neurons
The Broad Institute’s Human MitoCarta2.0 inventory of mitochondrial localized proteins was used to test for an enrichment of nuclear-encoded mitochondrial genes within the differentially localized transcripts in AXC (n = 958 genes after removing “possible” and “non-localized” proteins) (Calvo et al., 2016). Furthermore, we used Online Mendelian Inheritance in Man, OMIM database to filter for neurological disease-and/or axonal transport-related genes (Supplementary Table 3). We identified 6 genes related to genetic axonopathies, including BICD2 (bicaudal D homolog 2), DST (dystonin), KIF1C (kinesin family member 1C), KIF5C (kinesin family member 5C), MPZ (myelin protein zero) and MT-ATP6 (mitochondrially encoded ATP synthase 6). In order to validate those results, we performed quantitative real-time PCR analysis using cDNA from both somatodendritic and axonal compartments. Expression level changes between compartments observed in RNAseq experiments (Fig. 3A) were in agreement with relative expression levels detected in qPCR results (Fig. 3B), except for MPZ, which enrichment levels were not detected in two of the three controls and was therefore excluded from the enrichment analysis. Both the mitochondrial and neurological disease-associated genes were significantly enriched within the differentially localized axonal transcript set (Chi-square with Yate’s correction test, p-value ≤ 0.0001; Tables 1 and 2, respectively). MT-ATP6 gene is both mitochondria-and neurological disorder-associated gene appearing in the two tables. The read counts for all five neurological-disease-associated genes (Fig. 2D) (normalized by sequencing depth) show the consistent differential expression across compartments (Fig. 3A). Furthermore, we demonstrate that neurological disorders-associated transcripts (mitochondrial and axonal transport-related genes) had significantly higher levels in axons from human motor neurons when compared to motor-neuronal bodies at average Log2 fold-increases between 5.2 and 15.7 (Fig. 3C). To characterize the subcellular localization of axonal enriched mRNA we performed fluorescent in situ hybridization (FISH) in sorted motor neurons. Our results confirmed that KIF5C mRNA is localized in axons as well as in neuronal cell body and nucleus (Fig. 3E and F).
Fig. 3.
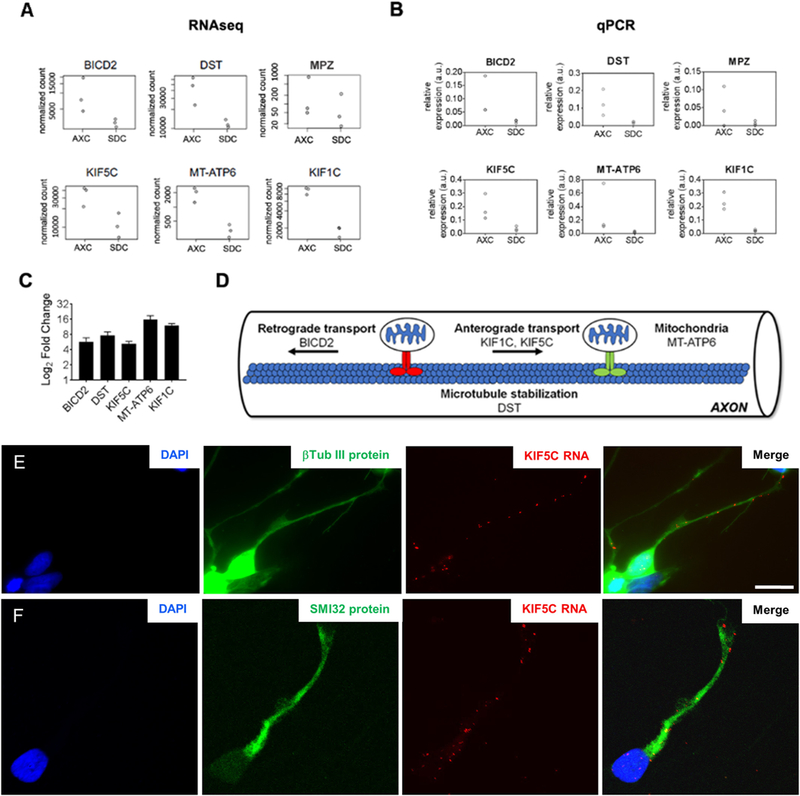
Differential localization of inherited axonopathies genes in axonal compartment. (A) Read counts, normalized by sequencing depth, of differentially localized transcripts from known mitochondria and inherited axonopathies-related genes. (B) Validation of RNAseq transcripts level changes between somatodendritic (SDC) and axonal compartment (AXC) by quantitative PCR (qPCR). (C) Mitochondria and inherited axonopathies-related genes are enriched in AXC of iPSC-derived motor neuron from 3 control lines, as shown by fold change graph. β-actin was used as internal control. Error bars are mean with SEM from at least two independent experiments. (D) Chart showing upregulated genes that are involved in microtubule-based axonal transport. (E &F) Subcellular localization of mRNA KIF5C by in situ hybridization showing mRNA KIF5C is present along the (E) β-Tubulin III positive neurites (green) by fluorescence microscopy and (F) SMI32 (NFH) positive axons by confocal microscopy in human motor neurons. Scale bar: 20 μm.
Table 1.
Mitochondrial genes overexpressed in axons.
| Overexpresseda | Not overexpressed | Total | |
|---|---|---|---|
| Mitochondrial gene | 12 | 947 | 959 |
| Non mitochondrial gene | 45 | 25,898 | 25,943 |
| Total | 57 | 26,845 | 26,902 |
| Chi-square (Yates correction) | 45.846 | ||
| P-value | <0.0001 | ||
| Odds ratio | 7.29 | ||
Fold change ≥ 1.5.
Table 2.
Neurological disease genes overexpressed in axons.
| Overexpresseda | Not overexpressed | Total | |
|---|---|---|---|
| Neurological disease genes | 5 | 162 | 167 |
| Non neurological disease genes | 52 | 26,683 | 26,735 |
| Total | 57 | 26,845 | 26,902 |
| Chi-square (Yates correction) | 65.519 | ||
| P-value | <0.0001 | ||
| Odds ratio | 15.84 | ||
Fold change ≥ 1.5.
3. Discussion
Uncovering disease pathways and developing treatments for neurodegenerative disorders have been hindered by the limited access to affected cell types in humans. The development of human cellular reprogramming in 2007 (Takahashi et al., 2007) allowed for the first time the use of human, patient-specific, induced pluripotent stem cell (iPSC)-derived neuronal cells to model neurological disorders. iPSCs-derived motor neurons are especially relevant for investigating disease mechanisms in neuromuscular disorders such as Amyotrophic Lateral Sclerosis (ALS), Spinal Muscular Atrophy (SMA), Charcot-Marie-Tooth disease and Hereditary Spastic Paraparesis, where motor neurons are the primarily affected cell type. Their use in disease model and drug screening efforts are now well established.
Active axonal mRNA transport and local translation of proteins at the distal axons have been recognized as fundamental for a variety of important neuronal functions, including axon maintenance and guidance, synaptogenesis, post and pre-synaptic plasticity and response to injury (Holt and Schuman, 2013; Lyles et al., 2006; Younts et al., 2016; Donnelly et al., 2010). On the other hand, disturbed axonal transport of mRNA and impaired local expression of proteins at the distal axons have been increasingly identified as an important disease mechanism in motor neuron disorders, including ALS (Perlson et al., 2009; Bilsland et al., 2010; De Vos et al., 2008), and SMA (Fallini et al., 2016). Therefore, understanding the role of axonal RNA trafficking and expression may reveal important pathways for axonal integrity and new therapeutic targets for diseases that preferentially affect axons (axonopathies).
Two recent studies investigated the axonal transcriptomes of rat embryonic motor neurons and human stem cell-derived cortical neurons. Rotem and colleagues (Rotem et al., 2017) identified specific transcriptome signatures in the axons of embryonic rat motor neurons using a similar system employed in our study. In keeping with our results, they demonstrated that approximately 12% of total mRNA species are enriched in the axons of rat embryonic motor neurons. These transcripts were enriched for functional groups related to processes such as “local translation”, the “cytoskeleton” and “metabolic” processes, confirming our findings and suggesting significant homology between human iPSC-derived motor neurons and embryonic rat motor neuron axonal transcriptomes. Interestingly, Rotem et al. identified three potential motor neuron-specific axonal targeting motifs found along the length of the differentially localized axonal mRNAs, providing a possible regulatory mechanism for axonal mRNA transport. Specific changes to the subcellular expression of RNAs in both the neuronal soma and axons of embryonic rat motor neurons induced by SOD1 and TDP43 mutations associated with familial Amyotrophic Lateral Sclerosis were identified, suggesting that axonal RNA dysregulation is a feature of SOD1-and TDP43-related familial ALS (Rotem et al., 2017).
In a similar study, Bigler et al. investigated the axonal transcriptome of glutamatergic cortical neurons, using compartmentalized microfluidic chamber devices to allow for effective separation of the somatodendritic and axonal compartments. They compared the axonal transcriptomes of human embryonic stem cell (ESC)-derived glutamatergic cortical neurons and rat cortical neurons and found significantly overlap between species, again suggesting homology between the axonal transcriptome of human and rat neurons and highlighting the evolutionary importance of axonal RNA expression (Bigler et al., 2017). Interestingly, the functional groups enriched in the axonal compartment of cortical glutamatergic neurons was slightly different from the one characterized for cholinergic motor neurons by our study and Rotem et al., with “secreted” and “extracellular” proteins, “DNA sequence-specific binding” proteins, “neurofilament” proteins and “voltage-gated” and “cation channels” being the groups found to be enriched in cortical neuron axons, which may be attributed to the specific requirements of highly complex and specialized synapses and neuronal networks characteristic of these neuronal subtype, compared to spinal cord motor neurons. Of note, even using a system that should be able to better isolate somatodendritic from axonal compartments, Bigler et al. still found significant overlap between the two transcriptomes, with over 80% of transcripts showing similar expression levels between compartments. Considering RNA sequencing’s high sensitivity, it is possible that mRNA species present even in very small amounts in the proximal axons and shared with the neuronal body could have “contaminated” the axonal compartment and limited the differential expression analysis. Nonetheless, several transcripts have been identified as enriched in axons of different neuronal subtypes and may provide important clues to specific cellular functions and pathways related to human disease.
To explore this potential use of axonal transcriptome mapping, we used the Online Mendelian Inheritance in Man (OMIM) database to identify, within our axonal RNA transcriptome, species that are associated with neurological diseases, specifically those presenting with axonal degeneration as their primary pathological feature. Interestingly, we determined that the axonal transcriptome of human motor neurons is not only enriched for nuclear-encoded mitochondrial genes, but also for mRNA species from genes associated with two inherited axonopathies, Charcot-Marie-Tooth disease and Hereditary Spastic Paraplegia. These include the genes BICD2, DST, KIF1C, KIF5C and MT-ATP6. Most of these genes have a direct function in microtubule-based axonal transport (Fig. 3D). BICD2 is an activating cargo adaptor that facilitates cargo trafficking in axons (Hoang et al., 2017), playing a key role in motor neuron development and maintenance (Oates et al., 2013). Autosomal dominant mutations in BICD2 have been implicated in spinal muscular atrophy lower extremity-predominant 2 (SMALED 2, OMIM 615290), and with overlapping clinical presentations with ALS, CMT and HSP (Drew et al., 2015; Lipka et al., 2013). DST encodes dystonin, a protein that bridges cytoskeletal filament networks, providing microtubule stabilization. Homozygous truncating mutations in DST have been associated with a severe complex inherited neuropathy (OMIM 614653). KIF1C and KIF5C are members of the kinesin family of proteins (KIFs), which are microtubule-based motor proteins. KIF1C interacts with BICDL1 to promote microtubule minus-end retrograde transport of secretory vesicles. Knockdown of Kif1C in mouse neurons reduced secretory vesicle number and neurite outgrowth (Schlager et al., 2010). Homozygous mutations in KIF1C have been identified in patients with autosomal recessive spastic-ataxia 2 (Dor et al., 2014; Novarino et al., 2014) and hereditary spastic paraplegia (Caballero Oteyza et al., 2014). KIF5C, on the other hand, has not been directly associated with inherited axonopathies, but with autosomal dominant complex cortical dysplasia with other brain malformations 2 (OMIM 615282), a central nervous system disorder. However, expression and knockout studies in mice revealed that KIF5C was also highly expressed in lower motor neurons in 2-week-old or older mice. Kif5C-null mice had relative loss of ventral spinal cord motor neurons when compared to sensory neurons (Kanai et al., 2000), suggesting an important role of KIF5C in the maintenance of motor neurons and axonal elongation. Our RNA FISH results demonstrating the presence of KIF5C mRNA in the soma and axon of human motor neurons provide further evidence to support this role. Taken together, the enrichment of transcripts from these genes in the axonal compartment seems to underscore the importance and dynamic requirements of local translation of microtubule-based transport-related proteins in axon maintenance.
In summary, we mapped for the first time the human motor neuron axonal transcriptome and identified several transcripts linked to human genetic axonopathies as enriched in axons of motor neurons. Using this same approach to investigate the axonal transcriptome of CMT and HSP motor neurons may help identify global or specific axonal mRNA dysregulation which may, in turn, reveal pathways associated with axonal degeneration in this group of genetic axonopathies. Lastly, our findings support the notion of local axonal protein translation as a key mechanism to meet the high metabolic and axonal transport demands of long motor axons.
4. Materials and methods
4.1. Spinal cord motor neuron differentiation
Human induced pluripotent stem cells (iPSC) from three healthy individuals were differentiated into spinal motor neurons using a dual SMAD inhibition modified protocol (Supplementary Fig. 1A), as described previously (Saporta et al., 2015). Briefly, iPSC were cultivated in Essential medium 8, on 100 mm plate coated with Geltrex until 90–95% confluency. On day 1 of differentiation, medium was changed to KSR medium (DMEM/F12 GlutaMAX, 15% Serum replacement medium, 1% NEAA, 110 μM 2-Mercaptoethanol, 1% Penicillin/Streptomycin). On day 4 medium was gradually changed to 70:30 KSR medium:N2Base medium (DMEM/F12 GlutaMAX, 1× N-2 Supplement, 0.16% D-Glucose, 0.2 mM L-Ascorbic Acid, 1% Penicillin/Streptomycin). On days 5 and 6 media were changed to 50:50 KSR medium:N2Base medium. From day 1 to day 6, media were supplemented with 1 μM dorsomorphine and 10 μM SB431542. On days 7 and 8 media were changed to 50:50 KSR medium:Maturation medium (DMEM/F12 GlutaMAX, 2× N-2 Supplement, 2× B-27 Supplement, 0.32% D-Glucose, 0.8 mM L-Ascorbic Acid, 1% Penicillin/Streptomycin), supplemented with 1 μM dorsomorphine, 10 μM SB431542, 1.5 μM retinoic acid and 200 nM SAG, 2 ng/mL BDNF, 2 ng/mL GDNF, 2 ng/mL CNTF. From day 9 to day 24 only Maturation medium with all supplements was used.
4.2. Magnetic sorting
On day 24, iPSC-differentiated cultures were dissociated and mature motor neurons were isolated using magnetic bead sorting based on L1CAM, as previously described (Saporta et al., 2015). Briefly, on day 24 of differentiation neurons were dissociated with Trypsin-EDTA 0.05% solution (Sigma) and counted. A portion of cells, called unsorted cell fraction, were separated and plated for further immunostaining characterization. The remaining neuronal culture was incubated for 1 h with anti-CD171 PE (L1CAM) antibody (Thermo Scientific) as primary antibody. Cells were centrifuged, washed once using Sorting Buffer (6% Fetal Bovine serum in 1× DPBS) and incubated with anti-PE microbeads (Miltenyi Biotec) for 15 min. After another centrifugation and washing step, cells were ressuspended in Sorting Buffer and applied onto prepared LS Column placed into MidiMACS separator (Miltenyi Biotec), following manufacturer’s instructions. The column was washed 3 times with 3 mL of Sorting Buffer, removed from the MidiMACS separator and placed in a separated conical tube. The positive cell fraction containing the purified motor neurons was eluted with 5 mL of Sorting Buffer by firmly pushing the plunger into the column. Neuronal cultures were maintained in motor neuron maturation medium until used in end-point experiments.
4.3. Axonal and somatodendritic mRNA isolation using filters
Immediately after sorting, motor neurons were counted and 2 million cells were plated on poly-L-ornithine/laminin-coated permeable inserts bearing a 1.0-μm porous membrane (filter) to obtain large amounts of enriched axonal material for RNA isolation (Unsain et al., 2014). To guide the axonal migration through the filter and into the axonal compartment (bottom) we used a gradient of growth factors, with 10 times higher concentration (20ng/mL) of GDNF, BDNF and CNTF supplemented on bottom compartment for two consecutive days. Axons grew for 10 days before RNA isolation. Axonal and somatodendritic RNA were isolated using the RNeasy Mini Kit or RNeasy Micro Kit (Qiagen). Filters were washed once with PBS and neuronal bodies were scraped using 200 uL of RNeasy buffer added to the top of the filter. To avoid any contamination to the axonal compartment, the top side of the filter was cleaned using cotton-tip before scrapping the axonal material. Then, the filter was removed from the plate and placed upside down and axonal material were collected using 100 uL of RNeasy Micro Kit buffer and a cell scraper. RNA integrity and quantity were measured on Agilent 2100 Bioanalyzer using the RNA 6000 Nano Assay (Agilent), and quantified using the RNA Quant-It assay for the Qubit Fluorometer (Invitrogen).
4.4. Immunostaining
Motor neuron were plated on top of permeable inserts as described above. Axons were allowed to grow for 10 days before immunostaining. Medium was removed, and inserts washed once in PBS before fixating the cells in 3.7% formaldehyde for 30 min. Top and bottom compartments were scrapped and cleaned with a cotton tip for the bottom and top filter immunostaining, respectively. Immunostaining was performed as described previously (Saporta et al., 2015). Primary antibodies used: anti-NFL (abcam), anti-βTubIII (Sigma), anti-HB9, anti-MAP2 (Thermo Scientific), anti-CHAT (Millipore), anti-SMI32 (NFH) (BioLegend). Alexa Fluor 488 and Alexa Fluor 594 (Jackson ImmunoResearch) were used as secondary antibodies. All nuclei were stained with DAPI. After immunostaining, filters were cut from inserts, placed on microscopy slides, and covered with a coverslip and mounting media.
4.5. RNA sequencing and data processing
Whole transcriptome sequencing was performed at the Sequencing Core of the John P. Hussman Institute of Human Genomics at the University of Miami. Following standard Illumina protocols, 80 to 100 ng of total RNA via Agilent Bioanalyzer were ribo-depleted using Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) (Illumina) and subsequent library construction was performed using the TruSeq Stranded Total RNA Library Prep Kit. Libraries were single-end sequenced on Illumina’s HiSeq2000 sequencer to produce 75 base pair reads. Quality control metrics for the raw reads were assessed using FastQC (Andrews, 2010). Total reads per sample showed low variance, ranging from 39.8–42.9 million reads. Reads were aligned to Ensembl’s human GRCh37.75 genome using STAR. HTSeq was used for gene quantification of the aligned reads against the GENCODE reference gtf file for the human GRCh37.75 genome (Anders et al., 2015).
4.6. RNA-seq analysis
Gene-level differential expression analysis was performed using DESeq2 (Love et al., 2014). Differentially localized transcripts were determined by an adjusted p-value (q-value) threshold of 0.1 and log fold change greater than or equal to 1.5 or less than or equal to 0.5. The DAVID Functional Annotation Tool was used for enrichment analysis of the differentially localized transcript sets (Huang et al., 2009b; Huang et al., 2009a). The intersection of known CMT disease genes and mitochondrial genes with the differentially localized transcripts was analyzed by Chi-square with Yate’s correction test.
4.7. Quantitative real-time PCR
qPCR was used to validate the expression of mitochondrial and CMT/HSP genes found to be enriched in axonal compartment. cDNA from axonal and somatodendritic compartments from three control lines were used in TaqMan Assays. Amplification was performed as duplex reactions using TaqMan Gene Expression Assays-FAM for target genes and β-actin-VIC as endogenous control. The cDNA was diluted 1:5 with water and used in quantitative real-time PCR with TaqMan Fast Advanced Master Mix (Thermo Scientific) on CFX96 Real-Time PCR Detection System (BIORAD). For each sample, the reaction was run in triplicates and expression levels of target genes were normalized by β-actin expression levels, within each sample.
4.8. RNA Fluorescence In Situ Hybridization (FISH)
RNA fluorescence in situ hybridization was performed using RNAscope probes following manufacturer’s procedures (Advanced Cell Diagnostics). Briefly, sorted motor neurons were plated on coverslips and axons were allowed to grow for 5 days before fixating with 3.7% formaldehyde for 30 min at room temperature. Cells were dehydrated with ethanol. After rehydration, neurons were permeabilized with PBS/ T 0.1% (PBS/Tween 20) for 10 min at RT. Protein digestion was performed using RNAscope Protease III at 1:3 dilution for 10 min at RT. Specific probe detecting KIF5C was incubated following the manufacturer’s instructions (RNAscope Fluorescent Multiplex Assay for Adherent Cells). All incubations were performed at 40 °C using the HybEZ hybridization oven. Hybridization with KIF5C mRNA probe was followed by immunofluorescent staining using anti-SMI32 (BioLegend) or anti-βTubIII (Sigma) antibodies. Images were acquired using EVOS Fluo equipped with a 100× oil objective, 1.28 Numerical Aperture and Olympus FV1000 Confocal equipped with a 60×/1.42 0il/0.16/ FN26.5 objective.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This work was supported by funding from the Department of Neurology and the Department of Human Genetics, University of MiamiMiller School of Medicine.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2018.06.008.
References
- Anders S, Pyl PT, Huber W, 2015. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31 (2), 166–169. Available at: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btu638 [Accessed October 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S, 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Baloh RH, et al. , 2007. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-tooth disease from Mitofusin 2 mutations. Journal of Neuroscience 27 (2), 422–430. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17215403 [Accessed August 4, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler RL, et al. , 2017. Messenger RNAs localized to distal projections of human stem cell derived neurons. Scientific Reports 7 (1), 611 Available at: http://www.nature.com/articles/s41598-017-00676-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, et al. , 2010. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. 107 (47), 20523–20528. Available at: 10.1073/pnas.1006869107 [Accessed August 30, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese M, et al. , 2015. Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res. 44 (4), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero Oteyza A, et al. , 2014. Motor protein mutations cause a new form of hereditary spastic paraplegia. Neurology 82 (22), 2007–2016. Available at: 10.1212/WNL.0000000000000479 [Accessed November 16, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK, 2016. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44 (D1), D1251–D1257. Available at: 10.1093/nar/gkv1003 [Accessed October 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ et al. , 2008. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci, 31(1), pp.151–173. Available at: 10.1146/annurev.neuro.31.061307.090711 [Accessed August 30, 2017]. [DOI] [PubMed] [Google Scholar]
- Donnelly CJ, Fainzilber M, Twiss JL, 2010. Subcellular communication through RNA transport and localized protein synthesis. Traffic 11 (12), 1498–1505. Available at: 10.1111/j.1600-0854.2010.01118.x [Accessed August 30, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor T, et al. , 2014. KIF1C mutations in two families with hereditary spastic paraparesis and cerebellar dysfunction. Journal of Medical Genetics 51 (2), 137–142. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24319291 [Accessed November 16, 2017]. [DOI] [PubMed] [Google Scholar]
- Drew AP, et al. , 2015. Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing. Molecular Genetics & Genomic Medicine 3 (2), 143–154. Available at: 10.1002/mgg3.126 [Accessed October 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ydewalle C, et al. , 2011. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1–induced Charcot-Marie-tooth disease. Nature Medicine 17 (8), 968–974. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21785432 [Accessed August 4, 2017]. [DOI] [PubMed] [Google Scholar]
- Fallini C, et al. , 2016. Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J. Neurosci. 36 (13), 3811–3820. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27030765%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4812137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil BJ, et al. , 2012. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-tooth disease. The FASEB Journal 26 (3), 1194–1203. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22155564 [Accessed August 4, 2017]. [DOI] [PubMed] [Google Scholar]
- Hoang HT, et al. , 2017. DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc. Natl. Acad. Sci. U. S. A. 114 (9), E1597–E1606. Available at: doi/10.1073/pnas.1620141114 [Accessed October 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Schuman EM, 2013. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80 (3), 648–657. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0896627313009884 [Accessed August 30, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA, 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1–13. Available at: doi/ 10.1093/nar/gkn923 [Accessed October 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA, 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4 (1), 44–57. Available at: 10.1038/nprot.2008.211 [Accessed October 19, 2017]. [DOI] [PubMed] [Google Scholar]
- Kanai Y, et al. , 2000. KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20 (17), 6374–6384. Available at. http://www.ncbi.nlm.nih.gov/pubmed/10964943 [Accessed November 16, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka J, et al. , 2013. Mutations in cytoplasmic dynein and its regulators cause malformations of cortical development and neurodegenerative diseases. Biochem. Soc. Trans. 41 (6), 1605–1612. Available at: 10.1042/BST20130188 [Accessed October 19, 2017]. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15 (12), 550 Available at: 10.1186/s13059-014-0550-8 (Accessed October 19, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles V, Zhao Y, Martin KC, 2006. Synapse formation and mRNA localization in cultured Aplysia neurons. Neuron 49 (3), 349–356. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0896627306000390 (Accessed August 30, 2017). [DOI] [PubMed] [Google Scholar]
- Misko A, et al. , 2010. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30 (12), 4232–4240. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20335458 Accessed August 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko AL, et al. , 2012. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32 (12), 4145–4155. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22442078 Accessed August 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, et al. , 2014. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343 (6170), 506–511. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24482476 [Accessed November 16, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates EC, et al. , 2013. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am. J. Hum. Genet. 92 (6), 965–973. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0002929713001791 Accessed October 19, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, et al. , 2009. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J. Neurosci. 29 (31), 9903–9917. Available at: 10.1523/JNEUROSCI.0813-09.2009 Accessed August 30, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon MM, et al. , 2006. Identification of process-localized mRNAs from cultured rodent hippocampal neurons. J. Neurosci. 26 (51), 13390–13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem N, et al. , 2017. ALS along the axons – expression of coding and noncoding RNA differs in axons of ALS models. Sci. Rep. 7, 44500 Available at: http://www.nature.com/articles/srep44500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta M, 2015. Cellular reprogramming and inherited peripheral neuropathies: perspectives and challenges. Neural Regeneration Research 10 (6), 894 Available at: http://www.nrronline.org/text.asp?2015/10/6/894/158345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, et al. , 2015. Axonal Charcot-Marie-tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electro-physiological properties. Exp. Neurol. 263, 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager MA, et al. , 2010. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. The EMBO Journal 29 (10), 1637–1651. Available. at. http://www.ncbi.nlm.nih.gov/pubmed/20360680 Accessed November 16, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM, 2006. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127 (1), 49–58. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17018276 Accessed August 4, 2017. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. , 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–872. [DOI] [PubMed] [Google Scholar]
- Taylor AM, et al. , 2009. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29 (15), 4697–4707. Available at: 10.1523/JNEUR0SCI.6130-08.2009 Accessed August 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre ER, Steward O, 1996. Protein synthesis within dendrites: glycosylation of newly synthesized proteins in dendrites of hippocampal neurons in culture. J. Neurosci. 16 (19), 5967–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsain N, et al. , 2014. Production and isolation of axons from sensory neurons for biochemical analysis using porous filters video link. J. Vis. Exp. 8910 (517953791–51795). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS, 2010. Spatially restricting gene expression by local translation at synapses. Trends in Neurosci. 33 (4), 173–182. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20303187 Accessed August 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younts TJ, et al. , 2016. Presynaptic protein synthesis is required for long-term plasticity of GABA release. Neuron 92 (2), 479–492. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0896627316306419 (Accessed August 30, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, et al. , 2001. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci. 21 (23), 9291–9303. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11717363 Accessed August 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.