

Abstract
Purpose
The purpose of this study was to determine if autoantibodies play a role in the immunopathogenesis of experimental dry eye disease.
Methods
Dry eye was induced by exposing female C57BL/6 wild-type mice or hen egg lysozyme B-cell receptor transgenic mice to desiccating stress (subcutaneous scopolamine [0.5 mg/0.2 mL] 3 times a day, humidity < 40%, and sustained airflow) for 3 weeks, allowing sufficient time for a humoral immune response. Serum or purified IgG isolated from dry-eye mice or untreated controls was passively transferred to nude recipient mice, which were evaluated for ocular surface inflammation 3 days after transfer. To determine if complement activation contributed to serum-induced dry eye disease, cobra venom factor was used to deplete complement activity.
Results
Autoantibodies against kallikrein 13 were identified in serum from dry-eye mice, but were undetectable in untreated controls. Autoantibody-containing serum or purified IgG from dry-eye mice was sufficient to mediate complement-dependent ocular surface inflammation. Serum or purified IgG caused marked inflammatory burden and tissue damage within the ocular surface tissues, including elevated Gr1+ neutrophil infiltration and proinflammatory cytokines/chemokines associated with goblet cell loss. Moreover, complement C3b deposition was found within the ocular surface tissues of mice receiving dry-eye serum, but not in recipients of control serum. Functionally, complement depletion attenuated the ability to transfer dry-eye–specific serum or IgG-mediated disease.
Conclusions
These data demonstrate for the first time a complement-dependent pathogenic role of dry-eye–specific autoantibodies, and suggest autoantibody deposition within the ocular surface tissues contributes to the predominantly T-cell–mediated immunopathogenesis of dry eye disease.
The authors demonstrate that B–cell–secreted autoantibodies play a role in the pathophysiology of autoimmune-based dry eye disease.
Introduction
Dry eye is a prevalent ocular surface disease with significant morbidity that regularly affects quality of life.1,2 Patients suffer from variety of symptoms, including ocular discomfort, fatigue, and chronic pain, accompanied by blurred and fluctuating vision that may persist over decades. In the most severe cases, recurrent corneal ulceration can culminate in reduced vision or blindness. Several lines of evidence strongly suggest that the signs and symptoms of dry eye disease are the consequence of chronic autoimmune-based inflammation within the lacrimal function unit (LFU: cornea, conjunctiva, lacrimal glands, and meibomian glands).3 Inflammatory cell infiltration within the LFU is associated with elevated proinflammatory cytokine levels, decreased tear production, increased epithelial cell apoptosis, and goblet cell loss in both animal models and patients with dry eye disease.4–7
For many years, the spotlight has been focused on the contribution of pathogenic CD4+ T cells on the immunopathogenesis of dry eye. Environmental and/or microbial stress, in combination with genetically predisposed factors, is hypothesized to trigger the immune response in a way that breaches the protective immunoregulatory mechanisms, leading to autoimmunity to self-antigens localized to the LFU.3 This paradigm suggests that activation of the innate immune response initiates a sequence that allows interaction between activated ocular surface-derived antigen presenting cells (APCs) bearing self-antigen and autoreactive CD4+ T cells, resulting in clonal expansion of LFU-specific lymphocytes. Indeed, activated CD4+ T cells are localized within the ocular surface tissues of patients with dry eye,5,8,9 and compounds that inhibit T cells are therapeutic in both animals and humans with dry eye disease.5,10 Moreover, dry eye disease can be adoptively transferred to nude recipient mice by CD4+ T cells isolated from the regional lymph nodes of mice with experimental dry eye disease induced by exposure to desiccating stress (DS).4 By contrast, local depletion of APCs in the conjunctiva of mice exposed to DS inhibited the generation of autoreactive CD4+ T cells and blocked the ability to adoptively transfer disease to T-cell–deficient nude recipient mice.11 In addition to T helper 1 (Th1) cells, Th17 cells are pathogenic during experimental dry eye and have also been implicated in human disease.6,12–14 Collectively, these data support the paradigm that dry eye is a self-antigen–driven autoimmune disease, and implies that other autoreactive lymphocytes (i.e., B cells) may be involved in disease.
B cells are also known to play a pathogenic role in several autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, and Sjögren's syndrome. B cells can contribute to disease by functioning as (1) APCs15–18; (2) cytokine-secreting cells (e.g., IL-2, IL-12, TNF-α, IFN-γ), which modulate T cells and/or exert direct pathogenic effects19; or (3) by differentiating into autoantibody-secreting plasma cells,20 which damage target tissues by recruiting inflammatory cells via Fcγ receptor signaling and/or by complement activation.21 The presence of autoantibodies in sera from patients with Sjögren's syndrome–mediated dry eye (Ro 52 and 60 kDa, La 48 kDa, alpha fodrin)22,23 provided the first line of evidence of a B-cell component in disease. In addition, work from the laboratory of the late Dr. Michael Humphreys-Beher and others established a link between autoantibodies to the type 3 muscarinic acetylcholine receptor (anti-M3R Ab) and the secretory response during the immunopathogenesis of Sjögren's syndrome; anti-M3R Abs are present in sera of patients and animal models,23–28 and passive transfer of IgG from patients with Sjögren's syndrome or rodent anti-M3R Abs are sufficient to induce exocrine dysfunction in recipient animals.29,30 More recently, autoantibodies to members of the kallikrein family of proteins (e.g., Klk1, Klk13) were identified in the serum of mice with Sjögren's-like disease31; however, the functional role of autoantibodies has not been addressed in the context of non-Sjögren's dry eye.
In this article, we evaluated the putative role of autoantibodies during the immunopathogenesis of experimental dry eye. Here we show for the first time that passive transfer of autoantibody-containing serum or purified IgG derived from mice with experimental dry eye mediates complement-dependent inflammation and tissue damage within the LFU of T-cell–deficient nude recipient mice. By contrast, serum from hen egg lysozyme B-cell receptor transgenic (HEL BCR Tg) mice exposed to DS was unable to induce disease. These results indicate that dry-eye–specific autoantibodies are sufficient to induce ocular surface inflammation in the context of the nude recipient mouse, and suggest that dry-eye–specific autoantibodies contribute to CD4+ T-cell–mediated immunopathogenesis of dry eye.
Materials and Methods
Mouse Model of Experimental Dry Eye Disease
Female C57BL/6 mice (6–8 weeks old) were purchased from Taconic Farms (Oxnard, CA) or Jackson Laboratories (Sacramento, CA). Female HEL BCR Tg mice (6–8 weeks old) were purchased from Jackson Laboratories. Experimental dry eye was induced as previously described.4,32 In brief, mice were exposed to DS in perforated cages with constant airflow from fans positioned on both sides and room humidity maintained at 30% to 35%. Injection of scopolamine hydrobromide (0.5 mg/0.2 mL; Sigma-Aldrich, St. Louis, MO) was administered subcutaneously, three times a day (8:00 AM, 12:00 noon, and 5:00 PM), on alternating hind-flanks to augment disease. Mice were exposed to DS for 3 weeks. Untreated control mice were maintained in a nonstressed environment at 50% to 75% relative humidity without exposure to forced air. All animal experiments were approved by the institutional animal care and use committees at Allergan, Inc. All animals were treated in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
RNA Isolation and Real-Time PCR
Total RNA from the cornea, conjunctiva, and lacrimal glands was isolated from untreated control (day 0) and dry-eye mice exposed to DS for 5 and 10 days using the TRIzol reagent, according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Subsequent steps were performed according to the manufacturer's specifications: ocular surface tissues from individual mice were pooled to increase RNA yield (n = 10 samples/time point) and treated with RNase-free DNAse to prevent genomic DNA contamination (Qiagen, Valencia, CA); first-strand cDNA was synthesized from 0.5 μg total RNA using random hexamers and reverse transcriptase (SABiosciences, Frederick, MD); real-time PCR was performed with a custom RT2 Profiler PCR array that included primers to murine Hprt1 and Klk13 (SABiosciences) and run on an Applied Biosystems 7500 Fast Real-Time PCR System (ABI, Carlsbad, CA). The Hprt1 gene was used as an endogenous reference for each reaction. The results of quantitative PCR were analyzed by the comparative cycle threshold (Ct) method, where target change = 2–ΔΔCt. The Ct was determined using the primary (fluorescent) signal as the cycle at which the signal crossed a user-defined threshold. The results were normalized by the Ct value of Hprt1 and the mean Ct of relative mRNA in the control.
Isolation of Serum and Purified IgG
Blood from each mouse was collected by cardiac puncture and transferred into a Microtainer serum separator tube (Becton Dickinson, San Diego, CA) and centrifuged at 6000g for 8 minutes. Serum was harvested and used for (1) passive transfer or (2) further purification of IgG. For IgG purification, Zeba Desalt Spin Column was used to de-salt the serum for optimal protein recovery according to manufacturer's specifications (Pierce Biotechnology, Rockford, IL). The desalted serum was added to Melon Gel column (Pierce Biotechnology) and centrifuged at 3000g for 1 minute. Purified antibody was quantified using BCA protein assay (Pierce Biotechnology).
Western Blot for Detecting Serum Autoantibodies
Western blot analysis was used to probe dry-eye–specific serum for autoantibodies specific for ocular surface proteins. MagicMark molecular weight marker (Invitrogen) and 1 μL of recombinant human Klk13 (Abcam, Cambridge, MA) were loaded into separate wells of a NuPAGE 12% Bis-Tris gel and run at 200 V, 120 mAmps for 50 minutes. Proteins were subsequently transferred to nitrocellulose for 60 minutes at 25 V, 160 mAmps, then probed overnight at 4°C with either whole serum from control or dry-eye mice (1:500) or rabbit anti-human recombinant Klk13 (1:10,000) as a positive control. Amersham RPN2108 chemiluminescent detection kit was used according to standard protocol to visualize antibody:protein complexes (GE Healthcare, Piscataway, NJ).
Adoptive Transfer
Serum or purified IgG was isolated from whole blood from mice exposed to DS for 3 weeks or matched controls and passively transferred to nude recipient mice via intraperitoneal (IP) injection. For serum transfer experiments, a 1:1 donor-to-recipient equivalent was used based on the average yield of serum per mouse (∼200 μL). For IgG transfer experiments, serum was pooled for each experimental group and IgG was purified as described previously; a 1:1 donor-to-recipient equivalent was used based on average yield of total IgG per dry-eye mouse. A separate cohort of nude recipient mice received the same amount of control IgG. Mice were killed 3 days after transfer and tears and ocular surface tissues were collected for analysis.
Complement Depletion with Cobra Venom Factor
Cobra venom factor (CVF) is a functional homolog of factor C3 that binds factor Bb in a complex resistant to regulation, consuming total plasma C3 and disabling the complement system,33–35 and has been used extensively to demonstrate the general role of complement in various models of autoimmunity. Nude recipient mice were injected IP with 18 μg (8 units) CVF (Calbiochem, San Diego, CA) resuspended in neutral PBS on day −3 and day −1 before receiving serum or purified IgG from mice exposed to DS for 3 weeks or matched naïve controls. Mice were killed 3 days after transfer and tears and ocular surface tissues were collected for analysis as described. A hemolysis assay modified for mouse serum using sheep erythrocytes sensitized with rabbit anti-sheep erythrocyte antibody confirmed the decomplementing activity of CVF36; serum isolated from nude mice treated with CVF displayed complete inhibition (97.7% ± 5.4%) of hemolytic complement activity, whereas serum from naïve mice displayed normal complement activity.
Cytokine and Chemokine Levels in Tears during Experimental Dry Eye
Relative levels of select cytokines and chemokines present in the tears were evaluated over the course of experimental dry eye using multiplex bead analysis (Luminex Corporation, Austin, TX) as previously described.37 In brief, tears were collected on the day mice were killed; 1.5 μL of Beadlyte assay buffer (Millipore, Bellerica, MA) was placed on each eye. Then 1 μL/eye of tear fluid was subsequently collected by capillary action from each eye with a 1-μL volume glass capillary tube (Drummond Scientific, Broomhall, PA) placed in tear meniscus at the lateral canthus, then combined with 8 μL of Beadlyte buffer. Samples were stored at −80°C and cytokine/chemokine levels were assessed using the appropriate Millipore beads analyzed on a Luminex 100 or Luminex FLEXMAP 3D (Luminex Corporation).
Histology
Whole eyes, including lids, were surgically excised, fixed in 10% formalin, and embedded in paraffin. Then, 8-μm sections were stained with hematoxylin-eosin (H&E) to evaluate gross inflammatory cell infiltration within the LFU or periodic acid-Schiff (PAS) reagent to quantify goblet cell density within the conjunctiva. Sections were viewed by light microscopy using an Eclipse E400 (Nikon, Melville, NY) and photographed with a DS-Fi1 digital camera (Nikon). H&E sections were evaluated for inflammatory cell infiltration within the ocular surface tissues on a scale from 0 to 3: 0 = no inflammatory cell infiltration, 1 = mild inflammatory cell infiltration, 2 = moderate inflammatory cell infiltration, and 3 = intense inflammatory cell infiltration. For quantification of goblet cells, a midline section of each eye was counted following the morphometric guideline of the entire superior and inferior conjunctiva starting at the limbus and spanning the entire length to the tarsal conjunctiva, including the conjunctival epithelium and stroma to a depth of 75 μM below the basement membrane, as previously reported.11 Evaluation of H&E sections and cell counts were performed by a masked observer. Data are expressed as average number of goblet cells per conjunctiva for each experimental group.
Immunohistochemistry
Whole eyes, including lids, were surgically excised, embedded, and flash frozen in optimal cutting temperature (OCT compound; VWR, Suwanee, GA). Then, 8-μm sagittal sections were cut with a cryostat (HM 500; Micron, Waldorf, Germany) and placed on glass slides that were stored at −80°C. Sections were stained for Klk13 with rabbit anti-human Klk13 polyclonal antibody (1:250) (Becton-Dickinson, Mountain View, CA), Gr1+ neutrophils with 1:100 rat anti-mouse Gr1 (NIMP-R14) (Hycult Biotech, Plymouth Meeting, PA), and complement factor C3b with 1:100 rat anti-mouse monoclonal C3b/iC3b/C3c antibody (clone 2/11) (Hycult Biotech). Polyclonal secondary antibodies were biotinylated and included, anti-rabbit and anti-rat and IgG (1:50) (BD Biosciences, San Jose, CA). Positive cells were visualized using the ABC Vectastain Kit in conjunction with NovaRED Substrate kit (Vector Laboratories, Burlingame, CA). For control, sections were also stained using the appropriate secondary antibodies alone or with the primary isotype antibodies in conjunction with the biotinylated secondary antibodies. Three sections from each animal were examined and photographed with a microscope equipped with a digital camera (Eclipse E400 with a DS-Fi1; Nikon). For quantification of Gr1+ neutrophils, a midline section of each eye was counted following a morphometric guideline of the entire superior and inferior conjunctiva starting at the limbus and spanning the entire length to the tarsal conjunctiva, including the conjunctival epithelium and stroma to a depth of 75 μM below the basement membrane. This method was previously validated for quantifying inflammatory cell infiltration in this model.4,11 All cell counts were performed by a masked observer. Data are expressed as average number of neutrophils per conjunctiva for each experimental group.
Statistics
Statistically significant differences between experimental groups were determined by one-way ANOVA or Student's t-test. P ≤ 0.05 was considered significant.
Results
Autoantibodies Are Present in the Serum from Mice with Dry Eye Disease
Members of the kallikrein family of proteins (e.g., Klk13 and Klk1b22) were previously implicated as autoantigens in rodent models of Sjögren's syndrome.31,38 To determine if Klk13 plays a similar role in non-Sjögren's dry eye, a combination of real-time PCR and immunohistochemistry (IHC) was used to determine if Klk13 was expressed within the LFU during the initiation and development of experimental dry eye (Fig. 1). Real-time PCR analysis revealed upregulation of Klk13 in the cornea, conjunctiva, and lacrimal gland by 5 to 10 days post-DS relative to control (Table 1). Klk13 was also detected by IHC within the cornea (Fig. 1A), conjunctiva (Fig. 1B), and lacrimal glands (Fig. 1C) of mice with dry eye.
Figure 1.

Klk13 is expressed in the LFU during experimental dry eye disease. C57BL/6 mice were exposed to DS, as previously described, and ocular surface tissues were collected at 5 and 10 days to evaluate Klk13 expression. Klk13 was noted in the (A) cornea, (B) conjunctiva, and (C) lacrimal gland of DS mice compared with controls using IHC. Arrows indicate examples of areas of positive Klk13 staining (brown). Images captured at ×20 magnification.
Table 1.
Fold Change in Klk 13 Gene Expression Relative to Naïve Control Mice
|
Tissue |
5 Days DS |
10 Days DS |
| Cornea | 4.6-fold ↑ | 2.8-fold ↑ |
| Conjunctiva | 3.2-fold ↑ | 3.9-fold ↑ |
| Lacrimal gland | NC | 3.7-fold ↑ |
| Meibomian gland | NC | NC |
Data are representative of pooled samples of n = 10, quantified as relative change in gene expression in tissues, normalized by the Ct value of Hprt1 and the mean Ct of relative mRNA in the control. NC, no change.
To further determine if expression of Klk13 within the LFU tissues was associated with an autoantibody response during the immunopathogenesis of dry eye, serum isolated from control mice or those exposed to DS for 3 weeks was probed for the presence of anti-Klk13 antibodies. Western blot analysis demonstrated that antibodies within serum from control mice did not cross-react with recombinant Klk13 (Fig. 2A); however, serum isolated from dry-eye mice exposed to DS contained autoantibodies specific for Klk13 (Fig. 2B). These results suggest that plasma cell–derived autoantibodies to an ocular surface antigen are generated following exposure to DS and could potentially play a pathogenic role in the development of experimental dry eye disease.
Figure 2.
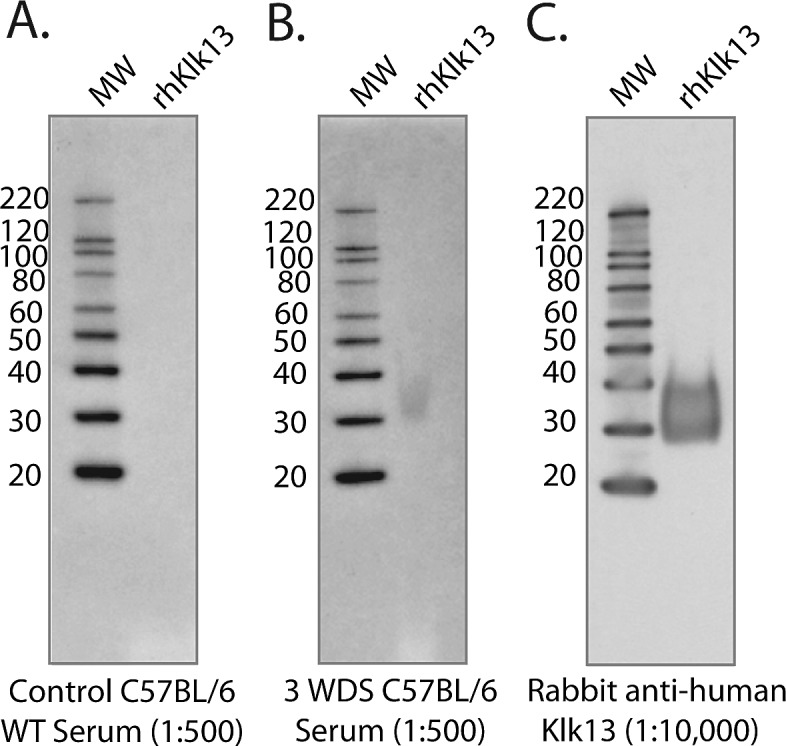
Klk13 autoantibodies are present in serum derived from dry-eye mice, but not naïve control mice. Serum was isolated from mice following a 3-week exposure to DS (3 WDS) and was used to probe 1 μg of recombinant human Klk13 that was previously transferred to nitrocellulose from an SDS-PAGE gel. (A) Serum from control mice did not cross-react with Klk13, but (B) serum from 3 WDS mice contained antibodies that recognized Klk13. (C) Anti-human Klk13 antibody was used as a positive control.
Passive Transfer of Serum or Purified IgG Mediates Dry Eye Disease in T-Cell–Deficient Nude Recipient Mice
We have previously reported that CD4+ T cells isolated from dry-eye mice exposed to DS for 5 to 10 days readily induce dry eye disease when adoptively transferred to nude recipient mice.4 To determine if self-reactive autoantibodies (e.g., against Klk13, M3R, and/or other unidentified autoantigens) present in the serum of dry-eye mice also contribute to ocular surface inflammation, (1) serum or (2) purified IgG was isolated from dry-eye mice and passively transferred to T-cell–deficient nude recipient mice. Donor mice were exposed to DS for 3 weeks to allow sufficient time to generate a humoral immune response. Passive transfer of whole serum from dry-eye donor mice yielded elevated proinflammatory cytokine levels within the tears of nude recipient mice by 3 days after transfer (Fig. 3). For example, nude mice receiving a 1:1 donor-to-recipient equivalent of dry-eye–specific serum displayed a significant (P < 0.05) increase in IL-1α (2425 ± 742 pg/mL) (Fig. 3A), IL-1β (3407 ± 1261 pg/mL) (Fig. 3B), and TNF-α (338 ± 109 pg/mL) (Fig. 3C) compared with recipients of control serum from naïve donor mice (244 ± 76 pg/mL, 112 ± 48 pg/mL, 36 ± 11 pg/mL, respectively). A trend toward increased IL-2 (9.0 ± 5.6 pg/mL vs. 1.2 ± 0.7 pg/mL) was also observed, although the increase was not significant (Fig. 3D). High tear cytokine levels were associated with decreased tear production in recipients of dry-eye–specific serum (62.1% ± 20.2% of baseline). By contrast, serum isolated from mice exposed to 10 days of DS, before there was sufficient time to mount a notable humoral response, did not induce elevated tear cytokine levels in the tears of nude mice (data not shown), nor did it induce decreased tear production (126.3% ± 35.8% of baseline). These data indicate that passive transfer of dry-eye–specific serum from mice exposed to DS for an extended 3-week period is sufficient to induce ocular surface inflammation in the context of T-cell–deficient nude recipient mice, and suggest that autoantibodies present in the serum are sufficient to mediate disease. Alternatively, it is possible that other soluble (e.g., cytokines) or cellular factors (e.g., contaminating inflammatory cells) present in the dry-eye serum were responsible for inducing ocular surface inflammation.
Figure 3.

Passive transfer of serum derived from dry-eye mice induces a proinflammatory cytokine response in the tears following adoptive transfer to nude recipient mice. Serum isolated from donor mice exposed to DS induced robust production of proinflammatory cytokines in the tears of nude recipient mice: (A) IL-1α, (B) IL-1β, (C) TNF-α, (D) IL-2. Data are shown as average concentration of protein (pg/mL) within a 2-μL tear volume ± SEM, and is representative of three independent experiments, with an n = 5 to 8 mice/group. Statistically significant values (*P ≤ 0.05) are indicated relative to tear values of nude mice receiving control serum isolated from naïve donor mice.
To resolve the alternative possibility that serum-derived proinflammatory cytokines or contaminating cells were responsible for inducing ocular surface inflammation resembling dry eye disease, total IgG was purified from serum isolated from mice exposed to 3 weeks of DS, then passively transferred to nude recipient mice. Purified IgG (1:1 donor-to-recipient equivalent) from DS mice resulted in a marked decrease in tear production (60.9% ± 10.0% of baseline) compared with recipients of IgG purified from control serum (143.2% ± 29.4% of baseline). In addition, an increase in the proinflammatory cytokine/chemokine response (e.g., IL-1β, TNF-α, IL-6, IL-12, CXCL10, IL-10, IFN-γ, and IL-17) in the tears of serum recipients (Figs. 4A–H) was associated with inflammatory cell infiltration within the ocular surface tissues of nude recipient mice that received DS-IgG compared with control-IgG (Fig. 5A); a significant (P < 0.05) increase in Gr1+ neutrophils accounted for most of the inflammatory cell burden (Figs. 5B, 5C). Furthermore, the inflammatory response also correlated with damage to ocular surface tissues, assessed by a significant (P < 0.01) reduction in goblet cell numbers within the conjunctiva of DS-IgG recipients (49.6 ± 7.6) compared with those that received control IgG (78.4 ± 4.3) or untreated nude mice (86.3 ± 7.9) (Figs. 5D, 5E). These data support the hypothesis that autoantibodies, and not cytokines or contaminating immune cells present in the dry-eye–specific serum, are sufficient to mediate inflammation-induced dry eye, and demonstrate that recruitment of neutrophils is a prominent feature of dry-eye–specific autoantibody-mediated disease in this model.
Figure 4.

Passive transfer of purified IgG derived from dry eye mice induces a robust proinflammatory tear cytokine response in tears following adoptive transfer to nude recipient mice. IgG purified from serum derived from dry-eye donor mice exposed to DS induced robust production of select acute response proinflammatory cytokines/chemokines in the tears of nude recipient mice, including (A) IL-1β, (B) TNF-α, (C) IL-6, (D) IL-12, (E) CXCL10, (F) IL-10, (G) IFN-γ, and (H) IL-17, compared with mice receiving IgG isolated from naïve donor mice (3 days after adoptive transfer). Data are shown as average concentration of protein (pg/mL) within a 2-μL tear volume ± SEM, and is representative of three independent experiments, with an n = 6 mice/group. Statistically significant values (*P ≤ 0.05) are indicated relative to tear values of nude mice receiving IgG from naïve donor mice as a control.
Figure 5.

Passive transfer of purified IgG derived from dry-eye mice induces inflammatory cell infiltration associated with ocular surface tissue damage. (A) H&E staining showed inflammatory cell infiltration within the ocular surface tissues of nude recipients of purified IgG from dry-eye donor mice compared with mice receiving IgG from naïve control mice (3 days after adoptive transfer). (B, C) A significant increase in neutrophils was noted using IHC, which was associated with (D, E) loss of PAS-positive goblet cells. (A) H&E staining; (B) Gr1+ neutrophil staining in the conjunctiva; (C) is average corneal and conjunctival Gr1+ cell counts ± SEM; (D) PAS+ goblet cell staining in the conjunctiva; (E) is average conjunctival goblet cell counts ± SEM. Open white/black arrowheads indicate examples of Gr1+ neutrophils (brown) and PAS+ goblet cells (pink); solid black arrowheads indicate areas of goblet cell loss. The data are representative of three independent experiments, with an n = 5 to 6 mice/group. Statistically significant values (*P ≤ 0.05, **P ≤ 0.01) are indicated relative to nude recipients of IgG purified from naïve donor mice. Images captured at ×20 magnification.
Serum from HEL BCR Tg Mice Does Not Induce Dry Eye Disease in Nude Recipient Mice
To confirm that autoreactive B-cell–derived plasma cells are involved in serum or IgG-induced dry eye disease, transgenic mice harboring HEL-restricted BCR were used as donor mice in a series of passive transfer experiments. DS did not induce an ocular specific humoral response in HEL-restricted BCR Tg mice (Fig. 6). Unlike passive transfer of serum or purified IgG from wild-type mice with experimental dry eye, passive transfer of serum from HEL BCR Tg mice did not result in reduced tear production between mice receiving DS-serum (93.3% ± 22.2%) compared with untreated controls (78.6% ± 14.4% of baseline) (Fig. 6A). Unaltered tear production between recipients of DS and control HEL BCR Tg serum was associated with similar, low-level proinflammatory cytokine levels within the tears of nude recipient mice (Fig. 6B); moreover, there were trace to no inflammatory cell infiltrates observed within the ocular surface tissues of nude recipient mice following passive transfer of serum from control or DS HEL BCR Tg mice (Figs. 6C, 6D), and no significant difference in the number of conjunctival goblet cells between recipients of serum from control (49.9 ± 9.2) or DS (54.9 ± 9.8) HEL BCR Tg mice (Figs. 6E, 6F). The overall absence of ocular surface inflammation following passive transfer of serum from HEL BCR Tg mice supports the paradigm that autoreactive B-cell–derived plasma cells secrete autoantibodies that may contribute to the immunopathogenesis of experimental dry eye disease.
Figure 6.

Passive transfer of serum from BCR-restricted donor mice exposed to DS does not mediate ocular surface inflammation. To determine if donor mice harboring irrelevant BCRs can induce a dry-eye–specific autoantibody response, serum from HEL BCR Tg mice exposed to desiccating was passively transferred to nude recipient mice. There was no difference in (A) tear production or (B) the proinflammatory cytokine/chemokine response in nude recipient mice of serum derived from HEL BCR Tg mice exposed to DS compared with serum from naïve mice (3 days after adoptive transfer). (C) H&E staining showed trace to no inflammatory cell infiltration within the ocular surface tissues, which also correlated with (E, F) no difference in the number of PAS-positive goblet cells between recipient of serum from HEL BCR Tg mice. Arrowheads indicate examples of PAS+ goblet cells (pink). (A) Tear production expressed as percentage of baseline; (B) average concentration of protein (pg/mL) within a 2-μL tear volume ± SEM; (C) H&E staining; (D) overall inflammatory score ± SEM (scale 0–3); (E) PAS+ goblet cell staining in the conjunctiva; and (F) is average conjunctival goblet cell counts ± SEM. Images captured at ×20 magnification.
Complement Depletion Attenuates Serum-Induced Dry Eye Disease
The complement system plays a fundamental role in facilitating antibody-mediated host protection, and may also contribute to disease. During antibody-dependent host protection, complement may (1) enhance effector function of pathogens/antigens via complement C3b/C5b deposition, (2) recruit effector cells via C3a/C5a, and (3) lyse target cells by formation of the membrane attack complex (MAC). Similar complement-mediated mechanisms are also thought to contribute to the immunopathological consequences of autoantibodies in a variety of autoimmune diseases. To begin to understand if serum-induced dry eye disease was associated with complement activation, we evaluated the extent of complement C3b deposition within the ocular surface tissues by IHC. Positive staining for C3b was detected in the conjunctiva and cornea of mice receiving serum from dry-eye mice (Fig. 7B), but not in recipients of control serum (Fig. 7A), or the same tissues stained with isotype controls (Figs. 7C, 7D). The presence of complement C3b deposition implies that the complement system contributes to autoantibody-mediated dry eye disease.
Figure 7.

Passive transfer of dry-eye–specific serum was associated with complement factor C3b deposition within the ocular surface tissues. IHC was used to evaluate the presence of complement C3b deposition in the cornea and conjunctiva of nude mice receiving serum isolated from (A) control naïve mice or (B) dry-eye mice; complement C3b deposition was noted in (B) recipients of dry-eye–specific serum, but not in (A) recipients of naïve control serum. (C, D) IHC using isotype antibody alone confirmed staining was specific for the anti-C3b antibody. Images captured at ×20 magnification.
To determine if complement activation contributes to serum-induced dry eye disease, CVF was used to deplete complement activity.33,34 Nude recipient mice were treated twice with CVF on day −3 and day −1 before receiving passive transfer of serum, and subsequently evaluated for inflammatory cell infiltration within the ocular surface tissues and proinflammatory cytokine production. Complement-deficient recipient mice of dry-eye–specific serum showed decreased inflammatory burden (Fig. 8A), which correlated with a significant reduction (P < 0.00001) in average Gr1+ cell infiltration per conjunctiva (11.8 ± 4.2) compared with mice with an intact complement system that were not treated with CVF (82.5 ± 11.6) (Figs. 8B, 8C). Decreased inflammatory cell accumulation was also associated with a trend toward decreased proinflammatory cytokines/chemokines, including IL-1β, TNF-α, IL-6, IL-12, CXCL10, IL-10, IFN-γ, and IL-17 within the tears of complement-depleted nude recipient mice (Figs. 9A–F), although only IL-1β, IL-10, and IFN-γ were significantly decreased (P < 0.05) in this experiment. In the context of CVF-treated mice, however, reduced inflammatory burden did not correlate with goblet cell preservation, as mice receiving dry-eye serum and treated with CVF displayed reduced goblet cell counts (48.0 ± 8.2) compared with DS serum recipients with an intact complement system (73.2 ± 10.0); decreased goblet cell counts were also noted in CVF-treated recipients of control serum (59.7 ± 11.5), suggesting that CVF may have a negative impact on the ocular mucosa. Similar overall findings were observed in complement-deficient nude mice receiving dry-eye–specific IgG purified from donor mice with dry eye disease (data not shown). Taken together, the results demonstrate dry-eye–specific serum does not induce neutrophil recruitment in the absence of a fully functional complement system, implying that autoantibody-mediated dry eye disease is dependent on complement activation.
Figure 8.

Complement depletion attenuates dry-eye–specific serum-induced inflammatory cell infiltration. To determine if complement activation contributes to serum-induced dry eye disease, CVF was used to deplete complement activity. Nude recipient mice treated with CVF on day −3 and day −1 before receiving dry-eye–specific serum from donor mice exposed to 3 WDS showed (A) decreased inflammatory burden, which correlated with (B, C) a significant reduction in Gr1+ neutrophil infiltration. Arrowheads indicate examples of Gr1+ neutrophils (brown). (A) H&E staining; (B) Gr1+ neutrophils; and (C) is average conjunctival Gr1+ cell counts ± SEM. The data are representative of two independent experiments, with an n = 5 to 6 mice/group. Statistically significant values (***P ≤ 0.00001) are indicated relative to nude recipients of serum purified from naïve donor mice. Images captured at ×20 magnification.
Figure 9.
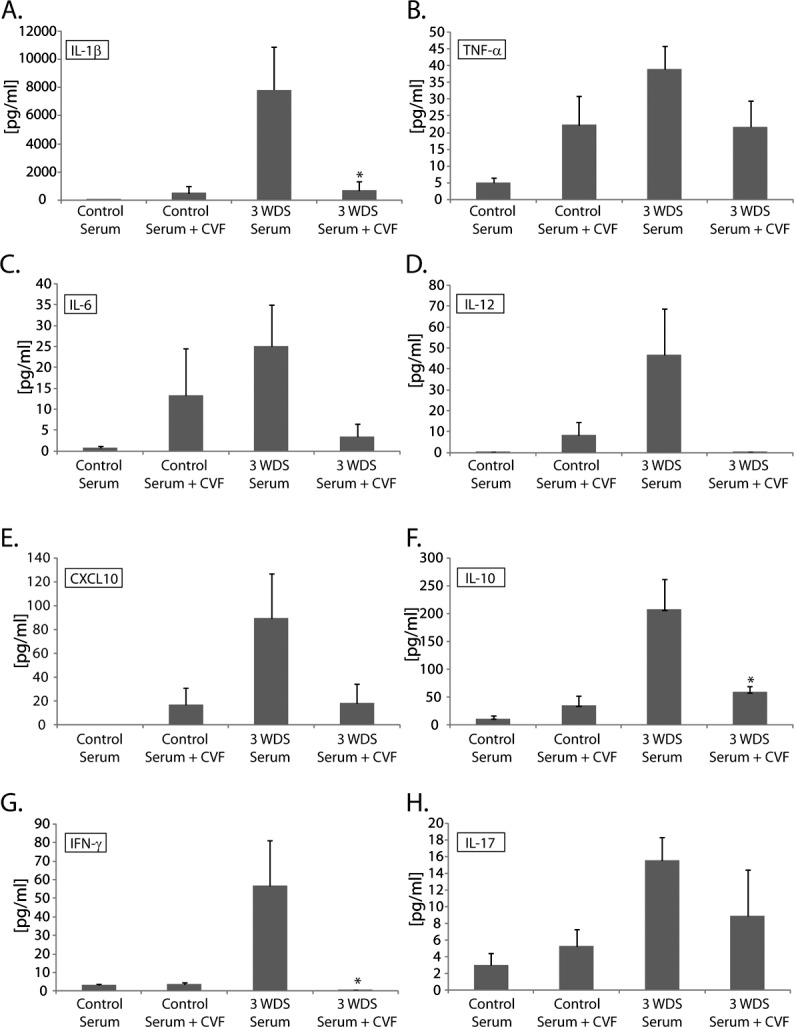
Complement depletion attenuates serum-induced proinflammatory cytokine response. Complement-depleted nude recipient mice of dry-eye–specific serum displayed a trend toward decreased proinflammatory cytokine/chemokine levels within the tears, including (A) IL-1β, (B) TNF-α, (C) IL-6, (D) IL-12, (E) CXCL10, (F) IL-10, (G) IFN-γ, and (H) IL-17, compared with nude recipient mice with an intact complement system, although only IL-1β, IL-10, and IFN-γ were significantly decreased (P < 0.05) in this experiment. The data are representative of two independent experiments, with an n = 5 to 6 mice/group. Statistically significant values (*P ≤ 0.05) are indicated relative to nude recipients of serum isolated from naïve donor mice.
Discussion
The immune system has evolved to protect the ocular surface from pathogenic challenge, while preserving tissue and maintaining tolerance to self-antigens and commensal microbial flora. Paradoxically, activation of the innate and adaptive response can also compromise tolerance, resulting in chronic autoimmune-based inflammation and tissue destruction on the ocular surface.3 Overwhelming evidence from both animal models and patients support the claim that dry eye is a chronic self-antigen–driven autoimmune disease.3 Previous studies using the DS-induced mouse model of dry eye have demonstrated that the initiation and development of dry eye requires coordinate interplay between the innate and adaptive immune systems, resulting in activation of autoreactive T cells and ensuing damage to the ocular surface tissues. Despite the strong contribution of pathogenic CD4+ T cells during of immunopathogenesis dry eye disease, self-antigen localized to the ocular surface tissues may also underlie activation and differentiation of autoreactive B cells, resulting in autoantibody production.
The current study is the first to our knowledge to show evidence that autoantibodies contribute to the immunopathogenesis of experimental dry eye disease. Western blot analysis with serum from 3-week DS mice revealed a faint, but definitive Klk13:Klk13 antibody complex, indicating that serum from dry-eye mice contained autoantibodies against Klk13 that were undetectable in serum from naïve control mice. Indeed, passive transfer of serum isolated from mice with experimental dry eye, but not naïve controls, was sufficient to induce marked inflammatory cell infiltration, elevated proinflammatory cytokine production, and goblet cell loss in nude recipient mice, independent of pathogenic T cells. An autoantigenic role for kallikrein protein family members has been reported by others. For instance, Takada et al.31 showed the presence of Klk1 and Klk13 autoantibodies in the serum of mice with Sjögren's-like disease, and more recently, Jiang et al.38 implicated Klk1b22 as a T-cell autoantigen in a model reminiscent of primary Sjögren's-like keratoconjunctivitis sicca (KCS). Lewis rats immunized with Klk1b22 displayed lymphocytic infiltration into the lacrimal and salivary glands, which was associated with decreased tear volume, corneal opacity, and ocular lesions, and further, CD4+ T cells from these mice were sufficient to cause KCS.38 At present, the signal transduction pathways leading to elevated Klk13 expression and its function in the LFU tissues during the course of experimental dry eye are unknown. Klk13 is a trypsin-like serine protease and was previously shown to be involved in degradation of major constituents of the extracellular matrix, including collagens (I–III), fibronectin, and laminin.39 In this way, it is possible that Klk13 is upregulated during DS-induced dry eye disease to facilitate inflammatory cell infiltration into the ocular surface tissues. It is interesting to speculate that Klk13 may play a dual role during dry eye, both in tissue remodeling and as a putative autoantigen. Taken together, the data suggest that kallikreins are autoantigenic during the immunopathogenesis of Sjögren's and non-Sjögren's dry eye disease, but do not exclude the contribution of other self-antigens in the initiation and progression of disease.
Autoantibodies, and not cytokines or contaminating immune cells present in dry eye-specific serum, were sufficient to mediate inflammation-induced dry eye in the context of the nude recipient mouse. This conclusion is supported by two major findings: (1) serum derived from HEL-restricted BCR Tg mice exposed to DS was not pathogenic, and (2) purified IgG from the dry-eye mouse serum maintained the ability to transfer ocular surface disease, whereas IgG from naïve control mice did not. Autoantibodies contribute to the immunopathogenesis of a variety of autoimmune diseases, including those that affect the ocular surface tissues, for example, Sjögren's syndrome and ocular cicatricial pemphigoid.3 As noted, autoantibodies, such as anti-Ro, anti-La, anti-alpha fodrin, anti-M3R, and anti-Klk1 and 13, have been implicated in secretory dysfunction of the salivary and lacrimal glands during the progression of Sjögren's syndrome.23,25,26,29,30,40,41 Moreover, passive transfer of human or rodent α-M3R Abs was sufficient to induce exocrine dysfunction resembling Sjögren's syndrome in recipient animals.29,30 Functionally, the immuopathogenic effect of autoantibodies may be facilitated by (1) Fcγ receptor-mediated activation/recruitment of neutrophils and/or macrophages that engulf target cells or release cytotoxic components (e.g., nitric oxide), and/or (2) activation of the complement system, which orchestrates tissue destruction by facilitating phagocytosis of target cells by neutrophils/macrophages or direct cell lysis via MAC formation.21
Passive transfer of dry-eye–specific serum or purified IgG resulted in neutrophil infiltration and ocular surface damage that was dependent on complement activation. Neutrophil recruitment following passive transfer of serum or IgG from dry-eye mice, coupled with tissue damage (i.e., goblet cell loss), is consistent with autoantibody-mediated immunopathology. Emerging evidence suggests that IgG can activate the complement system by way of the classical (e.g., IgG:C1q binding), mannan-binding lectin (e.g., IgG:glycan binding), or alternative (e.g., IgG:nascent C3b binding) pathways.21 In any case, activation of complement converges on C3 hydrolysis (e.g., C3→C3a and C3b→C5a and C5b+C6, 7, 8, 9→MAC formation), culminating in chemotaxis of inflammatory cells (C3a and C5a), opsonization of target cells/tissues (C3b and C4b), and/or cell lysis by MAC (C5b, 6, 7, 8, 9) formation.42 The presence of complement C3b deposition in the conjunctiva and cornea of mice receiving serum from dry-eye mice, but not in recipients of control serum, supports the hypothesis that the complement system is involved in dry-eye autoantibody-mediated induction of ocular surface inflammation.
Complement depletion attenuated the ability of dry-eye–specific serum or IgG to induce ocular surface inflammation in nude recipient mice. CVF is a functional homolog of factor C3 that binds factor Bb, forming a complex that is resistant to regulation, thereby consuming total plasma C3, and disabling the complement system.33–35 Therefore, CVF effectively blocks autoantibody-mediated effects that may be directed from either the classical, mannan-binding lectin, or alternative pathways. Mice receiving serum or IgG from dry-eye mice, and treated with CVF to deplete complement, displayed a marked reduction in Gr1+ neutrophil infiltration, which was associated with a trend toward decreased proinflammatory cytokine/chemokine levels. In the context of CVF, the reduction in inflammatory burden did not correlate with goblet cell preservation; the finding that CVF treatment alone was sufficient to reduce goblet cell counts, independent of elevated neutrophil infiltration/proinflammatory cytokines, suggests that systemic CVF affects goblet cell numbers. The increase in Gr1+ cells in CVF-treated mice receiving control serum also suggests that CVF treatment may be exerting an acute proinflammatory effect in the nude recipient mice, and may also explain, in part, why some of the decreases in cytokines assayed in the tears of complement-depleted mice receiving dry-eye–specific serum did not reach significance (e.g., TNF-α, IL-6, IL-17). Nonetheless, CVF has been used extensively to demonstrate the general role of complement in various models of autoimmunity. For example, CVF treatment delayed the onset and severity of experimental autoimmune encephalomyelitis,43 collagen-induced arthritis,44 and experimental autoimmune anterior uveitis.45 In addition, CVF was also used to show a central role of complement in the onset and development of spontaneous Sjögren's syndrome–like autoimmune exocrinopathy in NOD.B10-H2b mice.46
The presence of autoantibodies in the serum of mice with experimental dry eye implicates autoreactive B cells in disease, as is the case with other autoimmune diseases.47–49 During the progression of dry eye disease, Th2 cells may provide help to autoreactive B cells to promote clonal expansion, somatic hypermutation, isotype switching, affinity maturation, and differentiation into autoantibody-secreting plasma cells. Although Th1 and Th17 cells are considered to be the predominant CD4+ T-helper subsets during the immunopathogenesis of non-Sjögren's dry eye,4,12–14 activation of Th2 cells capable of driving B-cell differentiation has not been formally disputed. Alternatively, stimulation of autoreactive B cells via aberrant Toll-like receptor signaling and/or overexpression of the B-cell activation factor (BAFF) may account for T-cell–independent activation of autoreactive B cells. Increased B-cell survival and hyper-reactivity are hypothesized to promote hyper-gammaglobulinemia associated with relatively high titers of autoantibodies during Sjögren's syndrome. For example, elevated BAFF was associated with B-cell dysregulation50 and the presence of anti-SSA and anti-SSB autoantibodies in patients with Sjögren's syndrome.51 A modest increase in BAFF expression has also been noted in the conjunctiva and lacrimal glands of dry-eye mice as early as 5 days following induction of disease (Stern and Schaumburg, unpublished observations, 2009). During the development of dry eye, it is possible that elevated expression of Klk13, M3R, and/or other unidentified proteins exposes a higher burden of autoantigens, which may act locally to engender an autoantibody response derived from activation and differentiation of autoreactive B cells, or leak through the draining lymphatics to activate autoreactive B cells within the regional lymph nodes. Whether T-cell dependent or independent, however, the underlying mechanisms involved in autoantibody generation are currently unknown.
These data establish a role for autoantibodies in the immunopathogenesis of experimental dry eye disease. Certainly, CD4+ T cells play a dominant role in dry eye disease in both human and animal models, and, as the field advances, a new appreciation for the pathogenicity of different cell types in dry eye disease is emerging. For example, effector cells, such as Th17 cells,12 and more recently, NK cells,52 were shown to contribute to the initiation of experimental dry eye disease. We propose a working hypothesis in which autoantibodies contribute to CD4+ T-cell–mediated disease by complement-dependent neutrophil recruitment to the ocular surface. Future studies are needed to determine the relative contribution of the complement system in experimental autoantibody-mediated disease, and, more importantly, if autoantibody deposition within the ocular surface tissues also contributes to the predominantly T-cell–mediated immunopathogenesis of human dry eye disease.
Footnotes
Disclosure: M.E. Stern, Allergan, Inc. (E); C.S. Schaumburg, Allergan, Inc. (E); K.F. Siemasko, Allergan, Inc. (E); J. Gao, Allergan, Inc. (E); L.A. Wheeler, Allergan, Inc. (E); D.A. Grupe, Allergan, Inc. (E); C.S. De Paiva, None; V.L. Calder, Allergan, Inc. (C); M. Calonge, Allergan, Inc. (C); J.Y. Niederkorn, Allergan, Inc. (C); S.C. Pflugfelder, Allergan, Inc. (C)
References
- 1. Moss SE, Klein R, Klein BE. Prevalence of and risk factors for dry eye syndrome. Arch Ophthalmol. 2000;118:1264–1268. [DOI] [PubMed] [Google Scholar]
- 2. Pflugfelder SC. Prevalence, burden, and pharmacoeconomics of dry eye disease. Am J Manag Care. 2008;14:S102–S106. [PubMed] [Google Scholar]
- 3. Stern ME, Schaumburg CS, Dana R, Calonge M, Niederkorn JY, Pflugfelder SC. Autoimmunity at the ocular surface: pathogenesis and regulation. Mucosal Immunol. 2010;3:425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niederkorn JY, Stern ME, Pflugfelder SC, et al. Desiccating stress induces T cell-mediated Sjögren's syndrome-like lacrimal keratoconjunctivitis. J Immunol. 2006;176:3950–3957. [DOI] [PubMed] [Google Scholar]
- 5. Kunert KS, Tisdale AS, Stern ME, Smith JA, Gipson IK. Analysis of topical cyclosporine treatment of patients with dry eye syndrome: effect on conjunctival lymphocytes. Arch Ophthalmol. 2000;118:1489–1496. [DOI] [PubMed] [Google Scholar]
- 6. Reinoso R, Calonge M, Castellanos E, et al. Differential cell proliferation, apoptosis, and immune response in healthy and evaporative-type dry eye conjunctival epithelia. Invest Ophthalmol Vis Sci. 2011;52:4819–4828. [DOI] [PubMed] [Google Scholar]
- 7. Enriquez-de-Salamanca A, Castellanos E, Stern ME, et al. Tear cytokine and chemokine analysis and clinical correlations in evaporative-type dry eye disease. Mol Vis. 2010;16; 862–73.. [PMC free article] [PubMed] [Google Scholar]
- 8. Kunert KS, Tisdale AS, Gipson IK. Goblet cell numbers and epithelial proliferation in the conjunctiva of patients with dry eye syndrome treated with cyclosporine. Arch Ophthalmol. 2002;120:330–337. [DOI] [PubMed] [Google Scholar]
- 9. Stern ME, Gao J, Schwalb TA, et al. Conjunctival T-cell subpopulations in Sjögren's and non-Sjögren's patients with dry eye. Invest Ophthalmol Vis Sci. 2002;43:2609–2614. [PubMed] [Google Scholar]
- 10. Ecoiffier T, El AJ, Rashid S, Schaumberg D, Dana R. Modulation of integrin alpha4beta1 (VLA-4) in dry eye disease. Arch Ophthalmol. 2008;126:1695–1699. [DOI] [PubMed] [Google Scholar]
- 11. Schaumburg CS, Siemasko KF, de Paiva CS, et al. Ocular surface antigen presenting cells are necessary for autoreactive T cell-mediated experimental autoimmune lacrimal keratoconjunctivitis. J Immunol. 2011;187:3653–3662. [DOI] [PubMed] [Google Scholar]
- 12. Chauhan SK, El AJ, Ecoiffier T, et al. Autoimmunity in dry eye is due to resistance of Th17 to Treg suppression. J Immunol. 2009;182:1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lam H, Bleiden L, de Paiva CS, Farley W, Stern ME, Pflugfelder SC. Tear cytokine profiles in dysfunctional tear syndrome. Am J Ophthalmol. 2009;147:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng X, Bian F, Ma P, et al. Induction of Th17 differentiation by corneal epithelial-derived cytokines. J Cell Physiol. 2010;222:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–3918. [PubMed] [Google Scholar]
- 17. O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174:3781–3788. [DOI] [PubMed] [Google Scholar]
- 18. Rodriguez-Pinto D. B cells as antigen presenting cells. Cell Immunol. 2005;238:67–75. [DOI] [PubMed] [Google Scholar]
- 19. Lund FE. Cytokine-producing B lymphocytes—key regulators of immunity. Curr Opin Immunol. 2008;20:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manz RA, Arce S, Cassese G, Hauser AE, Hiepe F, Radbruch A. Humoral immunity and long-lived plasma cells. Curr Opin Immunol. 2002;14:517–521. [DOI] [PubMed] [Google Scholar]
- 21. Daha NA, Banda NK, Roos A, et al. Complement activation by (auto-) antibodies. Mol Immunol. 2011;48:1656–1665. [DOI] [PubMed] [Google Scholar]
- 22. Routsias J, Tzioufas A. Sjögren's syndrome—study of autoantigens and autoantibodies. Clin Rev Allergy Immunol. 2007;32:238–251. [DOI] [PubMed] [Google Scholar]
- 23. Bacman S, Berra A, Sterin-Borda L, Borda E. Muscarinic acetylcholine receptor antibodies as a new marker of dry eye Sjögren syndrome. Invest Ophthalmol Vis Sci. 2001;42:321–327. [PubMed] [Google Scholar]
- 24. Humphreys-Beher MG, Brinkley L, Purushotham KR, et al. Characterization of antinuclear autoantibodies present in the serum from nonobese diabetic (NOD) mice. Clin Immunol Immunopathol. 1993;68:350–356. [DOI] [PubMed] [Google Scholar]
- 25. Yamamoto H, Sims NE, Macauley SP, Nguyen KH, Nakagawa Y, Humphreys-Beher MG. Alterations in the secretory response of non-obese diabetic (NOD) mice to muscarinic receptor stimulation. Clin Immunol Immunopathol. 1996;78:245–255. [DOI] [PubMed] [Google Scholar]
- 26. Kovacs L, Marczinovits I, Gyorgy A, et al. Clinical associations of autoantibodies to human muscarinic acetylcholine receptor 3(213–228) in primary Sjögren's syndrome. Rheumatology (Oxford). 2005;44:1021–1025. [DOI] [PubMed] [Google Scholar]
- 27. Bacman S, Sterin-Borda L, Camusso JJ, Arana R, Hubscher O, Borda E. Circulating antibodies against rat parotid gland M3 muscarinic receptors in primary Sjögren's syndrome. Clin Exp Immunol. 1996;104:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borda E, Camusso JJ, Perez LC, et al. Circulating antibodies against neonatal cardiac muscarinic acetylcholine receptor in patients with Sjögren's syndrome. Mol Cell Biochem. 1996;163–164:335–341. [DOI] [PubMed] [Google Scholar]
- 29. Nguyen KH, Brayer J, Cha S, et al. Evidence for antimuscarinic acetylcholine receptor antibody-mediated secretory dysfunction in nod mice. Arthritis Rheum. 2000;43:2297–2306. [DOI] [PubMed] [Google Scholar]
- 30. Robinson CP, Brayer J, Yamachika S, et al. Transfer of human serum IgG to nonobese diabetic Igmu null mice reveals a role for autoantibodies in the loss of secretory function of exocrine tissues in Sjögren's syndrome. Proc Natl Acad Sci U S A. 1998;95:7538–7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takada K, Takiguchi M, Konno A, Inaba M. Autoimmunity against a tissue kallikrein in IQI/Jic mice: a model for Sjögren's syndrome. J Biol Chem. 2005;280:3982–3988. [DOI] [PubMed] [Google Scholar]
- 32. Dursun D, Wang M, Monroy D, et al. A mouse model of keratoconjunctivitis sicca. Invest Ophthalmol Vis Sci. 2002;43:632–638. [PubMed] [Google Scholar]
- 33. Van den Berg CW, Aerts PC, Van DH. In vivo anti-complementary activities of the cobra venom factors from Naja naja and Naja haje. J Immunol Methods. 1991;136:287–294. [DOI] [PubMed] [Google Scholar]
- 34. Cochrane CG, Muller-Eberhard HJ, Aikin BS. Depletion of plasma complement in vivo by a protein of cobra venom: its effect on various immunologic reactions. J Immunol. 1970;105:55–69. [PubMed] [Google Scholar]
- 35. Vogel CW, Fritzinger DC, Hew BE, Thorne M, Bammert H. Recombinant cobra venom factor. Mol Immunol. 2004;41:191–199. [DOI] [PubMed] [Google Scholar]
- 36. Andrews BS, Theofilopoulos AN. A microassay for the determination of hemolytic complement activity in mouse serum. J Immunol Methods. 1978;22:273–281. [DOI] [PubMed] [Google Scholar]
- 37. Siemasko KF, Gao J, Calder VL, et al. In vitro expanded CD4+CD25+Foxp3+ regulatory T Cells maintain a normal phenotype and suppress immune-mediated ocular surface inflammation. Invest Ophthalmol Vis Sci. 2008;49:5434–5440. [DOI] [PubMed] [Google Scholar]
- 38. Jiang G, Ke Y, Sun D, et al. A new model of experimental autoimmune keratoconjunctivitis sicca (KCS) induced in Lewis rat by the autoantigen Klk1b22. Invest Ophthalmol Vis Sci. 2009;50:2245–2254. [DOI] [PubMed] [Google Scholar]
- 39. Kapadia C, Ghosh MC, Grass L, Diamandis EP. Human kallikrein 13 involvement in extracellular matrix degradation. Biochem Biophys Res Commun. 2004;323:1084–1090. [DOI] [PubMed] [Google Scholar]
- 40. Brayer JB, Cha S, Nagashima H, et al. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjögren's syndrome. Scand J Immunol. 2001;54:133–140. [DOI] [PubMed] [Google Scholar]
- 41. Waterman SA, Gordon TP, Rischmueller M. Inhibitory effects of muscarinic receptor autoantibodies on parasympathetic neurotransmission in Sjögren's syndrome. Arthritis Rheum. 2000;43:1647–1654. [DOI] [PubMed] [Google Scholar]
- 42. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Terenyi N, Nagy N, Papp K, Prechl J, Olah I, Erdei A. Transient decomplementation of mice delays onset of experimental autoimmune encephalomyelitis and impairs MOG-specific T cell response and autoantibody production. Mol Immunol. 2009;47:57–63. [DOI] [PubMed] [Google Scholar]
- 44. Kerwar SS, Bauman N, Oronsky AL, Sloboda AE. Studies on type II collagen induced polyarthritis in rats. Effect of complement depletion. J Immunopharmacol. 1981;3:323–337. [DOI] [PubMed] [Google Scholar]
- 45. Jha P, Sohn JH, Xu Q, et al. The complement system plays a critical role in the development of experimental autoimmune anterior uveitis. Invest Ophthalmol Vis Sci. 2006;47:1030–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nguyen C, Cornelius J, Singson E, Killedar S, Cha S, Peck AB. Role of complement and B lymphocytes in Sjögren's syndrome-like autoimmune exocrinopathy of NOD.B10-H2b mice. Mol Immunol. 2006;43:1332–1339. [DOI] [PubMed] [Google Scholar]
- 47. Leadbetter EA, Rifkin IR, Marshak-Rothstein A. Toll-like receptors and activation of autoreactive B cells. Curr Dir Autoimmun. 2003;6:105–122. [DOI] [PubMed] [Google Scholar]
- 48. Shlomchik MJ. Activating systemic autoimmunity: B's, T's, and tolls. Curr Opin Immunol. 2009;21:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Groom J, Mackay F. B cells flying solo. Immunol Cell Biol. 2008;86:40–46. [DOI] [PubMed] [Google Scholar]
- 50. Mackay F, Groom JR, Tangye SG. An important role for B-cell activation factor and B cells in the pathogenesis of Sjögren's syndrome. Curr Opin Rheumatol. 2007;19:406–413. [DOI] [PubMed] [Google Scholar]
- 51. Mariette X, Roux S, Zhang J, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjögren's syndrome. Ann Rheum Dis. 2003;62:168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen Y, Chauhan SK, Saban DR, Sadrai Z, Okanobo A, Dana R. Interferon-{gamma}-secreting NK cells promote induction of dry eye disease. J Leukoc Biol. 2011;89:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]