

We develop a universal platform for CRISPR/HDR-engineering of hybridomas to obtain functionally diverse antibody panels.
Abstract
Hybridoma technology is instrumental for the development of novel antibody therapeutics and diagnostics. Recent preclinical and clinical studies highlight the importance of antibody isotype for therapeutic efficacy. However, since the sequence encoding the constant domains is fixed, tuning antibody function in hybridomas has been restricted. Here, we demonstrate a versatile CRISPR/HDR platform to rapidly engineer the constant immunoglobulin domains to obtain recombinant hybridomas, which secrete antibodies in the preferred format, species, and isotype. Using this platform, we obtained recombinant hybridomas secreting Fab′ fragments, isotype-switched chimeric antibodies, and Fc-silent mutants. These antibody products are stable, retain their antigen specificity, and display their intrinsic Fc-effector functions in vitro and in vivo. Furthermore, we can site-specifically attach cargo to these antibody products via chemoenzymatic modification. We believe that this versatile platform facilitates antibody engineering for the entire scientific community, empowering preclinical antibody research.
INTRODUCTION
Monoclonal antibodies (mAbs) have revolutionized the medical field, allowing the treatment of diseases that were previously deemed incurable (1). For mAb discovery, screening, and production, hybridoma technology has been widely used since its introduction in 1975 (2). Hybridomas are immortal cell lines capable of secreting large quantities of mAbs and have been proven to be essential for the development of new antibody-based therapies. Over the past decades, a large number of hybridomas have been generated, validated, and made available, including hybridomas used for preclinical research. Besides the antigen specificity, the mAb format and isotype are important determinants for performance in preclinical models. However, as the mAb heavy chain sequence is genetically fixed in the hybridoma, the possibilities to adjust antibody format and functionality without compromising target specificity are limited. Typically, genetically engineered mAbs are produced using recombinant technology. This requires sequencing of the variable domains, cloning of these variable domain sequences into the plasmids with the appropriate backbone, and expression of these plasmids in transient systems. These procedures are often time-consuming, challenging, and expensive and therefore often outsourced to specialized contract research companies, hampering academic early-stage antibody development and preclinical application.
The importance of the antigen binding variable regions is apparent. However, the constant domains that form the Fc domain are also central to the therapeutic efficacy of mAbs. Numerous studies have demonstrated that therapeutic efficacy of certain mAbs depends on the Fc domain and their engagement with specific Fc receptors (FcRs) (3–8). In a recent example, Vargas et al. (4) demonstrated that anti–cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) immunotherapeutic working mechanism revolves around depletion of regulatory T cells in the tumor microenvironment using Fc variants of the same antibody. The authors postulated that this could explain why patients with cancer treated with ipilimumab, a human immunoglobulin G1 (IgG1) CTLA-4–targeting mAb, greatly benefit from an activating FcγRIIIa polymorphism, which increases affinity for human IgG1 (4). This highlights the central role the Fc plays in antibody-based therapeutics and emphasizes the importance of evaluating novel mAbs in different Fc formats in early development.
The clustered regularly interspaced short palindromic repeat (CRISPR)–Cas9 (CRISPR-associated protein 09)–targeted genome editing technology opened up a plethora of exciting opportunities for gene therapy, immunotherapy, and bioengineering (9). Recently, CRISPR-Cas9 has been used to modulate mAb expression in hybridomas: Cheong et al. (10) forced class-switch recombination in IgM producing hybridomas to downstream immunoglobulin classes via deletion of the natural S regions and induced Fab′ fragment production by deletion of the entire Fc region. Pogson et al. (11) generated a hybridoma platform in which the variable domains can be substituted and affinity maturation can be simulated via homology-directed mutagenesis with single-stranded oligos (12). Khoshnejad et al. engineered a hybridoma to introduce a sortase recognition motif (sortag) (13) on the C terminus of the heavy chain constant domain 3 (CH3) for site-specific chemoenzymatic antibody modification purposes (14). However, to date, no platform has been described for versatile and effective Fc substitution in hybridomas with constant domains from foreign species, isotypes, or formats.
Here, we demonstrate genetic engineering of hybridomas via a one-step CRISPR/homology-directed repair (HDR)–based strategy enabling rapid generation of recombinant hybridomas secreting mAbs in the Fc format of choice with the freedom to install preferred tags or mutations. We generated hybridomas producing Fab′ fragments, C-terminally equipped with a sortag and a his-tag for site-specific chemoenzymatic modification and purification purposes. Furthermore, we incorporated preferred constant domains upstream of the native constant region to generate hybridomas, which secrete chimeric antibodies with the isotype and species of choice, as well as Fc mutant mAbs without compromising antigen specificity. The engineered antibodies are easily isolated from the hybridoma supernatant and display their expected biochemical and immunological characteristics, both in vitro and in vivo. Because we target the constant domains of the immunoglobulin locus, our CRISPR/HDR method is adaptable to hybridomas from any species or isotype. We believe that this versatile platform will empower preclinical antibody research by opening up antibody engineering for the entire scientific community.
RESULTS
Generation of recombinant hybridomas producing sortagable Fab′ fragments
Monovalent Fab′ fragments consist of a shortened heavy chain and light chain and, due to their smaller size, have certain advantages over their parental mAbs. Fab′ fragments have better tissue penetration, are not susceptible to Fc-mediated uptake, and do not exert immune effector functions. Therefore, Fab′ fragments have been used for treatment of certain autoimmune diseases (15), targeting of different payloads (16, 17), and generation of bispecific antibodies (18). The conventional method to obtain Fab′ fragments is papain cleavage of the parental mAb followed by purification of the Fab′ fragment. However, this does not allow for installment of useful tags or mutations. Alternatively, one could use recombinant production in transient systems; however, this requires prior knowledge of the variable heavy and light chain sequence, optimization of the transient system, and multiple rounds of production. Rapid conversion of hybridomas into Fab′ secreting cell lines via CRISPR/HDR engineering represents a simple and cost-effective alternative to obtain inexhaustible source of Fab′ fragment with the freedom to introduce mutations or tags of choice.
To obtain Fab′ secreting hybridomas via CRISPR/HDR, we selected the hybridoma clone NLDC-145 (19) as a first target (Fig. 1, A and B). This hybridoma secretes mAbs of the rat IgG2a (rIgG2a) isotype and is specific for murine DEC205. This endocytic receptor is abundantly expressed by dendritic cells and used as a target in various vaccination strategies (20, 21). To target the heavy chain locus of NLDC-145, we used an optimized guide RNA (gRNA-H), which directs Cas9 to the rIgG2a hinge region to generate double-stranded breaks (fig. S1). To repair the double-stranded break, we designed an HDR donor construct (table S1) that inserts a sortag and his-tag directly upstream of the cysteine involved in heavy chain dimer formation. Furthermore, as the efficiency of HDR is typically low (22, 23), we included the blasticidin-resistance gene (Bsr) to enrich for cells that integrated the donor construct. We electroporated NLDC-145 hybridoma cells with the HDR Fab′ donor construct and a Cas9 vector (PX459) containing gRNA-H. After 72 hours, we applied blasticidin pressure to the targeted cultures. Following blasticidin selection, we assessed knock-in of the donor construct by genomic polymerase chain reaction (PCR) with a forward primer that hybridizes upstream of the 5′ homologous arm and a reverse primer specific for the HDR construct. PCR products of the expected size [~1600 base pair (bp)] were exclusively observed for the transfected population, indicating the integration of the donor construct in the correct genomic location (Fig. 1C). After 1 week, we performed a limiting dilution to obtain monoclonal cell suspensions. We used the supernatant of these monoclonal cell suspensions to determine antigen specificity and phenotype of the secreted product. We first stained JAWSII, a DEC205-expressing cell line, with the supernatant from each clone and performed a subsequent secondary staining for the his-tag and rIgG2a heavy chain. Flow cytometry analysis revealed that a large fraction of monoclonal hybridomas was successfully engineered (Fig. 1D), as we observed DEC205 binding constructs harboring a his-tag in the supernatant of 47 of 76 clones. In contrast, only a single-cell suspension (clone 48) still produced the mAb DEC205 of the rIgG2a isotype. To validate the flow cytometry results, we performed a Western blot for the heavy and light chain on the supernatant of clones 47 to 52 (Fig. 1E). The four clones that were his-tagpos rIgG2aneg by flow cytometric analysis secreted a shortened heavy chain that was his-tag positive, confirming production of his-tagged Fab′ fragments by these clones. Clone 48 (his-tagneg, rIgG2apos) retained its expression of the rat heavy chain, while for clone 47 (his-tagneg, rIgG2aneg), only rat light chain could be observed.
Fig. 1. CRISPR/HDR engineering of hybridoma NLDC145 to obtain sortagable Fab′ fragments against DEC205.
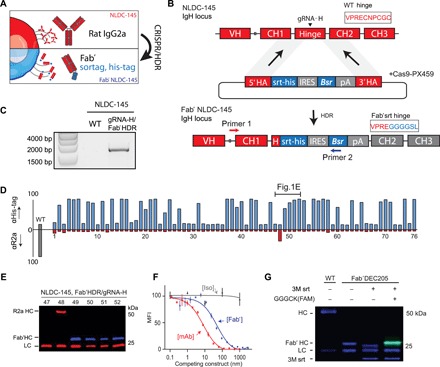
(A) Schematic representation of the CRISPR/HDR approach to convert wild-type (WT) hybridomas to Fab′ fragment–producing cell lines. (B) The targeted IgH locus of NLDC-145 with the variable region (VH) and constant regions (CH1, Hinge, CH2, and CH3) annotated is shown. Cas9 is guided by gRNA-H to hinge region and creates a double-stranded break before the first cysteine. The double-stranded break is subsequently repaired by HDR via the donor construct consisting of a sortag and his-tag motif (srt-his), an internal ribosomal entry site (IRES), blasticidin-resistance gene (Bsr), polyA transcription termination signal (pA), and homology arms (5′ HA and 3′ HA). The first 10 amino acids of the hinge before and after successful CRISPR/HDR are indicated. (C) Three days after electroporation, PCR was performed on genomic DNA from WT and CRISPR/HDR-targeted population using primers 1 and 2 (B). Agarose gel shows amplicon of expected size exclusively for the CRISPR/HDR-targeted population, indicating the correct integration of the donor construct within the population. (D) After limiting dilution of CRISPR/HDR-targeted cells, a flow cytometry screen was performed on the supernatant of monoclonal cell suspensions. To this end, DEC205-expressing cells (JAWSII) were incubated with clonal supernatants in combination with secondary antibodies against his-tag (blue) or rIgG2a (red). Exclusive his-tag signal indicates production of Fab′ fragments. (E) Immunoblotting of the supernatant of clones 47 to 52 for his-tag (blue) and rat heavy and light chain (red) confirms flow cytometry results. (F) In competition assay, JAWSII were incubated with a serial dilution (150 to 20 ng/ml) of either purified Fab′DEC205srt (blue), αDEC205 mAb (red), or an isotype control (gray) in combination with fluorescently labeled αDEC205 mAb (1 μg/ml). Decrease in mean fluorescent intensity (MFI) relates to the increase in competition for DEC205 binding with fluorescent-labeled αDEC205 mAb. n = 3, mean ± SEM. (G) Fab′DEC205srt can be C-terminally functionalized with a fluorescently labeled probe [GGGCK(FAM)] by using sortase (3M srt)-mediated ligation. LC, light chain.
For further characterization of the secreted Fab′DEC205–sortag–his-tag (hereafter referred as Fab′DEC205srt), we selected one clone (hereafter referred as Fab′ hybridoma) of which the heavy chain sequence was validated by Sanger sequencing. After expansion of the Fab′ hybridoma, Fab′DEC205srt was easily isolated from the supernatant with high purity via Ni-NTA gravity flow protein purification. To assess whether the Fab′DEC205srt retained antigen specificity, we performed a competitive binding assay. We used a fixed concentration of fluorescently labeled parental NLDC-145 mAb in combination with increasing concentrations of Fab′DEC205srt, unlabeled NLDC-145 mAb, and an isotype control antibody (Fig. 1F). Contrary to the isotype control, Fab′DEC205srt and NLDC-145 mAb competed for DEC205 binding with the fluorescent mAb in a dose-dependent manner, indicating that the CRISPR-engineered Fab′srt binds the same epitope and that antigen specificity is not affected by our CRISPR/HDR approach. The lower EC50 (half maximum effective concentration) of the Fab′ fragment compared to NLDC-145 is in line with the difference in avidity between monovalent Fab′ and bivalent mAb.
In contrast to conventional proteolytic cleavage to obtain Fab′ fragments, CRISPR/HDR engineering allows the inclusion of various tags. Besides the his-tag to facilitate purification, we introduced a C-terminal sortag (LPETGG) for chemoenzymatic conjugation using the sortase enzyme (fig. S2A). Unlike classical stochastic chemical conjugation strategies, the sortag facilitates site-specific coupling of cargo to mAbs without the danger of compromising the N-terminal binding region and results in a homogeneous final product (13, 24). To show sortag functionality of Fab′DEC205srt, we used a fluorescently labeled sortase substrate probe [GGGCK(FAM); fig. S2B] and (fluorescent) SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis. When Fab′DEC205srt was exposed to 3M sortase (a mutated sortase for higher conversion efficacy) (25) alone, the heavy chain C-terminal sortag, his-tag was hydrolyzed, resulting in a lower molecular weight heavy chain product (Fig. 1G). However, in the presence of the fluorescent sortase probe, the reaction gave conversion to a slightly higher molecular weight fluorescently labeled Fab′DEC205 heavy chain with high efficiency judged by SDS-PAGE, proving the functionality of the sortase recognition motif introduced using our CRISPR/HDR approach.
After successfully converting NLDC-145 to recombinant hybridomas producing fully functional Fab′DEC205srt, we applied the same strategy to other hybridomas of interest in the field of immuno-oncology. We generated Fab′ hybridomas against murine CD40 (clone FGK45.5), programmed death 1 (PD-1; clone RMP-14) and programmed death ligand-1 (PD-L1) (clone MIH5). The resulting Fab′ fragments were purified and site-specifically modified to obtain fluorescently labeled Fab′ fragments (fig. S2C). No decline in antibody production was observed for each CRISPR/HDR-engineered hybridoma over a long period (1 to 3 years). Furthermore, the antigen binding and sortagging capability of engineered antibodies between batches remained similar, indicating long-term stability of the CRISPR/HDR modified hybridomas. Together, we demonstrate that our CRISPR/HDR Fab′ hybridoma engineering approach is effective in generating an inexhaustible source of Fab′ fragments equipped with affinity and chemoenzymatic tags with retention of antigen specificity.
Isotype panel generation via CRISPR/HDR engineering of hybridomas
Encouraged by the success of our one-step CRISPR/HDR strategy to generate Fab′ hybridomas, we continued by further expanding the platform and engineered hybridomas to secrete a diverse set of Fc variants of the same antibody. We designed a CRISPR/HDR gene trap knock-in approach, which installs Fc constant domains [heavy chain constant domain 1 (CH1), hinge, CH2 and CH3, sortag, his-tag, and Bsr] upstream of the native rat domains (Fig. 2, A and B). To test our method, we genetically engineered the hybridoma MIH5, which secretes rIgG2a mAbs targeting mouse immune checkpoint PD-L1, to produce chimeric murine IgG1 (mIgG1), mIgG2a, mIgG2b, mIgG3, and mIgA antibodies. Furthermore, we included a mutant mIgG2a isotype with the amino acid substitutions L234A/L235A/N297A (mIgG2asilent), which is described to have reduced affinity for murine FcγRs (26). After selection of an optimal single guide RNA targeting a protospacer adjacent motif (PAM) sequence 100 bp upstream of the rIgG2a CH1 (gRNA-ISO), MIH5 cells were cotransfected with a Cas9 vector (PX458) containing gRNA-ISO and a construct from a panel for isotype HDR donor constructs (table S2). Next, we assessed on-target knock-in integration of blasticidin-selected cells by genomic PCR. Analysis showed the presence of PCR products of expected size (~2600 bp; Fig. 2C). Monoclonal cell lines were obtained, and supernatant was used for flow cytometric screening of the secreted mAbs. CT26 (a mouse colon carcinoma cell line) cells were stimulated with interferon-γ (IFN-γ) to induce up-regulation of PD-L1 before incubation with the supernatants followed by staining with secondary antibodies for the C-terminal his-tag and rIgG2a. For each antibody isotype, we identified multiple chimeric clones that expressed the designed MIH5 isotype variant. Notably, in the case of mIgG2a, mIgG2b, and mIgG2asilent, we always observed native rIgG2a isotype in the supernatant of chimeric hybridomas and in Ni-NTA gravity flow purified proteins, suggesting rat-mouse heavy chain heterodimer mAb formation (fig. S3A). We postulated that in these cases, gene elements downstream of the inserted murine heavy chain could still be transcribed due to incomplete transcription termination, facilitating splicing of the variable heavy chain (VH) region to the native rIgG2a isotype. Removal of this native splice acceptor in the HDR donor constructs of mIgG2a, mIgG2b, and mIgG2asilent to force linkage of the genetically engineered foreign CH domains to the native VH domains abrogated the production of the rIgG2a isotype. This completed the set of engineered isotype-switched chimeric antibody producing hybridomas, which were used for further characterization. The robustness of this CRISPR/HDR and selection approach was demonstrated by the identification of multiple chimeric clones that expressed the designed MIH5 isotype variant (fig. S3B). We selected a single clone for each isotype and validated the correct knock-in of the isotype of choice in the IgH locus by sequencing. Detailed flow cytometry analysis of the mAbs produced by the selected clones using IFN-γ–stimulated CT26 cells revealed that the engineered chimeric mAbs were positive for their respective murine isotypes and his-tag, but not for the native rIgG2a isotype (Fig. 2D). To validate antigen specificity of our MIH5-derived engineered antibodies, PD-L1 knockout CT26 cells [CT26PD-L1 KO; (27)] were exposed to the purified mAbs. Contrary to the wild-type (WT) CT26 cells, IFN-γ–stimulated CT26PD-L1 KO cells were not stained by our mAbs, proving retention of target specificity (fig. S3C). Western blot analysis of the purified antibodies confirmed substitution of the native rat heavy chain by the genetically introduced mouse heavy chain on protein level (fig. S3D).
Fig. 2. CRISPR/HDR engineering of hybridoma MIH5 to obtain panel of isotype variants.
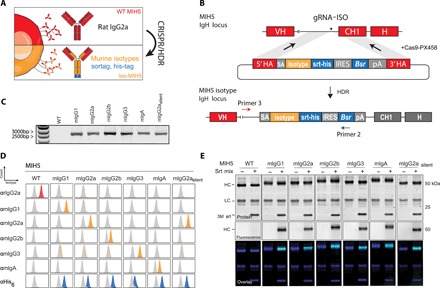
(A) CRISPR/HDR strategy to engineer rIgG2a hybridomas and obtain recombinant hybridomas secreting murine isotypes. (B) The targeted IgH locus of MIH5 with the variable region (VH), constant region 1 (CH1), and hinge annotated. Cas9 is guided by gRNA-ISO to the intronic region upstream of the CH1 is shown. The resulting double-stranded break is subsequently resolved via HDR through a donor construct, leading to an in-frame insertion of an splice acceptor (SA, gray), isotype of choice (yellow), a sortag and his-tag motif (srt-his, blue), an IRES, blasticidin-resistance gene (Bsr), and polyA transcription termination signal (pA) upstream of the native CH1. The insert is enclosed by homology arms (5′ HA and 3′ HA). (C) Three days after electroporation, DNA from CRISPR/HDR-targeted MIH5 populations is obtained for PCR with primer 3 and primer 2 (B). Agarose gel of the PCR product reveals amplicons of the correct size exclusively for the CRISPR/HDR-targeted populations. (D) After selecting monoclonal hybridoma for each isotype, the supernatant of each isotype modified clone was incubated with PD-L1–expressing target cells (CT26). Displayed plots demonstrate that supernatants exclusively contain the engineered isotype variant with a C-terminal his-tag, while the original rIgG2a mAbs is absent. (E) Purified MIH5 isotype variants were effectively labeled with the fluorescent probe GGGCK(FAM) using sortase-mediated ligation.
We equipped all our engineered chimeric mAbs with a C-terminal sortag to facilitate chemoenzymatic functionalization. To confirm functionality of C-terminal sortag, we performed a sortase ligation with the fluorescent peptide GGGCK(FAM) (Fig. 2E and fig. S4). Fluorescent SDS-PAGE analysis showed that all the heavy chains from chimeric mAbs were fluorescently tagged, whereas the parental MIH5 antibody was not. Modification of the isotype using our CRISPR/HDR genomic engineering approach is not restricted to the MIH5 hybridoma, as we were able to successfully perform the same strategy on the NLDC-145 hybridoma and obtained a panel of DEC205 isotype variant mAbs (fig. S5).
We extended the molecular characterization of our engineered mAbs by high-resolution native mass spectrometry analysis of the mIgG2asilent. Asparagine on position 297 (N297) is a conserved glycosylation site and is one of the major determinants of FcγR engagement and, hence, of therapeutic efficacy of certain mAbs. In mIgG2asilent, the N297A amino acid substitution is introduced, rendering it Fc silent (26). To validate the absence of glycans in this engineered mutant, we treated MIH5 WT and mIgG2asilent with the glycosidase peptide N-glycosidase F (PNGase F) to remove existing glycans. Subsequently, we performed high-resolution native mass spectrometry on treated and untreated samples to assess whether glycans were removed. For the WT samples, clear shifts in protein mass were observed, with a leading loss of 2889.6 Da corresponding to the loss of two glycans (G0F: asialo-agalacto core-fucosylated biantennary complex-type N-glycan, theoretical mass 2890 Da per mAb; Fig. 3A). In contrast, no difference in mass between the PNGase F–treated and PNGase F–untreated samples of mIgG2asilent could be measured, indicating the absence of glycans (Fig. 3B). Native mass spectrometry analysis of MIH5 mIgG2a variant before and after PNGase F treatment showed a mass difference of 2889.2 Da corresponding to two G0F glycans, analogous to the WT MIH5 mAb (fig. S6).
Fig. 3. Glycosylation profiling of MIH5 WT and Fc-silent variant using high-resolution native mass spectrometry.
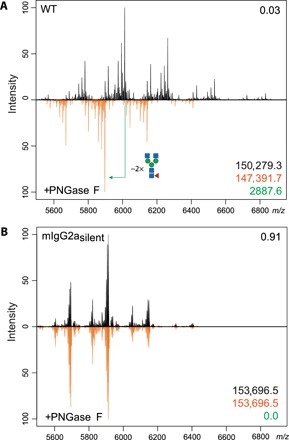
Purified samples of WT MIH5 (A) and mIgG2asilent (B) were treated overnight with PNGase F (orange) to remove existing glycans. Subsequently, these samples were compared to untreated samples (black) via high-resolution native mass spectrometry. The Pearson correlation coefficient between the two spectra over all ion signals is given in the top right corner. The molecule mass of untreated (black) and treated (orange) samples and the difference in leading mass (green) are given in the bottom right corner. m/z, mass/charge ratio.
Our data demonstrates the effective one-step generation of stable hybridomas producing Fc-engineered chimeric mAbs using our CRISPR/HDR approach. These mAbs retain their antigen specificity, and the addition of affinity and chemoenzymatic tags adds an additional layer of flexibility and utility.
Biochemical and functional characterization of the engineered antibodies
Having established and validated our hybridoma CRISPR/HDR genomic engineering approach, we set out to characterize the functional properties of our isotype-switched mAbs. We first evaluated the binding affinities of our MIH5 isotype panel for the activating murine FcγRI (mFcγRI) and mFcγRIV and for the inhibiting mFcγRIIb. We immobilized mFcγRI, mFcγRIIb, and mFcγRIV on a multiplex biosensor and assessed binding of our isotype variants to the different receptors via surface plasmon resonance (SPR). We monitored the association and dissociation over time (Fig. 4A) and generated affinity plots to calculate the dissociation constants (Kd) for each receptor concentration. Subsequently, these were interpolated to calculate the affinity of each antibody at receptor concentration giving Rmax = 500, as described before (Fig. 4B) (28). The obtained affinities of mIgG1, mIgG2a, and mIgG2b for the FcγRs fall well within the range of previously reported Kd values for recombinantly produced isotypes (table S3) (26, 28–33). In summary, mIgG1 did not bind mFcγRI and mFcγRIV but binds to mFcγRIIb. mIgG2a displayed binding to all tested mFcγRs with highest affinity for mFcγRI followed by mFcγRIV and mFcγRIIb. mIgG2b binds to mFcγRIIb and mFcγRIV, with stronger affinity for the latter. mIgA and the Fc-silent mutant mIgG2asilent did not show affinity for any of the mFcγRs. The WT rIgG2a displays an FcγR engagement profile similar to the one of mIgG1, binding to the FcγRIIb, but not to FcγRI and FcγRIV. Because of protein aggregation, a characteristic previously reported for mIgG3 (34, 35), we were not able to determine the Kd values for this isotype variant. On the basis of the differential affinity for activating and inhibiting receptors reported here, we predicted the capacity of CRISPR-engineered mAbs to evoke antibody-dependent cellular cytotoxicity (ADCC) (Fig. 4C), as others have done in the past (8).
Fig. 4. FcγR engagement of MIH5 isotype variants.
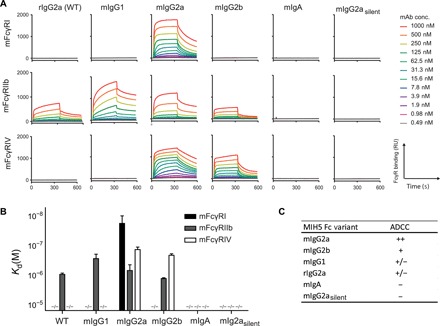
Representative sensograms (A) display interactions of MIH5 WT and MIH5-engineered mAbs (mIgG1, mIgG2a, mIgG2b, mIgA, and mIgG2asilent) for immobilized mFcγRI, mFcγRIIb, and mFcγRIV at increasing concentrations. Binding to FcγR is expressed in resonance units (RU). SPR was performed on four different concentrations of immobilized FcγRs (1, 3, 10, and 30 nM) to determine affinity [Kd(M)] of the Fc variants for the different FcRs (B). n = 3, mean ± SEM. (C) Predicted antibody-dependent cellular cytotoxicity (ADCC) activity for each murine isotype variant on the basis of their differential affinity for FcγRs.
To conclude the characterization of the generated isotype variants, we assessed their capacity to induce ADCC in vitro and in vivo. In vitro, we labeled MC38 (murine colon adenocarcinoma) cells with chromium-51, opsonized these with MIH5 Fc variants, and added whole blood from C57BL/6 mice for 4 hours. To assess specific lysis, we measured the chromium-51 release and used an aspecific mAb (AZN-D1) as a baseline (Fig. 5A). In vivo, we assessed the ability of MIH5 Fc variants to deplete B cells (Fig. 5B). We labeled isolated splenic B cells from C57BL/6 mice with Far Red tracer dye and opsonized these with mIgG2a, mIgG2b, or mIgA. Simultaneously, we used mIgG2asilent to opsonize splenic B cells labeled with Violet Blue tracer dye. Subsequently, we mixed the populations in equal ratio and injected these intravenously into lipopolysaccharide (LPS)-primed C57BL/6 mice. After 24 hours, we euthanized the mice and isolated the spleens to determine the ratio between the Violet Blue and Far Red population to assess relative B cell depletion via flow cytometry (Fig. 5C and fig. S7). Both in vitro and in vivo assays yielded similar results; mouse IgG2a displayed the strongest capacity to induce Fc-mediated killing of target cells followed by mIgG2b, which is in line with literature and the observed FcγR binding affinities (Fig. 4, A to C). Neither WT rIgG2a nor its CRISPR-engineered variants mIgG1, mIgG3, mIgA, and mIgG2asilent were able to mediate specific killing or depletion. As mIgG3, mIgG2asilent, and mIgA do not interact with murine FcRs, these are unable to mediate ADCC.The lack of effect for rIgG2a and mIgG1 Fc variants can be explained by their ability to bind only a single-activating FcγR (FcγRIII), whereas mIgG2a and mIgG2b bind respectively three and two activating FcγRs. Together, our in vitro and in vivo data indicate that chimeric and mutant mAbs through hybridoma engineering approach exhibit the same biochemical and immune effector characteristics as their natural and recombinantly produced counterparts.
Fig. 5. Effector function of MIH5 isotype variants.
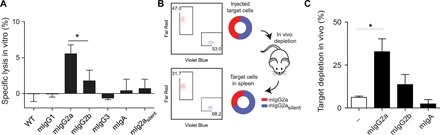
ADCC in vitro assay (A) to compare effector function of MIH5 isotype variants. MC38 cells were 51Cr-labeled, opsonized with different MIH5 isotype variants, and exposed to whole blood from C57BL/6 mice for 4 hours. Specific lysis was quantified by measuring 51Cr release. n = 3, mean ± SEM. *P < 0.05. (B) Experimental setup of in vivo depletion assay. Splenic B cells labeled with Violet and Red tracer dye were used as target cells for in vivo depletion. The violet B cells were opsonized with mIgG2asilent variant, while the red B cells were opsonized with mIgG2a, mIgG2b, or mIgA variant. Subsequently, B cells were mixed 1:1 (top plot and diagram) and injected intravenously into C57BL/6 mice. Twenty-four hours later, the spleens were isolated, and the ratios between violet and red B cells were determined via flow cytometry (bottom plot and diagram). The ratios before injection and after spleen isolation were used to quantify the isotype-specific depletion of the target cells. (C) Representative experiment indicating mIgG2a, mIgG2b, and mIgA specific depletion of target cells with mIgG2asilent opsonized cells as reference population. n = 3, mean ± SEM. *P < 0.05.
DISCUSSION
Here, we report the development of a versatile platform for easy and rapid generation of panels of Fc-engineered antibodies starting from their parental hybridomas. Using a one-step CRISPR/HDR approach, we obtained hybridomas producing either monovalent Fab′ fragments, Fc isotype variants from a foreign species (from rat anti-mouse to mouse anti-mouse), and Fc-silent mutants without effector functions within a period of 21 days. In addition, our method allows incorporation of useful protein tags for purification and site-specific conjugation purposes. Extensive characterization of the produced antibodies revealed that target specificity is uncompromised and that the CRISPR/HDR-engineered chimeric antibodies display their intrinsic Fc-effector functions. Currently, most researchers rely mainly on recombinant production methods to evaluate mAbs in different Fc formats. To do so, the variable heavy chain and light chain need to be sequenced, cloned into the appropriate backbones, and transfected multiple times into mammalian expression systems to obtain sufficient antibody yields. This process often requires extensive optimization, expertise, knowledge, and dedicated resources that are not available in most laboratories. In contrast, the CRISPR/HDR strategy outlined here offers a simple alternative approach requiring a single electroporation step to obtain an unlimited source of target antibody in the isotype format of choice.
Other investigators also described approaches to modulate heavy chain loci in hybridomas via genomic engineering. However, these approaches are either restricted in their engineering capabilities or require multiple sequential genome editing events. For example, Cheong et al. used viral CRISPR-Cas9 vectors to engineer isotype-switched mIgG1 and mIgG3 mAbs from IgM-producing hybridomas exploiting the class-switch recombination machinery present in hybridomas (10). Although elegant, the technique is restricted to the native heavy chain loci within the target hybridoma. Consequently, the generation of a complete isotype panel is restricted to scarcely available IgM-producing hybridomas. Furthermore, placement of recombinant tags, production of mutated isotypes with modulated effector functions, or isotypes from a foreign species to generate chimeric mAbs is impossible with this technique. Hashimoto et al. (36) described a multi-step method to introduce Fc variants with amino acid substitutions via recombinant-mediated cassette exchange (RMCE). However, this procedure relies on the prior incorporation of flanking Cre/lox sites required for RMCE, limiting the broad application in other hybridomas. Our single-step CRISPR/HDR method is easily adaptable to any hybridoma regardless of background species or isotype to obtain complete panels of engineered antibodies in the format of choice.
We demonstrate the ability to modulate the antibody isotype in hybridomas to a panel ranging from the activating Fc variant mIgG2a to the Fc-silent mIgG2asilent. It is widely recognized that mAb isotype is of major importance for the outcome of antibody-based therapies, especially in the field of cancer immunotherapy. Various studies have shown that the therapeutic efficacy of mAbs directed against immune checkpoints such as CTLA-4, CD25, PD-L1, GITR (glucocorticoid-induced TNFR-related protein), and OX-40 depends on their isotype (4, 5, 37–39). This is mainly due to the interaction of the Fc region with different components of the immune system, such as FcRs on the surface of immune cells and the C1q protein of the complement system. Substantial efforts have been made to modulate Fc affinity for these different components by making use of amino acid substitutions in the Fc region. This has led to a wide range of described mutants, which can be used to increase affinity for activating or inhibitory FcRs (40–42), ablate Fc-effector functions (43), or recruit complement (44, 45). Moreover, amino acid substitutions can also be used to control antibody half-life (46) or to construct bispecific antibodies (47, 48). All of these mutant Fc variants can be implemented in our versatile single-step CRISPR/HDR platform.
This method is accessible for any laboratory capable of hybridoma cell line culturing, thus providing a molecular toolbox for repurposing the wealth of established hybridoma cell lines available. Our universal CRISPR/HDR-based platform has a success rate of 100%, meaning that every genomic engineering experiment using a single specific HDR donor construct resulted in multiple correctly engineered hybridoma clones after selection. Since many hybridoma products are extensively used in preclinical in vivo studies, our method will empower preclinical antibody research by enabling screening of large panels of antibody variants early on in therapeutic antibody development. Moreover, the freedom to add affinity tags and amino acid recognition sequences for chemoenzymatic modification adds an additional layer of flexibility and utility. The ability to site-specifically functionalize the engineered antibody products will find application in the fields of biomolecular engineering, chemical biology, antibody-drug conjugates, multimodal imaging, and nanomedicine. We are convinced that this method will open up antibody engineering for the entire scientific community.
MATERIALS AND METHODS
General culture conditions
The following hybridoma clones were genetically engineered for expression of Fab′ fragments or murine isotypes: NLDC-145 [American Type Culture Collection (ATCC) HB-290], FGK-45, MIH5, and RMP1-14. Other cell lines used in this study were CT26.WT (ATCC CRL-2638), CT26.PD-L1KO (27), MC38, and JAWSII (ATCC CRL-11904). For cultivation, NLDC-145, FGK-45, MIH5, RMP1-14, CT26, CT26.PD-L1KO, and MC38 were grown in complete medium formulation consisting of RPMI 1640 medium (11875093, Thermo Fisher Scientific), 2 mM ultraglutamine (BE16-605E/U1, Lonza), 50 μM Gibco 2-mercaptoethanol (2-ME) (21985023, Thermo Fisher Scientific), 1× antibiotic-antimycotic (anti-anti) (15240062, Thermo Fisher Scientific), and 10% heat-inactivated fetal bovine serum (FBS). The JAWSII cell line was cultivated in α–minimum essential medium with ribonucleosides, deoxyribinuuleosides, 4 mM l-glutamine, 1 mM sodium pyruvate, and murine granulocyte-macrophage colony-stimulating factor (5 ng/ml). For expression of PD-L1, MC38 and CT26 were stimulated overnight with recombinant murine IFN-γ (100 ng/ml).
Cloning of CRISRP-Cas9 and donor constructs
The genomic sequence of the rIgG2a heavy chain locus was identified via the Ensembl rate genome build Rnor_6.0 (accession no. AC_0000741) and used for the design of the HDR donor templates. gRNA-H (GACTTACCTGTACATCCACA) and gRNA-ISO (TGTAGACAGCCACAGACTTG) were designed using the CRISPR tool from the Zhang laboratory (http://crispr.mit.edu) and ordered as single-stranded oligos from Integrated DNA Technologies (IDT) with the appropriate overhangs for cloning purposes. The oligos were phosphorylated with T4 PNK enzyme by incubation at 37°C for 30 min and annealed by incubation at 95°C for 5 min followed by gradually cooling to 25°C using a thermocycler. The annealed oligos of gRNA-H and gRNA-ISO were respectively cloned into the plasmids pSpCas9(BB)-2A-Puro (PX459) and pSpCas9(BB)-2A-GFP (PX458), which were obtained as gifts from F. Zhang (Addgene plasmids nos. 62988 and 48138) (49). Synthetic gene fragments containing homologous arms and desired insert for Fab′ hybridomas were obtained via IDT and cloned into the PCR2.1 TOPO TA vector (K-450001, Thermo Fisher Scientific). For the generation of the mIgG1 isotype variants, a vector containing the appropriate homology arms was synthesized by Invitrogen. The different genes for Fc domains were subsequently inserted via traditional cloning techniques. All CRISPR-Cas9 and HDR constructs were purified with the NucleoBond Xtra Midi Kit (740410.100, Machery-Nagel) according to the manufacturer’s protocol. Removal of the native splice acceptor in the 3′ homologous arm of HDR templates was achieved via site-directed mutagenesis (200523, Agilent Technologies).
Hybridoma nucleofection with donor constructs and CRISPR-Cas9
Nucleofection of the HDR template and CRISPR-Cas9 vectors was performed with the SF Cell Line 4D-Nucleofector X Kit L (V4XC-2024, Lonza). Before nucleofection, hybridoma cells were assessed for viability, centrifuged (90g, 5 min), resuspended in phosphate-buffered saline (PBS)/1% FBS, and centrifuged again (90g, 5 min). One million cells were resuspended in SF medium with 1 μg of HDR template and 1 μg of CRISPR-Cas9 vectors or 2 μg of GFP vector (control) and transferred to cuvettes for nucleofection with the 4D-Nucleofection System from Lonza (CQ-104, Program SF). Transfected cells were transferred to a six-well plate in 4 ml of complete medium. The following day, the cells were transferred to a 10-cm petri dish in 10 ml of complete medium supplemented with blasticidin (10 to 20 μg/ml; anti–bl-05, Invivogen). Antibiotic pressure was sustained until GFP-transfected hybridomas were dead and HDR transfections were confluent (typically between days 10 and 14). Subsequently, antibiotic-resistant cells were clonally expanded by seeding the hybridomas in 0.3 cells per well in U-bottom 96-well plates in 100 μl of complete medium. After 10 days, supernatant from wells with a high cell density were obtained for further characterization.
Genomic DNA isolation, PCR, and sequencing
One week after nucleofection with HDR and targeting constructs, a minimum of 10,000 cells of the surviving cell population were collected for genomic phenotyping. DNA was extracted using the Genomic DNA Isolate Kit from Bioline (BIO52067). The DNA pellet was resuspended in ultrapurified water and used for PCR analysis. For PCR confirmation of Fab′ hybridoma, we used primer 1 (TGTAGGAGCTTGGGTCCAGA), which hybridizes the upstream of the 5′ homolog arm of the HDR template, and primer 2 (ATACATTGACACCAGTGAAGATGC), which hybridized with the Bsr gene. For confirmation of the integration of isotype constructs, we used primer 3 (GGCGACCTGTAACAACTTGG), annealing the upstream of the 5′ end of the HDR, and primer 2. PCR products were visualized on a 1% (w/v) agarose gel containing Nancy-520 dye stain. DNA was excised, purified (740609, Macherey-Nagel), and validated via Sanger sequencing.
Flow cytometry
Ten days after limiting dilution, the supernatant from wells with high cell densities were used to stain target cells. Supernatants were transferred to wells in a V-bottom 96-well plate containing 20,000 target cells. After 20 min of incubation at 4°C, plates were centrifuged (90g, 2 min), and hybridoma supernatant was discarded by flicking. Plates were washed twice with PBS/5% FBS and incubated with anti-rIgG2a [phycoerythrin (PE)] (12-4817-8211, Thermo Fisher Scientific) or anti–his-tag (PE) (362603, BioLegend) to assess Fab′-secreting hybridomas. For isotype-grafted hybridomas, the isotype was determined by using the following panel of secondary antibodies on his-tag–positive supernatants: anti-mouse IgG1 (PE) (12-4015-82, Thermo Fisher Scientific), anti-mouse IgG2a (PE) (12-4817-82, Thermo Fisher Scientific), anti-mouse IgG2b [fluorescein isothiocyanate (FITC)] (11-4220-82), anti-mouse IgG3 (A488) (A21151, Thermo Fisher Scientific), and anti-mouse IgA (FITC) (559354, BD Biosciences). To assess antibody specificity, 50,000 JAWSII cells per well were seeded in a V-bottom 96-well plate and stained with Fab′ DEC205, NLDC-145, or AZN-D1 (αDC-SIGN) in concentrations ranging between 0.01 and 1000 nM. After 10 min of incubation at 4°C, 50 μl of the parental antibody NLDC-145 (PE) (1 μg/ml; 138213, BioLegend) was added to the wells. After 30 min of incubation at 4°C, the plate was acquired via fluorescence-activated cell sorting (FACS).
Western blot
Supernatants from Fab′-engineered hybridomas were analyzed for presence of Fab′ fragments by anti–his-tag staining and anti-rIgG (H + L) staining. The purified murine MIH5 isotypes were stained for presence of rat and his-tagged heavy chains. Samples were run under reducing conditions on 12% SDS-PAGE. Gels were transferred on polyvinylidene difluoride membrane and blocked with 3% bovine serum albumin in PBS-Tween 0.02% and stained with rabbit anti–his-tag antibody (ab137839, Abcam), goat anti-rabbit IgG (H + L) (IRD800) (926-32211, LI-COR), and goat anti-rIgG (H + L) (AF680) (A-21096, Thermo Fisher Scientific).
Antibody production and isolation
Engineered hybridomas selected for production were expanded in T175 flasks containing 20 ml of complete medium. Once confluent, hybridomas were cultivated for 7 to 10 days in RPMI 1640 supplemented with 2 mM ultraglutamine, 1× arachidonic acid (AA), 50 μM 2-ME, and 10% FBS before Ni-NTA purification. For larger antibody titers, 20 million cells were seeded in CELLine Bioreactor Flasks (900-05, Argos) and cultivated for 1 week. Antibody-containing supernatants were separated from cells via centrifugation (90g, 5 min), filtered through a 20-μm filter, and supplemented with 10 mM imidazol (I2399, Sigma-Aldrich). Subsequently, the hybridoma supernatant was run over an Ni-NTA column (30210, Qiagen). The column was subsequently washed with 10 column volumes of PBS/25 mM imidazole before antibody elution with PBS/250 mM imidazole. Buffer exchange to PBS [for mIgG3, 300 mM NaH2PO4 and 50 mM NaCl2 (pH 6.5)] was performed via ultracentrifugation with Amicon Ultra-15 centrifugal filter units (Z717185, Sigma-Aldrich). Antibodies from WT hybridomas were obtained by cultivating parental hybridoma using a CD Hybridoma medium (11279023, Thermo Fisher Scientific) supplemented with 2 mM ultraglutamine, 1× AA, and 50 μM 2-ME. Antibodies were purified from medium using Protein G GraviTrap (28-9852-55, Sigma-Aldrich) according to the manufacturer’s protocol. Antibody concentration was determined via absorption at 280 nm with an extinction coefficient of 1.4. Protein purity was assessed by SDS-PAGE using the SYPRO Ruby Protein Gel Stain (S12000, Thermo Fisher Scientific).
Sortase-mediated ligation
To assess sortase-mediated conjugation, a small nucleophilic peptide [GGGCK(FAM)] was constructed via solid-phase peptide synthesis. Antibodies were dialyzed into sortase buffer [150 mM tris, 50 mM NaCl, and 10 mM CaCl2 (pH 7.5)] via ultracentrifugation. Sortase-mediated reactions were carried out with 5 μg of substrate (mAb or Fab′) in 10-μl volumes containing 1 (Fab′) or 2 (mAb) M equivalents of trimutant sortase A Δ59 (3M srt) (25) and 50 (Fab′) or 100 (mAb) M equivalents of GGGCK(FAM) peptide.After 1 hour of incubation at 37°C, the reaction was stopped via addition of EDTA (10 mM final concentration). For each reaction, 500 ng was loaded on reducing SDS-PAGE and analyzed for fluorescence and protein using the SYPRO Ruby Protein Gel Stain on Typhoon Trio+.
Native mass spectrometry analysis
mAbs were treated overnight with 4 U of PNGase F (Roche, IN, USA) at room temperature. Subsequently, treated and untreated samples were buffer-exchanged into 150 mM aqueous ammonium acetate (pH 7.5) by ultrafiltration (Vivaspin 500, Sartorius Stedim Biotech, Germany) with 30-kDa cut-off filter. Sample concentrations were adjusted to 5 μM before 4 μl was used for native mass spectrometry analysis. Native mass spectrometry analysis was carried out with a modified Exactive Plus Orbitrap instrument with extended mass range as described before (50). Intact Mass software was used for zero-charge deconvolution of the spectra to determine the antibody mass (51).
FcγR engagement
SPR measurement were performed on an IBIS MX96 (IBIS Technologies) with a similar methodology as described before (28). In short, biotinylated mouse FcγRI, FcγRIIb, and FcγRIV were immobilized in duplicate ranging from 30 to 1 nM onto a single SensEye G-streptavidin sensor. Then, MIH5 Fc variants were injected over the IBIS in PBS supplemented with 0.075% Tween 80 in concentrations ranging from 0.49 to 1000 nM. Kd calculation was performed by fitting a 1:1 Langmuir binding model to the RU360sec values at each antibody concentration. For consistency, these fits were performed at each receptor concentration, and the final Kd values were calculated by interpolating to Rmax = 500. The measurements were performed in triplicate.
ADCC in vitro
Mouse whole blood ADCC with a 51Cr release assay was performed as described before (52). Briefly, M38 cells were stimulated overnight with IFN-γ (100 ng/ml) to up-regulate PD-L1 expression and used as target cells. Subsequently, the target cells (5000 cells per well) were labeled with 100 μCu 51Cr for 2 hours at 37°C and washed in complete medium. MIH5 isotype variants were added in a final concentration of 10 μg/ml followed by addition of whole blood (25 μl) derived from pegylated granulocyte colony-stimulating factor–stimulated C57BL/6 mice expressing human CD89. Cellular mix was incubated for 4 hours in 200 μl of RPMI 1640 and 10% FBS at 37°C. 51Cr release was counted in 50 μl of supernatant with a liquid scintillation counter. The percentage of specific lysis was calculated as follows: (experimental cpm − basal cpm)/(maximal cpm − basal cpm) × 100, with maximal lysis determined in the presence of 5% Triton X-100 and basal lysis in the present of a nonspecific antibody control (AZN-D1, αDC-SIGN).
B cell depletion in vivo
B cells were isolated from two C57BL/6 mice via magnetic-activated cell sorting (MACS) immunomagnetic depletion (CD43 MicroBeads, 130-049-801, MACS) and labeled with CellTrace Violet (C34557, Thermo Fisher Scientific) or CellTrace Far Red (C34564, Thermo Fisher Scientific). To increase PD-L1 expression, labeled B cells were treated with IFN-γ (100 ng/ml) and interleukin-4 (10 ng/ml) overnight at 37°C. The following day, 9 million Far Red–labeled B cells were incubated with MIH5 mIgG2a, mIgG2b, or mIgA (25 μg/ml) and 27 million violet-labeled B cells with MIH5 mIgG2asilent. Far Red–labeled B cells and violet-labeled B cells were mixed 1:1 and intravenously injected into C57BL/6 mice (6 million per mouse), which were stimulated 6 hours earlier with LPS (1 μg/g). Part of the cell suspension was analyzed to determine the ratio between Far Red and Blue B cells injection. Twenty-four hours after injection, isolated spleens were meshed through a 100-μm cell strainer using a syringe plunger. The resulting cell suspension was centrifuged (90g, 5 min) and incubated in ammonium chloride potassium solution to lyse erythrocytes. After 5 min of incubation, cells were washed with PBS two times and analyzed with flow cytometry to determine the ratio between violet B cells and Far Red B cells. To determine the relative depletion, the following calculation was used 1 − [Injected (Violet:Red)/Isolated (Violet:Red)] × 100. Mice were maintained under specific pathogen–free conditions and treated according to Institutional Animal Care and Use Committee guidelines.
Supplementary Material
Acknowledgments
We thank S. van Duijnhoven (Radboudumc, Nijmegen, The Netherlands) for providing the 3 M sortase A and K. A. Marijt for providing the CT26PD-L1 KO cell line. Funding: This work was supported by the Netherlands Organisation for Scientific Research (NWO; project no. 13770) and in part through the proteomics facility Proteins@Work (project 184.032.201) embedded in the Netherlands Proteomics Centre. C.G.F. is the recipient of Dutch Cancer Society KWO grant 2009-4402, the NWO Spinoza award, and ERC Advance Grant PATHFINDER (269019). M.Ve. is the recipient of ERC Starting grant CHEMCHECK (679921) and a Gravity Program Institute for Chemical Immunology tenure track grant by NWO. T.C. is a PhD student fellow of the Gravity Program Institute for Chemical Immunology. F.A.S. is the recipient of an LUMC fellowship. Author contributions: M.Ve. and F.A.S. conceived the project and contributed equally. J.H.W.L., A.J.R.H., G.V., C.G.F., M.Ve., and F.A.S. provided guidance and support. J.M.S.v.d.S., J.H.W.L., A.J.R.H., G.V., M.Ve., and F.A.S. designed experiments. J.M.S.v.d.S., F.L.F., M.Va., Y.D., A.C., T.C., J.A.C.v.B., M.E., A.E.H.B., M.N., and F.A.S. performed the experiments. I.M.H., M.F.F., A.M.D.B., C.M.L.G., and D.v.D. contributed experimentally. J.M.S.v.d.S., M.Ve., and F.A.S. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions are present in the paper and/or the Supplementary Materials. The described plasmids used in this study are deposited in plasmid repository of Addgene (www.addgene.org/). Additional data and materials related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw1822/DC1
Fig. S1. Genomic map and annotated base pair sequence of rIgG2a constant domains.
Fig. S2. Sortagging of Fab′ fragments derived from CRISPR/HDR-engineered hybridomas.
Fig. S3. Characterization of MIH5 Fc variants hybridomas.
Fig. S4. Raw images of sortagging of MIH5 isotype variants.
Fig. S5. Murine isotype panel generation of NLDC-145 via CRISPR/HDR.
Fig. S6. Glycosylation profile of MIH5 mIgG2a via native mass spectrometry.
Fig. S7. Gating and FACS plots of isotype-dependent depletion in vivo.
Table S1. Fab′ donor construct for HDR.
Table S2. Isotype donor constructs for HDR.
Table S3. FcγR affinity values of MIH5 Fc variants and comparison to literature.
REFERENCES AND NOTES
- 1.Kaplon H., Reichert J. M., Antibodies to watch in 2018. MAbs 10, 183–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Köhler G., Milstein C., Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 3.Turaj A. H., Hussain K., Cox K. L., Rose-Zerilli M. J. J., Testa J., Dahal L. N., Chan H. T. C., James S., Field V. L., Carter M. J., Kim H. J., West J. J., Thomas L. J., He L.-Z., Keler T., Johnson P. W. M., Al-Shamkhani A., Thirdborough S. M., Beers S. A., Cragg M. S., Glennie M. J., Lim S. H., Antibody Tumor Targeting Is Enhanced by CD27 Agonists through Myeloid Recruitment. Cancer Cell 32, 777–791.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arce Vargas F., Furness A. J. S., Litchfield K., Joshi K., Rosenthal R., Ghorani E., Solomon I., Lesko M. H., Ruef N., Roddie C., Henry J. Y., Spain L., Aissa A. B., Georgiou A., Wong Y. N. S., Smith M., Strauss D., Hayes A., Nicol D., O'Brien T., Mårtensson L., Ljungars A., Teige I., Frendéus B.; TRACERx Melanoma; TRACERx Renal; TRACERx Lung consortia, Pule M., Marafioti T., Gore M., Larkin J., Turajlic S., Swanton C., Peggs K. S., Quezada S. A., Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33, 649–663.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arce Vargas F., Furness A. J. S., Solomon I., Joshi K., Mekkaoui L., Lesko M. H., Rota E. M., Dahan R., Georgiou A., Sledzinska A., Aissa A. B., Franz D., Sunderland M. W., Wong Y. N. S., Henry J. Y., O'Brien T., Nicol D., Challacombe B., Beers S. A.; Melanoma TRACERx Consortium; Renal TRACERx Consortium; Lung TRACERx Consortium, Turajlic S., Gore M., Larkin J., Swanton C., Chester K. A., Pule M., Ravetch J. V., Marafioti T., Peggs K. S., Quezada S. A., Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 46, 577–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richman L. P., Vonderheide R. H., Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol. Res. 2, 19–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uchida J., Hamaguchi Y., Oliver J. A., Ravetch J. V., Poe J. C., Haas K. M., Tedder T. F., The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 199, 1659–1669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nimmerjahn F., Ravetch J. V., Immunology: Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 310, 1510–1512 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheong T.-C., Compagno M., Chiarle R., Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat. Commun. 7, 10934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pogson M., Parola C., Kelton W. J., Heuberger P., Reddy S. T., Immunogenomic engineering of a plug-and-(dis) play hybridoma platform. Nat. Commun. 7, 12535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mason D. M., Weber C. R., Parola C., Meng S. M., Greiff V., Kelton W. J., Reddy S. T., High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis. Nucleic Acids Res. 46, 7433–7449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L., Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Khoshnejad M., Brenner J. S., Motley W., Parhiz H., Greineder C. F., Villa C. H., Marcos-Contreras O. A., Tsourkas A., Muzykantov V. R., Molecular engineering of antibodies for site-specific covalent conjugation using CRISPR/Cas9. Sci. Rep. 8, 1760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandes J. C., Therapeutic application of antibody fragments in autoimmune diseases: Current state and prospects. Drug Discov. Today 23, 1996–2002 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Alibakhshi A., Kahaki F. A., Ahangarzadeh S., Yaghoobi H., Yarian F., Arezumand R., Ranjbari J., Mokhtarzadeh A., la Guardia M., Targeted cancer therapy through antibody fragments-decorated nanomedicines. J. Control. Release 268, 323–334 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Tang Q., Onitsuka M., Tabata A., Tomoyashu T., Nagamune H., Construction of anti-HER2 recombinants as targeting modules for a drug-delivery system against HER2-positive cells. Anticancer Res. 38, 4319–4325 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Wu X., Sereno A. J., Huang F., Lewis S. M., Lieu R. L., Weldon C., Torres C., Fine C., Batt M. A., Fitchett J. R., Glasebrook A. L., Kuhlman B., Demarest S. J., Fab-based bispecific antibody formats with robust biophysical properties and biological activity. MAbs 7, 470–482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swiggard W. J., Mirza A., Nussenzweig M. C., Steinman R. M., DEC-205, a 205-kDa protein abundant on mouse dendritic cells and thymic epithelium that is detected by the monoclonal antibody NLDC-145: Purification, characterization, and N-terminal amino acid sequence. Cell. Immunol. 165, 302–311 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Cruz L. J., Rosalia R. A., Kleinovink J. W., Rueda F., Löwik C. W. G. M., Ossendorp F., Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: A comparative study. J. Control. Release 192, 209–218 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Niezold T., Storcksdieck genannt Bonsmann M., Maaske A., Temchura V., Heinecke V., Hannaman D., Buer J., Ehrhardt C., Hansen W., Überla K., Tenbusch M., DNA vaccines encoding DEC205-targeted antigens: immunity or tolerance? Immunology 145, 519–533 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maruyama T., Dougan S. K., Truttmann M. C., Bilate A. M., Ingram J. R., Ploegh H. L., Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Z., Bozzella M., Seluanov A., Gorbunova V., Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 7, 1765–1771 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swee L. K., Guimaraes C. P., Sehrawat S., Spooner E., Barrasa M. I., Ploegh H. L., Sortase-mediated modification of αDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc. Natl. Acad. Sci. U.S.A. 110, 1428–1433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen I., Dorr B. M., Liu D. R., A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U.S.A. 108, 11399–11404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arduin E., Arora S., Bamert P. R., Kuiper T., Popp S., Geisse S., Grau R., Calzascia T., Zenke G., Kovarik J., Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol. Immunol. 63, 456–463 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Kleinovink J. W., Marijt K. A., Schoonderwoerd M. J. A., van Hall T., Ossendorp F., Fransen M. F., PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology 6, e1294299 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dekkers G., Bentlage A. E. H., Stegmann T. C., Howie H. L., Lissenberg-Thunnissen S., Zimring J., Rispens T., Vidarsson G., Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baudino L., Nimmerjahn F., Shinohara Y., Furukawa J.-I., Petry F., Verbeek J. S., Nishimura S.-I., Ravetch J. V., Izui S., Impact of a three amino acid deletion in the CH2 domain of murine IgG1 on Fc-associated effector functions. J. Immunol. 181, 4107–4112 (2008). [DOI] [PubMed] [Google Scholar]
- 30.White A. L., Chan H. T. C., Roghanian A., French R. R., Mockridge C. I., Tutt A. L., Dixon S. V., Ajona D., Verbeek J. S., Al-Shamkhani A., Cragg M. S., Beers S. A., Glennie M. J., Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J. Immunol. 187, 1754–1763 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Bruhns P., Jönsson F., Mouse and human FcR effector functions. Immunol. Rev. 268, 25–51 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Nimmerjahn F., Bruhns P., Horiuchi K., Ravetch J. V., FcγRIV: A novel FcR with distinct IgG subclass specificity. Immunity 23, 41–51 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Gavin A. L., Leiter E. H., Hogarth P. M., Mouse FcgammaRI: Identification and functional characterization of five new alleles. Immunogenetics 51, 206–211 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Grey H. M., Hirst J. W., Cohn M., A new mouse immunoglobulin: IgG3. J. Exp. Med. 133, 289–304 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaelsen T. E., Kolberg J., Aase A., Herstad T. K., Høiby E. A., The four mouse IgG isotypes differ extensively in bactericidal and opsonophagocytic activity when reacting with the P1.16 epitope on the outer membrane PorA protein of Neisseria meningitidis. Scand. J. Immunol. 59, 34–39 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto K., Kurosawa K., Murayama A., Seo H., Ohta K., B Cell-based seamless engineering of antibody Fc domains. PLOS ONE 11, e0167232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulliard Y., Jolicoeur R., Windman M., Rue S. M., Ettenberg S., Knee D. A., Wilson N. S., Dranoff G., Brogdon J. L., Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 210, 1685–1693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bulliard Y., Jolicoeur R., Zhang J., Dranoff G., Wilson N. S., Brogdon J. L., OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol. Cell Biol. 92, 475–480 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Dahan R., Sega E., Engelhardt J., Selby M., Korman A. J., Ravetch J. V., FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 28, 285–295 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Lazar G. A., Dang W., Karki S., Vafa O., Peng J. S., Hyun L., Chan C., Chung H. S., Eivazi A., Yoder S. C., Vielmetter J., Carmichael D. F., Hayes R. J., Dahiyat B. I., Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., Presta L. G., High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Mimoto F., Katada H., Kadono S., Igawa T., Kuramochi T., Muraoka M., Wada Y., Haraya K., Miyazaki T., Hattori K., Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIaR131 and FcγRIIaH131. Protein Eng. Des. Sel. 26, 589–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hezareh M., Hessell A. J., Jensen R. C., Van De Winkel J. G. J., Parren P. W. H. I., Effector Function Activities of a Panel of Mutants of a Broadly Neutralizing Antibody against Human Immunodeficiency Virus Type 1. J. Virol. 75, 12161–12168 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Jong R. N., Beurskens F. J., Verploegen S., Strumane K., van Kampen M. D., Voorhorst M., Horstman W., Engelberts P. J., Oostindie S. C., Wang G., Heck A. J. R., Schuurman J., Parren P. W. H. I., A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol. 14, e1002344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Idusogie E. E., Wong P. Y., Presta L. G., Gazzano-Santoro H., Totpal K., Ultsch M., Mulkerrin M. G., Engineered antibodies with increased activity to recruit complement. J. Immunol. 166, 2571–2575 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Robbie G. J., Criste R., Dall’Acqua W. F., Jensen K., Patel N. K., Losonsky G. A., Griffin M. P., A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob. Agents Chemother. 57, 6147–6153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labrijn A. F., Meesters J. I., de Goeij B. E. C. G., van den Bremer E. T. J., Neijssen J., van Kampen M. D., Strumane K., Verploegen S., Kundu A., Gramer M. J., van Berkel P. H. C., van de Winkel J. G. J., Schuurman J., Parren P. W. H. I., Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. U.S.A. 110, 5145–5150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Labrijn A. F., Meesters J. I., Bunce M., Armstrong A. A., Somani S., Nesspor T. C., Chiu M. L., Altintaş I., Verploegen S., Schuurman J., Parren P. W. H. I., Efficient Generation of Bispecific Murine Antibodies for Pre-Clinical Investigations in Syngeneic Rodent Models. Sci. Rep. 7, 2476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Čaval T., Tian W., Yang Z., Clausen H., Heck A. J. R., Direct quality control of glycoengineered erythropoietin variants. Nat. Commun. 9, 3342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bern M., Caval T., Kil Y. J., Tang W., Becker C., Carlson E., Kletter D., Sen K. I., Galy N., Hagemans D., Franc V., Heck A. J. R., Parsimonious Charge Deconvolution for Native Mass Spectrometry. J. Proteome Res. 17, 1216–1226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Haij S., Jansen J. H. M., Boross P., Beurskens F. J., Bakema J. E., Bos D. L., Martens A., Verbeek J. S., Parren P. W. H. I., van de Winkel J. G. J., Leusen J. H. W., In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res. 70, 3209–3217 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw1822/DC1
Fig. S1. Genomic map and annotated base pair sequence of rIgG2a constant domains.
Fig. S2. Sortagging of Fab′ fragments derived from CRISPR/HDR-engineered hybridomas.
Fig. S3. Characterization of MIH5 Fc variants hybridomas.
Fig. S4. Raw images of sortagging of MIH5 isotype variants.
Fig. S5. Murine isotype panel generation of NLDC-145 via CRISPR/HDR.
Fig. S6. Glycosylation profile of MIH5 mIgG2a via native mass spectrometry.
Fig. S7. Gating and FACS plots of isotype-dependent depletion in vivo.
Table S1. Fab′ donor construct for HDR.
Table S2. Isotype donor constructs for HDR.
Table S3. FcγR affinity values of MIH5 Fc variants and comparison to literature.