

Methionine oxidation by viperin is critical for the expression and function of helicases.
Abstract
Helicases play pivotal roles in fundamental biological processes, and posttranslational modifications regulate the localization, function, and stability of helicases. Here, we report that methionine oxidation of representative helicases, including DNA and RNA helicases of viral (ORF44 of KSHV) and cellular (MCM7 and RIG-I) origin, promotes their expression and functions. Cellular viperin, a major antiviral interferon-stimulated gene whose functions beyond host defense remain largely unknown, catalyzes the methionine oxidation of these helicases. Moreover, biochemical studies entailing loss-of-function mutations of helicases and a pharmacological inhibitor interfering with lipid metabolism and, hence, decreasing viperin activity indicate that methionine oxidation potently increases the stability and enzyme activity of these helicases that are critical for DNA replication and immune activation. Our work uncovers a pivotal role of viperin in catalyzing the methionine oxidation of helicases that are implicated in diverse fundamental biological processes.
INTRODUCTION
Helicases mediate a wide range of cellular processes involving nucleic acid such as replication, repair, recombination, transcription, and translation (1, 2). In recent years, it has become evident that posttranslational modifications (PTMs) of helicases have an essential impact on regulating the localization, stability, and function of helicases. However, researches about the PTMs of helicases are limited mostly to three types of PTMs, namely, phosphorylation, ubiquitination, and SUMOylation (3–5). With the development of unbiased mass spectrometry analysis, previously unidentified PTM sites in helicases continue to be found. In eukaryotes, a family of six homologous subunits—MCM2 (minichromosome maintenance complex component 2) to MCM7—form a complex designated MCM2-7 complex, which is regarded as cellular replicative helicase. During S phase, MCM2-7 complex becomes active to unwind DNA bidirectionally from a replication origin that is under regulation of Cdc45 and the tetrameric GINS (2, 6). Besides these known mechanisms governed by cofactor-mediated regulation, MCM2-7 complex is likely regulated by PTMs in the accurate transition from an inactive state to an active state, which is critical for the spatial and temporal control of DNA replication initiation. Recent studies have revealed key roles of SUMOylation and phosphorylation of MCM2-7 complex in controlling DNA replication initiation (7–9). It remains unclear whether other PTMs and how their corresponding physiological cues may regulate helicases and fundamental biological processes thereof.
Kaposi’s sarcoma–associated herpesvirus (KSHV), belonging to the human γ-herpesvirus family, is a large double-stranded DNA virus with a genome of ~165 kb. KSHV etiologically links to Kaposi’s sarcoma, the most common AIDS-associated malignancy, and two lymphoproliferative diseases: primary effusion lymphoma and multicentric Castleman’s disease (10). Similar to other herpesvirus, KSHV exists in two phases of its life cycle, a dominant latent phase and a transient lytic phase. Lytic replication is critical for viral progeny production and viral pathogenesis (11). Replicated viral genome templates late gene expression and is packaged into capsid to form infectious virion progeny. During lytic replication, KSHV encodes open reading frame 44 (ORF44) (KSHV helicase) to initiate viral DNA replication, although molecular detail of helicase regulation is missing (12). With the development of bacterial artificial chromosome (BAC) (13) technology, recombinant KSHV can be generated with scarless genetic alteration in KSHV genome (14, 15). Therefore, BAC technology provides a powerful tool to probe the function and regulation of KSHV helicase in viral DNA replication, which may further provide insight into the helicases of other species.
In the present study, we found that both KSHV helicase and human DNA helicase MCM7 interact with cellular viperin, which was reported to exhibit antiviral activity against a broad spectrum of viruses (16, 17). One mechanism for antiviral activity was recently revealed by the discovery that viperin catalyzes the conversion of cytidine triphosphate (CTP) to 3′-deoxy-3′,4′-didehydro-CTP (ddhCTP), which acts as a chain terminator for the RNA-dependent RNA polymerases (18). There is one contradictory observation that viperin was previously reported to promote rather than inhibit human cytomegalovirus (HCMV) infection and DNA replication in an iron-sulfur cluster–dependent manner (19, 20). However, no specific catalytic activity has been described for viperin that could explain this observation to date. Here, we demonstrate that viperin oxidizes methionine residues of KSHV helicase, human DNA helicase MCM7, and RNA helicase retinoic acid–inducible gene I (RIG-I). Moreover, triacsin C (T.C), a pharmacological inhibitor of acyl–coenzyme A synthetase that blocks triglyceride synthesis and disrupts lipid droplet formation (21, 22), reduces viperin enzymatic activity. Last, mutations abolishing methionine oxidation in helicases and the treatment with T.C result in reduced stability and activity of helicases. Hence, methionine oxidation potently increases the stability and enzyme activity of these helicases that are fundamental to an array of biological processes such as DNA replication and immune activation. Our work unveils a pivotal function of viperin in biology beyond its function in host defense, forging a potential link between host defense and other processes involving helicases.
RESULTS
KSHV helicase and cellular DNA helicase MCM7 are evolutionarily conserved and interact with viperin
To probe the evolutionary conservation of KSHV helicase and its associated proteins, we performed a phylogenetic analysis of helicases and helicase-associated proteins covering humans, mice, yeast, zebrafish, and herpesviruses involving 672 amino acid sequences that were obtained from the UniProt database and filtered using SWISS-PROT. Bioinformatics analysis demonstrated that KSHV helicase and its associated proteins show some interdependency with helicases of herpesviruses, mouse, and human as well as their associated proteins (Fig. 1A and fig. S1). We further assessed the evolutionary relationship between KSHV helicase and 13 human replicative DNA helicases. This phylogenetic analysis and the amino acid sequence alignment reveal that KSHV helicase is closely related to human DNA helicase MCM7 (Fig. 1B and table S1).
Fig. 1. KSHV helicase and cellular DNA helicase MCM7 are evolutionarily conserved and interact with viperin.
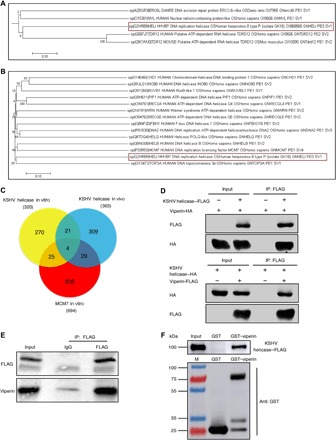
(A) Cladogram demonstrating the conservation between KSHV helicase and other helicases of the taxa in the evolutionary tree. Data represent a small portion of phylogenetic analysis of helicases and helicase-associated proteins covering human, mice, yeast, zebrafish, and herpesviruses. KSHV helicase is boxed in red. (B) Cladogram illustrating the evolutionary relationships between 13 human helicases and KSHV helicase amino acid sequences. The tree is drawn to scale, and its branch lengths represent evolutionary distances. KSHV helicase is boxed in red. (C) Venn diagram describing the overlap of all proteins identified via mass spectrometry in three different immunoprecipitation groups. (D) Reciprocal coimmunoprecipitation (co-IP) assays to examine physical interactions between viperin and KSHV helicase in 293T cells. HA, hemagglutinin. (E) In vivo co-IP assays to detect the interaction between KSHV helicase and viperin. RGB-FLAG cells were induced with doxycycline (2 μg/ml) for 72 hours. Whole-cell lysates (WCLs) were precipitated with anti-FLAG (KSHV helicase). Precipitated proteins and WCLs were analyzed by immunoblotting. IgG, immunoglobulin G. (F) Glutathione S-transferase (GST) pulldown, with GST or GST-viperin purified from Escherichia coli and the KSHV helicase–FLAG translated in vitro, was analyzed by immunoblotting.
Using BAC technology, we constructed a recombinant KSHV in which the FLAG tag is fused to the C terminus of KSHV helicase in the genome and established a stable cell line infected with this recombinant KSHV [RGB (Red-Green-Blue)-FLAG cells]. Molecular virology assays indicated the correct insertion of the FLAG sequence and the lack of alteration on KSHV lytic replication (fig. S2, A and B). We then probed KSHV helicase function in viral lytic replication using two pairs of small interfering RNAs (siRNAs). As shown in fig. S2 (C and D), the viral genome copy number was greatly reduced upon depletion of KSHV helicase. We also adopted the same recombination strategy to construct two KSHV containing helicase stop or helicase deletion via the insertion of a stop codon or deletion of the entire protein-coding sequence of KSHV helicase and then assessed their ability to produce viral progeny. Consistent with the siRNA results, knockout of KSHV helicase resulted in at least 10-fold reduction in the viral genome copy number compared to that of the parental FLAG-tagged recombinant KSHV (fig. S2, E and F). These results show that KSHV helicase is critical for viral DNA replication during lytic phase.
To better understand the function and regulation of KSHV helicase, we aimed to identify the cellular binding partners of KSHV helicase. Considering that KSHV helicase shows a high evolutionary similarity to human replicative DNA helicase MCM7, we used immunoprecipitation coupled with mass spectrometry to analyze proteins copurified with these helicases (fig. S2, G and H). With proteins identified by tandem mass spectrometry, we constructed a Venn diagram, which illustrates that 4 proteins are enriched in all three cell lines and 58 proteins are identified as potential interacting proteins of both KSHV helicase and cellular MCM7 (Fig. 1C). Viperin is ranked the first among 58 proteins, indicating that viperin is the most abundant binding protein of KSHV helicase. Coimmunoprecipitation (co-IP) assays showed that KSHV helicase formed complexes with viperin in transfected human embryonic kidney (HEK) 293T cells and KSHV-infected cells (Fig. 1, D and E). Furthermore, we also demonstrated that KSHV helicase directly interacted with viperin by in vitro glutathione S-transferase (GST) pulldown assay (Fig. 1F).
We next assessed the role of viperin in KSHV lytic replication. We examined viperin expression in cells in which KSHV lytic replication was induced with doxycycline and found that both the RNA and protein expression of viperin were increased in KSHV lytic replication (fig. S3, A and B). We then depleted viperin expression by siRNA and examined KSHV lytic replication. Viperin depletion greatly reduced both extracellular and intracellular viral DNA copy number, suggesting that viperin is essential for the replication of KSHV genomic DNA (fig. S3, C and D). Previous studies have shown that replication and transcription activator (23) encoded by KSHV ORF50 is the master molecular switch that drives KSHV from latency into lytic replication through transactivating its downstream viral and cellular genes (24, 25). Thus, we first tested whether replication and transcription activator (RTA) could up-regulate viperin expression in a SLK cell line that RTA expression can be induced by doxycycline (iSLK)–puro cells and 293T cells. As shown in fig. S3 (E to H), stable (iSLK) or transient (293T) expression of RTA greatly increased viperin RNA and protein, indicating that RTA is sufficient to elevate viperin expression. We next cloned a DNA fragment corresponding to the viperin promoter [from −1597 to +136 base pairs (bp)] into a luciferase reporter vector (26). Using this reporter, we found that RTA was sufficient to transactivate the viperin promoter (fig. S3I). To further determine whether RTA is directly recruited to the viperin promoter and to define the binding site(s) of RTA in the viperin promoter, we designed 17 pairs of primers encompassing approximately 1800 bp of the viperin promoter and performed a chromatin immunoprecipitation (ChIP) assay in transfected 293T cells. ChIP assay and subsequent quantitative polymerase chain reaction (qPCR) analysis revealed substantial enrichment of region 4 (from −1297 to −1147 bp) within the viperin promoter precipitated by RTA (fig. S3J). These results conclude that RTA, as a transcriptional factor, acts on viperin promoter to up-regulate its expression. Moreover, given that viperin is a well-known interferon-stimulated gene, we also discussed the possible contribution of interferon on the viperin expression in KSHV lytic replication during which type I interferon pathways are activated (27). We first showed that RTA depletion greatly reduced viperin expression during KSHV lytic replication, emphasizing the importance of RTA in viperin expression (fig. S3K). We then treated cells with BX795, an inhibitor to block TBK-1 (TANK-binding kinase 1) and downstream interferon-β (IFN-β) production (28). After assessing the effects of BX795 by detecting IFN-β mRNA expression, we then examined the expression of viperin with BX795 treatment during KSHV lytic replication (fig. S3, L to N). These results showed that both RTA and interferon are required for efficient viperin protein expression, while viperin up-regulation is mostly dependent on RTA during KSHV lytic reactivation.
Viperin promotes methionine oxidation of KSHV helicase
Viperin contains a conserved domain, from amino acids 71 to 182, comprising four radical SAM (S-adenosyl-l-methionine) enzymatic motifs. Each motif contains a CxxxCxxC sequence that is required for binding iron-sulfur (Fe-S) clusters. Despite the fact that viperin can bind Fe-S clusters via the CxxxCxxC motif, enzymatic activity of viperin on protein substrates has never been reported (29). Thus, we analyzed the PTM of KSHV helicase with viperin overexpression through mass spectrometry. This analysis identified five oxidized methionine residues that were only found with viperin expression: methionine-321 (M321), methionine-401 (M401), methionine-425 (M425), methionine-663 (M663), and methionine-754 (M754), indicating that viperin promotes methionine oxidation of KSHV helicase (Fig. 2A and fig. S4A). Methionine oxidation has been recognized as a naturally occurring PTM in native proteins and is implicated in the regulation of cell function via altering protein stability (30–32). Therefore, we first tested whether viperin affects KSHV helicase protein expression and found that viperin increased KSHV helicase protein in a dose-dependent manner, while it had no effect on its mRNA level (Fig. 2, B and C, and fig. S4, B and C). Conversely, depletion of viperin (via siRNA-mediated knockdown) in RGB-FLAG cells reduced KSHV helicase expression (Fig. 2D). To further explore the role of viperin in modulating KSHV helicase protein expression, we determined KSHV helicase half-life in 293T cells with the protein synthesis inhibitor cycloheximide (CHX). Notably, coexpression of viperin increased the half-life of KSHV helicase by more than 4 hours (Fig. 2E). Together, viperin promotes methionine oxidation, thereby stabilizing KSHV helicase protein.
Fig. 2. Viperin promotes methionine oxidation of KSHV helicase.
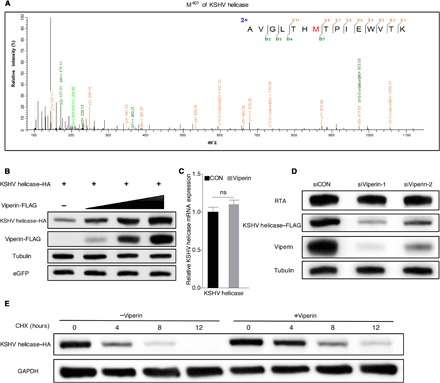
(A) Mass spectrometry analysis of methionine oxidation in 293T cells transfected with KSHV helicase with or without viperin expression. The M401 was oxidized (in red) under viperin overexpression. m/z, mass/charge ratio. (B) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were harvested and analyzed by immunoblotting. eGFP, enhanced green fluorescent protein. (C) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, RNA was extracted, and complementary DNA (cDNA) was prepared to determine KSHV helicase mRNA by qPCR analysis. The data are expressed as means ± SEM; n = 3; ns, not significant; CON, control. (D) RGB-FLAG cells were transfected with siRNA as indicated. At 6 hours after transfection, cells were induced with doxycycline (2 μg/ml) for 72 hours. WCLs were then analyzed by immunoblotting. (E) 293T cells were transfected with plasmids containing indicated genes. At 24 hours after transfection, cells were treated with cycloheximide (CHX) (10 μg/ml). WCLs were then analyzed by immunoblotting. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Viperin-induced methionine-401 oxidation enhances the stability and function of KSHV helicase
To further examine the effect of individual methionine oxidative site, we engineered (either individually or in combination) 11 mutants of KSHV helicase through site-directed mutagenesis and constructed 11 corresponding stable cell lines infected with this recombinant KSHV using BAC technology, as described previously (RGB-mutated FLAG cells). These 11 mutants consist of single point mutants (PMs), a mutant with mutations in all five sites (ALL Mut), and mutants containing four except one methionine (Re) (Fig. 3A). All methionine residues were changed to alanine. We then determined whether these mutations have an effect on KSHV helicase stability and function. Using transfected 293T cells, we demonstrated that, with the mutant containing methionine-401 (PM321, PM425, PM663, PM754, and Re401), viperin still increased KSHV helicase expression. By stark contrast, the M401A mutation (PM401, ALL Mut, Re321, Re425, Re663, and Re754) completely abolished the effect of viperin to increase KSHV helicase protein (Fig. 3, B and C). These results are consistent with those of stable KSHV helicase expression in recombinant virus–infected cell lines and those of dose-dependent manner of mutated plasmids (Fig. 3, D and E, and fig. S5, A to F). To explore the importance of viperin in modulating KSHV helicase stability, we examined stable KSHV helicase expression in KSHV-infected cell lines after viperin depletion. When viperin expression was depleted, the expression of KSHV helicase containing methionine-401 declined. However, viperin deletion had little effect on the expression of those helicase mutants containing M401A (Fig. 3, F and G, and fig. S5, G and H). These results demonstrate that methionine-401 is critical for the effect of viperin to increase KSHV helicase protein expression. Given that the methionine-401 site has the most effect on KSHV helicase stability, we then investigated whether this effect translates into the replication of KSHV genomic DNA. We detected the extracellular and intracellular viral genome copy number upon KSHV lytic induction and found that the viral genome copy number obviously declined in KSHV, with helicase containing M401A mutation comparing with those with M401 (Fig. 3H and fig. S5I). Consistent with this result, viperin depletion decreased viral DNA copy number of KSHV containing M401 but had a marginal effect on viral copy number in those containing M401A (Fig. 3I and fig. S5J). These results collectively show that methionine oxidation promotes helicase stability and the replication of viral genomic DNA during KSHV lytic replication.
Fig. 3. Viperin-induced methionine-401 oxidation enhances the stability and function of KSHV helicase.
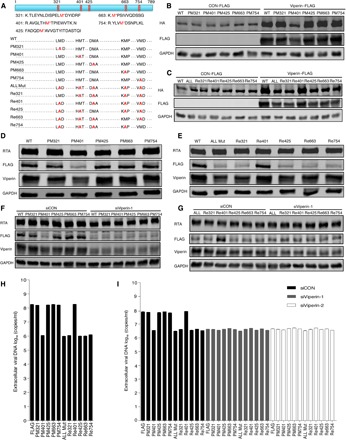
(A) Diagram illustrating five peptides of KSHV helicase examined to evaluate methionine oxidation (top) and depicting the 11 KSHV helicase mutants (bottom). WT, wild type. (B and C) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. (D and E) RGB-FLAG and RGB-mutated FLAG cells were induced with doxycycline for 72 hours. WCLs were then analyzed by immunoblotting. (F and G) RGB-FLAG and RGB-mutated FLAG cells were transfected with siRNA as indicated. At 6 hours after transfection, cells were induced with doxycycline for 72 hours. WCLs were then analyzed by immunoblotting. (H) RGB-FLAG and RGB-mutated FLAG cells were induced with doxycycline for 72 hours. Extracellular viral DNA was extracted and analyzed by qPCR. (I) RGB-FLAG and RGB-mutated FLAG cells were transfected with siRNA as indicated. At 6 hours after transfection, cells were induced with doxycycline for 72 hours. Extracellular viral DNA was extracted and analyzed by qPCR. The concentration of doxycycline is 2 μg/ml. For (H) and (I), the data are expressed as means ± SEM; n = 3.
Viperin associates with lipid droplets that are required to induce the methionine oxidation of KSHV helicase
Previous research has demonstrated that the N-terminal domain of viperin (amino acids 9 to 42) contains an amphipathic α helix that anchors viperin to the cytosolic face of the endoplasmic reticulum (ER) and lipid droplets (33, 34). Viperin localizes to the ER and is ultimately transported to lipid droplets, where it performs its main functions (35, 36). Hence, we reasoned that methionine oxidation of KSHV helicase induced by viperin requires lipid droplets. Thus, we performed an immunofluorescence assay in the iSLK-BAC16 cell line and found that viperin and helicase localized in lipid droplets during KSHV replication, indicating that KSHV helicase colocalizes with viperin in lipid droplets in vivo (Fig. 4A). Therefore, we treated RGB-FLAG cells with T.C, one of the few compounds that inhibit the formation of lipid droplets (37, 38). To assess the effect of T.C, we detected the expression of perilipin 2 that is a prominent protein associated with the surface of lipid droplets and participates in the formation and stabilization of lipid droplets (fig. S6, A and B) (39, 40). We demonstrated that both KSHV helicase and viperin expression in KSHV-infected cells were reduced by T.C treatment (Fig. 4B). Consistent with reduced protein expression, both viral extracellular and intracellular genome copy number also declined when cells were treated with T.C (Fig. 4C). Furthermore, we also detected stable KSHV helicase expression as well as viral extracellular and intracellular DNA copy number in 11 KSHV-infected cell lines with or without T.C treatment. These results showed that when cells were treated with T.C that suppresses viperin expression, both helicase expression and viral DNA copy number were impaired for helicase containing M401. However, T.C treatment had little effect on both helicase expression and viral DNA copy number in helicases containing M401A (Fig. 4, D to F, and fig. S6C). Similarly, T.C treatment impaired viperin enzyme activity to stabilize KSHV helicase in 293T cells (Fig. 4G). To further examine the site(s) of methionine oxidation in KSHV helicase, we adopted mass spectrometry to analyze the methionine oxidation of KSHV helicase after T.C treatment. As shown in fig. S6D, the methionine-754 site was deprived of oxidation, while oxidation of methionine-321, methionine-401, methionine-425, and methionine-663 was undetectable under T.C treatment. Together, these results demonstrate that the methionine oxidation of KSHV helicase induced by viperin requires lipid droplets.
Fig. 4. Viperin associates with lipid droplets that are required to induce the methionine oxidation of KSHV helicase.
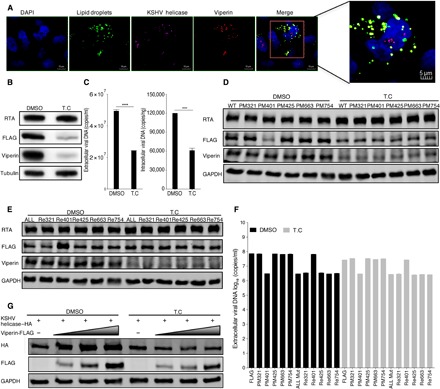
(A) In vivo immunofluorescence assay to detect KSHV helicase and viperin colocalization with lipid droplets in iSLK-BAC16 KSHV helicase-FLAG (GFP deletion) cells. Cells were induced with doxycycline for 72 hours and immunostained with a mouse anti-FLAG antibody against KSHV helicase (pink) and a rabbit anti-viperin antibody (red). Lipid droplets (green) and nuclei (blue) were labeled with BODIPY and 4′,6-diamidino-2-phenylindole (DAPI), respectively. Cells were analyzed by Leica confocal microscopy and representative images were shown. The magnified panel was derived from the red box in the merged panel. Scale bars, 10 μm (merged panel) and 5 μm (magnified panel). (B and C) RGB-FLAG cells were treated with 1 μΜ T.C or dimethyl sulfoxide (DMSO) as a negative control for 3 hours, and then cells were induced with doxycycline for 72 hours. (B) WCLs were analyzed by immunoblotting; (C) viral DNA was extracted and analyzed by qPCR. (D to F) RGB-FLAG and RGB-mutated FLAG cells were treated with 1 μΜ T.C or DMSO as a negative control for 3 hours, and then cells were induced with doxycycline for 72 hours. (D and E) WCLs were analyzed by immunoblotting; (F) extracellular viral DNA was extracted and analyzed by qPCR. (G) 293T cells were treated with 1 μΜ T.C or DMSO as a negative control for 3 hours, and then cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. The concentration of doxycycline is 2 μg/ml. For (C) and (F), the data are expressed as means ± SEM; n = 3; ****P < 0.0001.
Viperin-induced methionine oxidation stabilizes MCM7 and promotes DNA replication
Next, we assessed whether the human DNA helicase MCM7 undergoes methionine oxidation induced by viperin. Two-way co-IP assays showed that MCM7 interacted with viperin in transfected 293T cells (Fig. 5A). Furthermore, viperin stabilized MCM7 protein, as indicated by longer half-life, thus increasing MCM7 protein expression in a dose-dependent manner, but had no impact on MCM7 mRNA level (Fig. 5, B and C, and fig. S7A). To characterize the molecular action of viperin in MCM7 protein expression, we adopted mass spectrometry to analyze the methionine oxidation of MCM7 and identified three methionine oxidation sites: methionine-450 (M450), methionine-621 (M621), and methionine-639 (M639) (fig. S7B). Similarly, these triple methionine sites were all mutated to alanine (M450/621/639A, MCM7-TA), and we found that viperin demonstrated no effect on the protein expression of MCM7-TA (Fig. 5D). We noted that MCM7 was reported to directly bind to the c-Myc replication origin (41, 42), and we therefore performed ChIP in 293T cells with transient expression of MCM7 wild type or the MCM7-TA mutant to determine whether methionine oxidation affects the binding capacity of MCM7. The c-Myc replication origin bound by MCM7-TA was reduced by threefold than that of MCM7 wild type (Fig. 5E). Consequently, MCM7-TA resulted in threefold lower nascent (newly replicated) DNA driven by the c-Myc replication origin than MCM7 wild type (Fig. 5F). To further probe the role of viperin in mediating MCM7 methionine oxidation, we repeated these experiments upon viperin depletion. These results showed that viperin knockdown greatly reduced both the binding capacity and nascent DNA production of the c-Myc replication origin driven by MCM7 wild type. However, viperin depletion had little effect on both the DNA binding and nascent DNA production of MCM7-TA at the c-Myc replication origin (Fig. 5, G and H). Furthermore, the interaction between endogenous MCM7 and viperin was readily detected (Fig. 5I). We also determined the expression and function of endogenous MCM7 after viperin depletion (Fig. 5, J to L). These results examining endogenous proteins indicate the important role of viperin in the protein expression and function of MCM7. Together, these results demonstrate that viperin induces the methionine oxidation of MCM7 to promote its protein expression and subsequent DNA replication.
Fig. 5. Viperin-induced methionine oxidation stabilizes MCM7 and promotes DNA replication.
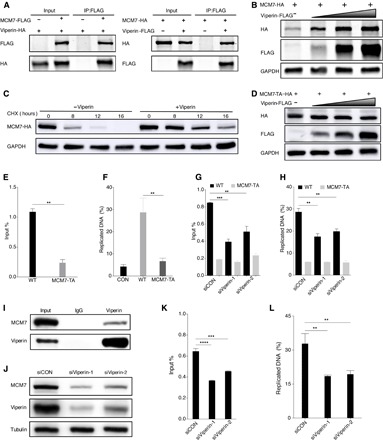
(A) Reciprocal co-IP assays to examine physical interactions between viperin and MCM7. (B) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. (C) 293T cells were transfected with plasmids containing indicated genes. At 24 hours after transfection, cells were treated with CHX (10 μg/ml). WCLs were then analyzed by immunoblotting. (D) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. (E) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, cells were harvested, cross-linked, and used for ChIP assay to determine relative binding of MCM7 wild type and MCM7-TA at cellular c-Myc replication origin. (F) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, cells were labeled with EdU (5-ethynyl-2′-deoxyuridine), total DNA was extracted, and Click-iT procedure was used to isolate DNA from the active sites of replication. DNA was then analyzed by qPCR at cellular c-Myc replication origin. (G) 293T cells were transfected with plasmids containing indicated genes after transfection for 6 hours with siRNA, as indicated. At 48 hours after transfection, cells were harvested, cross-linked, and used for ChIP assay to determine relative binding of MCM7 wild type and MCM7-TA at cellular c-Myc replication origin. (H) 293T cells were transfected with plasmids containing indicated genes after transfection for 6 hours with siRNA, as indicated. At 48 hours after transfection, cells were labeled with EdU, total DNA was extracted, and Click-iT procedure was used to isolate DNA from the active sites of replication. DNA was then analyzed by qPCR at cellular c-Myc replication origin. (I) In vivo co-IP assays to detect the interaction between MCM7 and viperin. The WCLs of 293T were precipitated with anti-viperin. Precipitated proteins and WCLs were analyzed by immunoblotting. (J) 293T cells were transfected with siRNA, as indicated. At 48 hours after transfection, WCLs were then analyzed by immunoblotting. (K) 293T cells were transfected with siRNA, as indicated. At 48 hours after transfection, cells were harvested, cross-linked, and used for ChIP assay to determine relative binding of endogenous MCM7 at cellular c-Myc replication origin. (L) 293T cells were transfected with siRNA as indicated. At 48 hours after transfection, cells were labeled with EdU, total DNA was extracted, and Click-iT procedure was used to isolate DNA from the active sites of replication. DNA was then analyzed by qPCR at cellular c-Myc replication origin. For (E) to (H), (K), and (L), the data are expressed as means ± SEM; n = 3; **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Considering that MCM7 is associated with MCM4 and MCM6 to form the MCM4/6/7 complex that assumes the function of replicative DNA helicase (43, 44). We therefore sought to determine whether viperin plays the same role for MCM4 and MCM6. We applied co-IP assays to examine the physical interaction between viperin and MCM4 or MCM6 (fig. S8, A and B). Viperin did not enhance the expression of either MCM4 or MCM6 (fig. S8, C and D). Moreover, DNA helicases are conserved among several species; we thus selected several key DNA helicases to examine whether viperin promotes methionine oxidation of these DNA helicases to affect their stability and functions. These selected DNA helicases include human DNA2 helicase and helicases of HCMV (α-herpesvirus) and murine γ-herpesvirus 68 (MHV68). The latter two viral helicases are crucial for DNA replication and productive infection of the corresponding virus (45, 46). Two-way co-IP assays showed that viperin interacted with human DNA2, HCMV, and MHV68 helicases in transfected 293T cells (fig. S8, E to G). Hence, viperin enhanced the protein expression of these helicases (fig. S8, H to J). Together, these results suggest that viperin-induced methionine oxidation ubiquitously exists in various DNA helicases.
Methionine oxidation catalyzed by viperin increases the stability and function of RNA helicase RIG-I
To this end, we showed that viperin-induced methionine oxidation exists in multiple DNA helicases, which stabilizes protein expression and increases their function in DNA replication. Hence, we asked whether this is true for RNA helicases. RIG-I is an RNA helicase that senses double-stranded RNA in the cytosol to induce innate immune activation (47, 48), and we therefore probed the effect of viperin on RIG-I. Similar to those DNA helicases, we found that viperin interacted with RIG-I by two-way co-IP assays (Fig. 6A) and that viperin increased RIG-I protein but not mRNA expression, which correlated with increased protein half-life (Fig. 6, B and C, and fig. S9A). Next, we then analyzed the PTM of RIG-I with viperin overexpression through mass spectrometry. This analysis identified five oxidized methionine residues that were only found with exogenous viperin expression: methionine-51 (M51), methionine-755 (M755), methionine-760 (M760), methionine-761 (M761), and methionine-923 (M923), indicating that viperin participates in RIG-I methionine oxidation (fig. S9B). Mutational analysis indicated that the quintuple mutation (RIG-I–Mut), but not single methionine to alanine mutations, abolished the viperin effect on RIG-I protein expression that was increased in a dose-dependent manner (Fig. 6D). To probe the effect of methionine oxidation on RIG-I function, we expressed exogenous RIG-I wild type (RIG-I–WT) or the quintuple mutant and analyzed CXCL10 (C-X-C motif chemokine ligand 10) and IFN-β mRNA expression by qPCR in response to polyinosinic:polycytidylic acid [poly(I:C)] stimulation. As shown in Fig. 6E, compared with RIG-I–WT, the quintuple RIG-I mutant demonstrated notably lower activity to induce CXCL10 and IFN-β. We then “reconstituted” RIG-I expression in Rig-i−/− MEF (mouse embryonic fibroblast) with RIG-I–WT or RIG-I–Mut and examined host immune responses and viral infection. Compared to RIG-I–WT, RIG-I–Mut obviously lost the capacity to reduce vesicular stomatitis virus (VSV) replication in Rig-i−/− MEFs (Fig. 6F). Furthermore, to assess the role of viperin in RIG-I–mediated signaling, we repeated the above experiments entailing CXCL10 and IFN-β mRNA induction by poly(I:C) and VSV replication in reconstituted Rig-i−/− MEFs under the condition of viperin depletion. These results showed that viperin depletion greatly reduced the CXCL10 and IFN-β induction by RIG-I–WT but had a marginal effect on the CXCL10 and IFN-β induction by the quintuple RIG-I mutant (Fig. 6G). Besides, after viperin depletion, the ability of RIG-I–WT to inhibit VSV replication decreased as much as that of RIG-I–Mut (Fig. 6H). Further, we examined the interaction between endogenous RIG-I and viperin (fig. S9C). We also determined the protein expression and function of endogenous RIG-I after viperin depletion (Fig. 6, I and J, and fig. S9D). These results derived from endogenous proteins also emphasized the importance of viperin in protein expression and function of RIG-I. Together, these results show that viperin induces the methionine oxidation of RNA helicase RIG-I, which increases RIG-I protein expression and signaling activity in innate immunity.
Fig. 6. Methionine oxidation catalyzed by viperin increases the stability and function of RNA helicase RIG-I.
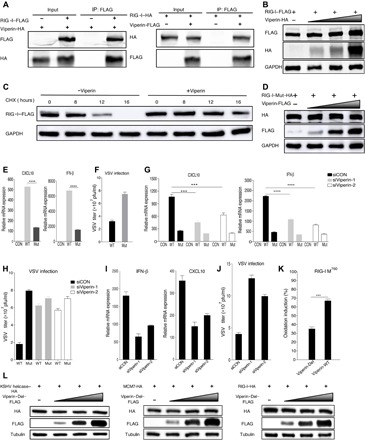
(A) Reciprocal co-IP assays to examine physical interactions between viperin and RIG-I. (B) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. (C) 293T cells were transfected with plasmids containing indicated genes. At 24 hours after transfection, cells were treated with CHX (10 μg/ml). WCLs were then analyzed by immunoblotting. (D) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. (E) 293T cells were transfected with plasmids containing indicated genes. At 24 hours after transfection, cells were transfected with poly(I:C) for 16 hours. RNA was extracted, and cDNA was prepared to determine CXCL10 and IFN-β mRNA expression by qPCR analysis. (F) Reconstituted MEF cells were infected with VSV [multiplicity of infection (MOI), 0.1], and viral titer in the supernatant was determined by plaque assay. pfu, plaque-forming units. (G) 293T cells were transfected with plasmids containing indicated genes after transfection for 6 hours with siRNA, as indicated. At 24 hours after transfection, cells were transfected with poly(I:C) for 16 hours. RNA was extracted, and cDNA was prepared to determine CXCL10 and IFN-β mRNA expression by qPCR analysis. (H) Reconstituted MEF cells were infected with VSV (MOI, 0.1) after transfection for 24 hours with siRNA, as indicated, and viral titer in the supernatant was determined by plaque assay. (I) 293T cells were transfected with siRNA, as indicated. At 24 hours after transfection, cells were transfected with poly(I:C) for 16 hours. RNA was extracted, and cDNA was prepared to determine CXCL10 and IFN-β mRNA expression by qPCR analysis. (J) MEF cells were infected with VSV (MOI, 0.1) after transfection for 24 hours with siRNA, as indicated, and viral titer in the supernatant was determined by plaque assay. (K) Viperin wild type (Viperin-WT), viperin-Del, and RIG-I were purified separately from 293T cells. Oxidative reaction was performed in vitro, and methionine oxidized peptides were quantitatively determined by mass spectrometry analysis. (L) 293T cells were transfected with plasmids containing indicated genes. At 48 hours after transfection, WCLs were analyzed by immunoblotting. For (E to K), the data are expressed as means ± SEM; n = 3; ***P < 0.001 and ****P < 0.0001.
Last, to further verify the enzymatic capacity of viperin, we purified viperin-WT (viperin wild type), viperin-Del (deletion of the central SAM enzymatic domain), RIG-I, MCM7, and KSHV helicases separately from 293T cells. We then performed oxidative reaction in vitro and analyzed helicases for possible methionine oxidation. Tandem mass spectrometry analysis indicated that viperin-WT, compared to the viperin-Del mutant, notably enhanced the oxidation of M760 of RIG-I (Fig. 6K). In addition, viperin-WT increased the oxidation of M51 of RIG-I and M450, M556, and M639 of MCM7 in reference to the viperin-Del mutant. However, viperin-WT slightly promoted the oxidation of M321 and M663 of KSHV helicase, largely due to the high level of oxidation in the presence of the viperin-Del mutant (fig. S9E). These results collectively show that viperin has intrinsic enzyme activity to oxidize helicases in vitro. Moreover, we also showed that viperin-Del was deprived of the ability to enhance the expression of helicases (Fig. 6L). These results emphasized the importance of the central SAM enzymatic domain in the functions of viperin and verified that viperin enhanced helicase expression through catalyzing their methionine oxidation.
DISCUSSION
In recent years, it has become evident that PTMs have a profound effect on the localization, function, and stability of helicases. However, researches about the PTMs of helicases are limited, mostly focusing on three types of PTMs including phosphorylation, ubiquitination, and SUMOylation. Our study found a previously unexplored PTM-methionine oxidation in helicases, which increases helicase stability and protein expression to promote their functions. Mechanistically, viperin catalyzes methionine oxidation of these DNA and RNA helicases of human and virus origin. On the basis of the recent research, Gizzi and colleagues (18) made a great contribution to find the nucleotide substrate of viperin, which suggests a unifying mechanism for the broad antiviral effects of viperin. Meanwhile, our work showed that viperin catalyzes helicases, which are crucial for DNA replication and immune activation. Our work also partially explains the mechanism that viperin promotes rather than inhibits HCMV infection and KSHV genomic DNA replication. To our knowledge, these helicases constitute the first family of protein substrates that are enzymatically modified by viperin. Along with the report of the Gizzi group that viperin catalyzes the conversion of CTP to ddhCTP, our work is among the first to show that viperin harbors intrinsic enzyme activity to modify proteins and small molecules (such as nucleotides), leading to protein methionine oxidation that enhances helicase stability and function. These studies represent the first work revealing the protein substrates for viperin. The biochemical property of viperin as an enzyme to oxidize proteins calls for more detailed investigation. Furthermore, the observation that viperin resides in the lipid droplets to catalyze methionine oxidation ascribes an unprecedented function of lipid droplets in PTM of proteins. Last, the ubiquitous existence of methionine oxidation in various DNA and RNA helicases implicates the role of viperin and methionine oxidation in a broad spectrum of fundamental biological processes, such as DNA replication and innate immune sensing, as shown in this study.
MATERIALS AND METHODS
Cell culture
Human embryonic kidney (HEK) 293T, MEF, iSLK-puro, iSLK-RGB, and related KSHV helicase mutation recombinant virus–infected cell lines were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), antibiotics (penicillin and streptomycin), and proper selective agents [hygromycin (0.5 mg/ml), puromycin (1.5 μg/ml), and G418 (0.5 mg/ml)]. All cells were cultured at 37°C in the presence of 5% CO2.
Antibodies and reagents
The anti-FLAG mouse antibody, anti-HA (hemagglutinin) rabbit antibody, and anti-tubulin mouse antibody were purchased from Sigma-Aldrich. The anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mouse antibody was purchased from Abmart. The anti-viperin rabbit antibody was purchased from Cell Signaling Technology. The anti-viperin mouse antibody was purchased from Santa Cruz Biotechnology. The anti-GST mouse antibody was purchased from ABclonal. The anti-MCM7 rabbit antibody and the anti–RIG-I rabbit antibody were purchased from Abcam, and the secondary antibodies were as follows: goat anti-mouse IRDye 800CW and goat anti-rabbit IRDye 800CW from LI-COR and goat anti-rabbit immunoglobulin G (IgG) (H+L) Alexa Fluor 488 and goat anti-mouse IgG (H+L) Alexa Fluor 555 from Invitrogen. The other reagents used in our experiments and their sources were as follows: anti-FLAG M2 affinity Sepharose from Sigma-Aldrich; recombinant protein A/G agarose from Invitrogen; Protease Inhibitor Cocktail Set III from Millipore; doxycycline hyclate, hygromycin, puromycin, and G418 disulfate salt from Sigma-Aldrich; Lipofectamine 2000 from Thermo Fisher Scientific; T.C and FuGENE HD Transfection Reagent from Promega; and CCK8 Kit from Yeasen Biotech.
Plasmids
The KSHV helicase (KSHV ORF44), MHV68 helicase, HCMV helicase, viperin, MCM4, MCM6, MCM7, RIG-I, and DNA2 proteins were all expressed in both the streptomycin-FLAG–tagged pCDH and HA-tagged pCMV vectors. The helicases of KSHV, MHV68, and HCMV were all cloned from the complementary DNA (cDNA) of their genomes. The viperin, MCM4, MCM6, MCM7, RIG-I, and DNA2 proteins were all cloned from the cDNA of the HEK293T genome. The KSHV helicase, MCM7, and RIG-I mutant plasmids were constructed via site-directed mutagenesis following the manufacturer’s protocol of the TIANGEN Fast Site-Directed Mutagenesis Kit. The luciferase reporter plasmid pGL3E-pViperin was constructed by inserting the amplified fragment of the viperin promoter (−1597 to +136 bp) from the HEK293T genomic library into the pGL3-Enhancer vector.
Genetic manipulation of the BAC-cloned KSHV genome
To generate a series of KSHV helicase mutant KSHV genomes, mutagenesis of BAC16-RGB (RGB) was performed using a recombinant system as previously described. Briefly, a linear DNA fragment was amplified via PCR using the pEPKan-S plasmid as a template, which contained a kanamycin cassette, an I-SceI restriction enzyme site, and duplicated flanking sequences derived from KSHV genomic DNA (approximately 40-bp copy). The PCR product was treated with Dpn I to remove the plasmid template, and the purified PCR fragments were electroporated into the RGB-containing GS1783 strain, which had been induced at 42°C for 15 min. The recombinant clones were selected at 32°C on LB plates containing 12.5 μg of chloramphenicol and kanamycin (50 μg/ml). Positives clones were treated with 1% l-arabinose, induced at 42°C, and plated on LB plates containing 1% l-arabinose for secondary recombination. The resultant BAC was further confirmed through DNA sequencing.
Constitution of the recombinant iSLK-RGB cell lines
The BAC-RGB plasmid was extracted using the NucleoBond Xtra Midi Kit. To generate KSHV helicase wild type or mutation recombinant virus–infected cells, 4 μg of the wild-type RGB or mutant RGB plasmids, respectively, was transfected into iSLK cells with 10 μl of the FuGENE HD Transfection Reagent. The medium of the iSLK cells was replaced with DMEM alone during transfection. At 1 hour after transfection, FBS was added to a final concentration of 10%. On the following day, the transfected iSLK cells were cultured in complete DMEM with a final concentration of hygromycin B (500 μg/ml) for selection. After 20 days of selection, hygromycin-resistant colonies were trypsinized, pooled, and subcultured. To induce viral lytic replication, the medium of BAC-carrying iSLK cells was replaced with fresh medium containing doxycycline (2 μg/ml) for 3 to 5 days.
Mass spectrometry
Mass spectrometry was used to identify KSHV helicase and MCM7 protein complexes. 293T cells transfected with the indicated plasmids using Lipofectamine 2000, and KSHV helicase-FLAG recombinant virus–infected cells were induced with doxycycline for 72 hours. M2-FLAG beads and 3× FLAG peptides were used to purify native protein complexes and elute beads, respectively, from 293T cells. Protein A/G beads and an anti-FLAG antibody were applied to related KSHV helicase–FLAG recombinant virus–infected cells. The eluate was monitored via SDS–polyacrylamide gel electrophoresis (PAGE) as well as silver staining kit and lastly subjected to mass spectrometry. Besides, mass spectrometry was also used to analyze methionine oxidation of KSHV helicase, MCM7, and RIG-I.
co-IP and Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer [50 mM tris (pH 7.6), 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 0.1 mM phenylmethylsulfonyl fluoride (PMSF)] for 30 min on ice with brief vortexing every 10 min, followed by centrifugation at 13,000 rpm at 4°C for 15 min to remove cell debris. Five percent of the cell lysates was retained at input, and the remainder was incubated with an antibody or affinity beads, as indicated, overnight at 4°C. The immunoprecipitates were washed three times with RIPA buffer and then boiled in SDS loading buffer for Western blotting (WB) analysis. For WB, protein samples were analyzed through SDS-PAGE and transferred to nitrocellulose membranes, followed by blocking and probing with the indicated antibodies for detection.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30 min, followed by permeabilization with 0.1% Triton X-100 for 10 min. After blocking with 5% normal goat serum for 1 hour, the cells were incubated with the indicated antibodies overnight at 4°C. The cells were then washed and incubated with the appropriate secondary antibodies in phosphate-buffered saline (PBS) for 1 hour. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole. Coverslips were mounted with antifade mounting medium, and photographs were taken using a digital camera and software from Leica.
Quantification of KSHV genomic DNA levels and RNA transcription in cells
Intracellular viral genomic DNA and extracellular virion DNA were extracted from induced related KSHV helicase wild type or mutation recombinant virus–infected cells or the cell medium, respectively. Copy numbers of viral genomic DNA were estimated via real-time PCR using primers for K9. Total RNA was extracted from harvested cells using the TRIzol reagent, and cDNA was obtained through reverse transcription with a genomic DNA eraser RT kit (TOYOBO). qPCR was performed with a SYBR green real-time PCR master mix kit. The reaction mixtures contained 5 μl of master mix plus ROX passive reference dye, 1 μM each primer, and 4 μl of diluted sample. The program of the 7900HT sequence detection system constitutes 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. Relative mRNA levels were normalized to the level of actin mRNA and calculated via the ΔΔCt method. A melting curve analysis was used to verify the specificity of the primers, and each sample was tested in triplicate.
RNA interference
Cells were transfected with control siRNA or siRNAs corresponding to the indicated genes from GenePharma Technology using Lipofectamine 2000 according to the manufacturer’s instructions. At 5 hours after transfection, the medium containing the siRNA was replaced with fresh medium, and the efficiency of the siRNA-mediated depletion was determined through immunoblotting analysis.
Chromatin immunoprecipitation
The ChIP protocol was adapted from the Rockland website. Cells were transfected with indicated plasmids. At 48 hours after transfection, cells were cross-linked with formaldehyde at a 1% final concentration for 30 min, and glycine was added to quench the formaldehyde at a final concentration of 125 mM. Cells were washed twice with PBS and suspended in 1 ml of buffer A [10 mM tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.2% Triton X-100, 1 mM dithiothreitol (DTT), 0.5 mM EDTA, and 0.2 mM PMSF] for 10 min at 4°C. The released nuclei were pelleted via centrifugation at 1300 rpm for 5 min and then incubated in SDS lysis buffer (50 mM Hepes, 1 mM EDTA, 1% SDS, and 1 mM PMSF) for 10 min on ice. The lysates were subjected to sonication to obtain approximately 200- to 500-bp fragments of DNA (cycle, two 6-s pulses; amplitude, 30 to 35%; Sonics) and then centrifuged at 13,000 rpm at 4°C for 15 min to obtain supernatant. Supernatant was diluted 1:10 in RIPA buffer. Next, protein A/G was pretreated with binding buffer containing 0.2 mg of salmon sperm DNA per milliliter for 6 hours, and the samples were precleared with pretreated protein A or G for 2 hours at 4°C. Then, 10% of the supernatant was retained as input, and the remainder was divided into two groups with pretreated protein A or G beads and 5 μg of the corresponding antibody (anti–mouse-FLAG antibody and mouse (IgG) overnight at 4°C. The beads were subsequently washed with RIPA buffer, wash buffer [10 mM tris-HCl (pH 8.0), 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, and 1 mM PMSF], and TE buffer [10 mM tris-HCl (pH 8.0) and 1 mM EDTA] three times each and then suspended in TE buffer. The suspended beads were subjected to ribonuclease A and proteinase K digestion following reverse cross-linking at 65°C for 8 to 10 hours. DNA was recycled with a DNA purification kit from TIANGEN.
Dual-luciferase reporter assay
All procedures were performed as described in the manual supplied with the luciferase assay system from Promega. HEK293T cells were seeded in a 12-well plate the day before transfection. The cells were subsequently cotransfected with the luciferase reporter pGL3E-pViperin, pRL-TK, and the FLAG-tagged RTA plasmid or the FLAG-tagged empty vector plasmid. The pRL-TK plasmid was used to normalize firefly luciferase activity based on the expression of Renilla luciferase activity. The cells were lysed in 200 μl of passive cell lysis buffer at 48 hours after transfection, and luciferase activity was detected. The results were expressed as the fold change relative to cells transfected with the FLAG-tagged empty vector plasmid.
Cycloheximide chase assay
293T cells were transfected with indicated plasmids. Twenty-four hours after transfection, these test proteins were treated with CHX, and the samples were processed at indicated times for protein extraction, SDS-PAGE, and immunoblotting with either anti-HA, anti-FLAG, or anti-GAPDH antibodies. The anti-GAPDH antibody was used to verify the uniformity of total protein loads.
Click-iT 5-ethynyl-2′-deoxyuridine immunoprecipitation
293T cells were transfected with indicated plasmids. Forty-eight hours after transfection, cells were labeled with 30 μM EdU (5-ethynyl-2′-deoxyuridine) for 40 min and then harvested for total DNA extraction using Hirt’s method. DNA was then sonicated to get an average length of 500 to 750 bp. Click-iT reaction was then performed by addition of Click-iT reaction components and incubation for 2 hours at room temperature. Increasing the sample volume to 500 μl, added streptavidin beads and the samples were rotated at room temperature for overnight. Next day, samples were washed twice with washing solution [10 Mm tris-HCL (pH 7.5), 1 mM EDTA, and 2 M NaCl] and once in distilled water. The samples were then eluted by resuspending beads in elution buffer (1% SDS and 0.1 M NaHCO3) and incubating at 65°C for 10 min, followed by centrifugation and transfer of supernatants to new tubes. DNA was precipitated with a DNA purification kit from TIANGEN and resuspended in 50 μl TE. This DNA was next used in qPCR analysis to assess levels of replicating DNA at cellular c-Myc replication origins.
Protein expression and purification
To obtain FLAG–RIG-I, FLAG-MCM7, FLAG–KSHV helicase, FLAG-viperin, and FLAG–viperin-Del proteins, we transfected HEK293T cells with indicated plasmids containing corresponding genes. Cells were harvested at 48 hours after transfection and lysed with Triton X-100 buffer [20 mM tris (pH 8), 137 mM NaCl, 2 mM EDTA, 10% glycerol, and 10 mM 2-mercaptoethanol, supplemented with 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Roche)]. Then, cell lysates were centrifuged, and the supernatant was incubated with anti-FLAG agarose beads at 4°C for 5 hours. Last, the agarose beads were washed extensively with Triton X-100 lysis buffer and eluted with oxidation reaction buffer [50 mM Hepes (pH 7.5) and 150 mM KCl] containing FLAG peptide (0.2 mg/ml), with 10 mM DTT for RIG-I, KSHV helicase, and MCM7 and without DTT for viperin and viperin-Del. Eluted proteins were analyzed by SDS-PAGE and Coomassie blue staining.
In vitro oxidation assay
In vitro oxidation assay was performed as described previously (18). Briefly, a reaction (100 μl total volume) containing 5 μg of substrates (purified RIG-I, KSHV helicase, or MCM7), 2 μg of enzymes (purified viperin or viperin-Del), 50 mM Hepes (pH 7.5), 150 mM KCl, 2 mM SAM, and 5 mM dithionite was incubated for 45 min at 37°C. Reaction was stopped by boil for 10 min. Reactions containing RIG-I, MCM7, and KSHV helicase were analyzed by mass spectrometry, and the ratio of oxidized peptides over total peptides was quantitatively determined.
Statistical analysis
GraphPad Prism software was used to perform the statistical analysis, and statistical significance was set at P < 0.05, as determined through the unpaired, two-tailed t test. Error bars represent SEM. Each experiment was carried out independently at least three times.
Supplementary Material
Acknowledgments
We thank X. Liang (Institut Pasteur of Shanghai, Chinese Academy of Sciences) and M. Luo (Wuhan Institute of Virology, Chinese Academy of Sciences) for the MHV68 and HCMV BAC plasmids, respectively. We are also grateful to H. Shu (Medical Research Institute, Wuhan University) for the MEF cell lines. Funding: This work was supported by grants from the Natural Science Foundation for Distinguished Young Scholars (81425017), the Ministry of Science and Technology of China (2016YFA0502100), and the NIH (7R01AI116442) to K.L.. It was also supported, in part, by NIH grants DE027556 and CA221521 to P.F. Author contributions: Conceptualization: K.L., P.F., L.B., and J.D.; methodology: L.B., J.D., and Y.R.; formal analysis: Z.L.; investigation: L.B. and J.D.; writing (original draft): L.B. and J.D.; writing (review and editing): K.L. and P.F.; funding acquisition: K.L. and P.F. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax1031/DC1
Fig. S1. KSHV helicase and cellular DNA helicase MCM7 are evolutionarily conserved.
Fig. S2. KSHV helicase is critical for viral DNA replication.
Fig. S3. Viperin is critical for viral DNA replication and KSHV-encoded RTA binds to viperin promoter to up-regulate viperin expression.
Fig. S4. Viperin promotes methionine oxidation of KSHV helicase.
Fig. S5. Viperin-induced methionine-401 oxidation enhances the stability and function of KSHV helicase.
Fig. S6. Viperin associates with lipid droplets that are required to induce the methionine oxidation of KSHV helicase.
Fig. S7. Viperin-induced methionine oxidation stabilizes MCM7 and promotes DNA replication.
Fig. S8. Viperin interacts with several DNA helicases.
Fig. S9. Methionine oxidation catalyzed by viperin increases the stability and function of RNA helicase RIG-I.
Table S1. Amino acid sequence alignment for each human DNA helicase with KSHV helicase.
REFERENCES AND NOTES
- 1.Schlierf M., Ha T., A helicase with an extra spring in its step. Cell 151, 244–246 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Ticau S., Friedman L. J., Ivica N. A., Gelles J., Bell S. P., Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 161, 513–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He S., Zhao J., Song S., He X., Minassian A., Zhou Y., Zhang J., Brulois K., Wang Y., Cabo J., Zandi E., Liang C., Jung J. U., Zhang X., Feng P., Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 58, 134–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merbl Y., Refour P., Patel H., Springer M., Kirschner M. W., Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell 152, 1160–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Psakhye I., Jentsch S., Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Chistol G., Walter J. C., Single-molecule visualization of MCM2-7 DNA loading: Seeing is believing. Cell 161, 429–430 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Cremona C. A., Sarangi P., Yang Y., Hang L. E., Rahman S., Zhao X., Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Mol. Cell 45, 422–432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randell J. C. W., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., Bell S. P., Mec1 is one of multiple kinases that prime the Mcm2-7 helicase for phosphorylation by Cdc7. Mol. Cell 40, 353–363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei L., Zhao X., A new MCM modification cycle regulates DNA replication initiation. Nat. Struct. Mol. Biol. 23, 209–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang Y., Cesarman E., Pessin M. S., Lee F., Culpepper J., Knowles D. M., Moore P. S., Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994). [DOI] [PubMed] [Google Scholar]
- 11.Hilton I. B., Simon J. M., Lieb J. D., Davis I. J., Damania B., Dittmer D. P., The open chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 87, 11831–11842 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu F. Y., Ahn J.-H., Alcendor D. J., Jang W.-J., Xiao J., Hayward S. D., Hayward G. S., Origin-independent assembly of Kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J. Virol. 75, 1487–1506 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grässer F. A., van Dyk L. F., Ho C. K., Shuman S., Chien M., Russo J. J., Ju J., Randall G., Lindenbach B. D., Rice C. M., Simon V., Ho D. D., Zavolan M., Tuschl T., Identification of microRNAs of the herpesvirus family. Nat. Methods 2, 269–276 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Brulois K., Toth Z., Wong L.-Y., Feng P., Gao S.-J., Ensser A., Jung J. U., Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 88, 9335–9349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brulois K. F., Chang H., Lee A. S.-Y., Ensser A., Wong L.-Y., Toth Z., Lee S. H., Lee H.-R., Myoung J., Ganem D., Oh T.-K., Kim J. F., Gao S.-J., Jung J. U., Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86, 9708–9720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stirnweiss A., Ksienzyk A., Klages K., Rand U., Grashoff M., Hauser H., Kröger A., IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J. Immunol. 184, 5179–5185 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Wang X., Hinson E. R., Cresswell P., The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2, 96–105 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Gizzi A. S., Grove T. L., Arnold J. J., Jose J., Jangra R. K., Garforth S. J., Du Q., Cahill S. M., Dulyaninova N. G., Love J. D., Chandran K., Bresnick A. R., Cameron C. E., Almo S. C., A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558, 610–614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo J.-Y., Yaneva R., Cresswell P., Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 10, 534–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seo J.-Y., Yaneva R., Hinson E. R., Cresswell P., Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332, 1093–1097 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Huang Y., He S., Li J. Z., Seo Y.-K., Osborne T. F., Cohen J. C., Hobbs H. H., A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. U.S.A. 107, 7892–7897 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omura S., Tomoda H., Xu Q. M., Takahashi Y., Iwai Y., Triacsins, new inhibitors of acyl-CoA synthetase produced by Streptomyces sp. J. Antibiot. 39, 1211–1218 (1986). [DOI] [PubMed] [Google Scholar]
- 23.Kattur Venkatachalam A. R., Szyporta M., Kiener T. K., Balraj P., Kwang J., Concentration and purification of enterovirus 71 using a weak anion-exchange monolithic column. Virol. J. 11, 99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byun H., Gwack Y., Hwang S., Choe J., Kaposi’s sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cells 14, 185–191 (2002). [PubMed] [Google Scholar]
- 25.Sun R., Lin S.-F., Gradoville L., Yuan Y., Zhu F., Miller G., A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U.S.A. 95, 10866–10871 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan Y.-L., Chang T.-H., Liao C.-L., Lin Y.-L., The cellular antiviral protein viperin is attenuated by proteasome-mediated protein degradation in Japanese encephalitis virus-infected cells. J. Virol. 82, 10455–10464 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dittmer D. P., Damania B., Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Invest. 126, 3165–3175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Z., Hopcraft S. E., Yang F., Petrucelli A., Guo H., Ting J. P.-Y., Dittmer D. P., Damania B., NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLOS Pathog. 13, e1006350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helbig K. J., Beard M. R., The role of viperin in the innate antiviral response. J. Mol. Biol. 426, 1210–1219 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Bertolotti-Ciarlet A., Wang W., Lownes R., Pristatsky P., Fang Y., McKelvey T., Li Y., Li Y., Drummond J., Prueksaritanont T., Vlasak J., Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Mol. Immunol. 46, 1878–1882 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Bigelow D. J., Squier T. C., Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta 1703, 121–134 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Ghesquière B., Jonckheere V., Colaert N., Van Durme J., Timmerman E., Goethals M., Schymkowitz J., Rousseau F., Vandekerckhove J., Gevaert K., Redox proteomics of protein-bound methionine oxidation. Mol. Cell. Proteomics 10, M110.006866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinson E. R., Cresswell P., The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic α-helix. Proc. Natl. Acad. Sci. U.S.A. 106, 20452–20457 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinson E. R., Cresswell P., The N-terminal amphipathic α-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J. Biol. Chem. 284, 4705–4712 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monson E. A., Crosse K. M., Das M., Helbig K. J., Lipid droplet density alters the early innate immune response to viral infection. PLOS ONE 13, e0190597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peña Cárcamo J. R., Morell M. L., Vázquez C. A., Vatansever S., Upadhyay A. S., Överby A. K., Cordo S. M., García C. C., The interplay between viperin antiviral activity, lipid droplets and Junín mammarenavirus multiplication. Virology 514, 216–229 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Dechandt C. R. P., Zuccolotto-dos-Reis F. H., Teodoro B. G., Fernandes A. M. A. P., Eberlin M. N., Kettelhut I. C., Curti C., Alberici L. C., Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J. Bioenerg. Biomembr. 49, 399–411 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Wright H. J., Hou J., Xu B., Cortez M., Potma E. O., Tromberg B. J., Razorenova O. V., CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. U.S.A. 114, E6556–E6565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beller M., Bulankina A. V., Hsiao H. H., Urlaub H., Jäckle H., Kühnlein R. P., PERILIPIN-dependent control of lipid droplet structure and fat storage in Drosophila. Cell Metab. 12, 521–532 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Bickel P. E., Tansey J. T., Welte M. A., PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 1791, 419–440 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominguez-Sola D., Ying C. Y., Grandori C., Ruggiero L., Chen B., Li M., Galloway D. A., Gu W., Gautier J., Dalla-Favera R., Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Swarnalatha M., Singh A. K., Kumar V., The epigenetic control of E-box and Myc-dependent chromatin modifications regulate the licensing of lamin B2 origin during cell cycle. Nucleic Acids Res. 40, 9021–9035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishimi Y., A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 272, 24508–24513 (1997). [DOI] [PubMed] [Google Scholar]
- 44.You Z., Komamura Y., Ishimi Y., Biochemical analysis of the intrinsic Mcm4-Mcm6-Mcm7 DNA helicase activity. Mol. Cell. Biol. 19, 8003–8015 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ligat G., Da Re S., Alain S., Hantz S., Identification of amino acids essential for viral replication in the HCMV helicase-primase complex. Front. Microbiol. 9, 2483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo J., Chen J., Yang E., Shen A., Gong H., Pei Z., Xiao G., Lu S., Liu F., Modulation of the cellular distribution of human cytomegalovirus helicase by cellular factor Snapin. J. Virol. 87, 10628–10640 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C.-S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S., Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Medzhitov R., Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax1031/DC1
Fig. S1. KSHV helicase and cellular DNA helicase MCM7 are evolutionarily conserved.
Fig. S2. KSHV helicase is critical for viral DNA replication.
Fig. S3. Viperin is critical for viral DNA replication and KSHV-encoded RTA binds to viperin promoter to up-regulate viperin expression.
Fig. S4. Viperin promotes methionine oxidation of KSHV helicase.
Fig. S5. Viperin-induced methionine-401 oxidation enhances the stability and function of KSHV helicase.
Fig. S6. Viperin associates with lipid droplets that are required to induce the methionine oxidation of KSHV helicase.
Fig. S7. Viperin-induced methionine oxidation stabilizes MCM7 and promotes DNA replication.
Fig. S8. Viperin interacts with several DNA helicases.
Fig. S9. Methionine oxidation catalyzed by viperin increases the stability and function of RNA helicase RIG-I.
Table S1. Amino acid sequence alignment for each human DNA helicase with KSHV helicase.