

A single mutation in protein kinase A remodels the intramolecular allostery, changing substrate specificity and regulation.
Abstract
Genetic alterations in the PRKACA gene coding for the catalytic α subunit of the cAMP-dependent protein kinase A (PKA-C) are linked to cortisol-secreting adrenocortical adenomas, resulting in Cushing’s syndrome. Among those, a single mutation (L205R) has been found in up to 67% of patients. Because the x-ray structures of the wild-type and mutant kinases are essentially identical, the mechanism explaining aberrant function of this mutant remains under active debate. Using NMR spectroscopy, thermodynamics, kinetic assays, and molecular dynamics simulations, we found that this single mutation causes global changes in the enzyme, disrupting the intramolecular allosteric network and eliciting losses in nucleotide/pseudo-substrate binding cooperativity. Remarkably, by rewiring its internal allosteric network, PKA-CL205R is able to bind and phosphorylate non-canonical substrates, explaining its changes in substrate specificity. Both the lack of regulation and change in substrate specificity reveal the complex role of this mutated kinase in the formation of cortisol-secreting adrenocortical adenomas.
INTRODUCTION
The cyclic adenosine monophosphate (cAMP)–dependent protein kinase A (PKA) plays a fundamental role in the function and replication of endocrine cells (1), and aberrant cAMP signaling has been linked to several endocrine diseases (2). The first mutations in PKA have been found in the regulatory (R) subunits, which have been associated with Carney complex, a multiple neoplasia syndrome manifesting via adrenocortical adenomas, cutaneous and neuronal tumors, cardiac myxomas, and pigmented lesions of the skin and mucosae (2). Only recently have somatic mutations been identified in the PRKACA gene coding for the catalytic α subunit of PKA (PKA-C) and discovered in cortisol-secreting adrenocortical adenomas responsible for Cushing’s syndrome (2) (Fig. 1A). Nearly all identified mutations lie adjacent to the active site cleft and the regulatory/catalytic subunit interface (Fig. 1, B and C). Among these mutations, the most frequently found (PKA-CL205R) is a point mutation leading to substitution of Leu at position 205 (or 206, depending on the convention used for numbering) with Arg and has been found in up to 67% of Cushing’s syndrome patients (3–7).
Fig. 1. Architecture of PKA-C and locations of the Cushing’s syndrome mutations.
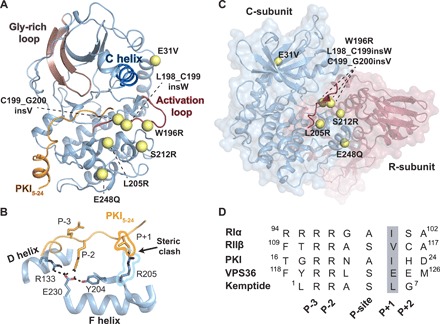
(A) Structure of PKA-C [Protein Data Bank (PDB) ID: 1ATP] in complex with pseudo-substrate PKI depicting the location of Cushing’s mutations (yellow spheres) in relation to structural elements of the kinase. The E248Q mutation includes an additional deletion (del243-247), and the S212R mutation includes an insertion (insIILR) not depicted. (B) X-ray structure of PKA-CL205R (PDB ID: 4WB6) with the overlay of PKI5–24 (PKIWT from PDB ID: 1ATP) describing the architecture of the peptide binding site and steric clash between kinase (PKA-CL205R) and pseudo-substrate. (C) Structure of the R/C complex (PDB ID: 2QCS) depicting locations of Cushing’s mutations in relation to the pseudo-substrate inhibitory sequence of the R-subunit. (D) Primary sequence comparison of common regulators (RIα, RIIβ, and PKI) and peptide substrates of the catalytic subunit (Kemptide and VPS36).
Inactive PKA exists as an inactive holoenzyme (R2C2) containing an R-subunit dimer bound to two catalytic (C) subunits. The inhibitory sequence of the R-subunit occupies the active site of the enzyme (Fig. 1D). Upon stimulation of membrane receptors coupled to the stimulatory Gs protein, produced cAMP binds to the R-subunits, unleashing active C-subunits (8). The spatiotemporal regulation of the kinase is provided by ancillary proteins such as A-kinase anchoring proteins that, via interactions with R-subunits, localize PKA-C in close proximity to its substrates (9). In addition, PKA-C is regulated by an endogenous inhibitor (PKI), whose function is to block access to substrates and recruit PKA-C to the nuclear export complex (CRM1 and RanGTP) (8). When this spatiotemporal regulation fails, PKA-C hyperphosphorylates its targets, leading to disease.
Structurally, PKA-C consists of two lobes. The N-lobe, smaller and more dynamic, comprises mostly β strands, as well as the αC helix, and harbors the adenosine 5′-triphosphate (ATP)–binding site (10). The C-lobe, larger and made up of α helices, is more rigid and harbors the substrate binding groove. PKA-C toggles between three major conformational states: open (apo), intermediate (nucleotide-bound), and closed (ternary complex with nucleotide/substrate-bound) (11). The L205R mutation is located at the interface of the N- and C-lobes of PKA-C in the P+1 loop, a highly conserved region of the enzyme that creates a hydrophobic pocket for substrate docking. On the basis of its positioning, the L205R mutation has been proposed and shown by independent laboratories to disrupt the binding of R-subunits and render the enzyme constitutively active (12, 13). In vivo studies revealed that the catalytic activity of PKA-CL205R is comparable to that of wild type (12). However, recent phosphoproteomic mapping showed a drastic change in the phosphorylation profile, suggesting that the mutation preserves the ability to phosphorylate downstream substrates, although with a different selectivity (14, 15). On the basis of these data, it has been proposed that disruption of the signaling network leading to the phosphorylation of non-canonical substrates may contribute to tumorigenesis. Notably, the x-ray crystal structures of the wild-type and mutant kinases in complex with nucleotide and pseudo-substrate, PKI5–24, are nearly superimposable [root mean square deviation (RMSD) = 0.49 Å] (16) and do not thoroughly explain the weaker binding for R-subunit, the lack of regulation by PKI, and the changes in the phosphoproteomic profile.
To gain mechanistic insights into the multifarious effects of this single mutation, we carried out solution nuclear magnetic resonance (NMR) spectroscopy in concert with binding thermodynamics, kinetic assays, and molecular dynamics (MD) simulations. We found that this single mutation, in addition to changing the protein-protein interface for substrate recognition, causes a disruption of the internal allosteric communication, reducing its affinity for nucleotide. The partial ablation of the intramolecular allosteric communication prevents the kinase from reaching a completely closed state, reducing the binding affinity for canonical substrates and pseudo-substrates, thus hindering endogenous regulation. Notably, kinetic assays using a substrate identified by phosphoproteomic analyses as a preferred substrate of PKA-CL205R (14) show a higher catalytic efficiency and a rearrangement of the internal structural dynamics and allosteric communication. Together, our results indicate that rewiring of the internal allosteric network and global changes in dynamics contribute to the complex dysregulation of the signaling network of PKA-CL205R within tumor cells.
RESULTS
PKA-CL205R shows a marked loss of binding cooperativity between ATP and PKI
To determine the thermodynamics of nucleotide binding (ATPγN) to both PKA-CWT and PKA-CL205R, we used isothermal titration calorimetry (ITC). We found that PKA-CL205R exhibits a threefold lower binding affinity compared to PKA-CWT [Kd (dissociation constant) of 215 ± 15 μM versus 83 ± 8 μM, respectively]. Fitting of the titration curves shows that the binding of ATPγN to PKA-CL205R is enthalpically favorable and entropically unfavorable, whereas both contributions are favorable for ATPγN binding to PKA-CWT. To evaluate the binding cooperativity between nucleotide and substrate (17, 18), we analyzed the binding thermodynamics of the pseudo-substrate peptide inhibitor, PKI5–24, to both PKA-CWT and PKA-CL205R in the presence and absence of nucleotide. ΔH, −TΔS, ΔG, and Kd, as well as the cooperativity coefficients (σ) derived from ITC titrations, are summarized in table S1. The binding thermodynamics of PKI5–24 to the apo forms of the two kinases is comparable, with similar enthalpy changes (favorable). When saturated with ATPγN, PKA-CWT binds PKI5–24 with higher affinity (Kd = 0.16 ± 0.02 μM) (18). In contrast, PKA-CL205R displays a 62-fold reduction in binding affinity (Kd = 10 ± 3 μM), with a net decrease in both ΔH and ΔS. The binding of PKI5–24 to the PKA-CWT/ATPγN complex is highly cooperative (σ = 106 ± 18), whereas its binding cooperativity to the PKA-CL205R/ATPγN complex decreases 18-fold (σ = 6 ± 2). Therefore, this single mutation affects both the affinity for ligands and the binding cooperativity between nucleotide and pseudo-substrate.
L205R mutation causes a reduction in phosphorylation kinetics
To evaluate the catalytic efficiency of PKA-CL205R, we carried out steady-state coupled assays using the standard substrate, Kemptide. As expected from the binding thermodynamics and previous kinetic assays, the Leu-to-Arg substitution at the P+1 loop increases the KM (Michaelis constant) 11-fold (table S2). The kcat for the mutant increases from 19 to 41 s−1, which results in an overall decrease in catalytic efficiency (kcat/KM) by over fivefold compared to PKA-CWT. These results are in quantitative agreement with experiments carried out by two other independent groups (16, 19).
PKI binding to PKA-CL205R falls short in shifting the enzyme to a fully closed state
To analyze the structural changes of PKA-C upon binding PKI and the effects of the L205R mutation, we mapped the amide backbone fingerprint of the enzyme using [1H, 15N]–TROSY (transverse relaxation-optimized spectroscopy)–HSQC (heteronuclear single-quantum coherence) experiments (20). The amide signatures of the kinases in different ligated forms are displayed in fig. S1. The binding of ATPγN to both PKA-CWT and PKA-CL205R gives rise to similar chemical shift perturbations (CSPs), with the exception of the residues in the Gly-rich, catalytic, and Mg2+ positioning loops that show larger chemical shift changes for PKA-CWT (fig. S2). To evaluate the global response to ligand binding for the kinase and its mutant, we used CONCISE (coordinated chemical shifts behavior), which performs a statistical analysis on linear chemical shift trajectories of each amide resonance to identify the position of each state along the conformational equilibrium (21). Following ligand binding, the amide resonances of PKA-C display linear chemical shift trajectories, with the amide resonances corresponding to the apo enzyme populating one extreme and those of the closed state (ternary complex with PKI) populating the opposite extreme of the linear correlations. This behavior exemplifies a conformational equilibrium along the three major conformational states of the kinase (open, intermediate, and closed), whose relative populations are modulated by ligand binding. A total of 55 residues exhibit linear chemical shift trajectories for both PKA-CWT and PKA-CL205R, indicating a fast exchange limit in the NMR chemical shift time scale. These residues are distributed throughout the kinase. We found that the binding of ATPγN and PKI shifts the conformational equilibrium of PKA-CWT toward a fully closed state. In contrast, these ligands drive the mutant to a partially closed state, which is located between the intermediate and closed conformation (table S3). This is supported by thermostability data obtained by circular dichroism (CD) measurements (table S4). CD melting curves show that the PKA-CWT/ATPγN/PKI complex has a higher melting temperature (Tm) than the corresponding complex with PKA-CL205R, further suggesting that this complex may adopt a more open and unstable conformation. Since the extent of the closed state is correlated with binding affinity and cooperativity (18), these results are consistent with the loss in allosteric cooperativity revealed by thermocalorimetric data.
L205R disrupts the allosteric network of the kinase upon PKI binding
To determine a possible correlation between the binding cooperativity and the intramolecular allosteric network, we analyzed the correlated CSPs for both PKA-CWT and PKA-CL205R using chemical shift covariance analysis (CHESCA) (22). This method identifies the allosteric networks of residues that are involved in concerted responses to ligand binding (23). The [1H, 15N]-TROSY-HSQC spectra of the three forms of wild-type and PKA-C mutant (apo, ATPγN bound, and ATPγN/PKI bound) were used for the analysis. The CHESCA matrices for PKA-CWT and PKA-CL205R reported in Fig. 2 (A and B) reveal remarkable differences in their allosteric networks. There is an overall reduction in the number of correlations throughout PKA-CL205R (Fig. 2, C and D), with specific domains of the enzyme more affected by this single mutation. In particular, residues belonging to the Gly-rich, activation loop and peptide-positioning loop experience the most substantial reductions in both number and extent of chemical shift covariation. In addition, several interresidue correlations within the activation loop (W196), αF helix (K217, V219, D220, G225, V226, and A233), and αG helix (Q245) are also ablated. The loss of interresidue correlations for the αF helix is especially important as this motif spans the hydrophobic core of the C-lobe and is conserved throughout the AGC kinase family. The function of the αF helix is to anchor all the hydrophobic motifs within the kinase core and to orchestrate catalysis (24, 25). As highlighted before, all Cushing’s driver mutations, with the exception of E31V, are located near L205R and are likely to affect the enzyme in a similar manner, disrupting these important allosteric nodes.
Fig. 2. Allosteric network of interactions observed upon pseudo-substrate binding.
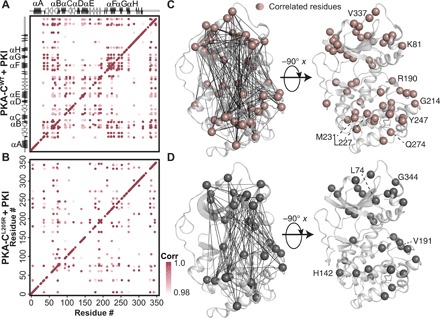
The CHESCA correlation matrix for (A) PKA-CWT upon binding PKI and (B) PKA-CL205R upon binding PKI. (C) Correlations corresponding to the binding of PKI to PKA-CWT plotted on the structure of PKA-C. Residues that are commonly correlated for both PKA-CWT and PKA-CL205R are highlighted. (D) Correlations corresponding to the binding of PKI to PKA-CL205R plotted on the structure of PKA-C. Specific residues that are correlated for only PKA-CL205R are highlighted. Only correlations with rij > 0.98 are shown throughout.
Binding of VPS36 substrate to PKA-CL205R rewires the intramolecular allosteric network and reestablishes binding cooperativity
In addition to affecting the regulation of PKA-C by the R-subunits, L205R changes the kinase’s downstream substrate specificity (14, 15). Specifically, it was found that in Escherichia coli, PKA-CL205R favors noncanonical substrates with negatively charged residues (Glu or Asp) at positions P+1, P+2, and P+3. From our own phosphoproteomic studies on endogenous substrates in human cells (14), we identified several substrates that are preferentially phosphorylated by the mutated kinase. Among those substrates, we selected the top target hyperphosphorylated by PKA-CL205R: the vacuolar protein-sorting–associated protein 36 (VPS36), a protein that plays a role in endosomal sorting of ubiquinated cargo proteins by the endosomal sorting complex. The recognition sequence of VPS36 (RRLSEEM) for PKA-C has two Glu residues in the P+1 and P+2 positions. Using steady-state coupled enzyme assays with Kemptide and a VPS36-derived peptide (residues 115 to 130 encompassing the PKA-C recognition sequence and denoted VPS36 from hereon), we found that PKA-CL205R reduces the kinase’s catalytic efficiency for Kemptide, which contains a hydrophobic residue at the P+1 position. In contrast, the mutation causes an increase in kcat (from 5 to ~18 s−1) for VPS36, leading to a threefold increase in catalytic efficiency (Fig. 3A and table S2). In addition, the ITC analysis carried out with VPS36 indicates that PKA-CL205R/ATPγN has an affinity typical of other substrates (Kd = 3.5 ± 0.1 μM), while the binding to wild type cannot be detected reliably, suggesting a notably lower binding affinity (table S1). A possible explanation is that the acidic residues in positions P+1 and P+2 of VPS36 hamper the intermolecular interactions between the peptide and residues lining the binding pocket of PKA-CWT, while they might favor the interactions with PKA-CL205R. These results support the proposed altered substrate specificity for PKA-CL205R detected in intact cells.
Fig. 3. Rewiring of the allosteric network of PKA-CL205R upon binding VPS36.
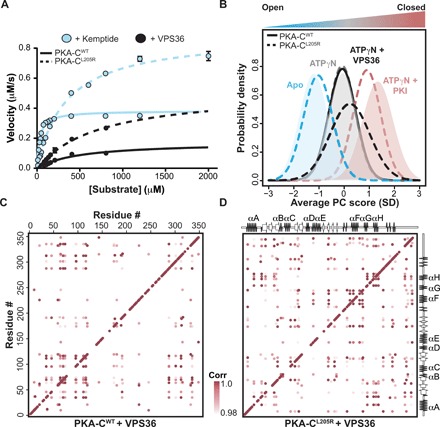
(A) Steady-state phosphorylation kinetics of Kemptide and VPS36 peptides for PKA-CWT and PKA-CL205R. Corresponding values can be found in table S2. (B) CONCISE analysis on the apo, ATPγN, ATPγN/PKI, and ATPγN/VPS36 states of PKA-CWT and PKA-CL205R. (C) The CHESCA correlation matrix for PKA-CWT upon binding VPS36. (D) The CHESCA correlation matrix for PKA-CL205R upon binding VPS36.
To further investigate the underlying mechanism for the change in substrate specificity of PKA-CL205R, we analyzed the trajectories of the amide resonances for the wild-type and mutated kinase upon binding VPS36. Although VPS36 binding causes linear chemical shift changes similar to PKI binding, the number of residues involved in the binding response is substantially higher for PKA-CL205R. In addition, VPS36 binding causes an overall attenuation of the peak intensities throughout the entire fingerprint of the enzyme, with some resonances broadened beyond detection (fig. S3). For instance, resonances associated with the αF helix and Gly-rich loop are noticeably broadened, suggesting an increase in protein dynamics in the microsecond to millisecond time scale. The CONCISE analysis shows that the conformation of the PKA-CWT/ATPγN/VPS36 complex only reaches the intermediate state. In contrast, the PKA-CL205R/ATPγN/VPS36 complex reaches a state lying between the intermediate and closed states (Fig. 3B). This position in between the intermediate and closed states was also observed in the crystal structure of PKA-CWT bound to a substrate peptide (26), suggesting that the ternary complex of PKA-CL205R with VPS36 adopts a catalytically committed state.
The CHESCA maps for VPS36 binding to PKA-CWT and PKA-CL205R are radically different (Fig. 3, C and D). While the PKA-CWT/ATPγN/PKI complex shows an intense cross-talk between the small and large lobes, the corresponding map for PKA-CWT/ATPγN/VPS36 reveals a complete loss of allosteric interactions. Notably, correlations between the nucleotide binding site and the substrate binding hub are completely missing. In particular, the interresidue correlations for the activation loop, peptide-positioning loop, and the αF and αG helices are either reduced in their number or completely missing. In contrast, a number of correlations between the N- and C-lobes for PKA-CL205R, which are absent for the complex with PKI, are regained upon VPS36 binding. Note that 10 of the resonances corresponding to residues located in and around the Gly-rich loop are broadened beyond detection and could not be included in the CHESCA analysis; therefore, the extent of coordinated changes in chemical shifts is largely underestimated. Nonetheless, the higher density of correlations found in the PKA-CL205R/ATPγN/VPS36 complex underscores an extensive rewiring of the internal allosteric network, supporting the reestablished binding cooperativity.
PKA-CWT and PKA-CL205R explore different conformational states
To determine the effects of the L205R mutation on the conformational energy landscape of PKA-C, we carried out parallel MD simulations of the ternary complexes in explicit water environments starting from their respective x-ray coordinates [Protein Data Bank (PDB) IDs: 4WB6 and 1ATP] (16). The dominant motion of the kinase core was imaged using principal components analysis (PCA) of the Cα atoms encompassing residues 50 to 300. PC1 depicts the global opening-closing motion of the small and large lobes (fig. S4A), while PC2 describes the shearing motion of the two lobes that can be envisioned as an “asymmetric bite.” To describe the opening-closing motion of the active site, we monitored the time dependence of the distance between the Cα atoms of S53 in the Gly-rich loop and G186 (fig. S4, B and C). The binary and ternary complexes of PKA-CWT and PKA-CL205R sample similar conformational energy basins; however, the apo form of PKA-CL205R explores a considerably broader basin than the wild type. The Gly-rich loop of PKA-CWT toggles between the open and closed conformations, featuring a transient hydrogen bond between S53 and the γ-phosphate of ATP, whereas the Gly-rich loop of PKA-CL205R exhibits larger motions with the S53-G186 distance ranging from 5 to 18 Å and, more frequently, samples a conformation that hinders nucleotide binding, which might explain its weaker binding affinity (Fig. 4A). R205 causes steric hindrance with the C-terminal tail of PKI, disrupting the hydrophobic pocket required to anchor the Ile residue in the P+1 position, which likely contributes to the loss of PKI binding affinity.
Fig. 4. Conformational dynamics of the activation loop of PKA-CWT and PKA-CL205R upon binding substrate.
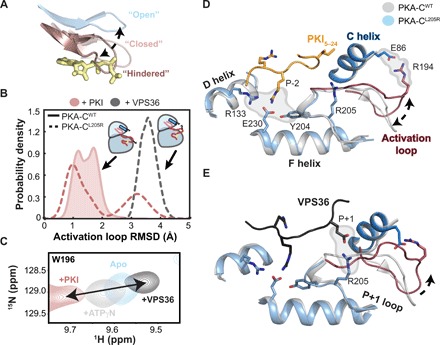
(A) Distinct opening-closing motions of the Gly-rich loop highlighting the hindered conformation that occludes the entering of ATP in PKA-CL205R. (B) Probability density describing the conformation of the activation loop in response to different substrates and pseudo-substrates. (C) [1H, 15N]-TROSY-HSQC spectra showing the backbone amide chemical shift changes of the W196 indole amide (located on the activation loop) in response to binding PKI or VPS36. (D) X-ray structure of PKA-CWT (gray) (PDB ID: 1ATP) with the overlay PKA-CL205R in complex with PKI (light blue) (PDB ID: 4WB6) describing the architecture of the peptide binding site and activation loop flip. (E) X-ray structure of PKA-CWT (gray) (PDB ID: 1ATP) with the overlay PKA-CL205R in complex with VPS36 (light blue) (MD simulations) describing the architecture of the peptide binding site and activation loop flip.
To visualize the alterations of the protein-protein interaction interface that may influence binding cooperativity, we analyzed the probability distribution of the inter-residue contacts between the substrate and the kinase binding pocket. Overall, PKA-CL205R shows a contact map with PKI similar to the wild-type enzyme (fig. S5, A and B). The high-affinity binding region (HAR) and the consensus binding sequences remain bound throughout the entire MD trajectories with a probability of contact greater than 0.8. However, the L205R mutation reduces the probability of interactions between the Ile (at position P+1 in PKI) and the P+1 loop from 0.5 to 0.2. In contrast, VPS36 shows stable interactions with the P+1 loop with a probability greater than 0.8 for Glu at the P+1 position and R205 (fig. S5C). Moreover, the N terminus of VPS36 forms transient interactions with the C-terminal tail located in the small lobe of PKA-CL205R, which are absent in the corresponding complex with PKI. As a result, the peptide is more dynamic, a feature that might explain the dramatic exchange-broadening in the NMR spectrum of the PKA-CL205R/ATPγN/VPS36 complex.
As observed previously (27), the backbone of the tertiary complex PKA-CWT/ATP/PKI is mostly rigid (fig. S5, D and F). The activation loop adopts a stable conformation as in the x-ray structure (PDB ID: 1ATP) (28) throughout the entire MD simulation with an RMSD of ~1.5 Å. In contrast, the activation loop and the adjacent loop that is anchored to the αF helix (residues 210 to 220) in the corresponding PKA-CL205R/ATP/PKI complex are more flexible, adopting two distinct conformations in which the most populated is similar to the x-ray structure (PDB ID: 1ATP) and a second minor flipped conformation (Fig. 4B). In this conformation, the activation loop no longer interacts with the C-lobe and forms a salt bridge with E86 of the αC helix (Fig. 4, D and E). For the PKA-CL205R/ATP/VPS36 complex, the activation loop becomes more ordered and the flipped configuration becomes dominant. The existence of these two conformations is corroborated by chemical shift trajectories observed for the indole resonance of W196 in the activation loop (Fig. 4C). Upon ligand binding, the indole resonance of W196 follows a linear chemical shift trajectory from the apo to the ternary form. This conformational change is highly correlated with the other major allosteric nodes (CHESCA plots), indicating that it follows the cooperative structural changes of the enzyme. In contrast, the CHESCA plots for PKA-CL205R show no correlations for these loops/residues, suggesting that the L205R mutation disrupts the internal allosteric network and may dislodge the activation loop from the large lobe.
The disruption of the allosteric network is mirrored by the pairwise mutual information plots (29) obtained from the analysis of the MD trajectories. For the wild-type enzyme, there are strong inter-residue correlations throughout the entire core of the kinase, especially among key catalytic motifs such as the Gly-rich loop (S53), the activation loop (R194, W196), and the αF (Y215), αG (D241), and αH (N283) helices, as well as the PIF motif at the C-terminal tail (F347) (fig. S6A). In contrast, PKA-CL205R lacks a number of allosteric interactions within the N-lobe, with only a few correlations between the activation loop and the C-lobe (fig. S6B). As for the CHESCA correlation maps, the mutual information plots reveal that the allosteric network between the N-lobe and C-terminal tail of PKA-CL205R is partially recovered upon VPS36 binding (fig. S6C) with the engagement of E91 in the αC helix and S53 of the Gly-rich loop.
DISCUSSION
The L205R mutation was discovered independently in our laboratory and in other laboratories as the dominant genetic alteration in cortisol-producing adrenocortical adenomas responsible for Cushing’s syndrome (3–7). Although it has a direct link with changes in cAMP/PKA signaling, adrenal Cushing’s syndrome has different molecular etiologies (2). Initially, rare germline mutations were found in the PRKAR1A gene encoding the regulatory Iα subunit of PKA in patients with Carney complex (30). Most of these mutations occur at the interface between the R- and C-subunits or near the cAMP-binding site in cyclic nucleotide binding (CNB) domain A and are thought to cause aberrant regulation of the kinase (31). Only recently has the focus shifted toward somatic mutations occurring in the C-subunit of the enzyme. Even in this case, most mutated residues reside at the interface of the R/C complex. However, the molecular mechanisms for the aberrant regulation of cAMP signaling by these mutations have been a matter of debate. Specifically, two independent studies suggested that the L205R mutation interferes with the formation of the holoenzyme, rendering the C-subunit constitutively active (3, 6). Sato et al. (4), on the other hand, proposed that this mutation does not interfere with the R-subunit regulation and that the development of the pathology is linked to intrinsic higher phosphoryl transfer activity of the mutant kinase. Recent studies both in vitro and in intact cells have put forward a possible new mechanism, suggesting that the mutated enzyme has altered substrate specificity (14, 15, 19). Notably, this arginine preceding the APE motif (residues 206-208) is characteristic in the CMGC kinase family and assumes a key role in substrate recognition and kinase activation (32). Perhaps PKA-CL205R might lead to promiscuity toward substrates of CMGC kinases.
Our studies reveal that the dysfunction of PKA-CL205R is multifaceted, whereby allosteric cooperativity is reduced, substrate specificity is altered, and canonical regulation is ablated. The L205R mutation disrupts the hydrophobic interactions between the enzyme and substrate, causing a dramatic decrease in PKI binding affinity. This is in line with the impaired binding of the R-subunits found in our previous study (12) as PKI and R-subunits share similar consensus sequences. Our CHESCA analyses reveal that L205R perturbs the allosteric network of PKA-C and disrupts specific allosteric nodes that connect the small and large lobes. The allosteric nodes are defined as the hotspots in the network with the highest number of CHESCA correlations (33). In particular, the allosteric communication between the node including L205 and encompassing the activation loop and αF and αG helices and the node surrounding the Gly-rich loop, β2, and β3 is ablated. Closer analysis of the CHESCA map of PKA-CWT reveals that each allosteric node harbors Cushing’s mutations located in the activation/αF/αG node except E31V, where the latter resides at the N-terminal αA helix, a unique regulatory motif in PKA-C (Fig. 5A) (34). Although spatially distinct, these allosteric nodes are highly correlated and coupled to one another to facilitate intramolecular communication and control binding cooperativity. Perturbation to one node, such as the case of L205R, has long-range effects on the other node. The latter suggests that Cushing’s mutations including E31V may disrupt the allosteric communication eliciting similar global responses in terms of binding cooperativity and regulation.
Fig. 5. Cushing’s mutations are located in allosteric nodes identified via CHESCA.
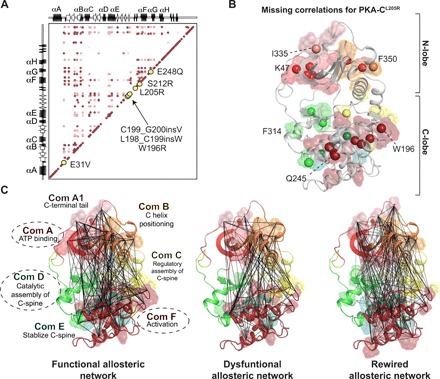
(A) Correlation matrix of PKA-CWT when bound to PKI emphasizing locations of Cushing’s mutation in relation to allosteric nodes. (B) Missing correlations for PKA-CL205R upon binding PKI colored according to the community map of PKA-CWT. (C) CHESCA correlation matrices for PKA-CWT + PKI, PKA-CL205R + PKI, and PKA-CL205R + VPS36, respectively, plotted on the structure of PKA-C with colors specific to the community map analyses completed previously for PKA-CWT (35). Specific communities are emphasized to highlight elements that experience the most dramatic changes in the number and extent of chemical shift covariance for PKA-CL205R upon binding PKI and VPS36.
The CHESCA analyses of the chemical shifts parallel the predictions obtained from MD simulations using the community analysis. Analyzing the different forms of the kinase, McClendon et al. (35) identified allosterically linked communities within PKA-C, each associated with a particular function or regulatory mechanism. Plotting correlated residues on PKA-C’s community maps reveals that the mutation dramatically reduces the allosteric communication between communities A, D, and F with respect to the wild-type enzyme (Fig. 5, B and C). In contrast, binding of VPS36 to PKA-CL205R reestablishes the intercommunity communication and the allosteric network.
The lack of regulation only partially explains the aberrant function of PKA-CL205R. Our thermodynamic and kinetic analyses reveal that this mutation shifts the specificity of PKA-CL205R toward substrates containing acidic residues in the P+1 and P+2 positions, with a dramatic effect on binding cooperativity. While the cooperativity is greatly abolished with the classical consensus sequence, the mutated enzyme regains cooperativity with complementary substrates such as VPS36, among others. The reason for this behavior is apparent from the atomic mapping of the cooperative global response of the enzyme to ligand binding. While the allosteric communication between the N-lobe, harboring the ATP binding site, and the C-lobe, with the substrate binding site, is abolished with the classical consensus sequence, the binding of the complementary substrate (VPS36) coincides with an extensive rewiring of the allosteric network of communication between the two lobes, altering substrate specificity. It should be noted that VPS36 lacks the HAR, which might affect its binding kinetics and thermodynamics. As with previous studies (17), VPS36 does not drive the enzyme to a completely closed state, perhaps facilitating product release. The reduction of the binding affinity for the nucleotide we observed under our experimental conditions was also detected by Luzi et al. (19). Taken with our NMR and thermodynamic studies, MD simulations suggest that the mutation changes the energy landscape of the kinase (Fig. 6). First, the mutation causes a reduction in binding affinity for nucleotide, which may be due to an increase in the population of the enzyme with the Gly-loop partially occluded. This conformation has been detected in other crystal structures and might prevent the nucleotide to intercalate in the C-spine of the enzyme (36). Moreover, a significant population of the enzyme spans a conformational space featuring a flipped conformation of the activation loop and a salt bridge formed between R194 and E86 in the αC helix. Notably, an increased plasticity of the activation loop, highlighted by W196, contributes to disruptions in the docking surface with the R-subunit (37). At the same time, the R194-E86 salt bridge suggests a different dynamic coupling between the activation loop and the αC helix. It is possible that this specific conformational state might dictate the selectivity of the substrate binding and the aberrant profile observed in phosphoproteomic assays.
Fig. 6. Schematic of the energy landscape for PKA-CWT and PKA-CL205R, combining thermodynamics and MD simulations data.
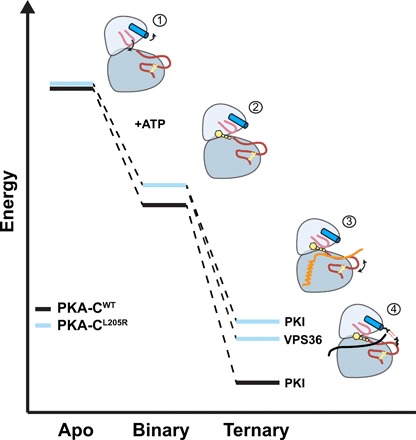
Relative free energy for the binding of ATPγN, PKI, and VPS36 to PKA-CWT and PKA-CL205R derived from ITC data. (1) Apo PKA-CL205R samples mostly uncommitted states, with the Gly-rich loop partially occluded and the αC helix turned outward. (2) Binary PKA-CL205R features a wired allosteric network for substrate binding, i.e., committed state. (3) Ternary PKA-CL205R complex with lower affinity for PKI (i.e., higher free energy relative to PKA-CWT). The conformation of the activation loop of PKA-CL205R is in equilibrium between an unflipped and a sparsely populated flipped conformation. (4) PKA-CL205R/ATPγN/VPS36 ternary complex features a flipped conformation of the activation loop with the electrostatic interactions between E86 and R194.
Given the ubiquitous nature of PKA and its involvement in numerous cell signaling events, its implication in specific diseases has been overlooked. Only recently, the attention of researchers has been directed to the PRKACA gene and its role in pathological lesions. A few years ago, Simon and co-workers discovered the presence of a chimeric construct of the PKA-C subunit that is the main driver of fibrolamellar hepatocellular carcinoma (38). Later on, our group and others discovered single mutations or insertions at the R/C interface that are linked to adrenal Cushing’s syndrome (3–7). More recently, Tseng et al. (39) reported the discovery of a new insertion in a similar region that is involved in the development of myxoma, a sporadic form of tumor in the atrium of the heart. It is possible that other defects in the PRKACA gene might be found to be implicated in the development of other tumors.
The analysis of the allosteric network carried out in this work reveals highly coupled allosteric nodes that harbor all Cushing’s mutations found in patients and may help to explain why mutations located in two spatially distinct regions of the enzyme result in the same phenotype. Our findings suggest that analysis of allosteric networks using CHESCA may prove useful in predicting mutations that perturb catalytic function for enzymes beyond PKA-C. Furthermore, our work reveals that rewiring of the intra- and intermolecular interactions leads to changes in cooperativity and selectivity. This suggests the opportunity to exploit small molecules or peptides that can change the internal communication and modulate the activity of this aberrant mutant selectively, i.e., without affecting the function of the wild type.
MATERIALS AND METHODS
Sample preparation
The recombinant human Cα subunit of cAMP-dependent PKA cDNA (PKA-CWT and PKA-CL205R) was cloned into a pET-28a vector. A tobacco etch virus (TEV) cleavage site was incorporated via mutagenesis into the vector between the cDNA coding for the kinase and a thrombin cleavage site. The kinase was expressed in E. coli BL21 (DE3) pLysS cells in M9 minimal media supplemented with 15NH4Cl. Protein overexpression was induced with 0.4 mM isopropyl β-d-thiogalactopyranoside and carried out overnight at 20°C. PKA-C purification was carried out using Ni2+ affinity chromatography. Cells were lysed using a French press in 50 mM tris-HCl, 30 mM KH2PO4, 200 mM NaCl, 200 μM ATP, 5 mM 2-mercaptoethanol, and lysozyme [lysis buffer (15 mg/100 ml)] (pH 8.0). After removing cell debris by centrifugation at 18,000 rpm for 45 min, the supernatant was incubated with Ni2+ nitrilotriacetic acid resin (Thermo Fisher Scientific) at 4°C overnight. The resin was washed with 50 mM tris-HCl, 30 mM KH2PO4, 200 mM NaCl, 10 mM imidazole, and 5 mM 2-mercaptoethanol (pH 8.0) and eluted with 50 mM tris-HCl, 30 mM KH2PO4, 200 mM NaCl, 250 mM imidazole, and 5 mM 2-mercaptoethanol (pH 8.0). Fractions containing PKA-C were cleaved overnight at 4°C with a sufficient amount of recombinant TEV while being dialyzed into 20 mM KH2PO4, 25 mM KCl, 0.1 mM phenylmethylsulfonyl fluoride, and 5 mM 2-mercaptoethanol (pH 6.5). The three isoforms of PKA-C (corresponding to the three phosphorylation states with identical catalytic parameters) were separated by chromatography on a HiTrap SP column (GE Healthcare Life Sciences) using a linear gradient from buffer A [20 mM KH2PO4 (pH 6.5)] to 30% buffer B [20 mM KH2PO4 and 1 M KCl (pH 6.5)] at a flow rate of 2 ml/min. Isoform II with pThr197, pSer338, and pSer10 was used for all NMR experiments. Peptides (Kemptide/PKI5–24/VPS36) were synthesized using standard Fmoc chemistry on a CEM Liberty Blue microwave synthesizer, cleaved with Reagent K (82.5% trifluoroacetic acid, 5% phenol, 5% thioanisole, 2.5% ethanedithiol, and 5% water) for 3 hours, and purified using a semipreparative Supelco C18 reverse-phase HPLC column at 3 ml/min. The VPS36 peptide sequence used for all studies is as follows: QIEFYRRLSEEMTQR. The molecular weight and quantity of the peptides were verified by liquid chromatography–mass spectrometry and/or amino acid analysis (Texas Tech Protein Chemistry Laboratory).
ITC measurements
ITC measurements were performed with a low-volume Nano ITC (TA Instruments). PKA-CWT and PKA-CL205R were dialyzed into 20 mM Mops, 90 mM KCl, 10 mM dithiothreitol (DTT), 10 mM MgCl2, and 1 mM NaN3 (pH 6.5). PKA-C concentrations for ITC measurements were between 100 and 130 μM as confirmed by A280 = 53,860 M−1 cm−1. All measurements with ATPγN-saturated PKA-CWT and PKA-CL205R were performed at 2 mM ATPγN and 4 mM ATPγN, respectively. ITC measurements were performed at 300 K in triplicate. Approximately 300 μl of PKA-C was used for each experiment, as well as 50 μl of 2 to 4 mM ATPγN, 0.6 to 4 mM PKI, or 2 mM VPS36 in the titrant syringe. The heat of dilution of the ligand into the buffer was taken into account for all experiments and subtracted accordingly. Binding was assumed to be 1:1, and curves were analyzed with the NanoAnalyze software (TA Instruments) using the Wiseman isotherm (40)
| (1) |
where d[MX] is the change in total complex with respect to change in total protein concentration and d[Xtot] is dependent on r (the ratio of Kd with respect to the total protein concentration) and RM (the ratio between total ligand and total protein concentration). The free energy of binding was determined using the following
where R is the universal gas constant and T is the temperature at measurement (300 K). The entropic contribution to binding was calculated using the following
Calculations for the cooperativity constant (σ) were calculated as follows
where Kd Apo is the Kd of PKI5–24 binding to the apoenzyme and Kd Nucleotide is the Kd of PKI5–24 binding to the nucleotide-bound enzyme.
Enzyme assays
Steady-state activity assays with Kemptide and VPS36 were performed under saturating ATP concentrations and spectrophotometrically at 298 K, as described by Cook et al. (41). The values of Vmax and KM were obtained from a nonlinear fit of the initial velocities to the Michaelis-Menten equation.
Circular dichroism
A change in ellipticity was measured at 220 nm on a Jasco J-815 spectrometer. A temperature scan between 20° and 70°C at a rate of 1°C/min with an equilibration time of 15 s was performed to unfold the protein. Spectra Manager Pro software was used to fit a two-state sigmoidal function. The inflection point was taken as the Tm (in °C), the point at which 50% of the protein is folded.
NMR spectroscopy
Uniformly 15N-labeled PKA-CWT and PKA-CL205R were overexpressed and purified as described above. NMR experiments were performed in 90 mM KCl, 20 mM KH2PO4, 10 mM DTT, 10 mM MgCl2, and 1 mM NaN3 at pH 6.5. Standard [1H, 15N]-TROSY-HSQC experiments were carried out for PKA-CL205R and PKA-CWT on 600-MHz and 900-MHz Bruker Avance III spectrometers equipped with TCI cryoprobes, respectively. Concentrations for samples were 0.2 to 0.3 mM as determined by A280 measurements, 12 mM ATPγN was added for the nucleotide-bound form, and 0.2 to 1.2 mM PKI or 0.4 to 1.8 mM VPS36 was added for the ternary complex. Full-length PKI was used for all NMR experiments and subsequent analyses. Spectra were collected at 300 K, processed using NMRPipe (42), and visualized using Sparky (43).
All [1H, 15N]-TROSY-HSQC experiments were acquired with 2048 (proton) and 256 (nitrogen) complex points. Spectra acquired in complex with VPS36 were acquired with substantially more scans (NS = 96) compared to other spectra [number of scans (NS) = 32 to 64]. Combined CSPs were calculated using 1H and 15N chemical shifts according to the following
| (2) |
Changes in CSPs were calculated according to the following for both nucleotide binding and pseudo-substrate binding
| (3) |
Chemical shift analyses
Coordinated chemical shifts behavior
CONCISE was used to monitor chemical shift trajectories and measure the change in equilibrium position using each PKA-C construct (apo, ATPγN, ATPγN/PKI, and ATPγN/VPS36). Using PCA, this method identifies sets of residues whose chemical shifts respond linearly to the conformational transition. Each residue provides a measure of the equilibrium position for every PKA-C construct in the form of scores along the first principal component (PC1). The equilibrium position for a given construct is given by the average of all principal component scores over all linear residues. To identify the residues whose chemical shifts follow a linear trajectory, a threshold of 3.0 for the ratio of the SDs of PC1 over PC2 was used, and residues not exhibiting a significant chemical shift were excluded based on linewidth. After this threshold was applied, a total of 55 residues formed the subset that was used to trace the equilibrium position of each state for PKA-CWT and PKA-CL205R.
Chemical shift covariance analysis
CHESCA was used to identify and functionally characterize allosteric networks of residues eliciting concerted responses to, in this case, nucleotide, pseudo-substrate, and substrate. A total of three states were used to identify inter-residue correlations: apo, ATPγN-bound, and either ATPγN/PKI- or ATPγN/VPS36-bound. Identification of inter-residue correlations by CHESCA relies on agglomerative clustering and singular value decomposition. Pairwise correlations between chemical shift variations experienced by different residues are analyzed to identify networks of coupled residues and, when plotted on a correlation matrix, allow for the identification of regions that are correlated to one another. A correlation coefficient cutoff of 0.98 was used to filter nonlinear residues. Residues not exhibiting a significant chemical shift (small shifts in parts per million) were excluded based on linewidth. Linewidth was calculated for each resonance in each of the three forms (apo, ATPγN, and ATPγN/PKI or ATPγN/VPS36). For each residue, the maximum change in chemical shift was calculated in both the 1H (x) and 15N (y) dimension (Δδx,y). Residues were included in CHESCA analysis only if they satisfied the following: Δδx,y > ½ ΔνxA,yA + ½ Δν xB,yB, where A and B correspond to two different forms analyzed (note that there is no dependence on which two forms satisfied this statement).
MD simulations
System setup on wild type and L205R
We used the crystal structure of PKA-CWT (PDB ID: 1ATP) (28) and PKA-CL205R (PDB ID: 4WB6) (16) as the template and chose a monomer (chain A, protein; chain I, PKI5–24) from the dimer. We further aligned the current structure with the full-length PKA-CWT and added the missing residues 1 to 12 at the N terminus. The protonation state of histidine residues followed our previous settings (27). The protein was solvated in a rhombic dodecahedron solvent box with a TIP3P water molecule layer extended approximately 10 Å away from the surface of the proteins. Counter ions (K+ and Cl−) were added to ensure electrostatic neutrality corresponding to an ionic concentration of ~150 mM. All protein covalent hydrogen bonds were constrained with the LINCS (linear constraint solver) algorithm, and long-range electrostatic interactions were treated with the particle-mesh Ewald method with a real-space cutoff of 10 Å. Parallel simulations on the apo form, the binary form with one Mg2+ ion and one ATP, and the ternary form with two Mg2+ ions, one ATP, and one PKI5–24 were performed simultaneously using GROMACS 4.6 (44) in CHARMM36a1 force fields (45). Each system was minimized using the steepest decent algorithm to remove the bad contacts and then gradually heated to 300 K at a constant volume over 1 ns, using harmonic restraints with a force constant 1000 kJ/(mol·Å2) on heavy atoms of both proteins and nucleotides. Over the following 12 ns of simulations at constant pressure (1 atm) and temperature (300 K), the restraints were gradually released. The systems were equilibrated for an additional 20 ns without positional restraints. A Parrinello-Rahman barostat was used to keep the pressure constant, while a V-rescale thermostat with a time step of 2 fs was used to keep the temperature constant. Each system was simulated for 1.05 μs, with snapshots recorded every 20 ps. A total of 3.15 μs and 157,500 conformations were used for the analyses.
Energy landscapes using PCA
Cartesian principal components of the backbone atoms were calculated using the GROMACS modules g_covar and g_anaeig to identify the large-scale, low-frequency conformational dynamics of the catalytic core. All the trajectories were aligned with the starting structure (minimization of the crystal structure to remove bad contacts) using helices E (residues 140 to 160) and F (residues 217 to 233) as a reference frame. Dominant principal components were computed from each resulting ensemble from the individual simulations. Moreover, the distance between the Cα atom of Ser53 and Gly186 was measured to characterize the opening and closing motions of the Gly-rich loop. Different trajectories were mapped onto the 2D projection along PC1 and S53-G186 distances.
Docking and simulation of the ternary complexes bound with VPS36 peptide and ATP
We used the unwound conformation of VPS36 and used HADDOCK (46) server for docking into the binding cleft of PKA-C for both wild type and L205R. Specifically, we used the easy interface and selected the active residues of PKA-C, i.e., 133, 168, 202, 198, 204, 205, 207, 230, and 330, as well as the active residues for VPS36, i.e., 3, 6, 7, 9, and 10. The passive residues were set automatically around the active residues by the server. The top-scored structures were further solvated for MD simulations following the same protocol as the ternary complexes with PKI.
Mutual information analysis and mapping of the allosteric network
To monitor the allosteric differences of the wild type and L205R, MutInf (29) was used to compute mutual information between all residues. MutInf is a python package that translates the distribution of dihedral angles of residues into their conformational entropy and identifies the correlated motions between residues. The time series of dihedral angles in the MD ensemble was computed using g_chi and divided into six overlapped blocks, and then, correlations of local motions were computed as the mutual information between selected residue pairs in each block. The results were averaged over these blocks to filter out the correlations that were not statistically significant. These matrices of mutual information and their differences were further mapped onto the crystal structure. Graph analysis was applied to detect hubs in the networks and key allosteric communication pathways in PKA-C.
Supplementary Material
Acknowledgments
NMR experiments were carried out at the Minnesota NMR Center, and MD calculations were performed at the Minnesota Supercomputing Institute. Funding: This work was supported by the National Institutes of Health (GM100310 and S10 OD021536 to G.V.) and the CRC/Transregio 166 (Project C1 to D.C.), as well the IZKF Würzburg (grant B-281 to D.C.). K.B. was partially supported by a fellowship through a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. Author contributions: C.W. prepared the PKA-C samples; designed, executed, and analyzed all NMR experiments; and contributed to the writing of the manuscript. Y.W. carried out all MD simulations and contributed to the writing of the manuscript. C.O. carried out triple-resonance NMR experiments for backbone assignment of PKA-C. A.K. carried out the initial NMR, thermodynamics, and kinetic experiments. J.C. assisted in the preparation of PKA-C samples. K.B. and D.C. contributed to the critical analysis of the data and writing of the manuscript. J.G. assisted Y.W. with setting up the MD and Markov model calculations. D.A.B. contributed to the critical analysis of the data and writing of the manuscript. S.S.T. contributed to the writing of the manuscript. G.V. designed the experiments and contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw9298/DC1
Fig. S1. [1H, 15N]-TROSY-HSQC spectra for PKA-CWT and PKA-CL205R in apo, ATPγN, and ATPγN/PKI-bound and ATPγN/VPS36-bound forms.
Fig. S2. CSPs observed upon ligand binding for PKA-CWT and PKA-CL205R.
Fig. S3. Intensity plot for the binding of VPS36 to ATPγN-saturated PKA-CL205R.
Fig. S4. PCA of the catalytic lobes in PKA-CWT and PKA-CL205R.
Fig. S5. Probability of the formation of inter-residue contact and ΔRMSF of PKA-C upon forming ternary complexes with PKI5–24 or VPS36.
Fig. S6. Allosteric changes upon peptide binding revealed by MD simulation and mutual information (MutInf) analysis.
Table S1. Changes in enthalpy, entropy, free energy, and dissociation constant of binding ATPγN, PKI5–24, and VPS36 for PKA-CWT and PKA-CL205R.
Table S2. Kinetic parameters of Kemptide and VPS36 phosphorylation by PKA-CWT and PKA-CL205R.
Table S3. PCA and SD of the CONCISE analysis of the structural states analyzed.
Table S4. Tm as determined using CD.
REFERENCES AND NOTES
- 1.Hunter T., Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2513–2516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lodish M., Stratakis C. A., A genetic and molecular update on adrenocortical causes of Cushing syndrome. Nat. Rev. Endocrinol. 12, 255–262 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Cao Y., He M., Gao Z., Peng Y., Li Y., Li L., Zhou W., Li X., Zhong X., Lei Y., Su T., Wang H., Jiang Y., Yang L., Wei W., Yang X., Jiang X., Liu L., He J., Ye J., Wei Q., Li Y., Wang W., Wang J., Ning G., Activating hotspot L205r mutation in PRKACA and adrenal Cushing's syndrome. Science 344, 913–917 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Sato Y., Maekawa S., Ishii R., Sanada M., Morikawa T., Shiraishi Y., Yoshida K., Nagata Y., Sato-Otsubo A., Yoshizato T., Suzuki H., Shiozawa Y., Kataoka K., Kon A., Aoki K., Chiba K., Tanaka H., Kume H., Miyano S., Fukayama M., Nureki O., Homma Y., Ogawa S., Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344, 917–920 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Goh G., Scholl U. I., Healy J. M., Choi M., Prasad M. L., Nelson-Williams C., Kunstman J. W., Korah R., Suttorp A.-C., Dietrich D., Haase M., Willenberg H. S., Stålberg P., Hellman P., Åkerström G., Björklund P., Carling T., Lifton R. P., Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 46, 613–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beuschlein F., Fassnacht M., Assié G., Calebiro D., Stratakis C. A., Osswald A., Ronchi C. L., Wieland T., Sbiera S., Faucz F. R., Schaak K., Schmittfull A., Schwarzmayr T., Barreau O., Vezzosi D., Rizk-Rabin M., Zabel U., Szarek E., Salpea P., Forlino A., Vetro A., Zuffardi O., Kisker C., Diener S., Meitinger T., Lohse M. J., Reincke M., Bertherat J., Strom T. M., Allolio B., Constitutive activation of pka catalytic subunit in adrenal Cushing's syndrome. N. Engl. J. Med. 370, 1019–1028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Dalmazi G., Kisker C., Calebiro D., Mannelli M., Canu L., Arnaldi G., Quinkler M., Rayes N., Tabarin A., Laure Jullié M., Mantero F., Rubin B., Waldmann J., Bartsch D. K., Pasquali R., Lohse M., Allolio B., Fassnacht M., Beuschlein F., Reincke M., Novel somatic mutations in the catalytic subunit of the protein kinase a as a cause of adrenal cushing's syndrome: A european multicentric study. J. Clin. Endocrinol. Metab. 99, E2093–E2100 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Taylor S. S., Ilouz R., Zhang P., Kornev A. P., Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langeberg L. K., Scott J. D., Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knighton D. R., Zheng J. H., Ten Eyck L. F., Ashford V. A., Xuong N. H., Taylor S. S., Sowadski J. M., Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 407–414 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Johnson D. A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S. S., Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Calebiro D., Hannawacker A., Lyga S., Bathon K., Zabel U., Ronchi C., Beuschlein F., Reincke M., Lorenz K., Allolio B., Kisker C., Fassnacht M., Lohse M. J., Pka catalytic subunit mutations in adrenocortical cushing’s adenoma impair association with the regulatory subunit. Nat. Commun. 5, 5680 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Rock R., Mayrhofer J. E., Bachmann V., Stefan E., Impact of kinase activating and inactivating patient mutations on binary pka interactions. Front. Pharmacol. 6, 170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bathon K., Weigand I., Vanselow J. T., Ronchi C. L., Sbiera S., Schlosser A., Fassnacht M., Calebiro D., Alterations in protein kinase a substrate specificity as a potential cause of Cushing’s syndrome. Endocrinology 160, 447–459 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Lubner J. M., Dodge-Kafka K. L., Carlson C. R., Church G. M., Chou M. F., Schwartz D., Cushing’s syndrome mutant PKAL205R exhibits altered substrate specificity. FEBS Lett. 591, 459–467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung J., Ginter C., Cassidy M., Franklin M. C., Rudolph M. J., Robine N., Darnell R. B., Hendrickson W. A., Structural insights into mis-regulation of protein kinase a in human tumors. Proc. Natl. Acad. Sci. U.S.A. 112, 1374–1379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masterson L. R., Mascioni A., Traaseth N. J., Taylor S. S., Veglia G., Allosteric cooperativity in protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 105, 506–511 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J., Li G., Walters M. A., Taylor S. S., Veglia G., Uncoupling catalytic and binding functions in the cyclic amp-dependent protein kinase A. Structure 24, 353–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luzi N. M., Lyons C. E., Peterson D. L., Ellis K. C., Kinetics and inhibition studies of the l205r mutant of cAMP-dependent protein kinase involved in Cushing's syndrome. FEBS Open Bio 8, 606–613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pervushin K., Riek R., Wider G., Wüthrich K., Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cembran A., Kim J., Gao J., Veglia G., NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 16, 6508–6518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selvaratnam R., Chowdhury S., VanSchouwen B., Melacini G., Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6133–6138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulton S., Akimoto M., Selvaratnam R., Bashiri A., Melacini G., A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci. Rep. 4, 7306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J., Ahuja L. G., Chao F.-A., Xia Y., McClendon C. L., Kornev A. P., Taylor S. S., Veglia G., A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 3, e1600663 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornev A. P., Taylor S. S., Ten Eyck L. F., A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masterson L. R., Cheng C., Yu T., Tonelli M., Kornev A., Taylor S. S., Veglia G., Dynamics connect substrate recognition to catalysis in protein kinase A. Nat. Chem. Biol. 6, 821–828 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masterson L. R., Shi L., Metcalfe E., Gao J., Taylor S. S., Veglia G., Dynamically committed, uncommitted, and quenched states encoded in protein kinase a revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 6969–6974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng J., Trafny E. A., Knighton D. R., Xuong N., Taylor S. S., Ten Eyck L. F., Sowadski J. M., 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. Sect. D 49, 362–365 (1993). [DOI] [PubMed] [Google Scholar]
- 29.McClendon C. L., Friedland G., Mobley D. L., Amirkhani H., Jacobson M. P., Quantifying correlations between allosteric sites in thermodynamic ensembles. J. Chem. Theory Comput. 5, 2486–2502 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horvath A., Bertherat J., Groussin L., Guillaud-Bataille M., Tsang K., Cazabat L., Libé R., Remmers E., René-Corail F., Faucz F. R., Clauser E., Calender A., Bertagna X., Carney J. A., Stratakis C. A., Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Hum. Mutat. 31, 369–379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruystens J. G. H., Wu J., Fortezzo A., Kornev A. P., Blumenthal D. K., Taylor S. S., PKA RIα homodimer structure reveals an intermolecular interface with implications for cooperative cAMP binding and Carney complex disease. Structure 22, 59–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kannan N., Neuwald A. F., Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2α. Protein Sci. 13, 2059–2077 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aoto P. C., Martin B. T., Wright P. E., NMR characterization of information flow and allosteric communities in the map kinase p38gamma. Sci. Rep. 6, 28655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herberg F. W., Zimmermann B., McGlone M., Taylor S. S., Importance of the a-helix of the catalytic subunit of cAMP-dependent protein kinase for stability and for orienting subdomains at the cleft interface. Protein Sci. 6, 569–579 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClendon C. L., Kornev A. P., Gilson M. K., Taylor S. S., Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. U.S.A. 111, E4623–E4631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonn S., Herrero S., Breitenlechner C. B., Erlbruch A., Lehmann W., Engh R. A., Gassel M., Bossemeyer D., Structural analysis of protein kinase a mutants with rho-kinase inhibitor specificity. J. Biol. Chem. 281, 24818–24830 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Wu J., Brown S. H. J., von Daake S., Taylor S. S., PKA type IIα holoenzyme reveals a combinatorial strategy for isoform diversity. Science 318, 274–279 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honeyman J. N., Simon E. P., Robine N., Chiaroni-Clarke R., Darcy D. G., Lim I. I. P., Gleason C. E., Murphy J. M., Rosenberg B. R., Teegan L., Takacs C. N., Botero S., Belote R., Germer S., Emde A.-K., Vacic V., Bhanot U., LaQuaglia M. P., Simon S. M., Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343, 1010–1014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tseng I. C., Huang W. J., Jhuang Y. L., Chang Y. Y., Hsu H. P., Jeng Y. M., Microinsertions in PRKACA cause activation of the protein kinase a pathway in cardiac myxoma. J. Pathol. 242, 134–139 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Wiseman T., Williston S., Brandts J. F., Lin L.-N., Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179, 131–137 (1989). [DOI] [PubMed] [Google Scholar]
- 41.Cook P. F., Neville M. E. Jr., Vrana K. E., Hartl F. T., Roskoski R. Jr., Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: Kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799 (1982). [DOI] [PubMed] [Google Scholar]
- 42.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRpipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Lee W., Tonelli M., Markley J. L., NMRfam-sparky: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hess B., Kutzner C., van der Spoel D., Lindahl E., Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., Mackerell A. D. Jr., Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dominguez C., Boelens R., Bonvin A. M. J. J., Haddock: A protein−protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw9298/DC1
Fig. S1. [1H, 15N]-TROSY-HSQC spectra for PKA-CWT and PKA-CL205R in apo, ATPγN, and ATPγN/PKI-bound and ATPγN/VPS36-bound forms.
Fig. S2. CSPs observed upon ligand binding for PKA-CWT and PKA-CL205R.
Fig. S3. Intensity plot for the binding of VPS36 to ATPγN-saturated PKA-CL205R.
Fig. S4. PCA of the catalytic lobes in PKA-CWT and PKA-CL205R.
Fig. S5. Probability of the formation of inter-residue contact and ΔRMSF of PKA-C upon forming ternary complexes with PKI5–24 or VPS36.
Fig. S6. Allosteric changes upon peptide binding revealed by MD simulation and mutual information (MutInf) analysis.
Table S1. Changes in enthalpy, entropy, free energy, and dissociation constant of binding ATPγN, PKI5–24, and VPS36 for PKA-CWT and PKA-CL205R.
Table S2. Kinetic parameters of Kemptide and VPS36 phosphorylation by PKA-CWT and PKA-CL205R.
Table S3. PCA and SD of the CONCISE analysis of the structural states analyzed.
Table S4. Tm as determined using CD.