

Abstract
An efficient CoIII-catalyzed three-component strategy to prepare homoallylic alcohols containing acyclic quaternary centers is disclosed. This transformation enables introduction of two C–C σ bonds via C–H bond activation and sequential addition to internally substituted dienes and a wide range of aldehydes and activated ketones. Isoprene and other internally monosubstituted dienes are effective inputs, with the reaction proceeding with high diastereoselectivity for those substrate combinations that result in more than one stereogenic center. Moreover, the opposite relative stereochemistry can be achieved by employing 1,2-disubstituted dienes. A mechanism for the transformation is proposed based upon the relative stereochemistry of the products and studies with isotopically labeled starting materials.
Keywords: C–H activation, homogeneous catalysis, multicomponent reactions, cobalt, diastereoselectivity
Graphical Abstract
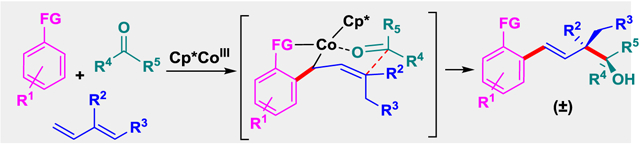
An efficient CoIII-catalyzed three-component synthesis of homoallylic alcohols containing acyclic quaternary centers is disclosed. This highly diastereoselective approach introduces two C–C σ bonds via C–H bond activation and sequential addition to internally substituted dienes and a wide range of aldehydes or activated ketones.
Transition metal-catalyzed C–H bond functionalization has become an effective strategy to access synthetically useful structural motifs.[1] While remarkable progress has been made for two-component C–H bond additions to diverse coupling partners, the sequential addition of C–H bonds across two different coupling partners to access complex motifs has only recently begun to be developed.[2,3] Recently, our lab reported the CoIII-catalyzed C–H bond addition to dienes and aldehydes [Figure 1, Eq. (1)].[2c,4] In this approach, two new C–C σ bonds are formed in a regio- and stereoselective manner as dictated by a CoIII-allyl chair-like transition state. To expand upon this work, we have focused on internally substituted dienes to introduce acyclic quaternary centers, which are synthetically challenging motifs to access due to their congested nature.[5,6] Recently Zhao et al. reported the first RhIII-catalyzed three-component C–H bond addition to a range of monosubstituted dienes and aldehydes [Eq. (1)].[2a] Although two examples of additions of internally substituted dienes were included, these reactions proceeded in modest yields and were demonstrated only for the highly activated aldehyde ethyl glyoxylate.
Figure 1.

Transition metal-catalyzed three-component addition for the synthesis of homoallylic alcohols.
Herein, we describe a three-component strategy to access homoallylic alcohols containing acyclic quaternary centers via CoIII-catalyzed C–H bond activation and sequential addition to internally substituted dienes and aldehydes or activated ketones [Figure 1, Eq. (2–4)]. For isoprene, secondary alcohol products are obtained by reactions of a variety of C–H bond substrates with both activated and unactivated alkyl and aryl aldehydes [Eq. (2)]. In addition, the first examples of three-component coupling with activated ketones[7] provide tertiary alcohols that incorporate contiguous tetrasubstituted carbons [Eq. (2)]. Upon extending the three-component reaction to internally monosubstituted dienes other than isoprene, two stereogenic centers are introduced with high diastereoselectivity [Eq. (3)].[5] Moreover, the opposite relative stereochemistry of the methyl and hydroxyl groups on the two vicinal stereogenic carbon centers can be obtained by employing previously unexplored 1,2-disubstituted dienes [Eq. (4)]. The catalytic cycle, which is supported by studies with isotope labeled starting materials, provides a rationale for the disparate stereochemical outcomes for the two different diene substitution patterns [Eqs (3) and (4)].
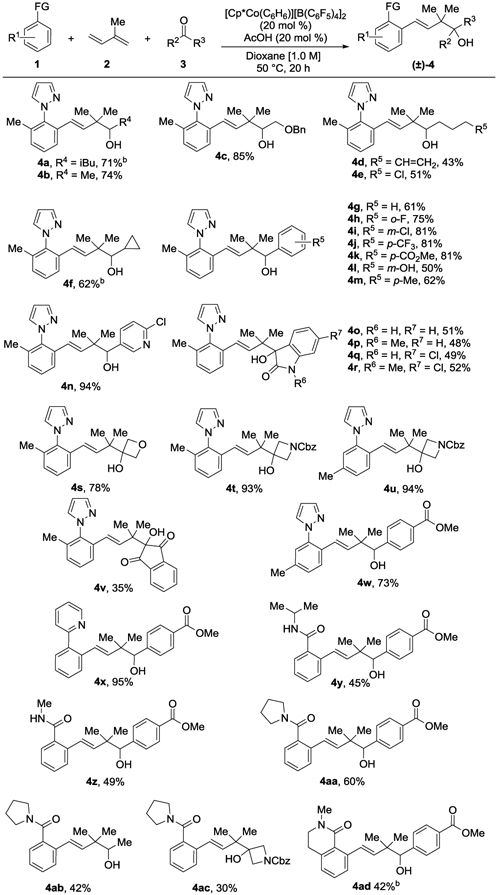
An extensive evaluation of different reaction conditions was conducted for the three-component coupling of phenyl pyrazole 1a, isoprene 2a and isovaleraldehyde 3a (Table 1; see Supplementary Table S1). The use of [Cp*Co(C6H6)][B(C6F5)4]2,[8] previously developed in our lab, was found to be crucial, with other preformed CoIII and RhIII catalysts, such as [Cp*Co(C6H6)][PF4]2, [Cp*Rh(MeCN)2][SbF6]2 and [Cp*Rh(MeCN)2][SbF6]2, resulting in little to no conversion. A catalytic amount of AcOH was found to be beneficial as opposed to LiOAc[2f] or PivOH,[9] which have been used as additives for other CoIII-catalyzed C–H bond addition reactions. Concentration was shown to have a critical role in this transformation, with 0.4 M leading to much lower conversion than 1.0 M.[2c,d,f] Heating at 50 °C was chosen as the standard conditions for isoprene addition, but 70 °C was found to be necessary for some dienes and/or carbonyl compounds bearing bulkier substituents (vide infra). In addition, while dioxane was selected for the standard reaction conditions, DCE and toluene were also effective solvents, and toluene was in fact superior to dioxane for some sterically hindered coupling partners (vide infra).
Table 1.
C–H bond functionalization with isoprene and aldehydes/ketones.
|
Conditions: 0.2 mmol of 1, 0.8 mmol of 2, 0.6 mmol of 3. Isolated yields of products after purification by chromatography are reported.
Reactions performed in toluene at 70 ° C.
Having determined the optimal reaction conditions, we next explored the scope for isoprene addition (Table 1). First, alkyl aldehydes were investigated (4a-4b), including those displaying benzyl ether (4c), terminal alkene (4d), and primary alkyl chloride (4e) functionality. The reaction is sensitive to sterics, with α-branched cyclopropanecarboxaldehyde requiring an elevated 70 °C reaction temperature and toluene as solvent to provide homoallylic alcohol 4f in 62% yield. Not surprisingly, the more sterically hindered α,α-dibranched pivaldehyde gave only a trace amount of product (data not shown). A series of aryl aldehydes were next examined. Benzaldehyde (4g), along with derivatives with electron-deficient groups at the ortho, meta, or para-positions (4h-4k) all coupled in high yields. Acidic m-hydroxybenzaldehyde (4l) and the moderately electron-rich p-tolualdehyde (4m) coupled in 50% and 62% yields, respectively. The presence of a basic pyridyl nitrogen did not impede the reaction, with homoallylic alcohol 4n obtained in excellent yield.
Along with alkyl and aromatic aldehydes, the first examples of three-component coupling of more challenging ketones proceeded to give homoallylic tertiary alcohols 4o-4v that incorporate contiguous tetrasubstituted carbons (Table 1). Electron-deficient isatin and N-methylisatin furnished tertiary alcohols 4o and 4p, respectively, in moderate yields. Chloro-substituted isatin and N-methylisatin also afforded the alcohols 4q and 4r in comparable yields, respectively. The highly strained cyclic ketone 3-oxetanone also displayed good reactivity, providing tertiary alcohol 4s in good yield. Additionally, N-carboxybenzyl (Cbz)-protected 3-azetidinone afforded tertiary alcohols 4t and 4u in >90% yields. Reaction with the indantrione furnished 4v, albeit in moderate yield. We also investigated the coupling of unactivated ketones such as acetone and acetophenone, but three-component coupling products were not obtained.
A number of different C–H bond substrates were also investigated. A methyl substituent at either the ortho (4t) or meta (4u and 4w) position prevented bis-functionalization for phenyl pyrazole substrates. Additionally, 2-phenylpyridine served as an excellent C–H bond substrate, furnishing product 4x in near-quantitative yield. C–H bond substrates with amide directing groups were also effective as demonstrated for secondary N-isopropyl (4y) and N-methyl (4z) benzamides as well as for tertiary pyrrolidine benzamide (4aa-4ac). Lastly, homoallylic alcohol 4ad was prepared by C–H functionalization of N-methyldihydroisoquinolin-2-one.
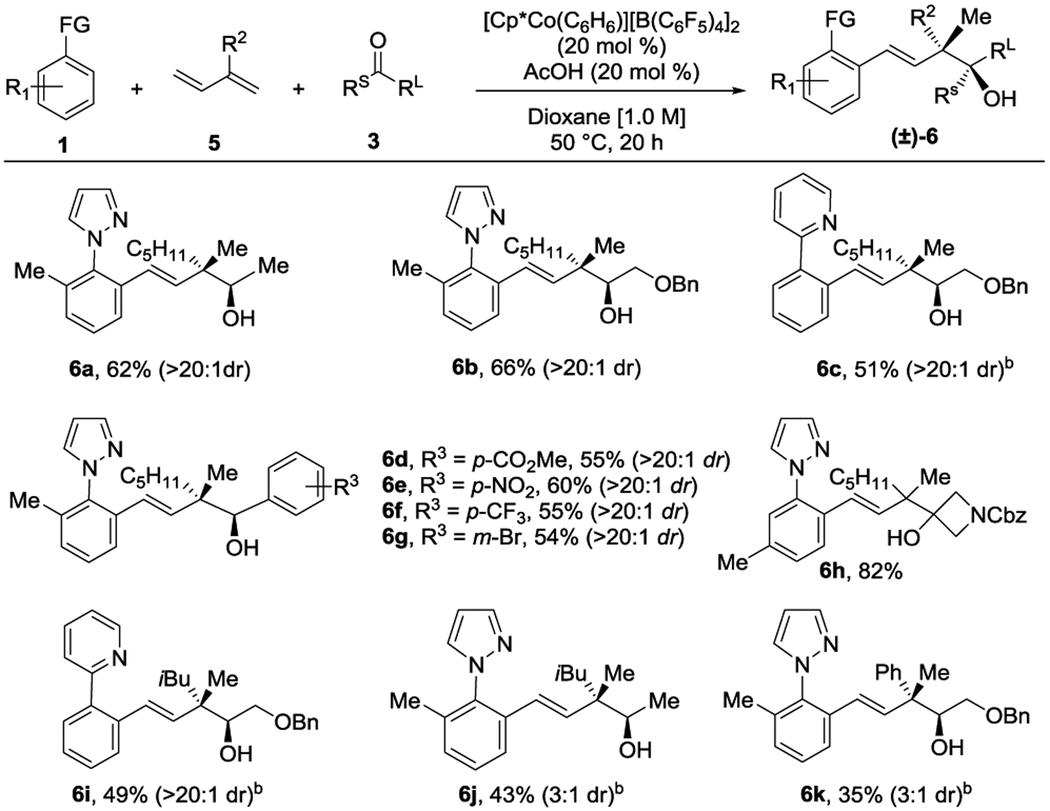
A series of internally substituted dienes 5 were next explored for preparing homoallylic alcohols 6 with stereogenic quaternary carbons (Table 2). Three-component addition to 3-methyleneoctene and acetaldehyde provided alcohol 6a in 62% yield and with >20:1 dr. The relative stereochemistry of 6a was confirmed by X-ray crystallographic determination of its 3,5-dinitrobenzoylated derivative, confirming a syn-relationship between the methyl and hydroxyl groups.[10] C–H bond substrates with both pyrazole and pyridine directing groups were effective for coupling with benzyloxyacetaldehyde to give alcohols 6b and 6c with high diastereoselectivities and 66% and 51% yields, respectively. Benzaldehyde derivatives with electron-withdrawing groups at the para position afforded the corresponding products 6d-6f in moderate yields. m-Bromobenzaldehyde was also an effective coupling partner to give alcohol 6g that incorporates a site for further elaboration. Contiguous tetrasubstituted carbons could also be introduced as demonstrated by coupling the activated ketone 1-Cbz-3-azetidinone to give alcohol 6h in good yield. More sterically demanding substituents on the diene were investigated as well. The isobutyl substituted diene coupled with 2-benzyloxyacetaldehyde to give the homoallylic alcohol 6i with good diastereoselectivity. In contrast, reaction with acetaldehyde provided product 6j in modest yield and with lower diastereoselectivity. The aryl substituted diene, 2-phenyl-1,3-butadiene, also furnished product 6k, albeit in low yield and with poor diastereoselectivity.
Table 2.
C–H bond functionalization with internally monosubstituted dienes and aldehydes/ketones.
|
Conditions: 0.2 mmol of 1, 0.4 mmol of 5, 0.6 mmol of 3. Isolated yields of products after purification by chromatography are reported.
Reactions performed at 70 °C for 40 h.
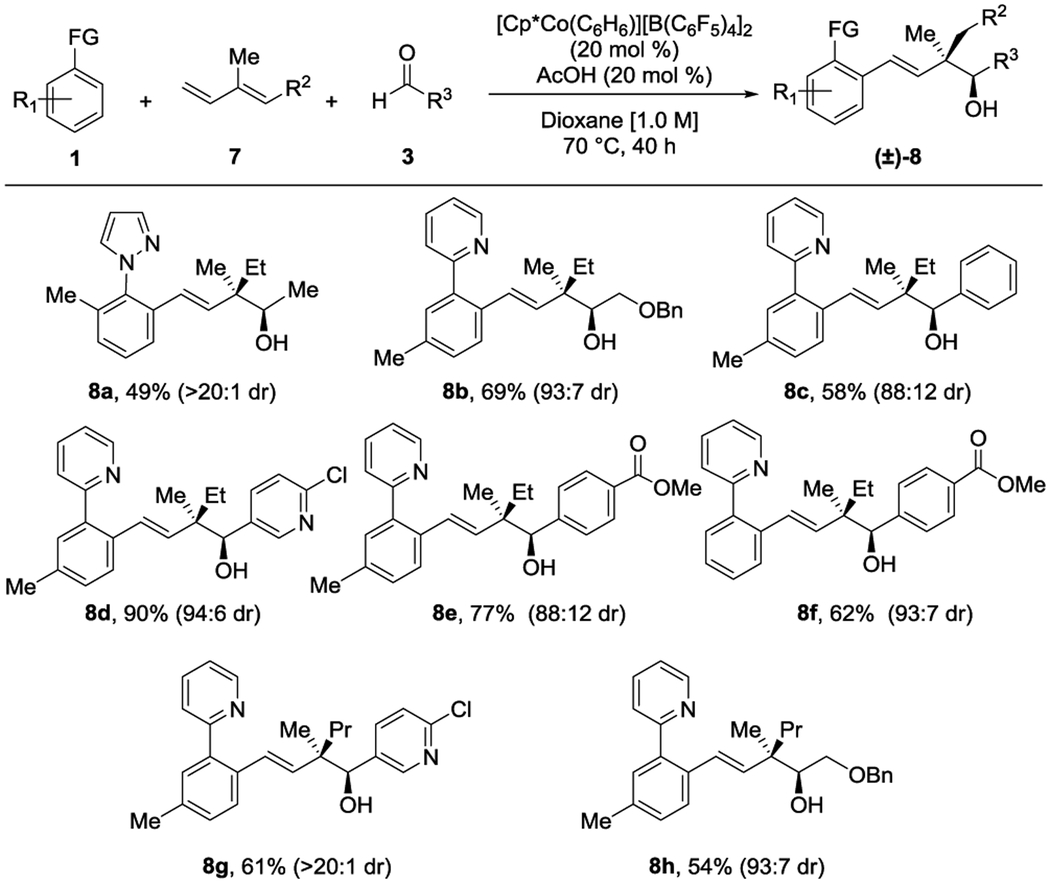
Next, we extended the scope to 1,2-disubstituted 1,3-dienes 7 (Table 3), which provide the opposite relative stereochemistry for the methyl and hydroxyl groups on the two contiguous stereogenic centers as compared to internally monosubstituted dienes 5 (Table 2). Three-component coupling with 3-methylpentadiene and acetaldehyde gave alcohol 8a in 49% yield and with high diastereoselectivity (>20:1 dr). The relative stereochemistry was confirmed by X-ray crystallographic determination of its 3,5-dintrobenzoylated derivative,[10] showing that the methyl and the hydroxyl groups are in an anti-relationship. Three-component coupling for C–H bond substrates with the pyridyl directing group provided moderate to excellent yields and good diastereoselectivity for alkyl, aromatic and heteroaromatic aldehydes (8b-8f). Finally, when the diene with R2 = Et was employed, both aryl (8g) and alkyl (8h) aldehydes gave homoallylic products that incorporated a propyl substituent at the quaternary carbon center.
Table 3.
C–H bond functionalization with 1,2-disubstituted dienes.
|
Conditions: 0.2 mmol of 1, 0.4 mmol of 7, 0.6 mmol of 3. Isolated yields of products after purification by chromatography are reported.
A series of experiments were conducted to obtain mechanistic information using isotopically labeled starting materials. When 2-(phenyl-D5)pyridine was subjected to the standard reaction conditions with isoprene 2 and 4-formylbenzoate as the coupling partners, an 18% conversion to the three-component coupling product was observed after a 30 min reaction time. Under these conditions, 58% of the C–H bond substrate was recovered and showed extensive H/D exchange at the ortho positions (Figure 2a). These results establish that the C–H activation step is reversible.
Figure 2.

Mechanistic experiments with deuterium labeled coupling partners.
When the terminally dideuterated isoprene 2-d2 was subjected to the standard reaction conditions, deuterium migration was observed on two different sites in the product 4u-d2 (Figure 2b). Only one out of the two methyl groups in 4u-d2 incorporated a significant amount of deuterium. Additionally, the ortho position of the aromatic ring incorporated 30% deuterium likely due to reversible C–H activation. The product incorporated 1.65 out of the two deuteriums present in 2-d2. Presumably, some deuterium loss occurs from CoIII–hydride intermediate C (Figure 3) during the catalytic cycle (vide infra).
Figure 3.

Proposed mechanism for the three-component transformation.
A mechanism is proposed based upon the studies with isotope labeled starting materials, the stereochemistry observed for the products, and prior mechanistic studies for three-component coupling of butadiene[2c] (Figure 3). In the presence of the CoIII-catalyst, a reversible ortho C–H bond activation of substrate 1 via concerted metalation deprotonation generates 5-membered metallocycle A.[11] Alkene coordination and migratory insertion of the diene affords the η3-Co-allyl species B.[2c] Syn β-hydride elimination provides species C, with the syn coplanar hydrogen Ha incorporated as the Co hydride. This intermediate undergoes reversible hydride reinsertion of Ha into the C-4 position of the diene, as evidenced by deuterium incorporation at the C-4 position (Figure 2b). Parallel reactions of 2 and 2-d2 proceeded at the same rate, establishing that neither the β-hydride elimination nor reinsertion is a rate limiting step (See Supplementary Information VI). Diastereoselective addition of intermediate D to aldehyde 3 through the chair-like transition state shown in E provides the observed relative stereochemistry in the final product for both the internally monosubstituted (Table 2) and 1,2-disubstituted (Table 3) dienes. Release of product F upon protonolysis regenerates the cobalt catalyst.
In summary, a CoIII-catalyzed C–H bond addition to form secondary and tertiary homoallylic alcohols containing quaternary carbons has been developed. The sequential C–H bond addition to internally substituted dienes and carbonyls shows broad scope, including not only alkyl and aryl aldehydes but also the first examples of additions to activated ketones for the formation of tertiary homoallylic alcohols. Additionally, this robust transformation proceeds with high diastereoselectivity for various internally monosubstituted dienes. Notably, with the first application of 1,2-disubstituted dienes, the opposite relative stereochemistry of the two stereogenic centers can be achieved. Both X-ray structural characterization of the products and mechanistic experiments support the proposed mechanism.
Supplementary Material
Acknowledgements
This work was supported by the NIH (R35GM122473). We gratefully acknowledge Dr. Brandon Mercado (Yale University) for solving the crystal structures reported in this manuscript. Helpful input provided by Jeffrey Boerth and Soham Maity is greatly appreciated.
References
- [1].Selected reviews on transition metal-catalyzed C–H bond funtionalization:; (a) Woźniak L, Cramer N, Trends in Chem 2019, 10.1016/j.trechm.2019.03.013; [DOI] [Google Scholar]; (b) Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L, Chem. Rev 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]; (c) Kim D-S, Park W-J, Jun C. -Ho., Chem. Rev 2017, 117, 8977–9015; [DOI] [PubMed] [Google Scholar]; (d) Hummel JR, Boerth JA, Ellman JA, Chem. Rev 2017, 117, 9163–9227; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J, Chem. Soc. Rev 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]; (f) Yang L, Huang H, Chem. Rev 2015, 115, 3468–3517; [DOI] [PubMed] [Google Scholar]; (g) Wencel-Delord J, Glorius F, Nat. Chem 2013, 5, 369–375; [DOI] [PubMed] [Google Scholar]; (h) Yamaguchi J, Yamaguchi AD, Itami K, Angew. Chem. Int. Ed 2012, 51, 8960–9009; Angew. Chem. 2012, 124, 9092–9142. [DOI] [PubMed] [Google Scholar]
- [2].(a) Li R, Ju C-W, Zhao D, Chem. Commun 2019, 55, 695–698; [DOI] [PubMed] [Google Scholar]; (b) Tang M, Li Y, Han S, Liu L, Ackermann L, Li J, Eur. J. Org. Chem 2019, 660–664; [Google Scholar]; (c) Boerth JA, Maity S, Williams SK, Mercado BQ, Ellman JA, Nat. Catal 2018, 1, 673–679; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Boerth JA, Ellman JA, Angew. Chem. Int. Ed 2017, 56, 9976–9980; Angew. Chem. 2017, 129, 10108–10112; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang S-S, Xia J, Wu J-Q, Liu X-G, Zhou C-J, Lin E, Li Q, Huang S-L, Wang H, Org. Lett 2017, 19, 5868–5871; [DOI] [PubMed] [Google Scholar]; (f) Boerth JA, Hummel JR, Ellman JA, Angew. Chem. Int. Ed 2016, 55, 12650–12654; Angew. Chem. 2016,128, 12840–12844; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Boerth JA, Ellman JA, Chem. Sci 2016, 7, 1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].A three-component approach initiated by Pd-catalyzed C-I oxidative addition followed by C−H functionalization:; Ye J, Lautens M, Nat. Chem 2015, 7, 863. [DOI] [PubMed] [Google Scholar]
- [4].Selected reviews on CoIII-catalyzed C–H bond functionalization:; (a) Planas O, Chirila PG, Whiteoak CJ, Ribas X, Adv. Organomet. Chem 2018, 69, 209–282; [Google Scholar]; (b) Santhoshkumar R, Cheng C-H, Beilstein J. Org Chem 2018, 14, 2266–2288; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoshino S, Matsunaga S, Adv. Synth. Catal 2017, 359, 1245–1262; [Google Scholar]; (d) Moselage M, Li J, Ackermann L, ACS. Catal 2016, 6, 498–525; [Google Scholar]; (e) Wei D, Zhu X, Niu J-L, Song MP, Chem. Cat. Chem 2016, 8, 1242–1262; [Google Scholar]; (c) Yoshikai N, Chem. Cat. Chem 2015, 7, 732–734. [Google Scholar]
- [5].Selected reviews on stereogenic quaternary carbon centers:; (a) Marek I, Pierrot D, Angew. Chem. Int. Ed 2019, 10.1002/anie.201903188; Angew. Chem. 2019 https://doi.org/10.1002/ange.201903188; [DOI] [Google Scholar]; (b) Liu Y, Han S-J, Liu W-B, Stoltz BM, Acc. Chem. Res 2015, 48, 740–751; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marek I, Minko Y, Pasco M, Mejuch T, Gilboa N, Chechik H, Das JP, J. Am. Chem. Soc 2014, 136, 2682–2694; [DOI] [PubMed] [Google Scholar]; (c) Quasdorf KW, Overman LE, Nature 2014, 516, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Recent examples of syntheses of homoallylic alcohols vicinal to acyclic quaternary centers:; (a) Xiong Y, Zhang G, Org. Lett 2016, 18, 5094–5097; [DOI] [PubMed] [Google Scholar]; (b) Nguyen KD, Herkommer D, Krische MJ, J. Am. Chem. Soc, 2016, 138, 14210–14213; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sam B, Luong T, Krische MJ, Angew. Chem. Int. Ed 2015, 54, 5465–5469; Angew. Chem. 2015, 127, 5555–5559; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vabre R, Island B, Diehl CJ, Schreiner PR, Marek I, Angew. Chem. Int. Ed 2015, 54, 9996–9999; Angew. Chem. 2015,127,10134–10137; [DOI] [PubMed] [Google Scholar]; (e) Alam R, Vollgraff T, Eriksson L, Szabó KJ, J. Am. Chem. Soc 2015, 137, 11262–11265; [DOI] [PubMed] [Google Scholar]; (f) Kliman LT, Mlynarski SN, Ferris GE, Morken JP, Angew. Chem. Int. Ed 2012, 51, 521–524; Angew. Chem. 2012, 124, 536–539; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dutta B, Gilboa N, Marek I, J. Am. Chem. Soc 2010, 132, 5588–5589. [DOI] [PubMed] [Google Scholar]
- [7].Intermolecular addition of unactivated C–H bonds to ketones to give tertiary alcohols has only been reported for a couple of very highly activated ketones.; (a) Jo H, Park J, Choi M, Sharma S, Jeon M, Mishra NK, Jeong T, Han S, Kim IS, Adv. Synth. Catal 2016, 358, 2714–2720. [Google Scholar]; (b) Zhang XS, Zhu QL, Luo FX, Chen GH, Wang X, Shi ZJ, Eur. J. Org. Chem 2013, 6530–6534. [Google Scholar]
- [8].(a) Hummel JR, Ellman JA, J. Am. Chem. Soc 2015, 137, 490–498; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshino T, Ikemoto H, Matsunaga S, Kanai M, Angew. Chem. Int. Ed 2013, 52, 2207–2211; Angew. Chem. 2013,125, 2263–2267. [DOI] [PubMed] [Google Scholar]
- [9].Tanaka R, Ikemoto H, Kanai M, Yoshino T, Matsunaga S, Org. Lett 2016, 18, 5732–5735. [DOI] [PubMed] [Google Scholar]
- [10].CDC1917393 (3,5-dinitrobenzoylated derivative of 6a) and CDC1917394 (3,5-dinitrobenzoylated derivative of 8a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. [Google Scholar]; See SI for additional details.
- [11].(a) Sen M, Rajesh N, Emayavaramban B, Premkumar JR, Sundararaju B, Chem. Eur. J 2018, 24, 342–346; [DOI] [PubMed] [Google Scholar]; (b) Sanjosé-Orduna J, Gallego D, Garcia-Roca A, Martin E, Benet-Buchholz J, Pérez-Temprano MH, Angew. Chem. Int. Ed 2017, 56, 12137–12141; Angew. Chem. 2017, 129, 12305–12309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.