

Abstract
Protein Arginine Deiminases (PADs) hydrolyze the side chain of arginine to form citrulline. Aberrant PAD activity is associated with rheumatoid arthritis, multiple sclerosis, lupus, and certain cancers. These pathologies established the PADs as therapeutic targets and multiple PAD inhibitors are known. Herein, we describe the first highly potent PAD1-selective inhibitors (1 and 19). Detailed structure-activity relationships indicate that their potency and selectivity is due to the formation of a halogen bond with PAD1. Importantly, these inhibitors inhibit histone H3 citrullination in HEK293TPAD1 cells and mouse zygotes with excellent potency. Based on this scaffold, we also developed a PAD1-selective activity-based probe that shows remarkable cellular efficacy and proteome selectivity. Based on their potency and selectivity we expect that 1 and 19 will be widely used chemical tools to understand PAD1 biology.
Keywords: Activity-based protein profiling (ABPP), Covalent protein modification, Halogen bonding, Histone citrullination, Protein arginine deiminase (PAD)
Citrullination, a post-translational modification (PTM) of arginine, plays pivotal roles in several processes, including the epigenetic regulation of gene expression, neutrophil extracellular trap (NET)-formation and DNA-damage induced apoptosis.[1] Protein citrullination is catalyzed by the protein arginine deiminases or PADs (Figure 1A). Of the five PAD isozymes (PAD1, 2, 3, 4 and 6), only four (PADs1–4) are catalytically active. PAD activity is tightly regulated by Ca2+ and PADs contain 4 (PAD1), 5 (PAD3, 4) or 6 (PAD2) Ca2+-binding sites.[2] Ca2+-binding reorients key catalytic residues into catalytically competent positions. Catalysis is mediated by a nucleophilic cysteine (C645 in PAD1, 4; C647 in PAD2; C646 in PAD3), a histidine (H471), and two aspartates (D350 and D473) that properly orient the substrate guanidium group for nucleophilic attack by the cysteine.[1b, 1d] In addition to their important physiological roles, aberrant protein citrullination leads to several autoimmune diseases (e.g., rheumatoid arthritis, lupus, and multiple sclerosis) as well as certain cancers. These disease links have established multiple PADs as promising therapeutic targets.[1b, 1d]
Figure 1.
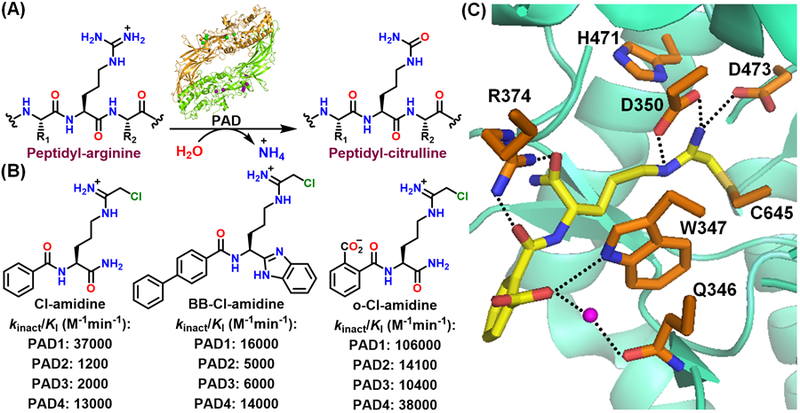
(A) Conversion of peptidyl-arginine to peptidyl-citrulline by the protein arginine deiminases (PADs). (B) Chemical structures and potencies of PAD inhibitors. (C) Crystal structure of the PAD4-o-F-amidine complex (PDB: 3B1U).
Cl-amidine and BB-Cl-amidine are two widely used pan-PAD inhibitors that possess a chloroacetamidine warhead that covalently modifies the active site cysteine (Figure 1B).[3] Although these inhibitors inhibit PADs1–4 similarly, BB-Cl-amidine shows superior efficacy in cells and animal models of disease owing to its higher cell permeability and metabolic stability.[3a] Interestingly, o-Cl-amidine, with a carboxylate substitution on the Cl-amidine scaffold, exhibits superior potencies for PAD inhibition (Figure 1B).[4] A structure of PAD4 bound to o-F-amidine, a fluoroacetamidine-containing analogue, indicates that the carboxylate hydrogen bonds with W347 (universally conserved amongst the PADs) and makes a water-mediated hydrogen bond with Q346 (Figure 1C). However, o-Cl-amidine exhibits poor cellular efficacy, likely due to the negatively charged carboxylate that limits cell permeability.
Recently, the use of halogens in medicinal chemistry has gained significant interest due to their high lipophilicity which boosts cell permeability.[5] Expecting this approach to increase the cell permeability of PAD inhibitors, we synthesized 1 with a 2-hydroxyl (which we hypothesized might form a hydrogen bond with W347), 3,5-diiodo substitutions in the N-terminal phenyl ring (to enhance permeability), and a C-terminal benzimidazole substitution (as in BB-Cl-amidine) (Figure 2A). Notably, this compound contains the less-reactive fluoroacetamidine warhead, which has less off-target toxicity.[6] Remarkably, 1 exhibits 74-fold selectivity for PAD1 over the other PADs with a kinact/KI value of 10,400 ± 2,380 M−1min−1 (Figure 2B). To our knowledge, this is the first example of a highly selective fluoroacetamidine-containing PAD1 inhibitor. By contrast, compound 2, which contains a chloroacetamidine warhead, is only 19-fold selective for PAD1 (Figure 2B and Table S1). The loss of selectivity observed with this warhead is consistent with previous studies showing that the fluoroacetamidine warhead can impart a higher degree of selectivity.[7]
Figure 2.
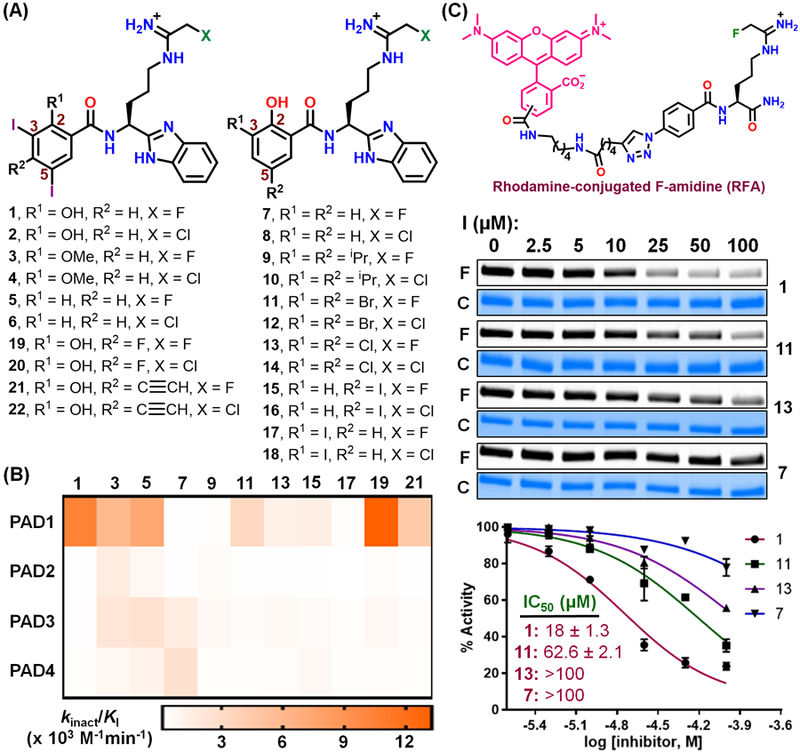
(A) Chemical structures of 1-22. (B) Potency heat-map of fluoroacetamidine-containing compounds for PADs1–4. The kinetics of inhibition of PADs by 1-22 are given in Figures S1–4. (C) Chemical structure of RFA and inhibition of RFA labelling of PAD1 by compounds 1, 11, 13 and 7. I, inhibitor; F, fluorograph; C, coomassie stain.
To investigate the origin of its potency and PAD1 selectivity, we established a structure-activity relationship (SAR) for 1. Since fluoroacetamidine-containing compounds afford better isozyme-selectivity, we only discuss these compounds in our analysis. The potency of the chloroacetamidine-containing compounds are, however, provided (Tables S1–S2) and they follow similar trends. Briefly, 3 and 5, which replace the 2-hydroxyl with a methoxy and hydrogen substitution, respectively, exhibit similar potency for PAD1 but a significant loss in selectivity (Figure 2B and Table S1). By contrast, 7, which lacks the iodo groups at 3- and 5-positions, shows an 87-fold drop in potency for PAD1 and a remarkable loss in selectivity. In fact, 7 is 21-fold selective for PAD4 (Figure 2B and Table S1). These results indicate that the 2-hydroxyl group is important for PAD1 selectivity, while the iodo groups are responsible for both potency and PAD1 selectivity. Given this data, we initially hypothesized that the iodo groups contribute to potency and selectivity via the formation of favourable hydrophobic interactions with PAD1. However, 9, which replaces the iodo groups with hydrophobic and isosteric isopropyl groups, exhibits almost identical activity as 7, suggesting that the iodo groups may be forming a halogen bond.
Owing to their anisotropic charge distribution, halogens in organic halides contain a significant amount of positive electrostatic potential (σ-hole) along the C-X (X = halogen) axis, and this potential increases in the order, Cl<Br<I. Therefore, in the presence of a suitable electron donor (D, often the backbone carbonyl or polar side chains of proteins), halogens act as an electrophile to form a halogen bond (D••X-C).[5c, 8] There are numerous examples where halogen bonds increase the affinity of small molecules for their proteins targets.[5a, 5c] For example, an iodophenol-based p53-cancer mutant (Y220C) stabilizer forms a halogen bond with the backbone carbonyl of L145.[9]
To validate the possible formation of halogen bonds between compound 1 and PAD1, we synthesized 11-14 with bromo- and chloro substitutions at the 3- and 5-positions. Since the positive potential on halogens decreases in the order: I>Br>Cl, the strength of a halogen bond follows the same order. Therefore, if a halogen bond is present, potency should decrease in that order. In fact, we observed a gradual decrease in both potency and selectivity upon replacement of the iodo groups. For example, 11 is 3-fold less potent than 1 and exhibits only 25-fold PAD1 selectivity, whereas 13 is 8-fold less potent than 1 and exhibits only 10-fold selectivity for PAD1 (Figure 2B and Table S1). Furthermore, the kinact and KI values for 1, 11 and 13 reveal that while these compounds share similar inactivation rates (kinact), there is a gradual decrease in binding affinity, as measured by an increase in KI, as one goes from 1 (12.5 µM) to 11 (54.8 µM) to 13 (124.5 µM) (Table S2). These observations clearly indicate that the potency and selectivity of 1 originates from the iodo groups that form halogen bonds with PAD1. Although different halogens are also expected to alter the pKa of the 2-hydroxyl, and thereby enhance hydrogen bond formation of this group to W347, the potency of the inhibitor is expected to increase as the electronegativity of the halogen atoms increase.
We further confirmed the halogen-dependent change in potency with an activity-based protein profiling (ABPP) assay that uses the PAD-targeted probe, rhodamine-conjugated F-amidine (RFA, Figure 2C).[10] RFA enables the covalent modification and fluorescent labelling of PADs. In the presence of a PAD inhibitor, however, RFA labelling is inhibited.[11] Using this assay, 1 inhibited RFA labelling of PAD1 with an IC50 of 18 µM (Figure 2C). Consistent with the kinact/KI data, compounds 11, 13 and 7 with bromo-, chloro- and hydrogen substitutions at the 3- and 5-positions, respectively, exhibit a gradual decrease in the potency (Figure 2C). Similar results were obtained with the chloroacetamidine-containing compounds 2, 12, 14 and 8 (Figure S5).
Having established their importance, we next questioned whether both iodo groups contribute to potency and selectivity. Although a structure of PAD1 in complex with 1 would definitively address this issue, several attempts to crystallize this complex were unsuccessful. Therefore, we synthesized 15-18 which contain an iodo group at either the 3- or 5-position (Figure 2A). Notably, 15 and 17 are 7- and 35-fold less potent than the parent compound 1. Furthermore, 15 and 17 are only 13- and 3-fold selective for PAD1 (Figure 2B and Table S1). These results indicate that both iodines are important for potency and PAD1 selectivity. Moreover the data indicate that the 3-iodine places the iodine at the 5-position in a suitable position to form a halogen bond. Since electron withdrawing substituents increase the halogen bond strength by increasing the positive potential on halogen, we synthesized 19 and 20 which contain a fluoro substitution in the parent scaffold (Figure 2A). 19 exhibited improved potency (kinact/KI = 13250 ± 2870 M−1min−1) and was 44-fold selective for PAD1 (Figure 2B and Table S1).
We next evaluated cellular potency in PAD1-overexpressing HEK293TPAD1 cells (see Figure S6 for PAD1 expression). As previously reported in mouse embryos,[12] we found that PAD1 citrullinates histone H3 at the 2, 8 and 17 positions in vitro and in HEK293TPAD1 cells in the presence of calcium (Figure S7). Gratifyingly, 1 and 19 inhibit histone H3 citrullination in a dose-dependent manner with EC50 values of 14.5 and 31.8 µM, respectively (Figure 3A–C). In contrast to the 6–7-fold higher in vitro potency, chloroacetamidine-containing compounds 2 and 20 exhibit only 1.3–2-fold higher activity in cells compared to 1 and 19 (Figure S8). These results indicate that despite having a lower in vitro potency, fluoroacetamidine-containing compounds exhibit similar cellular activity to chloroacetamidine-containing compounds, likely due to their lower off-target toxicity. Interestingly, the cellular activities of 1, 7, 11 and 13 for the inhibition of histone H3 citrullination in HEK293TPAD1 cells also follow the order 1>11>13>7 (Figures 3A–C), paralleling their in vitro potencies (Figures 1B). We do recognize, however, that this trend may also be due to a combination of potency and cell permeability as the lipophilicity of these compounds follows the same order (see log P values in Table S3). Notably, the Efficacy Index (the ratio of cytotoxicity to EC50) of 1 and 19 are 17 and 10, respectively, indicating that these compounds inhibit cellular PAD1 without causing significant toxicity (Figure 3C and S9).
Figure 3.
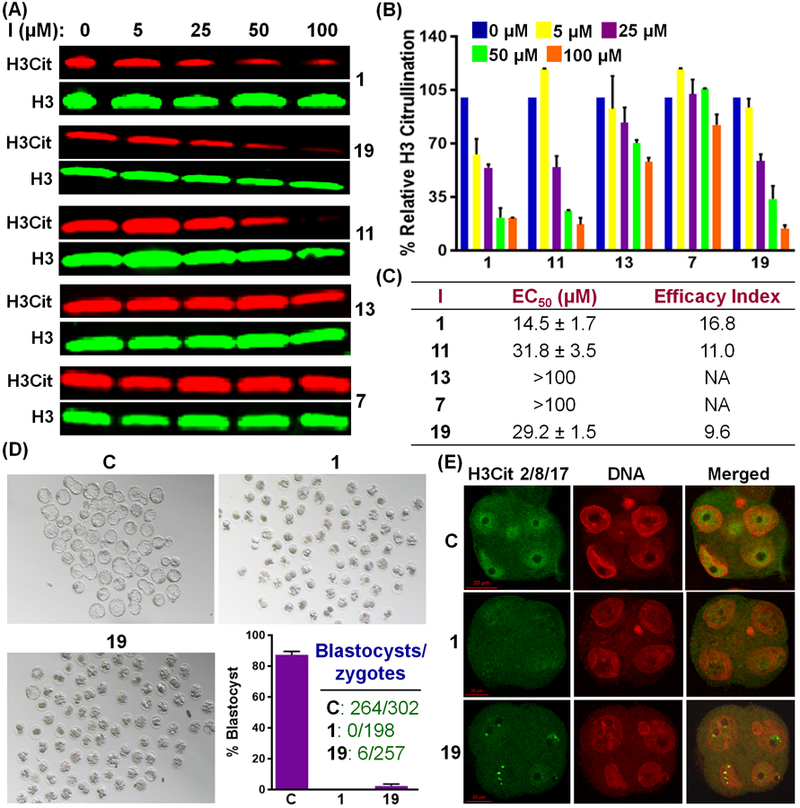
(A-C) Inhibition of histone H3 citrullination in HEK293TPAD1 cells by 1, 7, 11, 13 and 19. I, inhibitor; NA, Not applicable. (D) Inhibition of development of pronuclear mouse zygotes into blastocysts by 1 and 19. Embryo development was monitored by light microscopy. C, DMSO-treated control. (E) Confocal microscopy of mouse embryos at the 4-cell stage, indicating the inhibition of histone H3 citrullination by 1 and 19. anti-Histone H3Cit2/8/17 antibody and DAPI were used for immunofluorescence staining of citrullinated histone H3 and DNA, respectively. C refers to control.
PAD1 is the predominant nuclear PAD in early embryos and PAD1-catalyzed histone H3 citrullination is essential for early embryo development.[12] Depletion of PAD1 in mouse zygotes does not affect oocyte maturation or pronuclear formation but dramatically arrests early embryo development at the 4–8-cell stage. Encouraged by the inhibition of histone H3 citrullination in HEK293TPAD1 cells, we evaluated 1 and 19 in pronuclear (PN) zygotes and showed that these compounds dramatically arrest embryo development at the morula stage. By contrast, 87% of control zygotes progressed to the blastocyst stage (Figure 3D). Notably, the arrest in embryo development correlates strongly with the inhibition of histone H3 citrullination by 1 and 19 as revealed by immunofluorescence (Figures 3E and S10). These observations show that 1 and 19 inhibit endogenous levels of PAD1.
We also synthesized 21 and 22 (Figure 2A), which contain an alkyne handle that can be conjugated with fluorescent (TAMRA)- or biotin-containing reporter tags using the copper-catalyzed alkyne-azide cycloaddition (CuAAC) reaction for ABPP-based applications. Similar to the parent compounds, 21 and 22 exhibit almost 34- and 9-fold selectivity for PAD1 over the other PADs (Figure 2B and Table S1). 21 dose-dependently labels recombinant PAD1 with a limit of detection (LOD) of 4.3 pmol when using TAMRA-N3 as the reporter tag (Figure 4A, see top and bottom for dose-dependence and LOD, respectively). Similar results were obtained with 22 (Figure S11). Fluorescence intensities, normalized against the Coomassie stain intensities, for the labelling of PADs 1–4 by 21 and 22 indicate that 21 exhibits significant selectivity for PAD1, whereas 22 is less selective (Figure 4B). Therefore, we used 21 to label PAD1 in HEK293TPAD1 cells (Figure 4C). Gratifyingly, 21 labels PAD1 in these cells in a dose- and time-dependent manner (Figures 4D and S12). The cellular LOD is an impressive 72 pmol (Figure S12B). Encouraged by the selective labelling of PAD1, we used 21 to develop a target engagement assay to evaluate the cellular potency of PAD1 inhibitors. For this application, we tested whether compound 1 could block labelling of PAD1 by 21. As expected, compound 1 dose-dependently inhibits the labelling of PAD1 by 21 in HEK293TPAD1 cells (Figure 4E) with an EC50 of 12.7 µM, which is similar to that obtained for the inhibition of histone H3 citrullination (Figure 3C). We also coupled a 21-labeled HEK293TPAD1 proteome to Biotin-N3 and selectively captured the biotinylated proteins on streptavidin-agarose beads followed by an on-bead digestion of the captured proteins with trypsin (Figures 4C and S13). Reductive methylation (ReDiMe) was then performed on the trypsin digest with heavy (for the 21-treated samples) or light (for control samples) formaldehyde and sodium cyanoborohydride. Subsequent proteomic analysis afforded the enrichment ratio of the proteins that are covalently modified by 21. Impressively, PAD1 was the main protein isolated by 21 in HEK293TPAD1 cells, indicating the remarkable proteome-wide selectivity of this molecular scaffold. (Figure 4F).
Figure 4.
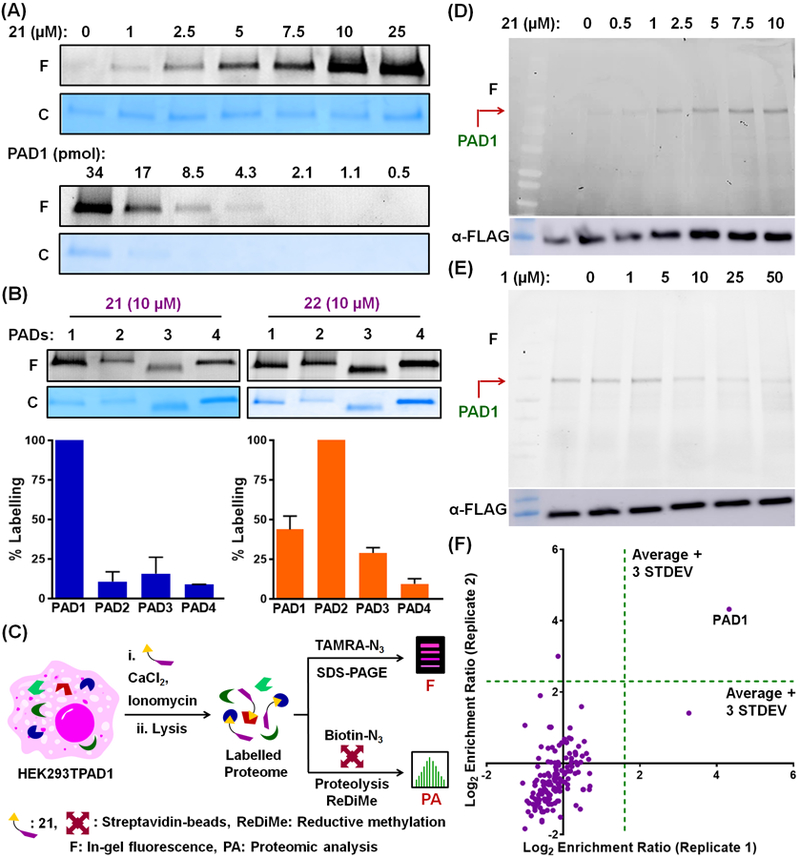
(A) Dose-response (top) and limit of detection (bottom) studies of labelling of recombinant PAD1 by 21. F, fluorograph; C, coomassie stain. (B) Selectivity of labelling of PADs1–4 by 21 and 22. (C) Schematic representation of labelling of PAD1 by 21 in HEK293TPAD1 cells. (D) Dose-response of labelling of PAD1 in HEK293TPAD1 cells by 21. An immunoblot using anti-FLAG antibody is used as a loading control. (E) Target engagement assay in HEK293TPAD1 cells using 21 (5 µM) and increasing concentrations of 1. (F) Selective enrichment of PAD1 in HEK293TPAD1 cells by 21. Enrichment ratio is determined from the proportion of heavy and light formaldehyde labelling of probe-treated and control samples, respectively.
In summary, we developed iodinated chemical probes with excellent potency and PAD1 selectivity. The high potency and selectivity arises mainly from the iodo groups, which form halogen bonding interactions with PAD1. These PAD1 inhibitors inhibit histone H3 citrullination in HEK293TPAD1 cells and mouse embryos with excellent efficacy. To the best of our knowledge, this is the first demonstration of a potent PAD1 inhibitor with a fluoroacetamidine warhead and the potential use of halogens to increase the potency and isozyme-selectivity of PAD inhibitors.
Supplementary Material
Footnotes
Experimental Section
Synthesis and characterization of inhibitors and probes (Figures S14–S57), in vitro PAD inactivation, cell-based assays, and the labelling of recombinant and cellular PAD1 are discussed in supporting information.
Conflict of Interest
P.R.T. founded Padlock Therapeutics and is entitled to payments from Bristol Myers Squibb if certain milestones are met. P.R.T. is a consultant for Celgene and Disarm Therapeutics. We are filing a patent based on these results.
References
- [1].a) Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A, Science 2004, 303, 1532–1535; [DOI] [PubMed] [Google Scholar]; b) Fuhrmann J, Clancy KW, Thompson PR, Chem. Rev 2015, 115, 5413–5461; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A, Elife 2017, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mondal S, Thompson PR, Acc. Chem. Res 2019, 52, 818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M, Nat. Struct. Mol. Biol 2004, 11, 777–783; [DOI] [PubMed] [Google Scholar]; b) Saijo S, Nagai A, Kinjo S, Mashimo R, Akimoto M, Kizawa K, Yabe-Wada T, Shimizu N, Takahara H, Unno M, J. Mol. Biol 2016, 428, 3058–3073; [DOI] [PubMed] [Google Scholar]; c) Slade DJ, Fang P, Dreyton CJ, Zhang Y, Fuhrmann J, Rempel D, Bax BD, Coonrod SA, Lewis HD, Guo M, Gross ML, Thompson PR, ACS Chem. Biol 2015, 10, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ, Ann. Rheum. Dis 2015, 74, 2199–2206; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR, J. Am. Chem. Soc 2006, 128, 1092–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Causey CP, Jones JE, Slack JL, Kamei D, Jones LE, Subramanian V, Knuckley B, Ebrahimi P, Chumanevich AA, Luo Y, Hashimoto H, Sato M, Hofseth LJ, Thompson PR, J. Med. Chem 2011, 54, 6919–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Mendez L, Henriquez G, Sirimulla S, Narayan M, Molecules 2017, 22; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ungati H, Govindaraj V, Mugesh G, Angew. Chem. Int. Ed. Engl 2018, 57, 8989–8993; [DOI] [PubMed] [Google Scholar]; c) Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM, J. Med. Chem 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- [6].Nemmara VV, Subramanian V, Muth A, Mondal S, Salinger AJ, Maurais AJ, Tilvawala R, Weerapana E, Thompson PR, ACS Chem. Biol 2018, 13, 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Muth A, Subramanian V, Beaumont E, Nagar M, Kerry P, McEwan P, Srinath H, Clancy K, Parelkar S, Thompson PR, J. Med. Chem 2017, 60, 3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Metrangolo P, Meyer F, Pilati T, Resnati G, Terraneo G, Angew. Chem. Int. Ed. Engl 2008, 47, 6114–6127; [DOI] [PubMed] [Google Scholar]; b) Mondal S, Manna D, Mugesh G, Angew. Chem. Int. Ed. Engl 2015, 54, 9298–9302; [DOI] [PubMed] [Google Scholar]; c) Mondal S, Mugesh G, Chem. Eur. J 2014, 20, 11120–11128; [DOI] [PubMed] [Google Scholar]; d) Mondal S, Mugesh G, Angew. Chem. Int. Ed. Engl 2015, 54, 10833–10837; [DOI] [PubMed] [Google Scholar]; e) Mondal S, Mugesh G, Cryst. Growth Des 2016, 16, 5896–5906; [Google Scholar]; f) Mondal S, Raja K, Schweizer U, Mugesh G, Angew. Chem. Int. Ed. Engl 2016, 55, 7606–7630. [DOI] [PubMed] [Google Scholar]
- [9].Wilcken R, Liu X, Zimmermann MO, Rutherford TJ, Fersht AR, Joerger AC, Boeckler FM, J. Am. Chem. Soc 2012, 134, 6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR, J. Am. Chem. Soc 2006, 128, 14468–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mondal S, Parelkar SS, Nagar M, Thompson PR, ACS Chem. Biol 2018, 13, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang X, Liu X, Zhang M, Li T, Muth A, Thompson PR, Coonrod SA, Sci. Rep 2016, 6, 38727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.