

Abstract
Background & Aims
Many differentiated epithelial cell types are able to reprogram in response to tissue damage. Although reprogramming represents an important physiological response to injury, the regulation of cellular plasticity is not well understood. Damage to the gastric epithelium initiates reprogramming of zymogenic chief cells into a metaplastic cell lineage known as spasmolytic polypeptide-expressing metaplasia (SPEM). The present study seeks to identify the role of xCT, a cystine/glutamate antiporter, in chief cell reprogramming after gastric injury. We hypothesize that xCT-dependent reactive oxygen species (ROS) detoxification is required for the reprogramming of chief cells into SPEM.
Methods
Sulfasalazine (an xCT inhibitor) and small interfering RNA knockdown were used to target xCT on metaplastic cells in vitro. Sulfasalazine-treated wild-type mice and xCT knockout mice were analyzed. L635 or DMP-777 treatment was used to chemically induce acute gastric damage. The anti-inflammatory metabolites of sulfasalazine (sulfapyridine and mesalazine) were used as controls. Normal gastric lineages, metaplastic markers, autophagy, proliferation, xCT activity, ROS, and apoptosis were assessed.
Results
xCT was up-regulated early as chief cells transitioned into SPEM. Inhibition of xCT or small interfering RNA knockdown blocked cystine uptake and decreased glutathione production by metaplastic cells and prevented ROS detoxification and proliferation. Moreover, xCT activity was required for chief cell reprogramming into SPEM after gastric injury in vivo. Chief cells from xCT-deficient mice showed decreased autophagy, mucus granule formation and proliferation, as well as increased levels of ROS and apoptosis compared with wild-type mice. On the other hand, the anti-inflammatory metabolites of sulfasalazine did not affect SPEM development.
Conclusions
The results presented here suggest that maintaining redox balance is crucial for progression through the reprogramming process and that xCT-mediated cystine uptake is required for chief cell plasticity and ROS detoxification.
Keywords: Cellular Plasticity, xCT, CD44, Reactive Oxygen Species, Sulfasalazine, Metaplasia, Chief Cell, Reprogramming, Oxyntic Atrophy, Autophagy, SPEM
Abbreviations used in this paper: ATPase, adenosine triphosphatase; Clu, clusterin; CD, cluster-of-differentiation; CD44v9, CD44 variant isoform 9; DHE, dihydroethidium; ESRP1, epithelial splicing regulatory protein 1; FITC, fluorescein isothiocyanate; GIF, gastric intrinsic factor; GSII, Griffonia simplicifolia; IFN, interferon; ImChief, Immortalized Chief; ImSPEM, Immortalized SPEM; LAMP2, lysosomal associated membrane protein-2; MAP1LC3B, Microtubule-associated proteins 1A/1B light chain 3B; mRNA, messenger RNA; Muc6, mucin 6; PAS, periodic acid–Schiff; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; PFA, paraformaldehyde; ROS, reactive oxygen species; siRNA, small interfering RNA; SLC7A11, solute carrier family 7 member 1; SPEM, spasmolytic polypeptide-expressing metaplasia; TEM, transmission electron microscopy; TFF2, trefoil factor 2; UEA1, Ulex Europaeus Agglutinin I; xCT, cystine/glutamate transporter; xCTKO, xCT knockout
Graphical abstract

See editorial on page 528.
Summary.
We have determined that the cystine/glutamate antiporter, solute carrier family 7 member 11 (SLC7A11; also known as xCT), is required for the reprogramming of chief cells into mucus-secreting metaplasia after parietal cell loss in the stomach. This study indicates that xCT is crucial for cystine uptake, reactive oxygen species detoxification, and survival of chief cells after gastric damage.
Severe injury to the lining of the gastrointestinal tract or other epithelial-lined organs leads to reprogramming of differentiated cells and the emergence of metaplastic cell lineages.1 Metaplastic lineages often are characterized by the expression of mucins, adding a protective barrier to the mucosa.2 In fact, metaplastic lineages often acquire the characteristics of mucus-secreting cells found in the distal stomach: antral/pyloric glands or Brunner’s glands.1 Similar reparative lineages are observed in the esophagus, stomach, small bowel, colon, and pancreas. In this study, we focus on the development of metaplastic lesions in the oxyntic region (body) of the stomach.
Zymogenic chief cells in the oxyntic region of the stomach produce enzymes required for digestion, such as pepsinogen and lipases.3 Chief cells also show high cellular plasticity after injury to the gastric epithelium.4, 5, 6, 7, 8 Damage to the epithelial cell barrier, including loss of acid-secreting parietal cells or ulceration, promotes the reprogramming of chief cells into a metaplastic cell lineage known as spasmolytic polypeptide-expressing metaplasia (SPEM).7, 9 SPEM expresses mucin 6 (Muc6) and trefoil factor 2 (TFF2, also known as the spasmolytic polypeptide) with a morphology similar to mucus-producing cells found deep within antral glands.10, 11 Although chief cell plasticity is a key mechanism to promote repair and tissue regeneration in the stomach, the maintenance of metaplasia in the presence of chronic injury and inflammation can predispose an individual to develop dysplasia or cancer in the gastric mucosa.10, 12
The epithelium that lines the stomach faces harsh conditions, such as ingested food and the bacteria resident in our gut, making the stomach more susceptible to damage.13, 14 Chronic infection with the bacterium Helicobacter pylori leads to the loss of acid-secreting parietal cells in the stomach.15 Gastric pathology can take months to develop in Helicobacter-infected mice, and years in human beings. To facilitate an accelerated model for induction of oxyntic atrophy, our laboratory has developed 2 acute models of parietal cell loss through the administration of the parietal cell toxic drugs L635 and DMP-777.16 Chief cell reprogramming is visualized within hours of the administration of a parietal cell toxic drug and includes the up-regulation of proteins required for the metaplastic transition.11
Cluster-of-differentiation (CD)44 is widely known as the receptor for hyaluronic acid that influences cell motility, survival, and proliferation.17 CD44 regulates proliferation of normal progenitors and metaplastic cells in the stomach.18, 19 Alternative splicing can produce variant isoforms of CD44 with unique functions.20 CD44 variant isoform 9 (CD44v9) is a cell surface glycoprotein not normally expressed in the stomach. However, it recently was discovered that CD44v9 is up-regulated early in the transition to SPEM and CD44v9 now is considered a SPEM marker.21 One of the functions of CD44v9 is to interact with and stabilize xCT (SLC7A11), a subunit of the heterodimeric cystine-glutamate antiporter.22 CD44v9 stabilization of xCT on the cell membrane increases cystine uptake into the cell. Increased intracellular cystine promotes glutathione synthesis, an important molecule for the defense against reactive oxygen species (ROS).23 The oxidative stress response, including up-regulation of nutrient transporters, plays an important role in many biological processes and the pathogenesis of a variety of diseases. Perturbations to the CD44v9-xCT system often result in redox imbalance.24
Sulfasalazine is a drug that was first synthesized in the 1940s from a combination of sulfapyridine (an antibiotic) and mesalazine (an anti-inflammatory agent) linked by an azo bridge.25 Recently, it was discovered that the parent compound sulfasalazine is a specific and potent inhibitor of xCT-mediated cystine transport.26 Several studies have used sulfasalazine treatment to target xCT activity on cancer stem cells.21, 27, 28, 29 Here, we used sulfasalazine as a tool to inhibit xCT-mediated cystine transport, an event that is required for chief cell reprogramming after gastric injury. We determined that xCT activity is required for cystine uptake and ROS detoxification by SPEM cells and metaplasia proliferation. Inhibition of xCT prevented zymogenic chief cell reprogramming in vivo. xCT-deficient mice showed increased levels of ROS and apoptosis and did not develop SPEM after acute parietal cell loss. Our results suggest that adaptation to oxidative stress and up-regulation of xCT activity is crucial for reprogramming of chief cells after gastric injury.
Results
Parietal Cell Loss Promotes SPEM and Up-Regulation of CD44v9-xCT
L635 is a parietal cell toxic drug that causes acute parietal cell necrosis.8 We used L635 treatment to elucidate the role of xCT in epithelial repair and chief cell reprogramming. Chief cell reprogramming can be visualized within hours of administering L635, and proliferative metaplasia subsequently develops within 3 days. Immunohistochemical analysis of tissue sections from wild-type C57Bl/6J mice treated with 3 days of L635 showed basolateral membrane expression of the metaplastic marker CD44v9 on chief cells at the base of glands. No CD44v9 staining was visualized in the stomach of untreated mice (Figure 1A). CD44v9 is known to interact and stabilize xCT, a subunit of a cystine-glutamate antiporter, at the plasma membrane.22, 23 Increased xCT expression on the membrane was observed in L635-treated mice, corresponding to CD44v9 expression on SPEM cells (Figure 1A). Alternative splicing of CD44 is regulated by epithelial splicing regulatory protein 1 (ESRP1).30, 31 Immunostaining showed increased nuclear expression of ESRP1 in gastric intrinsic factor (GIF)-positive chief cells from L635-treated mice (Figure 1C and D). In addition, we observed increased messenger RNA (mRNA) expression of Cd44v9, xCT, and Esrp1 after 3 days of L635 treatment (Figure 1B). These results suggest that alternative splicing of CD44 is up-regulated by ESRP1 after L635-induced injury to the stomach. Furthermore, increased CD44v9 stabilizes xCT on the plasma membrane.
Figure 1.

Increased expression of CD44v9, xCT, and ESRP1 in L635-treated mice. (A) Immunohistochemical staining of serial sections from the body of the stomach for CD44v9 (top) and xCT (bottom) in untreated and L635-treated (3 days) C57Bl/6J mice. Scale bar: 100 μm. Magnified inset from corresponding chief cell regions in top right corner. (B) Relative mRNA expression of Cd44v9, xCT, and Esrp1 in untreated and L635-treated (3 days) C57Bl/6J mice determined by reverse-transcription quantitative PCR (P = .006∗∗, .03∗, and .001∗∗, respectively). Statistical significance was determined by unpaired Student t test (n = 4 per group). (C) Immunofluorescent staining for the zymogenic granule marker GIF (red) and ESRP1 (green) in untreated and L635-treated (3 days) C57Bl/6J mice. Scale bar: 100 μm. Magnified inset of chief cell region with arrows indicating ESRP1 and GIF dual-positive cells (right). (D) Relative fluorescence intensity of nuclear ESRP1 in GIF-positive chief cells (P = .001∗∗). Statistical significance was determined by unpaired Student t test (n = 4 per group).
Metaplastic Cells Are Dependent on xCT for Cystine Uptake, ROS Detoxification, Proliferation, and Survival In Vitro
To target xCT activity on the plasma membranes of metaplastic (SPEM) cells, we used sulfasalazine, an inhibitor of xCT-mediated cystine transport, to treat previously characterized cell lines for chief cells (ImChief) and SPEM cells (ImSPEM) isolated from Immortomice.32 The relative expression of Cd44v9 and xCT were measured in ImChief and ImSPEM cells. ImSPEM cells showed increased expression of CD44v9 and xCT compared with ImChief cells (Figure 2A). ImChief and ImSPEM cells were immunostained for CD44v9 and xCT. No CD44v9 or xCT staining was visualized in ImChief cells, but CD44v9 and xCT were observed on the plasma membranes of ImSPEM cells, recapitulating what is observed in chief and SPEM cells in the stomach (Figure 2B–D).
Figure 2.

Inhibition of xCT with sulfasalazine blocks cystine uptake and ROS detoxification by metaplastic cells. (A) Relative mRNA expression of Cd44v9 and xCT in ImChief and ImSPEM cells determined by reverse-transcription quantitative PCR (P = .0002∗∗∗ and .0149∗, respectively). (B) ImChief and ImSPEM cells were immunostained for CD44v9 (red) with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar: 100 μm. (C) Percentage of cells per 20× field that are CD44v9-positive (P < .0001∗∗∗∗). (D) Diagram of ROS indicators CellROX green reagent and DHE. (E) Representative fluorescence images of cystine-FITC (green), glutathione (green), CellROX (green), and oxidized DHE (DHE-ox) (red) in ImSPEM cells ± sulfasalazine with nuclear counterstain Hoechst (blue). Scale bar: 100 μm. (F) Relative fluorescence intensity of cystine-FITC (P = .007∗∗), glutathione (P = .001∗∗), CellROX (P = .0019∗∗), and DHE-ox (P = .0003∗∗∗) in ImSPEM cells ± sulfasalazine. Statistical significance was determined by unpaired Student t test (n = 4 per condition).
To monitor xCT activity and cystine uptake into ImSPEM cells, we added fluorescently labeled cystine (cystine–fluorescein isothiocyanate [FITC]) to cultures.33 Abundant intracellular fluorescent signal was observed in ImSPEM cells 2 hours after the addition of cystine-FITC to culture. xCT blockade with sulfasalazine treatment significantly reduced the uptake of cystine-FITC by ImSPEM cells (Figure 2E and F). Through a multistep process, intracellular cystine can be converted to glutathione, a critical antioxidant for protection against ROS. We used an antibody that recognizes glutathione and ROS indicators (dihydroethidium [DHE] and CellROX) to measure glutathione production and ROS levels. Inhibition of xCT with sulfasalazine decreased glutathione in ImSPEM cells (Figure 2E and F). Similarly, sulfasalazine increased ROS in ImSPEM cells as shown by increased accumulation of oxidized DHE and CellROX (Figure 2E and F).
In addition, we observed that sulfasalazine treatment decreased the proliferation of ImSPEM cells by staining for the proliferation marker Ki67, a protein present in all active phases of the cell cycle, and monitoring cell number (Figure 3A–C). Sulfasalazine treatment also increased apoptosis of ImSPEM cells, as assessed by staining for the apoptosis marker, cleaved caspase-3 (Figure 3E and F). Caspase-3 is activated when it is cleaved by an initiator caspase and mediates cell death by apoptosis. These results indicate our in vitro culture system mimics the expression pattern observed in the stomach and that sulfasalazine inhibits cystine uptake, ROS detoxification, and proliferation of metaplastic cells in culture. Likewise, increased ROS levels led to SPEM cell death.
Figure 3.

Inhibition of xCT with sulfasalazine decreases proliferation and increases apoptosis of SPEM cells. (A) ImSPEM cells were seeded at the same density ± sulfasalazine for 3 days. Representative transmitted light images monitoring cell density over 3 days. Scale bar: 100 μm. (B) ImSPEM cell number ± sulfasalazine at 24, 48, and 72 hours (P = .001∗, .006∗, and .0002∗, respectively). (C) ImSPEM cells were treated with sulfasalazine for 48 hours and immunostained for proliferation marker Ki67 (red) with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar: 100 μm. (D) Percentage of cells per 20× field that are Ki67 positive (P = .01∗). (E) ImSPEM cells were treated with sulfasalazine for 72 hours and immunostained for apoptosis marker cleaved caspase-3 (CC3) (green) with nuclear counterstain DAPI (blue). Scale bar: 100 μm. (F) Percentage of cells per 20× field that are CC3 positive (P = .003∗∗). Statistical significance was determined by unpaired Student t test (n = 3 per condition).
Sulfasalazine is broken down to sulfapyridine and mesalazine through azo cleavage (Figure 4A). Therefore, we sought to confirm that the outcomes observed were owing to the specific inhibition of xCT by sulfasalazine, and not the anti-inflammatory properties of sulfasalazine metabolites. In contrast with sulfasalazine treatment, sulfapyridine or mesalazine treatment of ImSPEM cells in culture did not inhibit cystine uptake or proliferation (Figure 4B–E).
Figure 4.

Metabolites of sulfasalazine do not prevent cystine uptake, ROS detoxification, or proliferation of metaplastic cells. (A) Diagram of sulfasalazine metabolism. Sulfasalazine is broken down to sulfapyridine (an antibiotic) and mesalazine (an anti-inflammatory agent) through azo cleavage. (B) ImSPEM cells were seeded at the same density plus sulfapyridine or mesalazine for 3 days. ImSPEM cell number with sulfapyridine or mesalazine at 24, 48, and 72 hours. (C) Representative transmitted light images monitoring cell density over 3 days. Scale bar: 100 μm. (D) ImSPEM cells were treated with sulfapyridine or mesalazine and then treated with cystine-FITC. Live cell imaging of intracellular cystine-FITC (green) in ImSPEM cells treated with sulfapyridine or mesalazine with nuclear counterstain Hoechst (blue). Scale bar: 100 μm. (E) Relative fluorescence intensity of intracellular cystine-FITC in ImSPEM cells treated with sulfapyridine or mesalazine (n = 3 per condition).
To further examine if the effects of sulfasalazine are mediated by inhibition of xCT, we transfected ImSPEM cells with control or xCT small interfering RNAs (siRNAs). Similar to sulfasalazine treatment, ImSPEM cells transfected with xCT siRNA showed decreased cystine uptake and glutathione production, increased ROS, less proliferation, and more apoptotic cell death compared with ImSPEM cells transfected with control siRNA (Figure 5). Taken together, these results suggest that sulfasalazine effectively blocks xCT activity on SPEM cells and SPEM cells are dependent on xCT for control of ROS.
Figure 5.

xCT is required for cystine uptake and ROS detoxification in SPEM cells. (A) Relative mRNA expression of xCT in ImSPEM cells transfected with control or xCT siRNAs determined by reverse-transcription quantitative PCR (P = .0010∗∗∗ and < .0001∗∗∗). (B) Representative fluorescence images of cystine-FITC (green), CellROX (green), and oxidized DHE (DHE-ox) (red) in ImSPEM cells transfected with control or xCT siRNAs with nuclear counterstain Hoechst (blue). Scale bar: 100 μm. (C) Relative fluorescence intensity of cystine-FITC (P = .02∗ and .005∗∗), CellROX (P = .01∗ and .005∗∗), and DHE-ox (P = .002∗∗ and .0009∗∗∗) in ImSPEM cells transfected with control or xCT siRNAs. Statistical significance was determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test (n = 3 per condition).
Inhibition of xCT Blocks Chief Cell Reprogramming Into SPEM
To elucidate the role of xCT in chief cell reprogramming in vivo, we combined sulfasalazine treatment with the parietal cell toxic drug, L635. Chief cell reprogramming into SPEM occurs through a coordinated process that involves disassembly of the chief cell protein secretory apparatus and transcriptional changes including the up-regulation of mucus granule proteins Muc6 and TFF2.5, 9 We treated wild-type C57Bl/6J mice with sulfasalazine 2 days before and throughout 3 days of L635 treatment (Figure 6A). Stomachs were harvested from 4 experimental groups: (1) untreated, (2) sulfasalazine-treated, (3) L635-treated, and (4) L635 + sulfasalazine–treated mice for histologic analysis. Treatment with sulfasalazine by itself did not alter the gastric mucosa (Figure 7). To visualize L635-induced parietal cell loss, we performed immunostaining for the proton pump, H+/K+–adenosine triphosphatase (ATPase), an integral membrane protein responsible for gastric acid secretion by parietal cells (Figure 6B). In untreated mice, a large number of H+/K+-ATPase–positive parietal cells were detected throughout the corpus glands. Treatment with L635 reduced the number of parietal cells by almost 80%. Sulfasalazine treatment did not affect L635-induced parietal cell loss (Figure 6C). To detect chief cell reprogramming, we immunostained for the zymogenic granule marker GIF and the mucus granule marker Griffonia simplicifolia (GSII)-lectin, which binds to a sugar modification on Muc6 (Figure 6B). Chief cells reprogram to mucus-secreting metaplastic cells to protect and fuel repair of the stomach when there is injury or cell loss in the gastric mucosa. During this process, SPEM cells contain both zymogenic (GIF-positive) granules and mucus (GSII-positive) granules. Sulfasalazine treatment decreased the number of GIF and GSII dual-positive SPEM cells by greater than 80% after L635-induced parietal cell loss (Figure 6D). Similarly, periodic acid–Schiff (PAS) staining, a stain that detects mucins, showed decreased mucus at the base of glands in L635 + sulfasalazine–treated mice (Figure 7B). Collectively, these results suggest that xCT blockade prevents chief cell reprogramming after acute parietal cell loss.
Figure 6.

Inhibition of xCT blocks chief cell reprogramming in mouse models of acute gastric damage. (A) Diagram of drug treatments. L635 was administered to C57BL/6J mice for 3 days to induce acute gastric damage. Mice were treated with 10 mg of sulfasalazine per day, 2 days before and throughout L635 administration. Mice were killed 2 hours after the final dose of L635, and stomach tissue from untreated mice (n = 4), L635-treated mice (n = 4), and L635 + sulfasalazine–treated mice (n = 3) were harvested for histologic analysis. (B) Immunofluorescence staining for parietal cell marker H+/K+-ATPase (red), mucus granule marker GSII-lectin (green), and zymogenic granule marker GIF (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (C) Quantification of parietal cells as determined by number of H+K+ ATPase–positive (red) cells per 20× objective field. (D) Quantification of GSII (green) and GIF (blue) dual-positive (SPEM) cells per 20× objective field (P = .004∗∗). Statistical significance was determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test.
Figure 7.

Histologic analysis of sulfasalazine-treated mice. (A) H&E– and (B) PAS-stained sections from untreated (n = 4), sulfasalazine-treated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) mice. Scale bars: 100 μm. Dark purple–magenta color represents PAS-positive, mucus-producing cells. Glands containing PAS-positive cells at the base are indicated with arrows.
xCT Activity Is Required for Autophagy in Reprogramming Chief Cells
The process of reprogramming requires chief cells to downscale their mature characteristics. The basic helix-loop-helix transcription factor Mist1 (Bhlha15) governs the secretory architecture of chief cells including production of large protein-containing zymogen granules and cellular organization.3 Loss of Mist1 is a distinct feature of chief cell reprogramming into SPEM.34 Dual immunofluorescence staining for GIF and Mist1 allows for monitoring of Mist1 loss in zymogenic granule-containing chief cells (Figure 8A). In untreated mice, GIF-positive chief cells were positive for the transcription factor Mist1. L635 treatment resulted in almost complete loss of Mist1 expression in GIF-positive cells. Interestingly, treatment with L635 + sulfasalazine did not rescue Mist1 loss by chief cells (Figure 8B). This suggests that xCT inhibition with sulfasalazine does not influence the initiating step of Mist1 loss in reprogramming chief cells.
Figure 8.

xCT blockade inhibits autophagy of zymogenic granules in downscaling chief cells. (A) Immunostained sections from untreated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment for zymogenic granule marker GIF (red) and chief cell transcription factor Mist1 (green). Scale bars: 50 μm. (B) Quantification of Mist1-positive cells per 20× field. (C) Immunostained sections from untreated (n = 5), L635-treated (n = 5), and L635 + sulfasalazine–treated (n = 4) C57Bl/6J mice killed after 24 hours of L635 treatment for GIF (red) and autophagosome marker LC3B (green). Scale bars: 50 μm. Magnified inset of GIF-positive cell with arrows indicating puncta (right). (D) Average number of LC3B puncta per GIF-positive cell (P = .003∗∗). (E) Immunostained sections from untreated (n = 5), L635-treated (n = 5), and L635 + sulfasalazine–treated (n = 4) C57Bl/6J mice killed after 24 hours of L635 treatment for GIF (red) and lysosome marker LAMP2 (green). Scale bars: 50 μm. Magnified inset of GIF-positive cell with arrows indicating puncta (right). (F) Average number of LAMP2 puncta per GIF-positive cell (P = .0003∗∗∗). Statistical significance determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test. (G) Transmission electron micrographs of zymogenic chief cells 12 hours after L635 treatment ± sulfasalazine. Scale bars: 2 μm. Magnified inset of double-membrane autophagic structures (right). (H) Relative mRNA expression of autophagy-related proteins in L635-treated and L635 + sulfasalazine–treated mice (Atg4, Atg5, Atg7, Atg12, Atg16L1, Beclin1, Lamp1, Lamp2, and Lc3) (P = .02∗, .005∗, .03∗, .01∗, and .02∗, respectively). Statistical significance was determined by an unpaired Student t test.
In addition to loss of Mist1, autophagic and lysosomal pathways in chief cells are up-regulated acutely after injury to the stomach. In particular, rough endoplasmic reticulum, mitochondria, and secretory granules are targeted for degradation during early stages of SPEM development. Furthermore, mice with defects in autodegradative function (Gnptab-/- mice) are unable to develop SPEM after gastric injury.5 To investigate autophagic and lysosomal pathways, L635-treated mice were killed 12 or 24 hours after L635 treatment. We performed immunostaining for the autophagosome marker microtubule-associated proteins 1A/1B light chain 3B (MAP1LC3B or LC3B) and the lysosome marker lysosomal associated membrane protein-2 (LAMP2) (Figure 8C and E). Very few LC3B or LAMP2-positive puncta were observed in GIF-positive chief cells from untreated mice. As expected, however, gastric injury induced by L635 treatment resulted in increased LC3B and LAMP2-positive puncta in close proximity to GIF-positive zymogenic granules. By contrast, L635 + sulfasalazine–treated mice showed fewer LC3B and LAMP2-positive puncta in GIF-positive cells compared with L635 treatment only (Figure 8D and F). Notably, large/dense zymogen granules were maintained in L635 + sulfasalazine–treated mice compared with the L635-treated mice. Similarly, transmission electron microscopy (TEM) of tissue from L635-treated mice showed abundant double-membrane–bound (autophagic) structures engulfing granules and other cytosolic components in chief cells. Although similar double-membrane–bound structures were observed in L635 + sulfasalazine–treated mice, they were significantly less frequent (Figure 8G). Real-time quantitative polymerase chain reaction (PCR) analysis for several autophagy-related genes showed a significant decrease in the relative expression of Atg4, Atg7, Atg16L1, Lamp1, and Lc3 in sulfasalazine-treated mice (Figure 8H). Although there was Mist1 loss in L635 + sulfasalazine–treated mice, these findings suggest that xCT blockade decreases downscaling by autophagic and lysosomal pathways in reprogramming chief cells.
xCT Blockade Suppresses Proliferation of Reprogramming Chief Cells
Sulfasalazine treatment inhibited proliferation of ImSPEM cells in culture, so we sought to determine the effect of sulfasalazine treatment on proliferation after gastric injury in vivo. To do this, we immunostained for the proliferation marker Ki67. In the normal oxyntic mucosa, Ki67 labeled stem/progenitor cells approximately a third of the way down the gland in the gland isthmus. Upon gastric injury, chief cells reprogram and are capable of re-entering into the cell cycle and proliferating. In addition, surface mucus-producing (foveolar) cells located near the lumen also expand in response to injury and increases in gastrin. This gastric lesion is referred to as foveolar hyperplasia. Foveolar cells produce Muc5ac, a mucin recognized by Ulex Europaeus Agglutinin I (UEA1) lectin. Unlike reprogramming chief cells, foveolar cells do not express xCT. To classify the identity of the proliferating cells in each of our experimental groups we immunostained for Ki67, UEA1 lectin, and GIF (Figure 9A). To evaluate foveolar hyperplasia after L635 treatment, we measured the average thickness of the UEA1-positive foveolar region. L635-treated mice and L635 + sulfasalazine–treated mice showed similar levels of foveolar hyperplasia (Figure 9B). We also quantified the total number of Ki67-positive cells in the oxyntic mucosa of each experimental group. Although the total number of Ki67-positive proliferating cells was not significantly different between L635-treated mice and L635 + sulfasalazine–treated mice, the percentage of Ki67-positive cells that were dual-positive for GIF was decreased significantly in the sulfasalazine-treated group (Figure 9C and D). Together, these results show that proliferation of xCT-negative foveolar cells is not affected by xCT blockade. However, chief cells that express high levels of xCT as they reprogram do not proliferate after xCT blockade with sulfasalazine.
Figure 9.

Inhibition of xCT suppresses SPEM proliferation. (A) Immunostained sections from untreated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment for proliferation marker Ki67 (red), mucus-producing foveolar cell marker UEA1-lectin (green), and zymogen granule marker GIF (blue). Scale bars: 100 μm. Magnified inset of chief cell region with arrows indicating proliferative metaplasia (right). (B) Average thickness (μm) of UEA1-positive foveolar region. (C) Quantification of Ki67-positive (red) proliferating cells per 20× field. (D) Percentage of Ki67-only–positive cells or Ki67-positive cells dual-positive for UEA1 or GIF to determine distribution of proliferative cells (P = .002∗∗). Statistical significance was determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test.
Inhibition of xCT Leads to Oxidative Stress and Chief Cell Death After Gastric Injury
Next, we examined evidence of cell stress or cell death. Clusterin (Clu) is a heterodimeric protein up-regulated in many diseases related to oxidative stress and is associated with clearance of cellular debris and apoptosis.35 Previous studies have shown clusterin expression in SPEM lineages through gene microarray analysis. Clu protein expression in SPEM was validated in DMP-777, L635, and Helicobacter infection mouse models.11 In the normal oxyntic mucosa, clusterin is expressed in some mucous neck cells along with the spasmolytic polypeptide TFF2. In L635-treated mice, clusterin and TFF2 are up-regulated in chief cells at the base of oxyntic glands as they reprogram into SPEM. Interestingly, although sulfasalazine inhibited TFF2 up-regulation, chief cells at the gland base still showed clusterin expression after L635 treatment (Figure 10). Thus, it is likely that Clu is not up-regulated as part of the metaplastic process, but rather as a result of increased oxidative stress. Similarly, staining with ROS indicators (DHE and CellROX) showed increased levels of ROS in L635 + sulfasalazine–treated mice compared with L635-treated mice, especially in GIF-positive chief cells (Figure 11). Furthermore, L635 + sulfasalazine–treated mice showed a few GIF-positive chief cells that were dual-positive for the apoptosis marker cleaved caspase-3 (Figure 12A). Taken together, these findings indicate that inhibition of xCT prevents chief cell reprogramming, but chief cells still initiate a stress response owing to increased ROS and, in some cases, undergo apoptosis.
Figure 10.

Inhibition of xCT blocks reprogramming of chief cells and results in cellular stress. (A) Representative immunostained sections from untreated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment for cellular stress marker clusterin (red) and TFF2 (green), with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (B) Quantification of Clu-positive (red) cells per 20× field. (C) Quantification of Clu (red) and TFF2 (green) dual-positive cells per 20× field (P = .004∗∗). Statistical significance determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test.
Figure 11.

xCT blockade leads to increased ROS after acute gastric damage. Sections from untreated (n = 3), L635-treated (n = 3), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment were treated with ROS indicators DHE and CellROX. (A) Representative images of DHE staining with oxidized DHE (DHE-ox) (red), f-actin marker phalloidin (green), and unoxidized DHE (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (B) Relative fluorescence intensity of nuclear DHE-ox (P = .02∗). (C) Representative images of CellROX green staining with zymogenic granule marker GIF (red), CellROX Green Reagent (green), with nuclear counterstain Hoechst (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (D) Relative fluorescence intensity of CellROX in GIF-positive cells (P = .01∗). Statistical significance was determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test.
Figure 12.

Inhibition of xCT in reprogramming chief cells results in apoptosis. Immunostained sections from untreated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment for apoptosis marker cleaved caspase-3 (red), zymogenic granule marker GIF (green), with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 100 μm. Magnified inset of chief cell region with arrows indicating cleaved caspase-3 and GIF dual-positive apoptotic chief cells.
Next, we examined in each of our experimental groups the expression of CD44v9, a metaplastic cell marker protein that interacts and stabilizes xCT (Figure 13A). Immunostaining for CD44v9 confirmed no expression in untreated mice. Similarly, no CD44v9 staining was observed in the sulfasalazine-only treatment group. In contrast, mice treated with L635 showed high expression of CD44v9 on the basolateral membranes of cells at the base of oxyntic glands. The majority of these cells were dual-positive for the mucus granule marker GSII lectin. Interestingly, reduced expression of CD44v9 was observed in L635 + sulfasalazine–treated mice (Figure 13B). The remaining CD44v9 staining in mice treated with L635 and sulfasalazine appeared to be intracellular and not on the basolateral membrane. We used real-time PCR to measure the relative expression of Cd44v9 mRNA transcript in each of the experimental groups (Figure 13C). No significant difference in Cd44v9 mRNA was observed between L635-treated mice ± sulfasalazine treatment. Therefore, these results indicate that sulfasalazine may disrupt the interaction of CD44v9 and xCT on the membrane and may result in mislocalization and possible degradation of CD44v9 protein. On the other hand, it also is possible that maintenance of redox balance is required for up-regulation of CD44v9 protein.
Figure 13.

Basolateral membrane expression of CD44v9 is decreased after xCT inhibition with sulfasalazine. (A) Immunostained sections from untreated (n = 4), sulfasalazine-treated (n = 4), L635-treated (n = 4), and L635 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment for CD44v9 (red), mucus granule marker GSII-lectin (green), with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (B) Number of cells per 20× field with basolateral membrane expression of CD44v9 (P < .0001∗∗∗). (C) Relative mRNA expression of Cd44v9. Statistical significance was determined by 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test.
Metabolites of Sulfasalazine Do Not Alter Metaplasia Development
To validate further that our observations were owing to specific xCT blockade by sulfasalazine and not the anti-inflammatory properties of sulfasalazine metabolites, we treated mice with sulfapyridine or mesalazine 2 days before and throughout 3 days of L635 treatment (Figure 14A).36 Immunostaining for CD44v9, mucus granule marker GSII lectin, and zymogenic granule marker GIF showed similar high numbers of triple-positive cells in both sulfapyridine + L635–treated mice and mesalazine + L635–treated mice (Figure 14B and C). These results indicate that metabolites of sulfasalazine are not sufficient to block chief cell reprogramming to SPEM.
Figure 14.

Metabolites of sulfasalazine do not prevent metaplasia development after acute gastric damage. (A) Diagram of drug treatments. L635 was administered to C57BL/6J mice for 3 days to induce acute gastric damage. Mice were treated with sulfapyridine or mesalazine daily, 2 days before and throughout L635 administration. Mice were killed 2 hours after the final dose, and stomachs from sulfapyridine + L635–treated (n = 4) and mesalazine + L635–treated (n = 4) C57Bl/6J mice were harvested for histologic analysis. (B) Immunofluorescence staining for metaplastic cell marker CD44v9 (red), mucus granule marker GSII-lectin (green), and zymogenic granule marker GIF (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (C) Quantification of CD44v9, GIF, GSII-lectin triple-positive cells per 20× field.
xCT Knockout Mice Recapitulate the Phenotype of Sulfasalazine-Treated Mice
To examine further if the effects of sulfasalazine are mediated by inhibition of xCT we treated xCT knockout (xCTKO) mice with the parietal cell toxic drug L635. Stomachs were harvested from 2 experimental groups for histologic analysis: untreated xCTKO mice and L635-treated xCTKO mice (Figure 15A). H&E or PAS staining showed normal gastric histology in xCTKO mice (Figure 15B). To visualize L635-induced parietal cell loss, we performed immunostaining for the proton pump H+/K+-ATPase (Figure 15C). In untreated xCTKO mice, a large number of H+/K+-ATPase–positive parietal cells were detected throughout the corpus glands. Treatment with L635 in xCTKO mice reduced the number of parietal cells by more than 70% (Figure 15D). To detect chief cell reprogramming, we immunostained for the zymogenic granule marker GIF and the mucus granule marker GSII lectin (Figure 15C). L635-treated xCTKO mice did not show reprogramming chief cells, dual-positive for GIF and GSII (Figure 15E). Similarly, PAS staining showed no mucus staining at the bases of glands in L635-treated xCTKO mice (Figure 15B). Collectively, these results suggest that chief cells from xCT-deficient mice are unable to reprogram after acute parietal cell loss.
Figure 15.

Loss of xCT prevents the development of metaplasia after acute gastric damage. (A) Diagram of drug treatments. L635 was administered to xCTKO C57BL/6J mice for 3 days to induce acute gastric damage. Mice were killed 2 hours after the final dose of L635, and stomach tissue from untreated xCTKO mice (n = 4) and L635-treated xCTKO mice (n = 4) were harvested for histologic analysis. (B) H&E– and PAS-stained sections. (C) Immunostained sections for parietal cell marker H+/K+-ATPase (red), mucus granule marker GSII lectin (green), and zymogenic granule marker GIF (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (D) Quantification of parietal cells as determined by the number of H+/K+-ATPase–positive (red) cells per 20× objective field (P = .0002∗∗∗). (E) Quantification of GSII (green) and GIF (blue) dual-positive (SPEM) cells per 20× objective field. Statistical significance was determined by the unpaired Student t test.
In addition, we examined xCTKO mice to determine ROS levels using the ROS indicators DHE and CellROX. Although untreated xCTKO mice had low levels of ROS, after L635 treatment the xCTKO mice showed high levels of ROS (Figure 16). Similar to L635 + sulfasalazine–treated mice, GIF-positive chief cells from L635-treated xCTKO mice had increased ROS determined by CellROX Green Reagent staining (Figure 16D). Likewise, several GIF-positive chief cells from L635-treated xCTKO mice were dual-positive for cleaved caspase-3 (Figure 17A). Taken together, our results indicate that chief cells from xCT-deficient mice are unable to reprogram and experience oxidative stress that leads to apoptotic cell death.
Figure 16.

xCT is required for ROS detoxification. Sections from untreated xCTKO (n = 4) and L635-treated xCTKO (n = 4) C57Bl/6J mice killed after 3 days of L635 treatment were incubated with ROS indicators DHE and CellROX. (A) Representative images of DHE staining with oxidized DHE (DHE-ox) (red), f-actin marker phalloidin (green), and unoxidized DHE (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (B) Relative fluorescence intensity of nuclear DHE-ox (P < .0001∗∗∗∗). (C) Representative images of CellROX green staining with zymogenic granule marker GIF (red), CellROX Green Reagent (green), with nuclear counterstain Hoechst (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (D) Relative fluorescence intensity of CellROX in GIF-positive cells (P = .004∗∗). Statistical significance was determined by unpaired Student t test.
Figure 17.

Chief cells from xCT knockout mice apoptose after acute gastric damage. Immunostained sections from untreated xCTKO (n = 4) and L635-treated xCTKO (n = 4) C57Bl/6J mice killed after 3 days of L635 treatment for apoptosis marker CC3 (red), zymogenic granule marker GIF (green), with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 100 μm. Magnified inset of chief cell region with arrows indicating CC3 and GIF dual-positive apoptotic chief cells.
Inhibition of xCT Prevents DMP-777–Induced SPEM
To test the efficacy of xCT blockade on chief cell reprogramming in a different mouse model of acute parietal loss, we used another parietal toxic drug, DMP-777. Similar to L635, DMP-777 induces parietal cell necrosis. However, DMP-777 also functions as an elastase inhibitor and prevents inflammatory infiltration.16 With DMP-777 treatment, chief cell reprogramming and metaplasia development occur in the absence of inflammation. We treated mice with sulfasalazine 2 days before and throughout 10 days of DMP-777 treatment. Stomachs were harvested from 2 experimental groups for histologic analysis: DMP-777–treated and DMP-777 + sulfasalazine–treated mice (Figure 18A). H+/K+-ATPase staining showed similar levels of parietal cell loss in both groups (Figure 18B and C). We also performed immunostaining for the zymogenic granule marker GIF and mucus granule marker GSII lectin to detect chief cell reprogramming. Sulfasalazine treatment decreased the number of GIF and GSII dual-positive SPEM cells by greater than 75% after DMP-777–induced parietal cell loss (Figure 18B and D). Therefore, xCT inhibition with sulfasalazine prevents metaplasia development even in the absence of inflammation.
Figure 18.

Sulfasalazine inhibits metaplasia development in the absence of inflammation. (A) Diagram of drug treatments. A parietal cell toxic drug (DMP-777) was administered to C57BL/6J mice for 10 days to induce acute gastric damage. Mice were treated with 10 mg of sulfasalazine per day, 2 days before and throughout DMP-777 administration. Mice were killed 2 hours after the final dose of DMP-777, and stomachs from DMP-777–treated (n = 5) and DMP-777 + sulfasalazine–treated (n = 5) mice were harvested for histologic analysis. (B) Immunofluorescence staining for parietal cell marker H+/K+-ATPase (red), mucus granule marker GSII lectin (green), and zymogenic granule marker GIF (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (C) Quantification of parietal cells as determined by number of H+/K+-ATPase–positive (red) cells per 20× objective field. (D) Quantification of GSII (green) and GIF (blue) dual-positive (SPEM) cells per 20× objective field (P = .0002∗∗∗). Statistical significance was determined by unpaired Student t test.
We further examined DMP-777–treated mice with ROS indicators (DHE and CellROX) and apoptosis markers. DMP-777 + sulfasalazine–treated mice showed slightly increased levels of ROS compared with DMP-777–treated mice (Figure 19). Furthermore, very few GIF-positive chief cells were dual-positive for the apoptosis marker cleaved caspase-3 (Figure 20A). It is likely that the generation of ROS intrinsic to the reprogramming process accounts for the increased ROS observed in DMP-777 + sulfasalazine–treated mice. However, in the absence of inflammatory ROS, chief cells are able to endure the oxidative stress, at least over the 10-day time course.
Figure 19.

xCT blockade with sulfasalazine leads to increased ROS in chief cells without inflammation. Sections from DMP-777–treated (n = 3) and DMP-777 + sulfasalazine–treated (n = 3) C57Bl/6J mice killed after 3 days of L635 treatment were incubated with ROS indicators DHE and CellROX. (A) Representative images of DHE staining with oxidized DHE (DHE-ox) (red), F-actin marker phalloidin (green), and unoxidized DHE (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (B) Relative fluorescence intensity of nuclear DHE-ox (P = .02∗). (C) Representative images of CellROX green staining with zymogenic granule marker GIF (red), CellROX Green Reagent (green), with nuclear counterstain Hoechst (blue). Scale bars: 100 μm. Magnified inset of chief cell region (right). (D) Relative fluorescence intensity of CellROX in GIF-positive cells (P = .02∗). Statistical significance was determined by unpaired Student t test.
Figure 20.

Reprogramming chief cells survive oxidative stress without inflammatory ROS. Immunostained sections from DMP-777–treated (n = 5) and DMP-777 + sulfasalazine–treated (n = 5) C57Bl/6J mice killed after 10 days of DMP-777 treatment for apoptosis marker CC3 (red), zymogenic granule marker GIF (green), with nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 100 μm. Magnified inset of chief cell region with arrows indicating CC3 and GIF dual-positive apoptotic chief cells.
CD44v9 and xCT Are Up-Regulated in SPEM in Helicobacter-Infected Mice and Human Beings
Given the prominent changes observed in the acute models of parietal cell loss, we sought to examine whether CD44v9 and xCT also were up-regulated after chronic SPEM induction in Helicobacter felis–infected mice or in clinical specimens. Figure 21A shows that staining for both CD44v9 and xCT was observed in SPEM from the stomach of mice infected with H felis for 12 months. Similarly, Figure 21B shows that both CD44v9 and xCT staining was observed in SPEM from human patients. These studies confirm the up-regulation of ROS detoxification mechanisms in both chronic induction of SPEM and SPEM associated with acute gastric damage.
Figure 21.
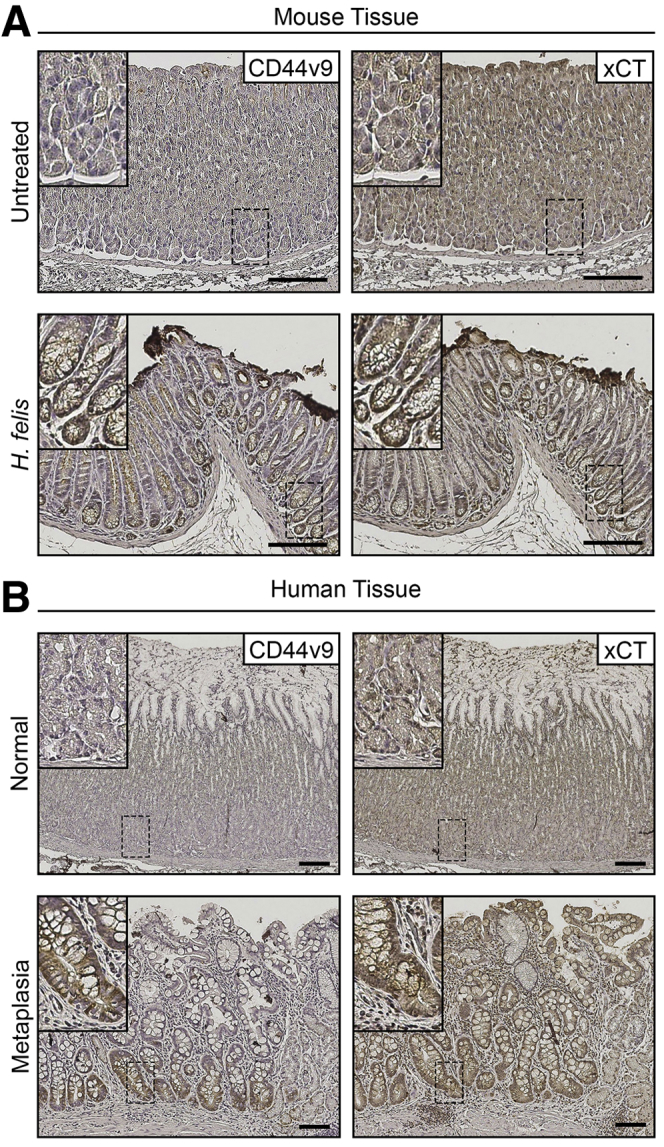
CD44v9 and xCT are expressed in Helicobacter-infected mice and human metaplasia. (A) Representative immunohistochemical stained serial sections from the body of the stomach for CD44v9 (left) and xCT (right) in untreated (n = 3) and 12-month H felis–infected (n = 3) C57Bl/6J mice. Scale bar: 100 μm. (B) Representative immunohistochemical stained serial sections from the body of the stomach for CD44v9 (left) and xCT (right) in normal (n = 6) and metaplastic (n = 3) human stomach. Scale bar: 100 μm. Magnified insets from corresponding basal gland regions in top left corner.
Discussion
After significant gastric injury, chief cells reprogram to mucous cell metaplasia or SPEM. The present study identifies that xCT is required for the reprogramming of digestive enzyme–secreting chief cells after gastric injury. Increased membrane expression of xCT is observed in reprogramming chief cells expressing CD44v9 after gastric damage with parietal cell toxic drugs. Inhibition of xCT, through sulfasalazine treatment, or xCT siRNA knockdown blocks cystine uptake, ROS detoxification, and the proliferation and survival of metaplastic cells in culture. In addition, chief cells from sulfasalazine-treated mice or xCT knockout mice were unable to reprogram after acute gastric injury. Importantly, we confirm that our findings were owing to specific inhibition of xCT by sulfasalazine, and not the anti-inflammatory characteristics of sulfasalazine metabolites. Although loss of xCT activity inhibited chief cell reprogramming, chief cells endured oxidative stress and in some cases apoptosis. Interestingly, the transcription factor Mist1 was down-regulated in chief cells after acute parietal cell loss, even with xCT blockade. In contrast, inhibition of xCT restricted the up-regulation of autosomal and lysosomal degradation machinery, a distinct step in the reprogramming process. Thus, although xCT inhibition does not alter initiation of reprogramming, it arrests the completion of the process. These results show that the process of reprogramming involves multiple discrete steps leading to metaplasia.
Recent studies have sought to understand the mechanisms used by mature postmitotic cells to change their differentiation state.37 Reprogramming promotes the regenerative capacity of differentiated cells. This is a critical process used to repair damage in tissues that lack an active adult stem cell compartment such as the pancreas.38 Still, in tissues with active adult stem cells such as the stomach and intestine, reprogramming differentiated cells can enable repair after tissue damage.37 It is evident that reprogramming of differentiated cells occurs through a stepwise process. First, differentiated cells must undergo autodegradation to downscale mature characteristics. Next, metaplastic genes are up-regulated, and mucus granules are formed. Finally, metaplastic cells have the capability of re-entering into the cell cycle and proliferating. Progression through this stepwise process can be blocked at intervening check points. The results presented here suggest that maintaining redox balance is crucial for progression through the reprogramming process in gastric chief cells and may represent a checkpoint for this process. It is likely that the downscaling of zymogen granules and up-regulation of mucus granule formation would engender the production of ROS. Furthermore, acute and chronic inflammation can add to increased ROS levels.39 Indeed, it is likely that decreased ROS levels are important for the undifferentiated status or stemness of reprogramming chief cells. CD44v9 and xCT also have been implicated as key players in malignant transformation in a variety of tissues and often are highly expressed by cancer stem cells.21, 27, 28, 29 Thus, up-regulation of xCT activity may be used by reprogramming cells or cancer stem cells to combat increased oxidative stress.
Oxidative stress refers to high levels of intracellular ROS that can cause damage to lipids, proteins, and DNA.40 Increased ROS act as signaling molecules that contribute to both physiological and pathologic conditions.41 Cells use several mechanisms to counteract oxidative stress by producing antioxidants such as glutathione.42 Increased expression of xCT is part of the oxidative stress response.43, 44 In addition, molecules that work to stabilize and increase the activity of xCT, such as CD44v9, are up-regulated as a consequence of oxidative stress. Increased xCT activity results in a build-up of intracellular cystine, which through a multistep process can be converted to glutathione.23 Disruption of xCT activity in cases of tissue injury leads to increased levels of ROS and oxidative damage. Excessive oxidative damage may cause cell death.45 The results here suggest that generation of ROS is inherent to the process of injury and reprogramming. Therefore, activation of antioxidant responses is required for the successful reprogramming of differentiated cells. Although the cell of origin for cancer in the stomach is still under debate, cellular reprogramming to metaplasia in the setting of chronic injury and inflammation may expose these cells to prolonged oxidative stress, which could promote neoplastic transformation.46
In summary, our investigations have shown that a response to ROS and up-regulation of xCT activity is crucial for reprogramming of zymogenic chief cells into SPEM. Targeting xCT may prove an effective tool for arresting metaplasia development in the stomach as well as mucous metaplasia in other epithelial tissues for the analysis of cellular plasticity and oxidative stress response.
Methods
Mouse Models
C57BL/6J mice approximately 8 weeks old were purchased from Jackson Labs (Bar Harbor, ME). xCTKO mice were generously donated by Dr David E. Featherstone, MS, PhD (University of Illinois, Chicago, IL).24, 47, 48 Genotype was confirmed for the xCTKO mice using PCR as previously described.48 Each experimental group consisted of at least 3–5 mice. L635 (synthesized by the Chemical Synthesis Core of the Vanderbilt Institute of Chemical Biology, Nashville, TN), dissolved in dH2O was administered by oral gavage (350 mg/kg) once a day for 3 consecutive days. Three time points were used to analyze chief cell reprogramming: 12, 24, and 72 hours after L635 treatment ± sulfasalazine. DMP-777 (a gift from DuPont-Merck Co. Wilmington, DE) dissolved in 1% methylcellulose was administered by oral gavage (350 mg/kg) once a day for 10 consecutive days. Intraperitoneal injection of sulfasalazine (10 mg/day) was administered 2 days before and throughout L635 and DMP-777 treatment. Sulfasalazine was freshly prepared each day in 0.1 mol/L NaOH adjusted with 1 mol/L HCl to pH 8. Intraperitoneal injection of the metabolites of sulfasalazine, sulfapyridine (3 mg/day), and mesalazine (5 mg/day) were administered 2 days before and throughout L635 treatment in the same manner as sulfasalazine.36 Archival sections of stomach from 12-month H felis–infected mice and human clinical specimens were obtained from previous investigations.8, 49 The care, maintenance, and treatment of animals in these studies adhere to the protocols approved by the Institutional Animal Care and Use Committee of Vanderbilt University.
Cell Culture
Cell lines were isolated from Immortomice as previously described.32 Immortomice are transgenic mice with interferon-γ (IFN-γ) inducible expression of temperature-sensitive T antigen. In cells that are isolated from Immortomice, addition of IFN-γ promotes the expression of immortalizing T antigen. At the permissive temperature (33°C), T antigen protein folds properly and immortalizes the cells. At the nonpermissive temperature (39°C) in the absence of IFN-γ, remaining T antigen protein misfolds and returns the cells to a differentiated state. Chief and SPEM cell lines were maintained at the permissive temperature in the presence of IFN-γ and were differentiated at 39°C for 48 hours with no IFN-γ before experiments were performed. Cells were cultured in ThermoFisher (Waltham, MA) Dulbecco’s modified Eagle medium/F12 (50:50) media supplemented with 10% Omega Scientific (Tarzana, CA) fetal bovine serum, 100 U/mL penicillin, and streptomycin (30-002-Cl; Corning, Corning, NY), 100 μg/mL MycoZAP Plus-PR (VZA-2021; Lonza, Basel, Switzerland), 1 ng/mL epidermal growth factor (AF-100-15; Peprotech, Rocky Hill, NJ), 1 ng/mL basic fibroblast growth factor (100-18B; Peprotech), 1 μg/mL hydrocortisone (H0888; Sigma, St. Louis, MO), 8 μg/mL insulin/transferring/selenium solution (41400045; ThermoFisher), and 5 U/mL IFN-γ (315-05; Peprotech). Cells were plated on collagen (PureCol Type 1 Advanced BioMatrix 5005, Carlsbad, CA)-coated plates (working solution, 0.03 mg/mL in phosphate-buffered saline [PBS]). For staining, cells were fixed in 4% paraformaldehyde (PFA) for 20 minutes. After fixation, cells were blocked and permeabilized in 1× PBS with 10% donkey serum and 0.3% Triton X-100 (Sigma) for 30 minutes. Cells were incubated in primary antibodies and diluted in 1× PBS with 1% donkey serum and 0.05% Tween-20 for 1 hour at room temperature. Cells then were incubated in fluorescent secondary antibodies, diluted in 1× PBS, for 1 hour at room temperature, before 4′,6-diamidino-2-phenylindole nuclear counterstain was added. A Zeiss (Oberkochen, Germany) Axio Imager M2 microscope with Axiovision digital imaging system was used to image fixed cells. For xCT blockade, cells were treated with 0.1 mmol/L sulfasalazine every day after 48 hours of differentiation. The pH level of media was monitored daily. To monitor cystine uptake, cells were treated with cystine-FITC (a gift from Dr Jeffrey Rathmell) for 2 hours.33 After 2 hours, Hoechst nuclear counterstain was added, and cells were washed 3 times with 1× PBS. The EVOS FL Cell Imaging System (ThermoFisher) was used to visualize intracellular cystine-FITC. To monitor proliferation, cell numbers were measured by a Bio-Rad (Hercules, CA) TC10 Automated Cell Counter. Three replicates were performed for each experiment.
siRNA Transfection
ImSPEM cells were plated on collagen-coated, 6-well plates the day before transfection to provide confluency of 60%–70% in 24 hours. ImSPEM cells were transfected with Slc7a11 Mouse siRNA Oligo Duplexes or Trilencer-27 Universal Scrambled Negative Control siRNA Duplex (SR416143; Origene, Rockville, MD) according to the manufacturer’s recommendations using siTran 1.0 siRNA transfection reagent (TT300001; Origene).
Quantitative Real-Time PCR Analysis
Total RNA from the stomach (oxyntic region) was extracted from paraformaldehyde-fixed, paraffin-embedded tissue of 3–5 mice per experimental group to examine the expression of mRNA transcripts (Cd44v9, xCT, Esrp1, Atg4, Atg5, Atg7, Atg12, Atg16L1, Beclin1, Lamp1, Lamp2, and Lc3). Five-micrometer sections were taken for H&E stain to identify the oxyntic region in tissue block. A 2-mm biopsy punch then was used to extract tissue. The standard Qiagen (Hilden, Germany) RNeasy FFPE Kit (73504) protocol was used for purification of total RNA. The ThermoFisher High-Capacity cDNA Reverse Transcription Kit (4368814) was used for complementary DNA synthesis. Quantitative real-time polymerase chain reaction was performed with the Bio-Rad SsoAdvanced Universal SYBR Green Super Mix (172-5270) and specific primers (Table 1) on the Bio-Rad CFX96 Touch Real-Time PCR Detection System. The expression of mRNA transcripts was normalized to TBP expression and shown as relative expression levels (2ΔCq). All graphs and statistics were completed in GraphPad Prism (San Diego, CA) using an unpaired Student t test to determine significance.
Table 1.
Catalog of Quantitative qPCR Primer Sequences
| Oligonucleotides |
|---|
| mCd44v (Cd44v9) primers |
| F: 5’ GGAGATCAGGATGACTCCTTCT 3’ |
| R: 5’ AGTCCTTGGATGAGTCTCGATC 3’ |
| mSlc7a11(xCT) primers |
| F: 5’ GGGGGTCTCCATCATCATCGGCA 3’ |
| R: 5’ TCTGCATAGGACAGGGCTCCAAA 3’ |
| mEsrp1 (Esrp1) primers |
| F: 5’ GCTTCACATTAGGCAGAT 3’ |
| R: 5’ CAGCACTTCTTGAACTCT 3’ |
| mAtg4 (Atg4) primers |
| F: 5’ GGGAACTGGCCCTACTTCAGA 3’ |
| R: 5’ TCCACCTCCAATCTCGACCTA 3’ |
| mAtg5 (Atg5) primers |
| F: 5’ GGACAGCTGCACACACTTGG 3’ |
| R: 5’ TGGCTCTATCCCGTGAATCAT 3’ |
| mAtg7 (Atg7) primers |
| F: 5’ GGCCTTTGAGGAATTTTTTGG 3’ |
| R: 5’ ACGTCTCTAGCTCCCTGCATG 3’ |
| mAtg12 (Atg12) primers |
| F: 5’ TGAATCAGTCCTTTGCCCCT 3’ |
| R: 5’ CATGCCTGGGATTTGCAGT 3’ |
| mAtg16l1(Atg16l1) primers |
| F: 5’ GCCCAGTTGAGGATCAAACAC 3’ |
| R: 5’ CTGCTGCATTTGGTTGTTCAG 3’ |
| mBeclin1 (Beclin1) primers |
| F: 5’ GGCCAATAAGATGGGTCTGA 3’ |
| R: 5’ GCTGCACACAGTCCAGAAAA 3’ |
| mLamp1 (Lamp1) primers |
| F: 5’ ACCTGTCGAGTGGCAACTTCA 3’ |
| R: 5’ GGGCACAAGTGGTGGTGAG 3’ |
| mLamp2 (Lamp2) primers |
| F: 5’ TAGGAGCCGTTCAGTCCAAT 3’ |
| R: 5’ GTGTGTCGCCTTGTCAGGTA 3’ |
| mMap1lc3b (Lc3) primers |
| F: 5’ ACTGCTCTGTCTTGTGTAGGTT 3’ |
| R: 5’ TCGTTGTGTGCCTTTATTAGTGCATC 3’ |
F, forward; R, reverse.
Immunohistochemical Staining
Mouse stomachs were fixed in 4% PFA overnight at 4°C and then were transferred into 70% ethanol for subsequent paraffin embedding. Five-micrometer sections were used for all immunohistochemistry studies. Deparaffinization, rehydration, and antigen retrieval were performed as previously described.50 Tissue sections were blocked in Dako (Santa Clara, CA) Peroxidase Blocking Solution at room temperature for 20 minutes followed by Dako Protein Block Serum-Free at room temperature for 1.5 hours. The primary antibodies were added overnight at 4°C in Dako Antibody Diluent with Background Reducing Components. Horseradish-peroxidase–conjugated secondary antibodies were added for 15 minutes at room temperature. 3,3′-Diaminobenzidine tetra hydrochloride chromogen was added for detection. A Leica (Wetzlar, Germany) SCN400 Slide Scanner in the Vanderbilt Digital Histology Shared Resource was used to image sections.
Immunofluorescence Staining
Five-micrometer tissue sections were blocked in Dako Protein Block Serum-Free at room temperature for 1.5 hours. For mouse primary antibodies, Mouse on Mouse blocking reagent (Vector Laboratories, Burlingame, CA) was added to slides for 20 minutes at room temperature. The primary antibodies (Table 2) were added overnight at 4°C in Dako Antibody Diluent with Background Reducing Components. Fluorescent donkey secondary antibodies were added in Dako Antibody Diluent at room temperature for 1 hour. A Zeiss Axio Imager M2 microscope with Axiovision digital imaging system, Zeiss LSM 710, or the Leica Aperio Versa 200 Fluorescent Slide Scanner in the Vanderbilt Digital Histology Shared Resource was used to image sections. Immunostaining for LC3 was performed at Washington University (St. Louis, MO) as previously described.5
Table 2.
Catalog of Primary Antibodies
| Antibodies | ||
|---|---|---|
| xCT (1:500) | Abcam | ab37185 |
| CD44v9 (1:25,000) | Cosmo Bio | CAC-LKG-M002 |
| ESRP1 (1:200) | Novus Biologicals | NBP1-82201 |
| Glutathione | Abcam | ab19534 |
| Ki67 (1:1000) | Cell Signaling Technology | 9129 |
| H+K+ATPase (1:10,000) | A gift from Dr Adam Smolka | N/A |
| GSII lectin (1:2000) | Invitrogen | L32451 |
| GIF (1:2000) | A gift from Dr David Alpers | N/A |
| Mist1 (1:1000) | A gift from Dr Jason Mills | N/A |
| LC3/MAP1LC3B (1:500) | Novus Biologicals | NB100-2220 |
| LAMP2 (GL2A7) (1:500) | Abcam | ab13524 |
| UEA1-lectin (1:2000) | Sigma | L9006 |
| Cleaved caspase-3 (Asp175) (1:200) | Cell Signaling Technology | 9661 |
| Clusterin-α antibody (M-18) (1:2000) | Santa Cruz Biotechnology | sc-6420 |
| TFF2 (1:500) | A gift from Dr Nicholas Wright | N/A |
Immunofluorescence Quantitation
Experimental groups contained 3–5 mice. Images were analyzed using CellProfiler (Cambridge, MA) to quantify objects and verified manually (nuclei, cells).51 At least 5 representative images (>150 glands) of proximal stomach corpus were taken from each mouse at 20× objective for quantification. Relative fluorescence intensity was determined using ImageJ (National Institutes of Health, Bethesda, MD) and was normalized to cell number.52 Relative fluorescence intensity for more than 250 cells per replicate was measured. To quantify autophagy of zymogenic granules, GIF-positive cells were selected. Images were analyzed using CellProfiler to quantify objects (puncta). More than 250 cells per mouse were measured. All graphs and statistics were completed in GraphPad Prism using an unpaired Student t test or 1-way analysis of variance with the Bonferroni post hoc multiple comparisons test to determine significance. n.s. P > .05, ∗P ≤ .05, ∗∗P ≤ .01, ∗∗∗P ≤ .001, ∗∗∗∗P ≤ .0001.
Electron Microscopy
For TEM, freshly excised stomach (oxyntic region) tissue was washed briefly in 0.1 mol/L cacodylate buffer. Samples then were fixed in 2.5% glutaraldehyde (in 0.1 mol/L sodium phosphate buffer, pH 7.4, 0.1 mol/L cacodylate buffer) for 1 hour at room temperature, followed by overnight fixation at 4°C. Subsequent tissue preparation and imaging for TEM was performed as previously described.53
ROS Detection
Fresh stomach tissue was trimmed, embedded in Tissue-Tek optimal cutting temperature compound, and snap frozen in an acetone bath on dry ice. Five-micrometer cryosections were obtained on glass slides. DHE (D1168; Invitrogen Carlsbad, CA) was diluted to a final concentration of 10 μmol/L in 1× PBS with Phalloidin-iFluor 488 (Abcam Cambridge, United Kingdom). DHE/phalloidin solution was applied to slides in a light-protected humidified chamber at 37°C for 30 minutes. Slides were washed with 1× PBS and imaged immediately using a Zeiss Axio Imager M2 microscope with an Axiovision digital imaging system. Relative fluorescence intensity (red) of oxidized DHE was determined using ImageJ and was normalized to area. CellROX Green Reagent (C10444; Invitrogen Carlsbad, CA) was diluted to a final concentration of 5 μmol/L in 1× PBS. CellROX solution was applied to slides in a light-protected humidified chamber at 37°C for 30 minutes. Slides were washed with 1× PBS and fixed in 4% PFA for 15 minutes. After fixation, to block/extract the tissue it was incubated in 1× PBS with 10% donkey serum and 0.3% Triton X-100 for 30 minutes. GIF primary antibody was diluted in 1× PBS with 1% donkey serum and 0.05% Tween-20 and incubated for 1 hour at room temperature. Fluorescent donkey anti-goat secondary antibody was diluted in 1× PBS and incubated for 1 hour at room temperature and Hoechst nuclear counterstain was added. A Zeiss Axio Imager M2 microscope with an Axiovision digital imaging system was used to image fixed tissue within 24 hours. DHE (10 μmol/L) or CellROX Green Reagent (5 μmol/L) was added to ImSPEM cells plated on glass coverslips and incubated at 39°C for 30 minutes. A Zeiss Axio Imager M2 microscope with an Axiovision digital imaging system was used to image cells immediately. The relative fluorescence intensity (green) of CellROX in GIF-positive cells was determined using ImageJ and was normalized to the cell number.
Footnotes
Author contributions Anne R. Meyer designed and performed experiments, analyzed data, and drafted the manuscript; Amy C. Engevik, Spencer G. Willet, and Janice A. Williams performed experiments, analyzed data, and revised the manuscript; Yong Zou and Pierre P. Massion designed experiments and revised the manuscript; and Jason C. Mills, Eunyoung Choi, and James R. Goldenring designed experiments, analyzed data, and revised the manuscript.
Conflicts of interest The authors disclose no conflicts.
Funding These studies were supported by grants from a Department of Veterans Affairs Merit Review Award IBX000930, National Institutes of HealthRO1 DK101332, and Department of DefenseCA160479 (J.R.G.); National Institutes of HealthR01s DK094989, DK105129, and DK110406 (J.C.M.); by Department of DefenseCA160399, American Association of Cancer Research-Debbie's Dream Foundation grant 17-20-41-CHOI, pilot grants from National Institutes of HealthP30 DK058404, and ACSIRG-15-169-56 (E.C.); by an American Association of Cancer Research-Debbie's Dream Foundation Grant (S.G.W.); by National Institutes of HealthT32 GM008554 and F31 DK117592 (A.R.M.); by National Institutes of HealthF32 DK111101 (A.C.E.); by National Cancer Institute grant CA102353 and Department of DefenseW81XWH-11-2-0161 (Y.Z. and P.P.M.); by core resources of the Vanderbilt Digestive Disease Center (National Institutes of HealthP30 DK058404); Washington University Digestive Disease Research Core Center (National Institutes of HealthP30 DK052574); Translational Pathology Shared Resource and Cell Imaging Shared Resource (National Cancer Institute/National Institutes of Health Cancer Center Support grant 2P30 CA068485); and imaging was supported by the Vanderbilt Digital Histology Shared Resource supported by a VA shared instrumentation grant (1IS1BX003097).
References
- 1.Goldenring J.R. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol. 2018;245:132–137. doi: 10.1002/path.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann W. TFF2, a MUC6-binding lectin stabilizing the gastric mucus barrier and more (review) Int J Oncol. 2015;47:806–816. doi: 10.3892/ijo.2015.3090. [DOI] [PubMed] [Google Scholar]
- 3.Ramsey V.G., Doherty J.M., Chen C.C., Stappenbeck T.S., Konieczny S.F., Mills J.C. The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development. 2007;134:211–222. doi: 10.1242/dev.02700. [DOI] [PubMed] [Google Scholar]
- 4.Leushacke M., Tan S.H., Wong A., Swathi Y., Hajamohideen A., Tan L.T., Goh J., Wong E., Denil S., Murakami K., Barker N. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol. 2017;19:774–786. doi: 10.1038/ncb3541. [DOI] [PubMed] [Google Scholar]
- 5.Willet S.G., Lewis M.A., Miao Z.F., Liu D., Radyk M.D., Cunningham R.L., Burclaff J., Sibbel G., Lo H.G., Blanc V., Davidson N.O., Wang Z.N., Mills J.C. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 2018;37 doi: 10.15252/embj.201798311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radyk M.D., Burclaff J., Willet S.G., Mills J.C. Metaplastic cells in the stomach arise, independently of stem cells, via dedifferentiation or transdifferentiation of chief cells. Gastroenterology. 2018;154:839–843 e2. doi: 10.1053/j.gastro.2017.11.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engevik A.C., Feng R., Choi E., White S., Bertaux-Skeirik N., Li J., Mahe M.M., Aihara E., Yang L., DiPasquale B., Oh S., Engevik K.A., Giraud A.S., Montrose M.H., Medvedovic M., Helmrath M.A., Goldenring J.R., Zavros Y. The development of spasmolytic polypeptide/TFF2-expressing metaplasia (SPEM) during gastric repair is absent in the aged stomach. Cell Mol Gastroenterol Hepatol. 2016;2:605–624. doi: 10.1016/j.jcmgh.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam K.T., Lee H.-J., Sousa J.F., Weis V.G., O'Neal R.L., Finke P.E., Romero-Gallo J., Shi G., Mills J.C., Peek R.M., Konieczny S.F., Goldenring J.R. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology. 2010;139:2028–2037. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozaki K., Ogawa M., Williams J.A., LaFleur B.J., Ng V., Drapkin R.I., Mills J.C., Konieczny S.F., Nomura S., Goldenring J.R. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology. 2008;134:511–521. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt P.H., Lee J.R., Joshi V., Playford R.J., Poulsom R., Wright N.A., Goldenring J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest. 1999;79:639–646. [PubMed] [Google Scholar]
- 11.Weis V.G., Sousa J.F., LaFleur B.J., Nam K.T., Weis J.A., Finke P.E., Ameen N.A., Fox J.G., Goldenring J.R. Heterogeneity in mouse spasmolytic polypeptide-expressing metaplasia lineages identifies markers of metaplastic progression. Gut. 2013;62:1270–1279. doi: 10.1136/gutjnl-2012-302401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saenz J.B., Mills J.C. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol. 2018;15:257–273. doi: 10.1038/nrgastro.2018.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beasley D.E., Koltz A.M., Lambert J.E., Fierer N., Dunn R.R. The evolution of stomach acidity and its relevance to the human microbiome. PLoS One. 2015;10:e0134116. doi: 10.1371/journal.pone.0134116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang I., Nell S., Suerbaum S. Survival in hostile territory: the microbiota of the stomach. FEMS Microbiol Rev. 2013;37:736–761. doi: 10.1111/1574-6976.12027. [DOI] [PubMed] [Google Scholar]
- 15.Roth K.A., Kapadia S.B., Martin S.M., Lorenz R.G. Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J Immunol. 1999;163:1490–1497. [PubMed] [Google Scholar]
- 16.Goldenring J.R., Ray G.S., Coffey R.J., Meunier P.C., Haley P.J., Barnes T.B., Car B.D. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology. 2000;118:1080–1093. doi: 10.1016/s0016-5085(00)70361-1. [DOI] [PubMed] [Google Scholar]
- 17.Isacke C.M., Yarwood H. The hyaluronan receptor, CD44. Int J Biochem Cell Biol. 2002;34:718–721. doi: 10.1016/s1357-2725(01)00166-2. [DOI] [PubMed] [Google Scholar]
- 18.Khurana S.S., Riehl T.E., Moore B.D., Fassan M., Rugge M., Romero-Gallo J., Noto J., Peek R.M., Jr., Stenson W.F., Mills J.C. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J Biol Chem. 2013;288:16085–16097. doi: 10.1074/jbc.M112.445551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertaux-Skeirik N., Feng R., Schumacher M.A., Li J., Mahe M.M., Engevik A.C., Javier J.E., Peek R.M., Jr., Ottemann K., Orian-Rousseau V., Boivin G.P., Helmrath M.A., Zavros Y. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015;11:e1004663. doi: 10.1371/journal.ppat.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prochazka L., Tesarik R., Turanek J. Regulation of alternative splicing of CD44 in cancer. Cell Signal. 2014;26:2234–2239. doi: 10.1016/j.cellsig.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Wada T., Ishimoto T., Seishima R., Tsuchihashi K., Yoshikawa M., Oshima H., Oshima M., Masuko T., Wright N.A., Furuhashi S., Hirashima K., Baba H., Kitagawa Y., Saya H., Nagano O. Functional role of CD44v-xCT system in the development of spasmolytic polypeptide-expressing metaplasia. Cancer Sci. 2013;104:1323–1329. doi: 10.1111/cas.12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishimoto T., Nagano O., Yae T., Tamada M., Motohara T., Oshima H., Oshima M., Ikeda T., Asaba R., Yagi H., Masuko T., Shimizu T., Ishikawa T., Kai K., Takahashi E., Imamura Y., Baba Y., Ohmura M., Suematsu M., Baba H., Saya H. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 23.Nagano O., Okazaki S., Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene. 2013;32:5191–5198. doi: 10.1038/onc.2012.638. [DOI] [PubMed] [Google Scholar]
- 24.Sato H., Shiiya A., Kimata M., Maebara K., Tamba M., Sakakura Y., Makino N., Sugiyama F., Yagami K., Moriguchi T., Takahashi S., Bannai S. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 25.Peppercorn M.A. Sulfasalazine. Pharmacology, clinical use, toxicity, and related new drug development. Ann Intern Med. 1984;101:377–386. doi: 10.7326/0003-4819-101-3-377. [DOI] [PubMed] [Google Scholar]
- 26.Gout P.W., Buckley A.R., Simms C.R., Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 27.Chung W.J., Lyons S.A., Nelson G.M., Hamza H., Gladson C.L., Gillespie G.Y., Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagane M., Kanai E., Shibata Y., Shimizu T., Yoshioka C., Maruo T., Yamashita T. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS One. 2018;13:e0195151. doi: 10.1371/journal.pone.0195151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zavros Y. Initiation and maintenance of gastric cancer: a focus on CD44 variant isoforms and cancer stem cells. Cell Mol Gastroenterol Hepatol. 2017;4:55–63. doi: 10.1016/j.jcmgh.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yae T., Tsuchihashi K., Ishimoto T., Motohara T., Yoshikawa M., Yoshida G.J., Wada T., Masuko T., Mogushi K., Tanaka H., Osawa T., Kanki Y., Minami T., Aburatani H., Ohmura M., Kubo A., Suematsu M., Takahashi K., Saya H., Nagano O. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- 31.Warzecha C.C., Shen S., Xing Y., Carstens R.P. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009;6:546–562. doi: 10.4161/rna.6.5.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weis V.G., Petersen C.P., Mills J.C., Tuma P.L., Whitehead R.H., Goldenring J.R. Establishment of novel in vitro mouse chief cell and SPEM cultures identifies MAL2 as a marker of metaplasia in the stomach. Am J Physiol Gastrointest Liver Physiol. 2014;307:G777–G792. doi: 10.1152/ajpgi.00169.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siska P.J., Kim B., Ji X., Hoeksema M.D., Massion P.P., Beckermann K.E., Wu J., Chi J.T., Hong J., Rathmell J.C. Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes. J Immunol Methods. 2016;438:51–58. doi: 10.1016/j.jim.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lennerz J.K., Kim S.H., Oates E.L., Huh W.J., Doherty J.M., Tian X., Bredemeyer A.J., Goldenring J.R., Lauwers G.Y., Shin Y.K., Mills J.C. The transcription factor MIST1 is a novel human gastric chief cell marker whose expression is lost in metaplasia, dysplasia, and carcinoma. Am J Pathol. 2010;177:1514–1533. doi: 10.2353/ajpath.2010.100328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang F., Kumano M., Beraldi E., Fazli L., Du C., Moore S., Sorensen P., Zoubeidi A., Gleave M.E. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014;5:5775. doi: 10.1038/ncomms6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nashed M.G., Ungard R.G., Young K., Zacal N.J., Seidlitz E.P., Fazzari J., Frey B.N., Singh G. Behavioural effects of using sulfasalazine to inhibit glutamate released by cancer cells: a novel target for cancer-induced depression. Sci Rep. 2017;7:41382. doi: 10.1038/srep41382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills J.C., Sansom O.J. Reserve stem cells: differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal. 2015;8:re8. doi: 10.1126/scisignal.aaa7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jensen J.N., Cameron E., Garay M.V., Starkey T.W., Gianani R., Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Mittal M., Siddiqui M.R., Tran K., Reddy S.P., Malik A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ni J., Mei M., Sun L. Oxidative DNA damage and repair in chronic atrophic gastritis and gastric cancer. Hepatogastroenterology. 2012;59:671–675. doi: 10.5754/hge12177. [DOI] [PubMed] [Google Scholar]
- 41.Vaahtera L., Brosche M., Wrzaczek M., Kangasjarvi J. Specificity in ROS signaling and transcript signatures. Antioxid Redox Signal. 2014;21:1422–1441. doi: 10.1089/ars.2013.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rushworth G.F., Megson I.L. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141:150–159. doi: 10.1016/j.pharmthera.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Habib E., Linher-Melville K., Lin H.X., Singh G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015;5:33–42. doi: 10.1016/j.redox.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishii T., Mann G.E. Redox status in mammalian cells and stem cells during culture in vitro: critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol. 2014;2:786–794. doi: 10.1016/j.redox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarro-Yepes J., Burns M., Anandhan A., Khalimonchuk O., del Razo L.M., Quintanilla-Vega B., Pappa A., Panayiotidis M.I., Franco R. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014;21:66–85. doi: 10.1089/ars.2014.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardbower D.M., de Sablet T., Chaturvedi R., Wilson K.T. Chronic inflammation and oxidative stress: the smoking gun for Helicobacter pylori-induced gastric cancer? Gut Microbes. 2013;4:475–481. doi: 10.4161/gmic.25583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCullagh E.A., Featherstone D.E. Behavioral characterization of system xc- mutant mice. Behav Brain Res. 2014;265:1–11. doi: 10.1016/j.bbr.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 48.De Bundel D., Schallier A., Loyens E., Fernando R., Miyashita H., Van Liefferinge J., Vermoesen K., Bannai S., Sato H., Michotte Y., Smolders I., Massie A. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792–5803. doi: 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sousa J.F., Ham A.J., Whitwell C., Nam K.T., Lee H.J., Yang H.K., Kim W.H., Zhang B., Li M., LaFleur B., Liebler D.C., Goldenring J.R. Proteomic profiling of paraffin-embedded samples identifies metaplasia-specific and early-stage gastric cancer biomarkers. Am J Pathol. 2012;181:1560–1572. doi: 10.1016/j.ajpath.2012.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen C.P., Weis V.G., Nam K.T., Sousa J.F., Fingleton B., Goldenring J.R. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology. 2014;146:1727–1738 e8. doi: 10.1053/j.gastro.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones T.R., Kang I.H., Wheeler D.B., Lindquist R.A., Papallo A., Sabatini D.M., Golland P., Carpenter A.E. CellProfiler analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics. 2008;9:482. doi: 10.1186/1471-2105-9-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weis V.G., Knowles B.C., Choi E., Goldstein A.E., Williams J.A., Manning E.H., Roland J.T., Lapierre L.A., Goldenring J.R. Loss of MYO5B in mice recapitulates microvillus inclusion disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell Mol Gastroenterol Hepatol. 2016;2:131–157. doi: 10.1016/j.jcmgh.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]