

Abstract
Objective.
Genetic variants affect both the development and severity of rheumatoid arthritis (RA). Recent studies have expanded the number of RA susceptibility variants. We tested the hypothesis that these associated with disease severity in a clinical trial cohort of patients with early, active RA.
Methods.
We evaluated 524 patients with RA enrolled in the Combination Anti-Rheumatic Drugs in Early RA (CARDERA) trials. We tested validated susceptibility variants — 69 single-nucleotide polymorphisms (SNP), 15 HLA-DRB1 alleles, and amino acid polymorphisms in 6 HLA molecule positions — for their associations with progression in Larsen scoring, 28-joint Disease Activity Scores, and Health Assessment Questionnaire (HAQ) scores over 2 years using linear mixed-effects and latent growth curve models.
Results.
HLA variants were associated with joint destruction. The *04:01 SNP (rs660895, p = 0.0003), *04:01 allele (p = 0.0002), and HLA-DRβ1 amino acids histidine at position 13 (p = 0.0005) and valine at position 11 (p = 0.0012) significantly associated with radiological progression. This association was only significant in anticitrullinated protein antibody (ACPA)-positive patients, suggesting that while their effects were not mediated by ACPA, they only predicted joint damage in ACPA-positive RA. Non-HLA variants did not associate with radiograph damage (assessed individually and cumulatively as a weighted genetic risk score). Two SNP — rs11889341 (STAT4, p = 0.0001) and rs653178 (SH2B3-PTPN11, p = 0.0004) — associated with HAQ scores over 6–24 months.
Conclusion.
HLA susceptibility variants play an important role in determining radiological progression in early, active ACPA-positive RA. Genome-wide and HLA-wide analyses across large populations are required to better characterize the genetic architecture of radiological progression in RA.
Keywords: RHEUMATOID ARTHRITIS, RADIOGRAPHY, OUTCOME MEASURES, GENETICS, HLA ANTIGENS
Identifying biomarkers that predict rheumatoid arthritis (RA) prognosis at first presentation is an important research goal. They could guide decisions on initial treatment intensity, facilitating stratified medicine. The progression of radiological damage that characterizes severe RA is 45% to 58% heritable1. This suggests that genetic variants are potential prognostic markers. Current knowledge of the genetic architecture of radiograph progression is limited, with up to half of its heritability unexplained2.
The concept that RA susceptibility variants also influence disease severity is an intuitive one. Several studies have tested the hypothesis that RA risk variants associate with radiographic damage. They have several limitations. First, many associations are not replicated3,4. Second, not all risk loci have been studied (the largest analysis examined 37 independent variants)5. Third, they represent observational studies assessing patients with established RA of variable activity6,7. Because randomized controlled trials (RCT) of early, active RA8,9,10 provide the evidence base for intensive therapy, for genetic variants to guide treatment intensity decisions, they also require evaluation in patients with early, active RA.
Metaanalyses of genome-wide association studies (GWAS) have expanded the number of RA susceptibility variants. These span single-nucleotide polymorphisms (SNP), HLA-DRB1 alleles, and HLA molecule amino acid polymorphisms11,12. We tested the hypothesis that they were also associated with disease severity by evaluating their relationship with radiographic progression, disease activity, and disability over 2 years in an RCT cohort of patients with early, active RA.
MATERIALS AND METHODS
Subjects.
We studied the combined data of patients in two 24-month trials of Combination Anti-Rheumatic Drugs in Early RA (CARDERA-1 and CARDERA-2) that recruited 467 and 159 patients with RA, respectively, from 42 rheumatology units in England13,14. Patients had less than 2 years of RA duration and active disease, defined as 3 of (1) ≥ 3 swollen joints, (2) ≥ 6 tender joints, (3) ≥ 45-min morning stiffness, or (4) erythrocyte sedimentation rate (ESR) ≥ 28 mm/h.
CARDERA-1 patients were randomized to 4 treatment groups: (1) methotrexate (MTX), (2) MTX and cyclosporine, (3) MTX and prednisolone, and (4) MTX, cyclosporine, and prednisolone. CARDERA-2 patients were randomized to 2 treatment groups: MTX, and MTX and anakinra.
Disease outcomes.
In CARDERA-1, modified Larsen scores, Disease Activity Scores on a 28-joint count (DAS28), and Health Assessment Questionnaire (HAQ) scores were recorded 6-monthly13. In CARDERA-2, modified Larsen scores were recorded 12-monthly, and DAS28 and HAQ scores were measured at 0, 6, 12, and 24 months. In both studies, the DAS28-ESR was used. Mean scores for each outcome at each timepoint are shown in Figure 1.
Figure 1.
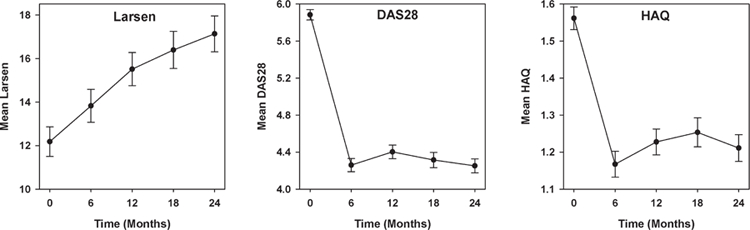
Mean Larsen, DAS28, and HAQ scores over 2 years. Standard error bars for each score shown. DAS28: Disease Activity Score at 28 joints; HAQ: Health Assessment Questionnaire.
Genotyping.
Archived DNA was genotyped on the Illumina ImmunoChip15. Quality control (QC) procedures are described in Supplementary Table 1 (available online at jrheum.org). From 560 genotyped individuals, 524 were included in our analysis. From 196,524 genetic markers, 138,873 passed QC procedures (genotyping rate 0.999).
Classical HLA-DRB1 alleles and amino acid polymorphisms in HLA-DRβ1, HLA-B, and HLA-DPβ1 were imputed using SNP2HLA (Beagle imputation method16, reference data from Type 1 Diabetes Genetics Consortium panel17). Imputed allele/amino acid dosages were analyzed. These probabilistic representations of the number of predicted alleles correlate better with true genotypes when compared with “best-guess” genotypes16. Imputed markers with INFO scores < 0.5 were excluded18.
SNP.
We evaluated validated European ancestry RA susceptibility SNP from the recent GWAS metaanalysis of European and Asian populations by Okada, et al11. To maximize study power, we evaluated only loci attaining genome-wide significance in Europeans. This omitted small effect size loci (requiring the larger transethnic sample size to achieve significance) and loci that had a stronger effect in Asians.
Of 77 potential susceptibility SNP, 69 were available in CARDERA (including 31 proxy SNP, r2 > 0.8; Supplementary Table 2, available online at jrheum.org). As neither the validated HLA region SNP (rs9268839) in the Okada, et al metaanalysis nor any proxy SNP were present in CARDERA, we used the validated HLA region SNP from the previous metaanalysis by Eyre, et al (rs660895)19. This ensured that a validated HLA susceptibility SNP was tested. Missing data in 9 SNP (7 SNP missing in 1 patient, 1 SNP in 4 patients, 1 SNP in 21 patients) were imputed by assigning them the expected allele count (twice the allele frequency)20,21.
HLA alleles and amino acids.
Four-digit HLA-DRB1 alleles attaining PGWAS < 5 × 10−8 in Europeans from the RA metaanalysis of association in the HLA region12 were evaluated. Of 17 potential alleles, 2 were unavailable in CARDERA (*11:03, *14:04) because of low INFO scores. Amino acids at positions 11, 13, 71, and 74 in HLA-DRβ1; position 9 in HLA-B; and position 9 in HLA-DPβ1 were assessed. In the HLA metaanalysis, these explained most of the region’s association with RA12. Positions 11 and 13 in HLA-DRβ1 were both included because their strong linkage disequilibrium (LD) meant causality could not be assigned12.
Associations with radiological progression.
The association between Larsen score progression and each genetic variant was examined using a linear mixed-effects (LME) model. This assessed all repeated Larsen scores simultaneously, and through incorporating correlated random effects, accounted for within-individual correlations in Larsen scores over time. This repeated measures modelling approach was preferable to evaluating associations with the 24-month change in Larsen scores (∆Larsen) because it (1) better evaluated within-individual changes over time22, (2) optimized the analysis power22, and (3) could accommodate missing radiographic data by assuming these observations were missing-at-random23.
Larsen scores were log-transformed with a constant added (because of zero unit scores) to approximate a normal distribution. Loge (Larsen score + 1) was the response variable. Time (yrs), clinical variables, genotype, and a genotype*time interaction term were fixed-effects predictor variables. Clinical variables (chosen using stepwise backward elimination) included treatment (coded categorically with MTX as the reference group), age, disease duration, and rheumatoid factor (RF) status. Their associations with Larsen scores are provided in Supplementary Table 3 (available online at jrheum.org). Genotype was coded additively; β values were back-transformed to the original Larsen scale. The genotype*time term β provided information on the annual increase in Larsen score per risk allele copy relative to the reference genotype; p values for this term provided information on the variant’s role in radiological progression. The X-chromosome SNP (rs2075596) was assessed separately in men and women.
RF was used as a covariate instead of anticitrullinated protein antibodies (ACPA) because of missing ACPA data (25 patients). Significant variants were (1) reanalyzed in a model with ACPA as a covariate and (2) evaluated in ACPA-positive and ACPA-negative patients separately (omitting serology as a covariate); this established whether their effects were independent of ACPA. Examination of residuals from a model containing time and clinical variables confirmed a good model fit.
To ensure the LME model results were not biased by missing radiographic data, a supplementary approach was undertaken. This involved a 3-way ANOVA model including the DLarsen as the response variable and treatment, RF status, and genotype (coded additively) as predictor variables (chosen through stepwise backward elimination). Because the ∆Larsen remained non-normally distributed despite log transformation, a rank-based inverse normal transformation was undertaken24. Residual plots confirmed a good model fit. The 19 individuals without 24-month radiograph data were excluded from this analysis.
Associations with DAS28 and HAQ scores.
The associations between each variant and DAS28 and HAQ scores were assessed using latent growth curve models (GCM). In contrast to Larsen scores, longitudinal changes in DAS28/HAQ scores were nonlinear: in months 0–6, an improvement was seen; subsequently a worsening then improvement was observed (Figure 1). A 2-piece GCM (Figure 2) was therefore used, composing of an intercept, slope 1 (0 to 6–mo change in DAS28/HAQ), and slope 2 (6 to 24–mo change in DAS28/HAQ). The GCM estimated the effect of each genotype on the means of the intercept, slope 1, and slope 2, corrected for the covariates sex and age (chosen through stepwise backward elimination).
Figure 2.
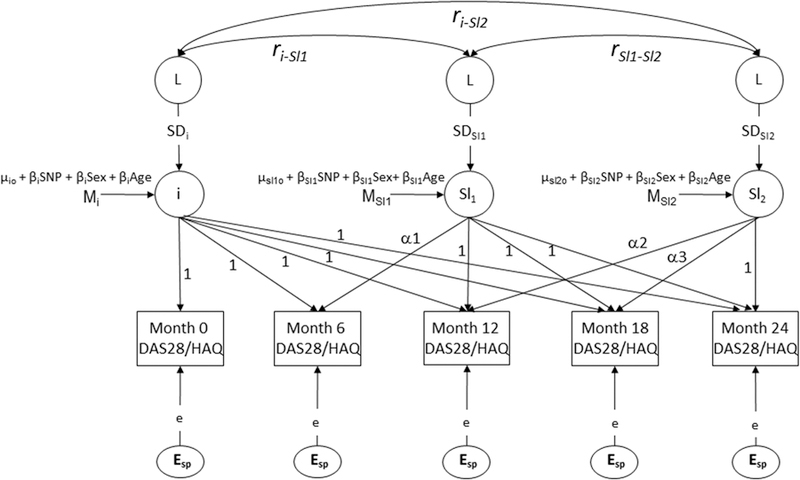
Multiphase latent growth curve factor model for DAS28 and HAQ scores. i: intercept. Sl1: slope 1 (identifies change in DAS28/HAQ from month 0–6). Sl2: slope 2 (identifies change in DAS28/HAQ from month 6–24). Esp: residual error terms (constrained to be equal). Mi, Msl1, and Msl2: intercept, slope 1, and slope 2 means, respectively. µio, µsl1o, and µsl2o: overall mean estimate of the intercept, slope 1, and slope 2 factors, respectively. βiSNP, βSl1SNP, and βSl2SNP: SNP dependent intercept, slope 1, and slope 2 means, respectively. βiSex, βSl1Sex, and βSl2Sex: sex-dependent intercept, slope 1, and slope 2 means, respectively. βiAge, βSl1Age, and βSl2Age: age-dependent intercept, slope 1, and slope 2 means, respectively. ri-sl1, ri-sl2 are the correlations between the intercept and slopes. rsl1-sl2 the correlation between slope 1 and slope 2. SDi, SDsl1, and SDsl2 are the SD of the latent factors. The overall variance-covariance structure and the overall means of the HAQ/DAS28 scores, which are optimized, are modeled by the variance-covariance structure and the means of the latent factors weighted by the factor loadings. DAS28: Disease Activity Score at 28 joints; HAQ: Health Assessment Questionnaire; SNP: single-nucleotide polymorphisms.
Weighted genetic risk score (wGRS) associations with disease outcomes.
The associations between a wGRS combining susceptibility loci, and each outcome were tested. Three wGRS were evaluated: (1) a full wGRS (all SNP except the X-chromosome marker), (2) an HLA wGRS (all HLA-DRB1 alleles), and (3) a non-HLA wGRS (as per the full wGRS, but excluding the HLA SNP). The wGRS replaced the genotype term in the LME/ANOVA/GCM models.
wGRS were constructed using the R package, REGENT25,26, which uses data on allelic OR from reference metaanalyses11,12 to generate a genotype relative risk (GRR) score across loci within a multiplicative model. The log (GRR) represented the wGRS. SNP allelic OR were obtained from the reference metaanalysis’ combined stage11.
Significance thresholds.
For SNP, HLA allele, and amino acid association tests, Bonferroni corrected p value thresholds of 0.0007 (69 markers), 0.0033 (15 markers), and 0.0019 (27 markers), respectively, were considered significant. For the 4 HLA markers tested separately in ACPA subsets, a p value threshold of 0.0125 was used.
Power calculations.
CARDERA had 81% power to detect an SNP accounting for 3.4% of the variance in each outcome27.
Statistical programs.
Analyses were performed in R version 3.0.2 (LME/ANOVA), OpenMx version 1.4 (latent GCM), and PLINK version 1.0728.
Ethical approval.
CARDERA-1 [Research Ethics Committee (REC) reference: MREC (1) 99/04] and CARDERA-2 (REC reference: MREC 02/1/089) were ethically approved. Approval was obtained to genotype archived DNA (REC reference: 11/EE/0544). All patients provided consent.
RESULTS
Subjects.
The 524 patients (Table 1) were mostly women (69%) and ACPA-positive (70%). Their mean age was 54.7 years. Median baseline disease duration was 1 month; 40 patients (8%) had disease durations > 12 months. Their high initial mean DAS28 (5.88) confirmed active RA. Some had radiological damage (median Larsen score 6.50) at baseline, which was expected in patients with active RA with disease durations up to 2 years.
Table 1.
CARDERA genetics cohort patient baseline data. Values are n (%) unless otherwise specified.
| Characteristics | Summary Statistic | |
|---|---|---|
| Demographic | Female | 359 (69) |
| Age, yrs, mean (95% CI) | 54.7 (53.6–55.8) | |
| RA-specific | RA duration, mos, median (IQR) | 1.0 (0.0–4.0) |
| ACPA-positive* | 350 (70) | |
| RF-positive | 352 (67) | |
| Larsen score, median (IQR) | 6.50 (2.00–16.50) | |
| DAS28, mean (95% CI) | 5.88 (5.77–5.99) | |
| HAQ, mean (95% CI) | 1.56 (1.50–1.62) | |
| Treatment | Receiving MTX | 161 (31) |
| Receiving MTX and cyclosporine | 108 (21) | |
| Receiving MTX and prednisolone | 102 (19) | |
| Receiving triple therapy | 107 (20) | |
| Receiving MTX and anakinra | 46 (9) |
ACPA status missing in 25 patients. RA: rheumatoid arthritis; IQR: interquartile range; ACPA: anticitrullinated protein antibodies; RF: rheumatoid factor; DAS28: Disease Activity Score at 28 joints; HAQ: Health Assessment Questionnaire; MTX: methotrexate; triple therapy: MTX, cyclosporine, and prednisolone; CARDERA: Combination Anti-Rheumatic Drugs in Early RA trials.
SNP.
Only rs660895 (p = 0.0003) had a p value for an association with radiographic progression that passed the pre-defined significance threshold when tested using the LME model (Table 2). This SNP tags *04:01; its β value of 1.07 (95% CI 1.03–1.12) indicated a 1.07-fold greater annual increase in Larsen scores per copy of the risk (G) allele carried, relative to the reference (A) allele (Figure 3). This SNP remained significant in a model including ACPA as a covariate in place of RF (p = 0.0003).
Table 2.
Genetic variants attaining a significant association with radiological progression.
| SNP (Gene) or HLA-DRB1 Allele or HLA Amino Acid |
All RA, n = 524 | ACPA+, n = 350 | ACPA−, n = 149 | |||
|---|---|---|---|---|---|---|
| β (95% CI) | p | β (95% CI) | p | β (95% CI) | p | |
| rs660895 (HLA-DRB1*04:01 tag) | 1.07 (1.03–1.12) | 0.0003 | 1.07 (1.02–1.13) | 0.0037 | 1.03 (0.96–1.11) | 0.4571 |
| HLA-DRB1*04:01 | 1.10 (1.05–1.15) | 0.0002 | 1.08 (1.02–1.14) | 0.0105 | 1.04 (0.94–1.16) | 0.4344 |
| Histidine at position 13 in HLA-DRβ1 | 1.07 (1.03–1.12) | 0.0005 | 1.07 (1.02–1.13) | 0.0060 | 1.02 (0.95–1.10) | 0.5590 |
| Valine at position 11 in HLA-DRβ1 | 1.07 (1.03–1.11) | 0.0012 | 1.07 (1.01–1.12) | 0.0111 | 1.02 (0.95–1.10) | 0.6130 |
p values from the linear-mixed effects model genotype*time interaction term. ACPA status missing in 25 patients. Bonferroni-corrected p value thresholds for SNP, HLA-DRB1 alleles, and HLA amino acid polymorphisms in all RA are 0.0007, 0.0033, and 0.0019, respectively. Bonferroni-corrected p value threshold for the 4 markers tested in ACPA-positive and ACPA-negative RA is 0.0125. SNP: single-nucleotide polymorphism; RA: rheumatoid arthritis; ACPA: anticitrullinated protein antibodies.
Figure 3.
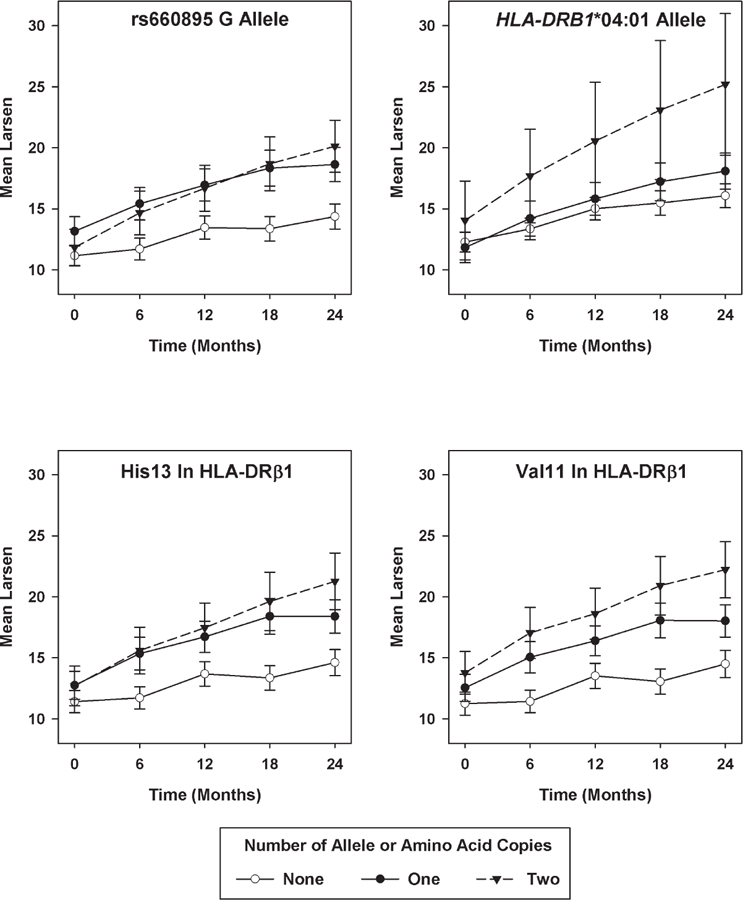
Significant genetic associations with Larsen score progression. Mean Larsen scores with standard error bars shown at each timepoint stratified by genotype (“best-guess” genotypes used for imputed data in this figure, but not in the statistical analysis). His13: histidine at position 13; Val11: valine at position 11.
Restricting analyses to ACPA-positive patients (Table 2) provided similar findings for this SNP (β = 1.07, p = 0.0037). No significant association was seen in ACPA-negative patients (β = 1.03, p = 0.4571), although the sample size was substantially smaller. This suggested that rs660895’s effect on radiological progression was not mediated by ACPA; if rs660895 drove ACPA formation, which itself caused joint destruction, then no association between this SNP and radiographic progression would be seen in ACPA-positive patients. Results for all SNP are given in Supplementary Table 4 (available online at jrheum.org).
Using the ANOVA model to test the association between each SNP and the ∆Larsen, the 2 most strongly associated SNP — rs660895 (p = 0.0002) and rs7579944 (p = 0.0140) — were the same as those identified by the LME model. Only rs660895 was significant.
HLA-DRB1 alleles.
Only *04:01 (p = 0.0002) had a p value that passed the predefined significance threshold using the LME model (Table 2). Its β value of 1.10 (95% CI 1.05–1.15) indicated a 1.10-fold greater annual increase in Larsen scores per *04:01 allele copy carried, relative to a non-carrier. This allele remained significant in a model including ACPA as a covariate instead of RF (p = 0.0006). Restricting analyses to ACPA-positive patients revealed similar findings (β = 1.08, p = 0.0105). No significant effect was seen in ACPA-negative patients (β = 1.04, p = 0.4344). Results for all alleles are given in Supplementary Table 5 (available online at jrheum.org). Using the ANOVA model to test the association between HLA-DRB1 susceptibility alleles and the ∆Larsen, *04:01 remained significant (p = 0.0009).
HLA amino acids.
Two amino acid polymorphisms — histidine at position 13 (His13; β = 1.07, p = 0.0005) and valine at position 11 (Val11; β = 1.07, p = 0.0012) in HLA-DRβ1 — had p values that passed the predefined significance threshold using the LME model (Table 2). Repeating the analysis with ACPA as a covariate instead of RF revealed similar results (His13 p = 0.0006, Val11 p = 0.0012). Restricting analyses to ACPA-positive patients showed similar findings; no significant association was seen in ACPA-negative patients (Table 2). Using the ANOVA model, His13 (p = 0.0002) and Val11 (p = 0.0002) in HLA-DRβ1 remained the most significant markers. Results for all amino acids are given in Supplementary Table 6 (available online at jrheum.org).
Because His13 and Val11 were in tight LD (r2 = 0.943), a conditional analysis was undertaken. Conditioning on His13 eliminated the effect of Val11 (LME p = 0.5547, ANOVA p = 0.4851). Conditioning on Val11 eliminated the effect of His13 (LME p = 0.1699, ANOVA p = 0.8697). This indicated the significance at both these markers was driven by their high LD, although it was not possible to establish which variant drove their association with Larsen scores.
None of the tested polymorphisms in the shared epitope (SE) region (positions 71 and 74) were significant (Supplementary Table 6 available online at jrheum.org); the lowest p value was for lysine at position 71 (p = 0.0293). However, the SE itself (sequences QRRAA, RRRAA, and QKRAA in positions 70–7429) significantly associated with radiological progression when the classical SE alleles (*01:01, *01:02, *04:01, *04:04, *04:05, *04:08, *10:01) were grouped together (LME p = 0.0003, ANOVA p = 0.0001).
Although His13 and Val11 are not in LD with positions 71 and 74, there is a marked correlation between their presence and carrying an SE sequence. His13 and Val11 are encoded by the SE alleles *04:01, *04:04, *04:05, *04:08, and *10:0112. A conditional analysis incorporating the SE as a modeling covariate was therefore undertaken. Conditioning on the SE eliminated the effect of His13 (LME p = 0.1996, ANOVA p = 0.1346) and Val11 (LME p = 0.3593, ANOVA p = 0.1224). Conditioning on His13 (LME p = 0.08301, ANOVA p = 0.0842) and Val11 (LME p = 0.05345, ANOVA p = 0.1023) eliminated the effect of the SE. This suggests that the association of His13, Val11, and the SE with joint destruction was driven by a high correlation between them, although it was not possible to establish which variant drove the association.
wGRS.
The full wGRS (LME p = 0.0428) and HLA wGRS (LME p < 0.0001), but not the non-HLA wGRS (LME p = 0.6720), associated with radiological progression (Supplementary Table 7, available online at jrheum.org).
Associations with disease activity.
Ten SNP, 1 HLA-DRB1 allele, and 4 HLA amino acid polymorphisms had p values < 0.05 for an association with the intercept and/or slope 1 and/or slope 2 DAS28 latent growth factor means (Supplementary Table 8, available online at jrheum.org). None were significant after Bonferroni correction.
The HLA wGRS associated with the intercept DAS28 latent growth factor means (β = 0.09, p = 0.041), but not those for slope 1 or slope 2 (Supplementary Table 7, available online at jrheum.org). No associations were seen between the full wGRS or non-HLA wGRS and DAS28.
Associations with disability.
From all tested variants, 2 SNP had p values for an association with HAQ score slope 2 latent growth factor means that passed the predefined significance threshold (Supplementary Table 9, available online at jrheum.org). These were composed of rs11889341 in STAT4 (β = 0.142, p = 0.0001) and rs653178 in SH2B3-PTPN11 (β = –0.109, p = 0.0004). Although the rs11889341 RA risk (T) allele associated with greater HAQ score increases over months 6–24, mean HAQ scores were lower at all timepoints in T allele carriers (Figure 4). Similarly, although the rs653178 RA risk (C) allele associated with an improvement in HAQ scores over months 6–24, at all timepoints except 24 months, mean HAQ scores were higher in CC homozygotes (Figure 4).
Figure 4.
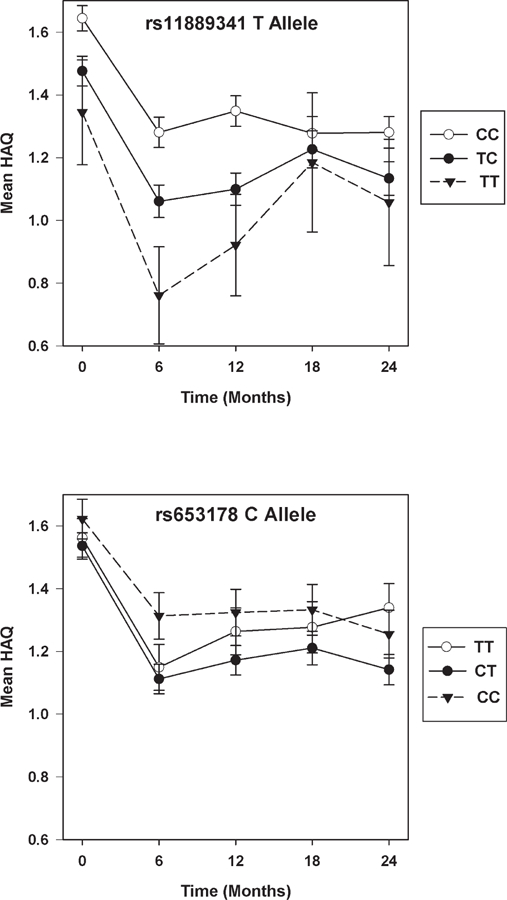
Significant genetic associations with HAQ scores over 6–24 months. Mean HAQ scores with standard error bars shown at each timepoint stratified by genotype. HAQ: Health Assessment Questionnaire.
A significant association was observed between the full wGRS (β = −0.08, p = 0.015) and HLA wGRS (β = −0.05, p = 0.026) and latent growth factor means of HAQ score slope 1. The non-HLA wGRS did not associate with HAQ scores (Supplementary Table 7, available online at jrheum.org).
DISCUSSION
Our study represents a comprehensive analysis of the association between RA susceptibility variants and disease severity in early active RA, using LME and latent growth curve modeling to evaluate whether risk variants predict radiograph, DAS28, and HAQ score progression. There are 3 main findings. The first and most important is that HLA susceptibility variants have a major influence on radiological progression with the main risk variants (*04:01 and amino acid polymorphisms Val11 and His13 in HLA-DRβ1) and an HLA wGRS associating with greater joint destruction. By contrast, no association was observed between non-HLA susceptibility variants and radiographic progression when evaluating these variants individually and cumulatively using a wGRS. Our results suggest that non-HLA susceptibility loci do not have a clinically relevant influence on joint damage in early, active RA, because CARDERA was powered to detect markers accounting for 3.4% of radiographic progression variance and identified known joint destruction predictors (RF, ACPA, SE30,31).
The associations between *04:01 and Val11 and radiographic damage have been reported in other RA populations, suggesting that the HLA associations in CARDERA are true-positive findings. Two large studies of established RA have reported associations between *04:01 and erosive damage or cross-sectional radiographic scores30,32. The largest report — a metaanalysis of 29 studies (3240 patients) — demonstrated a significant association between the SE and erosions; a subanalysis of European studies with 4-digit HLA-DRB1 data available demonstrated this association was only significant for the *04:01 allele. The effect of HLA-DRβ1 amino acid polymorphisms on radiographic damage has been reported in a conference abstract. In that analysis of patients with early RA from the Norfolk Arthritis Register (NOAR) and Early RA Study (ERAS), Val11 had a significant, SE-independent association with radiographic damage33,34. Although we were unable to establish whether Val11, His13, or the SE were driving their association with Larsen scores, when our results are considered alongside the NOAR/ERAS findings, it is likely that Val11 represents an independent predictor of joint destruction. This link has biological plausibility; Val11 is located in a peptide-binding groove and could mediate articular damage through influencing arthritogenic peptide presentation.
It is known that certain HLA variants predispose to ACPA35, which itself associates with erosive damage36. We found that *04:01, His13, and Val11 only associated with radiographic damage in ACPA-positive RA. This suggests that their effects are not mediated by ACPA; if they caused joint destruction by driving ACPA formation, they would not associate with radiographic progression in ACPA-positive patients because they all harbor these autoantibodies. It also indicates that these 3 variants only predict radiographic progression in ACPA-positive disease. This finding has importance for 2 reasons. First, it suggests that patients at a substantially increased risk of radiographic progression can be identified by combining ACPA and HLA status. In CARDERA, ACPA-positive patients carrying 2 Val11 copies had a mean DLarsen of 9.05 units (95% CI 5.94–12.15); ACPA-negative patients carrying no Val11 copies had a mean DLarsen of 2.77 units (95% CI 1.72–3.83). The clinical implication is that the former group may benefit from more intensive treatment than the latter group. Second, it indicates that as with gene-environment susceptibility factors19,37,38, ACPA-positive and ACPA-negative RA may also differ in their genetic risks for radiographic damage, further supporting the concept that they represent distinct disease entities.
Our second finding is that 2 SNP — rs11889341 (STAT4) and rs653178 (SH2B3-PTPN11) — associated with HAQ scores over 6–24 months. The relevance of these associations is, however, uncertain. Although the rs11889341 RA risk (T) allele associated with greater HAQ score increases at all timepoints, mean HAQ scores were lower in T allele carriers. Similarly, although the rs653178 RA risk (C) allele associated with improving HAQ scores at all timepoints except 24 months, mean HAQ scores were higher in CC homozygotes. Because of the uncertain clinical relevance of these associations, we did not seek to replicate them in alternative cohorts. A significant association was also seen between an HLA-wGRS and HAQ score over 0–6 months, although no associations were seen with individual HLA variants.
Our third finding is that RA susceptibility variants did not associate with disease activity. Although a significant association between an HLA-wGRS and baseline DAS28 was seen, this did not persist over time and no significant associations were seen with individual HLA-DRB1 alleles. To our knowledge, this is the first analysis of genetic associations with DAS28 after adjusting for treatment effects. Although several GWAS have attempted to identify genetic predictors of DAS28-defined anti-tumor necrosis factor responses18,39,40, only 1 definite association was identified (rs6427528 with etanercept response)18. One explanation for these largely negative findings is that DAS28 is not genetically determined. Heritability studies are needed to establish whether genetic markers could provide DAS28-defined prognostic data.
Our study has several unique features that strengthen existing evidence on genetic associations with RA severity. First, we examined genetic associations across many RA outcomes; previous studies have focused on radiographic damage alone2,3,5,6,7,41. Second, we evaluated genetic predictors in patients with early, active RA. Because the evidence supporting intensive combination therapy comes from RCT in such patients, our positive HLA findings have potential relevance to guiding decisions on treatment intensity. Third, to our knowledge, it is the first study of genetic associations with RA outcomes in an RCT; the standardized patient assessments increase the reliability of our findings. Finally, it evaluates all European RA susceptibility variants identified to date.
Our study has several other strengths. First, it was well powered to detect clinically relevant predictors of each outcome. Second, the inclusion of individuals with severely active disease meant any findings are relevant to patient populations in whom current UK RA guidelines are focused42. Third, the randomization to treatment groups ensured treatment effects could be adjusted for. Fourth, high levels of radiographic progression were observed (39% of patients had clinically relevant 2-yearly Larsen score increases of ≥ 4 units that is comparable to observational study data in early RA43), further optimizing the study’s power. Finally, our use of repeated-measures latent GCM meant we evaluated predictors of DAS28/HAQ scores at specific timepoints in an individual’s disease course; in contrast, previous studies of DAS28/HAQ score predictors have mainly evaluated associations with these outcomes only at the study endpoint44,45,46,47.
There are also several limitations to our study. First, we were unable to exclude genetic associations of a small effect size, with our sample size being substantially smaller than the GWAS metaanalyses undertaken to identify susceptibility loci. However, such associations would be of too small an effect to influence clinical decision making. Second, disease outcomes were only evaluated over 24 months; genetic variants could exert their effects over longer time periods. Third, we did not replicate previously validated associations between TRAF148, IL2RA6, and C5orf3049 and radiographic progression. One probable explanation for this is that CARDERA is composed of patients with early, active RA, in contrast to the observational studies reporting these associations. Fourth, there was evidence of a modest correlation between DAS28 and HAQ. The Pearson r correlation coefficient comparing the 24-month change in DAS28 and HAQ scores was 0.51 (p < 0.001); to a certain extent, the analyses of genetic associations with DAS28 and HAQ were therefore not independent. Finally, imputed HLA data were used that could not be internally validated (because of an absence of tissue typing data). However, the imputation method used (SNP2HLA) has been extensively validated16,50 and has documented high accuracy at imputing ImmunoChip genotypes (97% and 99% at imputing 4-digit HLA alleles and amino acids, respectively) in 918 individuals from the 1958 Birth Cohort16. Additionally, *04:01, His13, and Val11 had high INFO scores (0.8764, 0.9925, and 0.9810, respectively), indicating that their imputation was robust.
Further work is required to characterize the genetic architecture of radiological progression in RA. Our study has demonstrated that while HLA susceptibility variants have an important effect on joint destruction, non-HLA susceptibility variants do not associate with this trait in early, active RA. We consider that further candidate gene studies are unlikely to adequately characterize this trait and the metaanalysis of GWAS of radiographic progression is required, as undertaken for RA susceptibility. With large sample sizes, such an analysis would have sufficient power to detect salient non-HLA variants and to determine which HLA amino acids have SE-independent effects. Although any identified loci are likely to have a small effect size, if used in combination with known clinical predictors such as ACPA, they could deliver clinically meaningful prognostic data, enabling stratified medicine in RA. Additionally, defining which loci drive articular damage could identify novel therapeutic targets facilitating drug discovery.
Supplementary Material
Acknowledgments
Supported by Arthritis Research UK (Grant Reference Number 19739 to ICS) and the National Institute for Health Research (NIHR; Clinical Lectureship to ICS). We also acknowledge support from the NIHR Biomedical Research Centre at Guy’s and St. Thomas’ National Health Service Foundation Trust in partnership with King’s College London.
Footnotes
ONLINE SUPPLEMENT
Supplementary data for this article are available online at jrheum.org.
Contributor Information
Ian C. Scott, Department of Medical and Molecular Genetics, and Academic Department of Rheumatology, Centre for Molecular and Cellular Biology of Inflammation, King’s College London, and Guy’s Hospital.
Frühling Rijsdijk, SGDP Centre, Institute of Psychiatry, King’s College London.
Jemma Walker, Department of Medical and Molecular Genetics, King’s College London, and Guy’s Hospital.
Jelmar Quist, Department of Medical and Molecular Genetics, King’s College London, and Guy’s Hospital.
Sarah L. Spain, Department of Medical and Molecular Genetics, King’s College London, and Guy’s Hospital.
Rachael Tan, Academic Department of Rheumatology, Centre for Molecular and Cellular Biology of Inflammation, King’s College London.
Sophia Steer, Department of Rheumatology, King’s College Hospital.
Yukinori Okada, Department of Human Genetics and Disease Diversity, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, and Laboratory for Statistical Analysis, RIKEN Center for Integrative Medical Sciences.
Soumya Raychaudhuri, Division of Genetics, Brigham and Women’s Hospital, Harvard Medical School.
Andrew P. Cope, Academic Department of Rheumatology, Centre for Molecular and Cellular Biology of Inflammation, King’s College London.
Cathryn M. Lewis, Department of Medical and Molecular Genetics, and SGDP Centre, Institute of Psychiatry, King’s College London, and Guy’s Hospital.
REFERENCES
- 1.Knevel R, Gröndal G, Huizinga TW, Visser AW, Jónsson H, Víkingsson A, et al. Genetic predisposition of the severity of joint destruction in rheumatoid arthritis: a population-based study. Ann Rheum Dis 2012;71:707–9. [DOI] [PubMed] [Google Scholar]
- 2.van Steenbergen HW, Tsonaka R, Huizinga TW, le Cessie S, van der Helm-van Mil AM. Predicting the severity of joint damage in rheumatoid arthritis; the contribution of genetic factors. Ann Rheum Dis 2015;74:876–82. [DOI] [PubMed] [Google Scholar]
- 3.Knevel R, de Rooy DP, Gregersen PK, Lindqvist E, Wilson AG, Grondal G, et al. Studying associations between variants in TRAF1-C5 and TNFAIP3-OLIG3 and the progression of joint destruction in rheumatoid arthritis in multiple cohorts. Ann Rheum Dis 2012;71:1753–5. [DOI] [PubMed] [Google Scholar]
- 4.Marinou I, Maxwell JR, Wilson AG. Genetic influences modulating the radiological severity of rheumatoid arthritis. Ann Rheum Dis 2010;69:476–82. [DOI] [PubMed] [Google Scholar]
- 5.de Rooy DP, Zhernakova A, Tsonaka R, Willemze A, Kurreeman BA, Trynka G, et al. A genetic variant in the region of MMP-9 is associated with serum levels and progression of joint damage in rheumatoid arthritis. Ann Rheum Dis 2014;73:1163–9. [DOI] [PubMed] [Google Scholar]
- 6.Knevel R, de Rooy DP, Zhernakova A, Gröndal G, Krabben A, Steinsson K, et al. Association of variants in IL2RA with progression of joint destruction in rheumatoid arthritis. Arthritis Rheum 2013;65:1684–93. [DOI] [PubMed] [Google Scholar]
- 7.Krabben A, Wilson AG, de Rooy DP, Zhernakova A, Brouwer E, Lindqvist E, et al. Association of genetic variants in the IL4 and IL4R genes with the severity of joint damage in rheumatoid arthritis: a study in seven cohorts. Arthritis Rheum 2013;65:3051–7. [DOI] [PubMed] [Google Scholar]
- 8.Boers M, Verhoeven AC, Markusse HM, van de Laar MA, Westhovens R, van Denderen JC, et al. Randomised comparison of combined step-down prednisolone, methotrexate and sulphasalazine with sulphasalazine alone in early rheumatoid arthritis. Lancet 1997;350:309–18. [DOI] [PubMed] [Google Scholar]
- 9.Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, van Zeben D, Kerstens PJ, Hazes JM, et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): A randomized, controlled trial. Arthritis Rheum 2008;58 Suppl 2:S126–35. [DOI] [PubMed] [Google Scholar]
- 10.Grigor C, Capell H, Stirling A, McMahon AD, Lock P, Vallance R, et al. Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet 2004;364:263–9. [DOI] [PubMed] [Google Scholar]
- 11.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012;44:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy EH, Smith CM, Farewell V, Walker D, Hassell A, Chau L, et al. ; CARDERA (Combination Anti-Rheumatic Drugs in Early Rheumatoid Arhritis) Trial Group. Factorial randomised controlled trial of glucocorticoids and combination disease modifying drugs in early rheumatoid arthritis. Ann Rheum Dis 2008;67:656–63. [DOI] [PubMed] [Google Scholar]
- 14.Scott DL, Choy E. Effect of anakinra (soluble interleukin-1 receptor antagonist) as combination therapy: second UK combination therapy in early rheumatoid arthritis King’s College Lindon; [Internet. Accessed April 15, 2015] Available from: www.isrctn.com/ISRCTN15819795?q=Effect%20of%20anakinra%20(soluble%20interleukin-1%20receptor%20antagonist)%20as%20combination%20therapy:%20second%20UK%20combination%20therapy%20in%20early%20rheumatoid%20arthritis%20&filters=&sort=&offset=1&totalResults=1&page=1&pageSize=10&searchType=basic-search [Google Scholar]
- 15.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet 2013;14:661–73. [DOI] [PubMed] [Google Scholar]
- 16.Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 2013;8:e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown WM, Pierce J, Hilner JE, Perdue LH, Lohman K, Li L, et al. ; Type 1 Diabetes Genetics Consortium. Overview of the MHC fine mapping data. Diabetes Obes Metab 2009;11 Suppl 1:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui J, Stahl EA, Saevarsdottir S, Miceli C, Diogo D, Trynka G, et al. Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet 2013;9:e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 2012;44:1336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karlson EW, Chibnik LB, Kraft P, Cui J, Keenan BT, Ding B, et al. Cumulative association of 22 genetic variants with seropositive rheumatoid arthritis risk. Ann Rheum Dis 2010;69:1077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarwood A, Han B, Raychaudhuri S, Bowes J, Lunt M, Pappas DA, et al. A weighted genetic risk score using all known susceptibility variants to estimate rheumatoid arthritis risk. Ann Rheum Dis 2015;74:170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Y, Logan HL, Glueck DH, Muller KE. Selecting a sample size for studies with repeated measures. BMC Med Res Methodol 2013;13:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higgins JPT, Green S; The Cochrane Collaboration. Cochrane handbook for systematic reviews of interventions [Internet. Accessed April 9, 2015] Available from: handbook.cochrane.org/chapter_16/16_1_2_general_principles_for_dealing_with_missing_data.htm
- 24.Beasley TM, Erickson S, Allison DB. Rank-based inverse normal transformations are increasingly used, but are they merited? Behav Genet 2009;39:580–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crouch DJ, Goddard GH, Lewis CM. REGENT: a risk assessment and classification algorithm for genetic and environmental factors. Eur J Hum Genet 2013;21:109–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott IC, Seegobin SD, Steer S, Tan R, Forabosco P, Hinks A, et al. Predicting the risk of rheumatoid arthritis and its age of onset through modelling genetic risk variants with smoking. PLoS Genet 2013;9:e1003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–50. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205–13. [DOI] [PubMed] [Google Scholar]
- 30.Gorman JD, Lum RF, Chen JJ, Suarez-Almazor ME, Thomson G, Criswell LA. Impact of shared epitope genotype and ethnicity on erosive disease: a meta-analysis of 3,240 rheumatoid arthritis patients. Arthritis Rheum 2004;50:400–12. [DOI] [PubMed] [Google Scholar]
- 31.van der Woude D, Syversen SW, van der Voort EI, Verpoort KN, Goll GL, van der Linden MP, et al. The ACPA isotype profile reflects long-term radiographic progression in rheumatoid arthritis. Ann Rheum Dis 2010;69:1110–6. [DOI] [PubMed] [Google Scholar]
- 32.Mewar D, Marinou I, Coote AL, Moore DJ, Akil M, Smillie D, et al. Association between radiographic severity of rheumatoid arthritis and shared epitope alleles: differing mechanisms of susceptibility and protection. Ann Rheum Dis 2008;67:980–3. [DOI] [PubMed] [Google Scholar]
- 33.Viatte S, Plant D, Han B, Fu B, Yarwood A, Thomson W, et al. OP0190 Personalized genetic medicine: amino acid positions 11, 71 and 74 in HLA-DRB1 predict disease severity, treatment response and mortality in rheumatoid arthritis; multi-centre prospective cohort studies. Ann Rheum Dis 2014;73 Suppl 2:134. [Google Scholar]
- 34.Viatte S, Plant D, Han B, Fu B, Yarwood A, Thomson W, et al. Abstract session: emerging concepts in genomics and genetics — personalized genetic medicine: amino acid positions 11, 71 and 74 in HLA-DRB1 predict disease severity, treatment response and mortality in rheumatoid arthritis EULAR; 2014. Congress Presentations [Internet. Accessed April 9, 2015] Available from: www.pathlms.com/eular/events/147/thumbnail_video_presentations/2538 [Google Scholar]
- 35.Scott IC, Steer S, Lewis CM, Cope AP. Precipitating and perpetuating factors of rheumatoid arthritis immunopathology: linking the triad of genetic predisposition, environmental risk factors and autoimmunity to disease pathogenesis. Best Pract Res Clin Rheumatol 2011;25:447–68. [DOI] [PubMed] [Google Scholar]
- 36.Seegobin SD, Ma MH, Dahanayake C, Cope AP, Scott DL, Lewis CM, et al. ACPA-positive and ACPA-negative rheumatoid arthritis differ in their requirements for combination DMARDs and corticosteroids: secondary analysis of a randomized controlled trial. Arthritis Res Ther 2014;16:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott IC, Tan R, Stahl D, Steer S, Lewis CM, Cope AP. The protective effect of alcohol on developing rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology 2013; 52:856–67. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, et al. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res Ther 2006;8:R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Batliwalla F, Li W, Lee A, Roubenoff R, Beckman E, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol Med 2008;14:575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG, et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum 2011;63:645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knevel R, Klein K, Somers K, Ospelt C, Houwing-Duistermaat JJ, van Nies JA, et al. Identification of a genetic variant for joint damage progression in autoantibody-positive rheumatoid arthritis. Ann Rheum Dis 2014;73:2038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deighton C, O’Mahony R, Tosh J, Turner C, Rudolf M; Guideline Development Group. Management of rheumatoid arthritis: summary of NICE guidance. BMJ 2009;338:b702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanmartí R, Gómez-Centeno A, Ercilla G, Larrosa M, Viñas O, Vazquez I, et al. Prognostic factors of radiographic progression in early rheumatoid arthritis: a two year prospective study after a structured therapeutic strategy using DMARDs and very low doses of glucocorticoids. Clin Rheumatol 2007;26:1111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jansen LM, van Schaardenburg D, van Der Horst-Bruinsma IE, Bezemer PD, Dijkmans BA. Predictors of functional status in patients with early rheumatoid arthritis. Ann Rheum Dis 2000;59:223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Combe B, Cantagrel A, Goupille P, Bozonnat MC, Sibilia J, Eliaou JF, et al. Predictive factors of 5-year health assessment questionnaire disability in early rheumatoid arthritis. J Rheumatol 2003;30:2344–9. [PubMed] [Google Scholar]
- 46.Wiles NJ, Dunn G, Barrett EM, Harrison BJ, Silman AJ, Symmons DP. One year followup variables predict disability 5 years after presentation with inflammatory polyarthritis with greater accuracy than at baseline. J Rheumatol 2000;27:2360–6. [PubMed] [Google Scholar]
- 47.Gremese E, Salaffi F, Bosello SL, Ciapetti A, Bobbio-Pallavicini F, Caporali R, et al. Very early rheumatoid arthritis as a predictor of remission: a multicentre real life prospective study. Ann Rheum Dis 2013;72:858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viatte S, Plant D, Lunt M, Fu B, Flynn E, Parker BJ, et al. Investigation of rheumatoid arthritis genetic susceptibility markers in the early rheumatoid arthritis study further replicates the TRAF1 association with radiological damage. J Rheumatol 2013;40:144–56. [DOI] [PubMed] [Google Scholar]
- 49.Teare MD, Knevel R, Morgan MD, Kleszcz A, Emery P, Moore DJ, et al. Allele-dose association of the C5orf30 rs26232 variant with joint damage in rheumatoid arthritis. Arthritis Rheum 2013;65:2555–61. [DOI] [PubMed] [Google Scholar]
- 50.Han B, Diogo D, Eyre S, Kallberg H, Zhernakova A, Bowes J, et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am J Hum Genet 2014;94:522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.