

Abstract
Iron is an essential requirement for the survival and virulence for bacteria. The bacterial ferrous iron transporter protein FeoB functions as a major iron transporter in prokaryotes and has an N-terminal domain (NFeoB) with homology to eukaryotic G-proteins. Its GTPase activity is required for ferrous ion uptake, making it a potential target for anti-virulence therapies. Here, two assay strategies relying on different spectroscopic readouts are described for the monitoring of NFeoB GTPase activity. The first one is the colorimetric-based platform which utilizes a malachite green reagent to monitor phosphate production from GTP hydrolysis. The change in absorbance directly relates to the GTPase activity of NFeoB. The assay was further improved by the addition of Tween-20 and miniaturized in a 384-well plate format with a 10 µL assay volume. The second format is a luminescence-based platform that measures the substrate (GTP) depletion by using a modified GTPase-Glo assay from Promega Corporation. In this platform, the luminescence signal correlates to the amount of GTP remaining, which allows for the direct calculation of GTP hydrolysis by NFeoB. The colorimetric platform was tested in a high throughput manner against a custom assembled library of approximately 2,000 small molecules and was found to be simple, cost-effective, and robust. Additionally, the luminescence-based platform demonstrated its capability as an orthogonal assay to monitor GTPase activity, providing a valid and convenient method to filter false hits. These two assay platforms are proven to offset the limitations of each platform while enhancing overall quality and success rates.
Keywords: NFeoB, GTPase, malachite green, inhibitor, luminescence, high throughput screening
INTRODUCTION
Iron is an essential requirement for the survival and virulence for nearly all bacteria1. To shuttle iron, bacteria commonly use iron transport proteins which allow them to obtain free or chelated iron from their environments. Among these, the ferrous iron transporter protein B (FeoB), which is a transmembrane protein, functions as a major iron transporter in prokaryotes, particularly in anoxic environments. Notably, FeoB contains a C-terminal transmembrane anchor and an N-terminal domain that closely mimic eukaryotic G-proteins2,3. The cytosolic G-protein domain is required for ferrous iron uptake in vivo, but it is not clear whether the GTPase activity is providing the energy for iron transport4, or is playing a role in regulating ferrous iron transport in response to cellular iron status5. Because FeoB plays a role in the virulence of some pathogenic bacteria and is widely conserved in most prokaryotes, the N-terminal cytosolic domain (NFeoB), presents a good target for therapeutic intervention. Finding new small molecules that selectively target the GTPase activity of NFeoB may disrupt iron transport sufficiently to block growth and virulence within the host. However, there are no reported high-throughput screening platforms for monitoring NFeoB activity, and there are no reported specific inhibitors of NFeoB activity. Thus, we sought to develop a new platform that is capable of identifying novel inhibitors of the GTPase activity of NFeoB in a high throughput manner.
As illustrated in Figure 1, NFeoB GTPase activity can be determined by either the production of phosphate or GDP, or by the depletion of GTP. Commonly, the detection of inorganic phosphate is performed with a traditional malachite green (MG)-based colorimetric assay6 (denoted as “Colorimetric platform”). While the colorimetric platform has been widely employed in compound screening, sensitivity and miniaturization have been the major bottleneck to advancing this technology into high-throughput screening (HTS) campaigns. The other significant drawback of utilizing the colorimetric platform is the assay interference due to the inherent spectroscopic characteristics of small compound libraries, resulting in false hits. Here, we describe two assay platforms to monitor the GTPase activity of NFeoB, which can be employed in a complementary manner to allow for a cost-effective primary screen, with a sensitive and robust counterscreen. First, optimization of a simple mix-and-read colorimetric platform in a 384-well format by tweaking buffer composition and miniaturization for robust HTS application are presented. This optimized platform was validated in a pilot screen of approximately 2,000 small molecules.
Figure 1. Assay principle of NFeoB GTPase Activity.
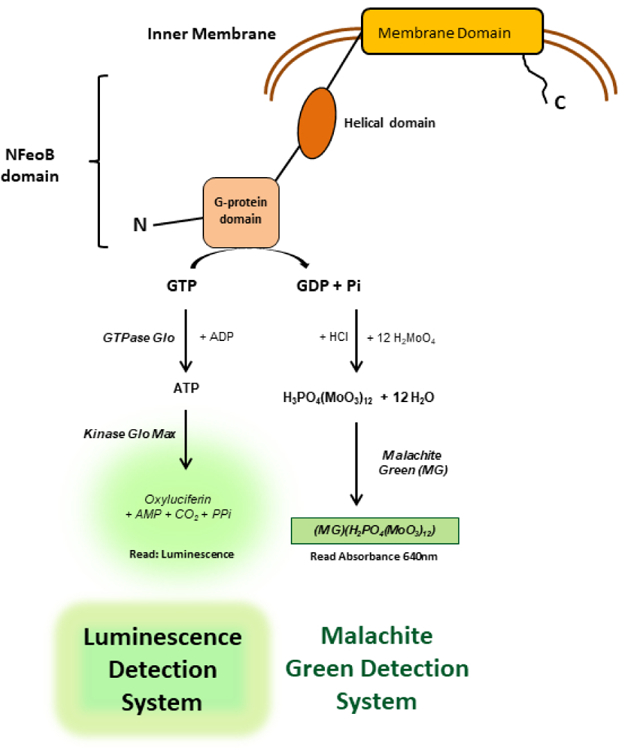
The amount of GTP hydrolyzed after the GTPase activity is measured by either a) production of free phosphate via complexation with malachite green reagent colorimetrically or b) depletion of GTP via coupled enzymatic reaction which generates luminescence. The malachite green signal is proportional to NFeoB activity while the luminescence signal is inversely proportional to NFeoB activity.
Next, we present a new luminescence-based platform that monitors the depletion of GTP, as an alternative to the colorimetric platform to resolve compound interference. The assay platform utilizes commercial GTPase Glo7 (denoted as “Luminescence platform” in Figure 1), originally designed to monitor GTP up to 5 µM. The luminescence readout is known to be much more sensitive than a colorimetric detection platform and a major benefit is its instant adaptation to biochemical assays. However, the protocol of GTPase Glo required revision to meet the dynamic range of the GTPase activity of NFeoB, and was subsequently validated for compound screening. This new luminescence platform demonstrated its potential to monitor GTPase activity of NFeoB with extended dynamic range. It also proved its capability as an orthogonal assay to discriminate false hits from the colorimetric platform. In summary, we present a convenient approach to the high-throughput investigation of compounds that modulate NFeoB GTPase activity, which will be useful in developing a new approach to combating bacterial pathogenesis.
MATERIALS AND METHODS
Reagents and Materials
All reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Ultrapure GTP (catalog# R0461), malachite green oxalate (Catalog# 41349–0250), and ammonium molybdate (catalog# 448540050) were purchased from Thermo Fisher Scientific (Waltham, MA).
NFeoB was expressed and prepared as described in Marlovits et al4 with modification. In detail, pET21a-based expression vectors containing the cytosolic N-terminus of FeoB from V. cholerae (NFeoB) with a hexa-histidine tag was generated and expressed in E. coli BL21(DE3) cells (Novagen; Madison, WI). Cells were grown in LB medium at 37 °C in a rotary shaker at 200 rpm and induced with 0.5 mM IPTG at 0.4~0.5 OD650. After overnight induction, the cells were harvested by centrifugation at 7,500 g for 5 min at 4 °C. For purification of NFeoB, the cell pellet was suspended in 2 mL of 25 mM Tris-Cl (pH 7.5) containing 100 mM NaCl and protease inhibitor cocktail (Complete Mini, EDTA free, Roche; Indianapolis, IN) per gram wet weight. The suspended cells were lysed by sonication and supernatant was collected by centrifugation at 30,000 g for 30 min at 4 °C. The histidine-tagged proteins were purified on a Ni2+-charged His-Bind resin (Novagen; Madison, WI) by using 20 mM imidazole, as the wash buffer, and gradually increasing imidazole (40 mM to 250 mM), as the elution buffer, in 25 mM Tris-Cl (pH 7.5) containing 100 mM NaCl, 10 % (v/v) glycerol, and 10 mM beta-mercaptoethanol.
Colorimetric GTPase activity assay
The GTPase activity assay was initially established using NFeoB purified from V. cholerae and GTP in a 96well plate (Nunc, catalog # 260836) with 40 µL assay volume. Enzymatic assays were carried out in an assay buffer (20 mM Tris-HCl pH 7.5, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 1 µg/mL BSA, and 0.01% Tween-20) using 1 µM NFeoB at ambient temperature. It is noted that 200 mM KCl was added to the assay buffer since the GTPase activity of FeoB is activated by potassium8. GTP was added to initiate the reaction, and reaction mixtures were allowed to incubate for 120 min, unless otherwise specified. Reactions were quenched using 150 µL malachite green detection solution (denoted as “MG” and prepared by dissolving 1 mM malachite green, 50 mM ammonium molybdate, 0.01% Tween-20 in 1 M HCl and filtering). The color was allowed to develop for 10 min before reading the absorbance at 640 nm on a Synergy H4 Multimode Plate Reader (BioTek, Winooski, VT). For comparison, a potassium phosphate standard curve was performed alongside the reactions, and the slope and y-intercept were used to calculate phosphate liberation in molar concentration. The assay was then miniaturized to 10 µL assay volume in clear 384-well microplates (Nunc catalog# 262160) and were quenched using 40 µL MG solution.
The room temperature stability of the assay was evaluated by varying enzymatic reaction time. In detail, the assay mixture after adding GTP was incubated for 0, 20, 60, 90, and 120 min before adding MG solution. Enzyme kinetics were evaluated by varying GTP concentrations and measuring the amount of phosphate generated after 120 min incubation. Unless otherwise stated, further assay validation was performed in an assay buffer using 1 µM NFeoB and 390 µM GTP for 120 min.
Validation of colorimetric GTPase activity assay
A full plate assay validation was conducted by plating columns 2–23 of 384-well plates with positive controls which contained NFeoB and GTP in an assay buffer, whereas plating columns 1 and 24 with negative control containing only substrate in an assay buffer. First, 5 µL of protein solution containing 2 µM NFeoB in assay buffer (columns 2–23) or assay buffer alone (columns 1 and 24) was dispensed using a Janus Automated Workstation (Perkin Elmer, Waltham, MA). Second, 100 nL of 100% DMSO was dispensed using an ECHO 550 Acoustic Dispenser (LabyCyte, Sunnyvale, CA). The reagents were allowed to mix for one hour at room temperature. Third, 5 µL of 780 µM GTP diluted in assay buffer, was dispensed with the MicroFlo Select Bulk Dispenser (BioTek, Winooski, VT). The assay was performed and read as described above while the final DMSO concentration was 1 percent. Inter-plate variation was measured as described above, but in two separate plates on two separate experiments, using freshly prepared assay mixtures.
Luminescence-based GTPase activity assay
A luminescence-based platform was developed by modifying GTPase Glo Assay (Promega Inc., Madison, WI, catalog# V7681) with Kinase Glo Max Assay (Promega, Madison, WI, catalog# V6071). The assays were carried out in white 384-well flat bottom low volume microplates (Corning #3825) with 10 µL assay volume. The dynamic range of GTPase Glo was investigated by varying the concentration of GTP up to 1 mM, diluted in an assay buffer according to manufacturer’s protocol. For the GTPase activity of NFeoB, enzymatic reaction was conducted as described above in a 10 µL assay volume with 1 µM NFeoB and 390 µM GTP. After 120 min incubation at room temperature, 10 µL modified GTP Glo I reagent containing 6 mM ADP was added to stop the reaction. This reaction was allowed to incubate for 60 min at room temperature, before adding 20 µL Kinase Glo Max detection solution. The DMSO tolerance was examined by varying the fractional percent of DMSO up to 10%. Percent conversion of GTP was calculated based on a control assay that included 390 µM ATP in an assay buffer.
Primary compound screening
Compound screening was performed using the colorimetric assay as described in Assay Validation above, but with compounds instead of DMSO. Over 2,000 bioactive compounds, which were custom-assembled were chosen and compounds were screened at a final concentration of either 10 µM or 100 µM with the DMSO concentration maintained at 0.1% or 1%. This unique collection is comprised of small molecules with known activities against over 100 kinases and other important therapeutic targets, representing a useful panel of compounds for developing leads against novel NTP-dependent proteins based on their off-target activities.
Hit validation
A total of 11 compounds selected from the primary screen were screened again at 100 µM using both colorimetric and luminescence-based GTPase activity assays. Compound interference with MG was also conducted by assaying them in the presence of 60 µM potassium phosphate instead of NFeoB and GTP in the colorimetric assay. Two compounds that exhibited comparable inhibition activity in both the colorimetric and the luminescence assays were chosen for dose response validation using colorimetric assay at final concentrations of 0, 25, 50, and 100 µM. Additionally, substrate dependent inhibition activity of those two hits were examined at 3 concentrations of GTP (50, 390, and 700 µM) using the colorimetric assay.
Statistical analysis
Initial velocities and turnover rate were calculated according to Eqs. (1) and (2), respectively, where vo is the initial velocity, vmax is the maximum velocity, [S] is the concentration of substrate, Km is the concentration of substrate at ½ the maximum velocity, kcat is the rate of turnover, and [ET] is the total enzyme concentration.
z’, a common statistical tool used to measure the quality of HTS assays,9 was calculated according to the Eq. (3), where σp and σn are the standard deviations of the positive and negative controls, respectively, and µp and µn are the means of the positive and negative controls, respectively.
Compound’s potency was calculated according to Eq. (4), Standard Curves, Four Parameter Logistic Curve from SigmaPlot 13.0, where Ai is the activity in the presence of inhibitor, Ao is the activity in the absence of the inhibitor, [I] is the concentration of the inhibitor, IC50 is the inhibitor concentration necessary to achieve 50% inhibition, and h is the Hill coefficient.
| Eq.(1) |
| Eq.(2) |
| Eq.(3) |
| Eq.(4) |
RESULTS AND DISCUSSION
Colorimetric GTPase activity assay optimization
Initially, the enzymatic assay was performed in a 40 µL reaction in 96-well plate, using an assay buffer without Tween-20. The reaction was quenched using 150 µL of MG, prepared in the absence of Tween-20. A phosphate standard curve was used to back calculate GTP conversion as a guide of assay performance. Since detergents are useful in reducing non-specific interactions and improving liquid handling performance, the effect of detergent on the colorimetric platform was first investigated by monitoring the absorbance spectra of MG with phosphate standard solution in the presence and absence of 0.01 % Tween-20. As shown in Figure 2A, the peak at 640 nm corresponding to a complex of MG with phosphate increased ~40 % in the presence of 0.01 % Tween-20, while the peak at 440 nm corresponding to MG itself was not affected. This result is accordance with the previous reports that the use of polyvinyl alcohol10 or detergents11 improved solubility of MG complex and ultimately signal stability. A dose response curve with phosphate standard in Figure 2B also showed the addition of Tween-20 did not alter the linearity up to 250 µM of phosphate in addition to the signal enhancement. Since compounds are often dissolved in DMSO, we examined whether the presence of 1% DMSO had any interference on the colorimetric platform. As shown in Figure 2C, the addition of 1 % DMSO did not alter the linearity even up to 500 µM of phosphate. These results supported that any assay additives did not alter the colorimetric assay protocol. Instead, the addition of Tween-20 enhanced the signal strength, improving the dynamic range.
Figure 2. Effect of assay additives on malachite green-based colorimetric platform.
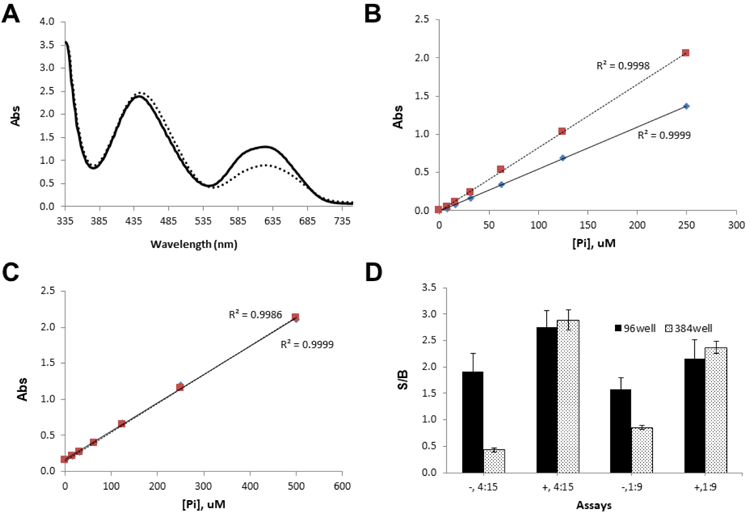
A) Absorbance spectra of malachite green reagent:phosphate complex in the presence (solid line) and absence (dotted line) of 0.01% Tween-20. Peaks at 440 and 640 nm correspond to MG:MoO7 complex and MG:MoO7:PO4 complex, respectively. B) Phosphate standard curves in the presence (dotted line) and absence (solid line) of 0.01% Tween-20. The r2 values at ~ 1 indicates best linearity for both regressions. Each data points represent averaged value of three replicates. C) Phosphate standard curves in the presence (solid line) and absence (dotted line) of 1% DMSO. The r2 values are ~1 for both regressions, indicating best linearity. Each data points represent averaged value of three replicates. D) Effect of Tween-20, assay format, and Pi/MG ratio on S/B. “−” and “+” signs represent the absence and presence of 0.01 % Tween-20, respectively. Ratios of 4:15 and 1:9 represent the volume ratio of enzymatic reaction to malachite green reagent, respectively. Both 96 (bar with solid fill) and 384well (bar with dotted fill) represent assay format.
Due to the relatively narrow dynamic range of the colorimetric platform, we explored whether the ratio of assay mixture to MG could expand the dynamic range further. Using a phosphate standard solution, it was observed that the ratio of phosphate standard-to-malachite-green (Pi/MG) altered the linear detection range but not the dynamic range. Conclusively, the linearity increased to 1 mM phosphate when a ratio of 1:9 was used, while the absorbance signal was linear between 0 to 250 µM phosphate at a 1:4 ratio (data not shown). Furthermore, we tested the effect of both Pi/MG and the presence of Tween-20 when the assay was miniaturized from 96 well to 384well format with assay volumes from 40 to 10 µL. As shown in Figure 2D, the signal-to-background ratio (S/B) was higher in the presence of Tween-20 regardless of Pi/MG. Also, the S/B was consistent regardless of miniaturization if the Tween-20 was present. However, the 384-well format with lower assay volume exhibited at least a 2x decreased S/B in the absence of Tween-20, indicating that Tween-20 is critical for assay miniaturization. Because NFeoB’s GTPase activity resulted in approximately 60 µM of phosphate in 2h, which corresponds to ~15 % conversion of GTP to GDP, a 1:4 ratio was chosen for further experiments in order to linearly detect phosphate generation.
The room temperature stability of the GTPase activity was evaluated by varying incubation time before the addition of MG in the presence of Tween-20. The phosphate production from NFeoB was proportional to assay time (Figure 3A) in the presence of Tween-20 at room temperature, resulting in an S/B ratio of approximately 2.0 after 2h of enzymatic reaction. This result demonstrated that GTPase activity of NFeoB is tolerant at room temperature at least up to 2h. Additionally, we confirmed that the presence of Tween-20 or DMSO in assay buffer did not alter the GTPase activity (data not shown). Based on these results, 0.01% of Tween-20 in both the MG detection reagent and assay mixture was chosen with enzymatic reaction time of 2h for further assay development.
Figure 3. Colorimetric GTPase activity assay performance.
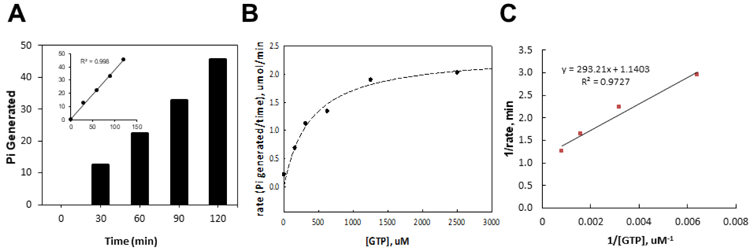
A) Reaction progress plot of NFeoB GTPase activity. The production of phosphate, [Pi], was calculated using the phosphate standard curve and the absorbance value measured at each time points. A subset graph with trendlines fit showed the linearity. B) Effect of substrate concentration on the rate of an enzyme–catalyzed reaction. Initial velocity data were fitted against corresponding substrate concentrations using a Hyperbola, Single Rectangular, 2-Parameter equation in SigmaPlot 13.0 (Systat Software, San Jose, CA). C) Lineweaver–Burk plot. The kinetic parameters are as follows: KmGTP = 357 ± 148 µM, Vmax = 0.877 ± 0.128 µM/min, kcat = 0.877 min-1.
Following the serial optimization and miniaturization, assay kinetics was next examined. As shown in Figures 3B and 3C, the Michaelis-Menten constant, Km, for GTP was found to be 357 ± 148 µM, and the Vmax was found to be 0.877 ± 0.128 µM/min (27.9 nmol/min/mg of NFeoB). The kcat was calculated to be 0.877 min−1. Substrate concentration at 390 µM was finalized for assay validation and compound screening.
To ensure that the assay is robust enough for HTS, a full plate validation using a 10 µL assay volume in a 384-well plate was conducted. Two validation experiments were conducted on different days with freshly prepared assay mixtures for each experiment. The averaged values for S/B, S/N, and z’ factor were 1.5, 53.3, and 0.69, respectively, proving the robustness of the assay. These results prove that the assay gives excellent separation between positive and negative controls across the plate (intra-plate) and between the plates (inter-plate), and is suitable for high-throughput compound screening.
Luminescence-based GTPase activity assay
Because many compounds may interfere with colorimetric absorbance detection, or may interact with the MG complex, it is important to have an orthogonal detection strategy to identify false positives. A luminescence-based platform was developed to visualize the remaining GTP as illustrated in Figure 1, where the luminescence signal is inversely proportional to the NFeoB’s GTPase activity. Promega’s GTPase-Glo assay was first considered, but it was found that this technology couldn’t be easily adapted to measure NFeoB’s GTPase activity because the detection limit of the Glo kit (5 µM GTP)7 was much lower than the NFeoB’s calculated Km for GTP. The GTPase-Glo assay measures GTP via two steps of enzymatic reactions as shown in Figure 4A and thus, it was feasible to alter the detection limit by adjusting the assay components in each step. In detail, the manufacture’s protocol was revised in both steps as follows; step 1) increased ADP concentration from 5 µM to 6 mM to ensure all remaining GTP from the enzymatic assay is converted to ATP and step 2) switched to Kinase Glo Max which has a detection limit of ATP up to 1 mM instead of using the original detection reagent, in order to convert all ATP to a luminescence signal. As shown in Figures 4B and 4C, the modified protocol successfully extended the detection limit of GTP by 200-fold; from 5 µM to 1 mM.
Figure 4. Luminescence-based detection platform for the monitoring NFeoB GTPase activity.

A) Assay principle of GTPase-Glo assay. GTPase-Glo reagent in step 1 includes ADP and nucloeside kinase to convert remaining GTP to ATP. Detection reagent in step 2 contains D-luciferin and luciferase to convert ATP to oxyluciferin and luminescence signal. Linear dependency of GTPase Glo using the original protocol (B) vs modified protocol (C). Dotted lines present trendlines fit to the entire range of doses while solid lines represent trendlines fit to best r2 values. D) DMSO tolerance by varying concentrations of DMSO up to 10 %.
Next, the modified GTP-Glo assay was validated for the detection of GTPase activity of NFeoB using the same enzymatic reaction protocol optimized in the colorimetric assay. The resulting luminescence signal corresponded to the depletion of ~ 65 µM GTP. This value is comparable with the value measured by the colorimetric platform, proving the reliability of the luminescence-based platform to monitor the NFeoB GTPase activity. The S/B and z’ calculated based on Pi production resulted in 13.31 and 0.83, supporting the potential of this assay to validate compounds’ activity. The tolerance test of this new detection platform against DMSO showed its stability up to 10 % of DMSO (Figure 4D). While further modification of this new luminescence-based strategy can improve the robustness of this platform to be suitable for the practical application to HTS campaigns, this assay still provides a convenient alternative to the colorimetric detection to monitor GTPase activity of NFeoB. While surface plasmon resonance (SPR) and thermal melt assay are good alternative methods to verify binding kinetics of the inhibitors or protein stability change by the inhibitors binding, they would not provide direct information on the GTP hydrolysis activity of FeoB. The distinctive advantages of the luminescence-based GTPase-Glo kit as an orthogonal assay over the other binding assays are: 1) it provides direct information on GTP hydrolysis activity; 2) it can be readily adapted to an HTS platform; and 3) its application allows for a miniaturized scale and produces less waste. In this regard, colorimetric and luminescence-based detection platforms were chosen for primary screening and hit validation, respectively.
Primary Screening and Hit validation
A primary screen was performed using the colorimetric detection platform with a custom-assembled library. The averaged S/B, S/N, and z’ factor were 1.90 ± 0.03, 55.1 ± 0.02, and 0.75 ± 0.04, respectively. Initial screening at 10 µM did not identify any potent hits. Rescreening at a final concentration of 100 µM identified 11 compounds over 35 % inhibition against the GTPase activity of NFeoB.
Eleven compounds were tested again in both the colorimetric and luminescence detection platforms in parallel at 100 µM. As summarized on Figure 5A, 6 compounds out of 11 failed to reproduce their activity in the colorimetric platform, indicating that they were false hits due to the systematic error. Among the remaining 5 compounds (Compounds 2, 6, 7, 8, and 11), three compounds were inactive in the luminescence assay, suggesting that they were false hits due to assay interference in primary screening. Two compounds (Compounds 2 and 8) were confirmed as reproducible using the colorimetric assay, while comparable activities in both platforms were observed.
Figure 5. Hit validation.
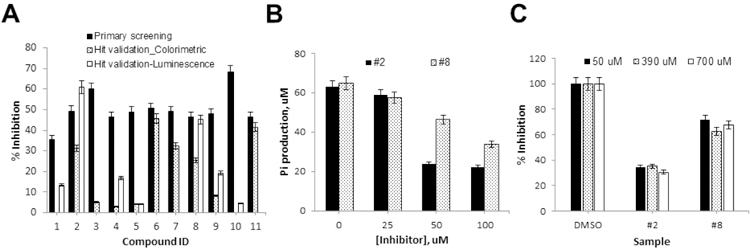
A) Activity profiles of 11 primary hits tested in colorimetric and luminescence platforms. Compounds’ % inhibition observed in primary screening was included to examine the reproducible activity. Compounds that showed reproducible activity in the colorimetric platform, and demonstrated comparable activity in the luminescence platform were selected for further validation. Data points represent mean ± standard deviation from 3 replicates. B) Dose-dependent response of compounds #2 and #8. Data points represent mean ± standard deviation from 3 replicates. C) Substrate dependency of compounds #2 and #8 examined at three different concentrations of GTP (50, 390, and 700 µM). Data points represent mean ± standard deviation from 3 replicates.
As shown in Figure 5B, a dose-response experiment using the colorimetric assay demonstrated that both compounds showed dose-dependent inhibition activities with IC50 values estimated at ~30 and 100 µM for Compounds 2 and 8, respectively. The mode of inhibition mechanism was examined at low, medium, and high substrate concentrations using the colorimetric detection platform. As shown in Figure 5C, their inhibitory activity was not altered at any substrate level, indicating that their inhibitory mechanism is non-competitive and both compounds inhibit the GTPase activity allosterically. CID1067700 and 3-methoxybenzamide, which are known inhibitors against Ras GTPases or FtsZ GTPase, respectively, did not inhibit the GTPase activity of VcNFeoB12,13 and data not shown). Non-hydrolyzable GTP-gamma-S would be used as a control compound, although it only represents a competitive inhibition14. The compounds 2 and 8 have been known as an inhibitor or an agonist of specific signaling cascades in humans. However, there are no reports regarding an effect of these compounds on a bacterial system. Further evaluation using in vitro cell-based and in vivo assays is undergoing.
To initially eliminate false hits due to colorimetric assay interference, a counter screen can be performed by replacing substrate and enzyme with 60 µM of phosphate standard. However, a different detection platform is more desirable to securely discriminate false hits considering the weak sensitivity and absorbance that may arise from the compounds themselves. Here, we have presented two detection platforms that measure either the product formation (MG) or substrate depletion (luminescence), that are individually optimized to a convenient mix-and-read protocol that can be read on a plate reader. The advantages of establishing two platforms for NFeoB relying on different detection strategies are 1) the same enzymatic assay protocol can be adapted instantly and 2) efficiently eliminate false hits due to spectroscopic interference. We demonstrated that the commercial GTPase-Glo assay can be amended for the biochemical GTPase assay where the Km of the substrate GTP concentration is much higher than its original detection limit. However, this luminescence-based detection platform still has some limitations for its application in HTS campaign: 1) the high cost associated with using two commercial kits, and 2) a low S/B of below 2. Nevertheless, this luminescence-based detection platform uses a simple mix-and-read format with endpoint detection, and proved its superior capability as an excellent alternative to the traditional colorimetric platform to assay the GTPase activity of NFeoB. This luminescence platform can be further optimized by adjusting quantity of essential components in each step to improve the S/B ratio, and the assay volume can be further miniaturized to save the cost. In conclusion, we have developed 2 complementary assay platforms to monitor the GTPase activity of NFeoB, and demonstrated their utility in an HTS application.
ACKNOWLEDGEMENTS
This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT), grant RP160657 to KND and by NIAID grant R01AI091957 to SMP.
REFERENCES
- 1.Payne SM; Mey AR; Wyckoff EE Vibrio Iron Transport: Evolutionary Adaptation to Life in Multiple Environments. Microbiol. Mol. Biol. Rev 2016, 80, 69–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilfoyle A; Maher MJ; Rapp M; et al. Structural Basis of GDP Release and Gating in G Protein Coupled Fe2+ Transport. EMBO J 2009, 28, 2677–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hantke K Is the Bacterial Ferrous Iron Transporter FeoB a Living Fossil? TRENDS Microbiol 2003, 11, 192–195. [DOI] [PubMed] [Google Scholar]
- 4.Marlovits TC; Haase W; Herrmann C; et al. The Membrane Protein FeoB Contains an Intramolecular G Protein Essential for Fe(II) Uptake in Bacteria. Proc. Natl. Acad. Sci 2002, 99, 16243–16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung K-W; Chang Y-W; Eng ET; et al. Structural Fold, Conservation and Fe(II) Binding of the Intracellular Domain of Prokaryote FeoB. J. Struct. Biol 2010, 170, 501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hohenwallner W; Wimmer E The Malachite Green Micromethod for the Determination of Inorganic Phosphate. Clin. Chim. Acta 1973, 45, 169–75. [DOI] [PubMed] [Google Scholar]
- 7. GTPase-Glo™ Assay Instructions for Use of Products V7681 and V7682.
- 8.Ash M-R; Guilfoyle A; Clarke RJ; et al. Potassium-Activated GTPase Reaction in the G Protein-Coupled Ferrous Iron Transporter B. J. Biol. Chem 2010, 285, 14594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang; Chung; Oldenburg. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 10.Cogan EB; Birrell GB; Griffith OH A Robotics-Based Automated Assay for Inorganic and Organic Phosphates. Anal. Biochem 1999, 271, 29–35. [DOI] [PubMed] [Google Scholar]
- 11.Baykov AA; Evtushenko OA; Avaeva SM A Malachite Green Procedure for Orthophosphate Determination and Its Use in Alkaline Phosphatase-Based Enzyme Immunoassay. Anal. Biochem 1988, 171, 266–70. [DOI] [PubMed] [Google Scholar]
- 12.Hong L; Guo Y; BasuRay S; et al. A Pan-GTPase Inhibitor as a Molecular Probe. PLoS One 2015, 10, e0134317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohashi Y; Chijiiwa Y; Suzuki K; et al. The Lethal Effect of a Benzamide Derivative, 3-Methoxybenzamide, Can Be Suppressed by Mutations within a Cell Division Gene, FtsZ, in Bacillus Subtilis. J. Bacteriol 1999, 181, 1348–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison C; Traynor JR The [35S]GTPgammaS Binding Assay: Approaches and Applications in Pharmacology. Life Sci 2003, 74, 489–508. [DOI] [PubMed] [Google Scholar]