

Abstract
Klotho is considered as an anti-aging factor inducing insulin resistance and involved in type 2 diabetes. However, mechanisms by which klotho induces insulin resistance remain to be understood. Thus, in this study, we aimed to evaluate possible interference points of klotho with insulin signaling pathways in 3T3-L1 adipocyte cells by focusing on phosphorylation levels of Akt, GSK3β, PFK-fβ3, and GLUT4 translocation. Differentiation of 3T3-L1 cells to the adipocyte-like cells were performed using specific differentiation kit and confirmed by mRNA expression assay of PPARγ using qRT-PCR, and Sudan black staining of lipid droplets. Then cells were co-treated with klotho and insulin. Expression and translocation of GLUT4 mRNA were evaluated using qRT-PCR and Alexa flour 488 conjugated GLUT4 antibody, respectively. P-Akt/Akt, p-GSK3β/GSK3β, and p-PFKfβ3/PFKfβ3 ratios were determined in insulin and klotho/insulin treated cells using western blot. Our result indicated that GLUT4 expression were decreased to 0.72 ± 0.16 fold in insulin treated cells, however it was calculated 1.12 ± 0.25 fold in klotho/insulin treated cells. In addition, klotho prevented GLUT4 membrane translocation by 27.2% in comparison with insulin-treated cells (P < 0.05). Interestingly, in insulin/klotho co-treated cells, phospho-levels of Akt, GSK3β, and PFKfβ3 proteins was decreased to 2.34 ± 0.14, 2.29 ± 0.63, and 1.95 ± 0.37 fold in comparison with the insulin cells, (P < 0.05). In conclusion, our study indicated that klotho induces insulin resistance in adipocytes possibly through prevention of GLUT4 translocation, and interfere with phosphorylation of Akt, GSK3β, and PFKf3β intracellular signaling mediators by insulin.
Keywords: Adipocyte, Insulin resistance, Klotho, 3T3-L1 cell
INTRODUCTION
It has been shown that klotho, as a new peptide discovered at the last decade, is involved in type 2 diabetes and insulin resistance (1,2,3). The klotho gene encodes a type I single-pass transmembrane protein with 1,014 amino acids. Its extracellular domain, named secreted klotho (s-klotho), is cleavable enzymatically and has 130 kDa, which can be detected in the body fluids such as blood and cerebrospinal fluid (4,5). The exact function of klotho is not well known; however, some in vitro and in vivo reports have n shown its relationship with insulin/glucose metabolism. It has been shown that an impaired glucose metabolism in obese mice was correlated with decreased klotho expression (6,7). Klotho-deficient mice kl/kl manifested with reduced pancreatic insulin content and hypoinsulinemia, contradictory, these mice experience hypoglycemia because of insulin hypersensitivity (8,9).
In addition, many studies have shown that klotho can be considered as a significant humoral factor associated with type 2 diabetes (10). Analyses of glucose and insulin tolerance showed that klotho may have inhibitory effect on insulin activity, however, its interference molecular mechanisms have not elucidated, yet (2).
Interestingly, klotho-overexpressing mice are resistant to insulin but not diabetes (11). It appears essence of this resistance differ from pathologic forms presented in type 2 diabetes (12). Intracellular signaling of insulin is mediated by stimulation of its tyrosine kinase receptor and subsequent activation of insulin receptor substrate-1 (IRS-1), protein kinase B (PKB, also known as Akt), and glycogen synthase kinase 3 beta (GSK3β) (13). In adipose and skeletal muscle cells activation of this pathway translocate glucose transporter type 4 (GLUT4) to the cell membrane to provide a path for entering of glucose. In general, insulin resistance is associated with either decreased number of insulin receptor (IR) or defect in optimal activation of subsequent signaling mediators (14,15). Kurosu et al. suggested that in rat myoblast cell line (L6) klotho interferes with autophosphorylation of insulin and insulin-like growth factor (IGF1) receptors (2). In addition, they mentioned that klotho interference is not mediated by changing affinity of these receptors towards their ligands.
Adipose tissue, by secretion of leptin, resistin, adiponectin, IL1-β, and other inflammatory mediators, plays a critical role in regulation of metabolism, insulin resistance, and aging (12). Since, adipose tissue is one of the main targets of insulin, agents that intervene with insulin signaling pathway can induce insulin resistance. For example, retinol-binding protein 4 prevents phosphorylation of Akt, IRS-1, and extracellular signal-regulated kinase 1/2 in insulin signaling pathway and cause insulin resistance (16,17).
Although klotho induces insulin resistance in adipose tissue, mechanisms by which klotho interferes with insulin activities in adipocytes remains to be determined. Thus, in this study, we aimed to evaluate possible interference mechanisms of klotho with insulin signaling pathways in adipocyte cells. Indeed, we evaluated the phosphorylation levels of Akt, GSK3β, and phosphofructokinase (PFK)-fβ3, GLUT4 translocation in klotho/insulin co-treated cells. According to our results, klotho interfere with downstream signaling of insulin receptor to induce mild insulin resistance.
MATERIAL AND METHODS
Chemicals and assay kits
3T3-L1 differentiation and Sudan black kits were purchased from Sigma-Aldrich Merck (St. Louis, Michigan, USA) company. PrimeScript RT reagent kit was from TAKARA company (TAKARA BIO INK Japan). Klotho was from R&D Systems (McKinley NE Minneapolis, USA). Rabbit anti-phospho-Akt (Ser-473), rabbit anti Akt, rabbit anti-phospho-GSK3β (Ser-9), rabbit anti GSK3β, and anti-rabbit IgG horseradish peroxidase (HRP)-linked antibody were purchased from Cell Signaling Technology company (Danvers, Massachusetts, USA). Rabbit anti-phospho-PFK-Fβ3 (Ser-461) and rabbit anti-PFK-Fβ3 were purchased from Abcam (USA). Glut4 fluorescence antibody (Alexa flour 488, Cat: SC-53566), mouse anti β-actin antibody, and rabbit anti-mouse IgG-HRP were provided from Santa Cruz Biotechnology company (Dallas, Texas, USA). Amersham Hyper film, polyvinylidene difluoride (PVDF), and enhanced chemiluminescence (ECL) start western blot analysis detection reagent were from Amersham Biosciences (Piscataway, Newjersey, USA) company.
Cell culture
Murine 3T3-L1 cell line (Cat: IBRC C10152) purchased from the Iranian Biological Resource Center, Tehran, I.R. Iran. According to the IBRC protocol, cells were cultured in Dulbecco’s modification of eagle medium (DMEM) high glucose (25 mM), 15% fetal bovine serum (FBS), and 1% penicillin/streptomycin. Cells incubation condition was humidified with air containing 5% CO2. All evaluation performed on cells between 15 and 35 passages (18,19,20).
Differentiation of 3T3-L1 cells to adipocyte
3T3-L1 cells, 3 × 105/well, cultured in the 6-well plate to reach 90% confluency. Then, cells differentiated into adipocytes using 3T3-L1 differentiation kit and according to the manufactures instructions. Briefly, 3T3-L1 cells culture medium was replaced with differentiation medium (DMEM/F12,1:1) with 10% FBS, insulin 1.5 µg/mL, dexamethasone 1 µM, IBMX 500 µM, and rosiglitazone 1 µM) and incubated for 3 days. Next, differentiation medium replaced with maintenance medium (1 µM of insulin to 1 mL of DMEM/F12 (1:1) with 10% FBS). According to the manufacturer’s instructions, maintenance medium changed every 2-3 days.
Sudan black staining
3T3L1 cells differentiation to the adipocytes confirmed by Sudan black staining of lipid droplets. Briefly, maintenance medium removed and then cells fixed in 10% formalin for 5 min. Afterward, cells washed with phosphate-buffered saline (PBS) three times. Then Sudan black (0.7g/100 mL propylene glycol) added to the cells for 7 min. Finally, cells rinsed with PBS and observed with light microscope.
Quantitative reverse transcription polymerase chain reaction
For further confirmation of 3T3-L1 differentiation, peroxisome proliferator-activated receptor gamma (PPAR-γ) gene expression levels also evaluated by semi-quantitative real-time polymerase chain reaction (qRT-PCR). As previously described (21), total RNA was extracted from 3 × 105 3T3-L1 differentiated cells using RNX-Plus solution (Sinaclon, I.R. Iran). Then, cDNA was synthesized and PPAR-γ amplicon was extended using specific primers (Table 1) following thermal stages: holding (1× at 95 °C for 10 min), cycling (40× at 95 °C for 10 sec and 55 °C for 30 sec), and melting (1× at 95 °C for 15 sec, 55 °C for 1 min, and 95 °C for 15 sec). Thermal conditions were provided using ABI 7500 Real-Time PCR System (Foster City, USA).
Table 1.
Primer sequences used for real time polymerase chain reaction.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| PPAR-γ | CTATGGAGTTCATGCTTGTG | GTACTGAGTACTGACATTTAT |
| GLUT4 | GTAACTTCATTGTCGGCATGG | AGCTGAGATCTGGTCAAACG |
| GAPDH | CGGAGTCAACGGATTTGGTC | AGCCTTCTCCATGGTGGTGA |
Klotho effects on GLUT4 gene expression also evaluated using qRT-PCR. Briefly, 3 × 105 3T3-L1 differentiated were subdivided to three groups: klotho-insulin group (800 pM klotho, incubated for 1 h and then added insulin 20 nM for 12 h), klotho group (800 pM klotho, incubated for 12 h) and insulin group (20 nM for 12 h). Then, GLUT4 amplicon was extended using abovementioned protocol. GLUT4 primer sequences are given in Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene expression used as an endogenous control. The results presented as relative gene expression level, 2-ΔΔCT.
Assessment of GLUT4 translocation using flow cytometry
3T3-L1 differentiated cells (3 × 105) pretreated with klotho (100-1000 pM, 1 h). Next, cells treated with GLUT4 fluorescence antibody and insulin (20 nM) for 30 min, simultaneously. Detection of plasma membrane embedded GLUT4 positive cells were evaluated as described previously (22). Briefly, cells were fixed in the dark for 20 min by adding 1% paraformaldehyde to each well. Cells harvested using a cell scraper and washed with the PBS three times. Finally, cells re-suspended in ice-cold PBS and subjected to the flow cytometer instrument (FACSscan caliber flow cytometer, USA) for immediate data acquisition.
Western blotting
3×105 3T3-L1 differentiated cells were pretreated with klotho (800 pM, 1 h) and then treated with insulin (20 nM insulin for 30 min). Then, as previously described (23), cells were harvested and lysed in ice-cold radio immunoprecipitation assay buffer, 0.5 mM phenylmethylsulfonyl fluoride, phosphatase inhibitor, and 0.5% protease mixture, as previously described (24). Total protein harvested by centrifuging of each sample at 10,000 g for 10 min at 4 °C. The protein concentrations determined by Bradford protein assay with a microplate reader at 595 nm. Total proteins (30 μg) from each sample separated at 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with 100 mV power. Separated proteins then transferred to the PVDF membrane at 300 mA power. Then, membranes blocked in 5% nonfat dry skim milk-Tween® 0.1% solution for 2 h. For the detection of protein of interes, membranes incubated with specific monoclonal antibodies, overnight at 4 °C. Next, membranes were washed three times with PBS-Tween® and then incubated with HRP-conjugated secondary anti-rabbit IgG antibody for 2 h. The desired protein blots visualized with ECL detection reagent after three times washing. The protein bands recorded on radiographic films. Densitometry of the examined protein bands were determined for each treatment and normalized to corresponding β-actin. Finally, results are presented as fold changes in comparison with the control group (25).
Statistical analysis
To evaluate and compare statistical significance of data, one-way analysis of variance (ANOVA) performed using Statistical Package for the Social Sciences (SPSS). P values < 0.05 were considered significant. All quantitative and semi-quantitative results presented as means ± SD.
RESULTS
Confirmation of 3T3-L1 cells differentiation to adipocytes
To evaluate klotho effects on insulin signaling pathways in adipocytes, first, we used 3T3-L1 cells and differentiated them to the adipocyte-like cells. Differentiation of cells was confirmed by evaluation of PPARγ mRNA expression using qRT-PCR. Our results indicated, after 7 days, expression of this gene was considerably induced in 3T3-L1 cells after differentiation (7.7 ± 0.67 folds) in comparison with intact cells (Fig. 1a).
Fig. 1.

Confirmation of 3T3-L1 differentiation to the adipocytes. (a) The PPARγ expression levels in 3T3-L1 cells (before and after differentiation to the adipocytes) were evaluated using qRT-PCR. Sudan black staining of (b) un- and (c) differentiated 3T3-L1 cells. Differentiation process was prolonged 7 days and then cells were fixed and stained with Sudan black dye. Arrows indicate some of stained lipid droplet in differentiated 3T3-L1 cells. PPARγ expression assay was performed three times in duplicated form (n = 6) and Sudan black staining was repeated three times (n = 3), independently. PPARγ, peroxisome proliferator-activated receptors gamma; qRT-PCR; quantitative real-time polymerase chain reaction.
Lipid droplets accumulate in 3T3-L1 cells after differentiation. Thus, to confirm further conversion of these cells to the adipocyte-like cells, we used Sudan black dye to stain these droplets, specifically. Our results showed that lipid droplets accumulated in these cells after 7 days of starting differentiation process (Fig. 1b).
Altogether, these results confirmed that 3T3-L1 cells successfully differentiated to the adipocyte-like cells. We used these differentiated cells to evaluate klotho effects on main intracellular mediators of insulin signaling pathways.
Effects of Klotho on membrane translocation of GLUT4 and insulin inhibitory effects on its mRNA expression
Following stimulation of adipocytes with insulin, localized GLUT4 translocated to the cell surface to facilitate glucose entrance into the cells (26). To evaluate klotho effects on GLUT4 translocation, differentiated 3T3-L1 cells pretreated with different concentrations of klotho for 1 h and then insulin added to cells for promotion of GLUT4 translocation. As mentioned in Fig. 2a and 2b, klotho in a dose-dependent manner decreased membrane translocation of GLUT4, which for 1000 pM was calculated to be 27.2% in comparison with positive control (P < 0.05). Taken together, these results indicated that klotho interfered with insulin effects on preventing GLUT4 translocation.
Fig. 2.

Klotho decreased GLUT4 plasma membrane translocation and prevented suppressory effects of insulin on mRNA expression of GLUT4 in differentiated 3T3-L1 cells. Differentiated 3T3-L1 cells were pretreated with different concentrations of klotho (100-1000 pM) for 1 h and then treated with insulin (20 nM) and anti-GLUT4 Alexa flour 488 antibody. Then, cells were fixed and GLUT4 positive cells were counted using a flow cytometer. (a) Flow cytometer charts indicate cell distribution and (b) percent of GLUT4 positive cells in comparison with control. Negative control just treated with anti-GLUT4 Alexa flour 488 antibody, but positive control also received insulin (20 nM). For gene expression assays, cells were treated with klotho (800 pM) and insulin (20 nM) for 12 h. Then total RNA was extracted and GLUT4 mRNA expression was evaluated using qRT-PCR. (c) Results were presented as fold of change of GLUT4 expression in comparison with untreated cells. GLUT4 translocation assay was repeated three times (n = 3), independently. GLUT4 mRNA expression analysis was performed three times in duplicated forms (n = 6).* P < 0.05 and ** P < 0.01 indicate significant differences compared to the positive control and control groups in parts b and c, respectively. GLUT4, glucose transporter type 4; qRT-PCR; quantitative real-time polymerase chain reaction.
Insulin suppressed GLUT4 mRNA expression in differentiated 3T3-L1 cells (27). To evaluate effects of klotho on GLUT4 mRNA expression, we treated 3T3-L1 differentiated cells with klotho/insulin and then measured gene expression of this transporter using qRT-PCR. Based on flow cytometry results (Fig. 2a and 2b), 800 pM of klotho was chosen to evaluate its effects on gene expression. According to our results (Fig. 2c), GLUT4 expression were calculated to be 0.72 ± 0.16 folds in insulin treated cells, which was increased to the 1.12 ± 0.25 folds in klotho/insulin treated cells, in comparison with control (P < 0.05). However, klotho per se has no significant effect on GLUT4 mRNA expression (P > 0.05). These results indicated that klotho decrease insulin-inhibitory effects on mRNA expression of GLUT4 transporter in these cells.
Phosphorylation of Akt, PFKfβ3, and GSK3β in differentiated 3T3-L1 cells
Akt, PFKfβ3, and GSK3β are considered as the main signaling mediators of insulin in adipocytes that control biochemical functions of these cells (28). To establish a timeline response of these mediators to the insulin in our differentiated 3T3-L1 cells, we treated these cells with this hormone in 0-120 min intervals and then evaluated phosphorylation levels using western blotting. As mentioned in Fig. 3, immediately after insulin treatment, p-Akt/Akt, p-GSK3β/GSK3β, and p-PFKfβ3/PFKfβ3 ratios elevated. This pattern continued until 30 min and semiquantified 4.98 ± 0.37, 4.87 ± 0.72, and 6.38 ± 0.75 folds in comparison with untreated cells (time of zero). After 30 min, these ratios returned to the base levels about 120 min. Altogether, these results indicated that insulin, in a time-dependent manner, increased phosphorylation levels of Akt, GSK3β, and PFKfβ3.
Fig. 3.

Insulin phosphorylated PFKFB3, Akt, and Gsk3β in differentiated 3T3-L1 cells at a dose dependent manner. These cells were treated with 20 nM insulin at a 0-120 min time intervals. Then, total protein was extracted and the levels of protein of interestwere determined using western blotting. These assay was repeated three times (n = 3). Bands were semiquantified using Image J software and protein phosphorylation compared with untreated groups of cells. * P < 0.05 and ** P < 0.01 indicate significant differences compared to the control (0 min). Akt, protein kinase B; GSK3, glycogen synthase kinase-3; PFK, phosphofructokinase.
Effects of Klotho on signaling extension of insulin in differentiated 3T3-L1 cells
It has been suggested that klotho induces insulin resistance with unknown mechanisms (12). To evaluate its possible interference with insulin intracellular signaling in differentiated 3T3-L1 cells, we co-treated these cells with klotho and insulin for 30 min. Then phosphorylation levels of Akt, GSK3β, and PFKfβ3 were determined using western blotting assays. According to our results presented in Fig. 4, insulin increased phosphorylation levels of these proteins to 3.97 ± 0.53, 3.41 ± 0.22, and 3.1 ± 0.25 folds of untreated cells, respectively. Interestingly, klotho per se did not show significant effects (P > 0.05). However, when co-treated with insulin, klotho attenuated the phospho-levels of Akt, GSK3β, and PFKfβ3 proteins to the 2.34 ± 0.14, 2.29 ± 0.63, and 1.95 ± 0.37 folds in comparison to untreated cells, which was significant statistically (P < 0.05). Taken together, these results indicated that klotho modulated and somehow decreased insulin signaling in differentiated 3T3-L1 cells.
Fig. 4.

Klotho decreased insulin signaling extension in differentiated 3T3-L1 cells. These cells were treated with either klotho or klotho/insulin for 30 min. Then, total protein was extracted and phosphorylation levels of protein of interes proteins were determined using western blot. * P < 0.05 and ** P < 0.01 indicate significant differences compared to the control (untreated cells).Akt, protein kinase B; GSK3, glycogen synthase kinase-3; PFK, phosphofructokinase.
DISCUSSION
It has been shown that klotho, through induction of a mild and non-pathologic insulin resistance increase longevity (12). Previous studies implicated that in rat myoblast cells, klotho specifically interferes with autophosphorylation of both insulin and IGF-1 receptors (2). However, its interference with insulin signaling in adipocytes has not elucidated yet. In the present study, we aimed to evaluate the effects of klotho on intracellular signaling of insulin in differentiated 3T3-L1 cells.
In general, our results indicated that klotho mildly, but significantly, prevented suppressor effects of insulin on GLUT4 expression. However, membrane embedding and translocation of this transporter reduced in klotho/insulin co-treated cells. In addition, herein we mentioned that klotho moderately decreased insulin-dependentphosphorylation of Akt, GSK3β, and PFKfβ3 in these cells, which indicated klotho somehow interrupted insulin signaling in adipocytes.
Previous study showed that insulin suppresses GLUT4 mRNA expression in 3T3-L1 mouse adipocytes (27). This suppression impairs glucose entrance into adipocytes and induces insulin resistance. Our result indicated that klotho, however, induces with this suppressor effect of insulin on GLUT4 expression and prevents its vigorous suppression. Interestingly, our results showed that klotho decreased insulin-induced translocation of GLUT4 in differentiated 3T3-L1 cells. These contradictory effects of klotho in GLUT4 expression and translocation may explain differences between klotho-induced insulin resistance and insulin-independent type 2 diabetes. Klotho-induced insulin resistance is not associated with GLUT4 depletion, but in insulin-independent type 2 diabetes its expression may be completely suppressed (27,29). It has been accepted that mild and non-pathologic insulin resistance leads to longevity (12), thus klotho through decrease of GLUT4 translocation, but not its expression, may involve in aging.
Our results showed that klotho attenuated insulin signaling extension in adipocytes through decreased phosphorylation of Akt, GSk3β, and PFKfβ3. Previous study by Kurosu et al. implicated that klotho may interfere with autophosphorylation of insulin and IGF1 receptors decrease IRS-1 activation to induce insulin resistance in rat myoblast cell line (L6) (2). Contradictory, these observations were argued later by Lurenzi and others which showed no direct effects of klotho on insulin resistance (30). They suggested this resistance might be secondary to the hypophosphatemia induced by klotho through coactivation of FGF23 receptor. Although, no independent receptor has been introduced for klotho (11), but it appears that membrane integrated klotho controls adipocyte differentiation and obesity through coactivation of FGF23 receptor (31,32,33). At least in 3T3-L1 cells, our results implicated that klotho through unknown mechanisms decrease insulin effects on signaling pathways (Fig. 5). Akt is a master regulator of GSK3β, PFKfβ3, and even GLUT4 translocation in adipocytes (13,34). Thus, it is possible that klotho’s effective point located in pathways activate Akt.
Fig. 5.
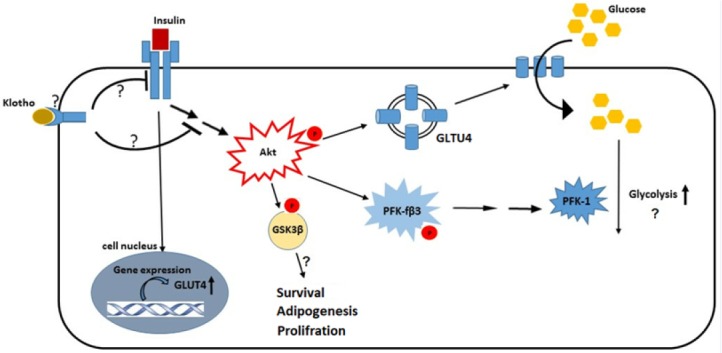
Schematic picture of possible interference mechanisms of klohto with insulin signaling pathway in 3T3-L1 differentiated cells. The exact interconnection points of klotho with insulin receptor or intracellular signaling mediators have not been elucidated; however, our results indicated inhibitory effects of klotho on Akt, GSK3β, and PFK-3β phosphorylation. These factors are involved in the adipocytes metabolism and survival and it appears that klotho affects most of this processes. Akt, protein kinase B; GSK3, glycogen synthase kinase-3; PFK, phosphofructokinase; GLUT4, glucose transporter type 4.
CONCLUSION
Taken together, our study indicated that klotho induced insulin resistance in adipocytes possibly through two mechanisms (Fig. 5): first, prevention of insulin effects on promotion of GLUT4 plasma membrane translocation, and second, attenuation of intracellular insulin signaling through main mediators, such as: Akt, GSK3β, and PFKf3β. These mediators exert insulin effects in adipocytes and control biochemical functions of these cells. Thus, klotho possibly has profound, but still unknown effects on these cells.
ACKNOWLEDGEMENTS
This work, submitted by Mohamad Hasannejad, was financially supported (Grant No. 396389) by the Vice Chancellery of Research of Isfahan University of Medical Sciences,Isfahan, I.R. Iran.
REFERENCES
- 1.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309(5742):1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson K, Nasca C, Aasly L, McEwen B, Rasgon N. Insulin resistance, an unmasked culprit in depressive disorders: Promises for interventions. Neuropharmacology. 2018;136(Pt B):327–334. doi: 10.1016/j.neuropharm.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, et al. Secreted Klotho protein in sera and CSF: implication for post‐translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565(1-3):143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- 5.Le Tilly E, Ong T, Abadie J, Guicheux J, Beck L, Vinatier C. Involvement of the anti-aging protein Klotho in chondrocyte autophagy and apoptosis during osteoarthritis. Osteoarthr Cartil. 2018;26:S20–S21. [Google Scholar]
- 6.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104(50):19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Razzaque MS. The role of Klotho in energy metabolism. Nat Rev Endocrinol. 2012;8(10):579–587. doi: 10.1038/nrendo.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utsugi T, Ohno T, Ohyama Y, Uchiyama T, Saito Y, Matsumura Y, et al. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism. 2000;49(9):1118–1123. doi: 10.1053/meta.2000.8606. [DOI] [PubMed] [Google Scholar]
- 9.Quarles LD. Fibroblast growth factor 23 and α-Klotho co-dependent and independent functions. Curr Opin Nephrol Hypertens. 2019;28(1):16–25. doi: 10.1097/MNH.0000000000000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inci A, Olmaz R, Sarı F, Coban M, Ellidag HY, Sarıkaya M. Increased oxidative stress in diabetic nephropathy and its relationship with soluble Klotho levels. Hippokratia. 2016;20(3):198–203. [PMC free article] [PubMed] [Google Scholar]
- 11.Kuro-o M. Klotho and aging. Biochim Biophys Acta. 2009;1790(10):1049–1058. doi: 10.1016/j.bbagen.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unger RH. Klotho-induced insulin resistance: a blessing in disguise? Nat Med. 2006;12(1):56–57. doi: 10.1038/nm0106-56. [DOI] [PubMed] [Google Scholar]
- 13.Rhodes CJ, White MF, Leahy JL, Kahn SE. Direct autocrine action of insulin on β-cells: does it make physiological sense? Diabetes. 2013;62(7):2157–2163. doi: 10.2337/db13-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, et al. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105(2):199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106(2):165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ost A, Danielsson A, Lidén M, Eriksson U, Nystrom FH, Stralfors P. Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEB J. 2007;21(13):3696–3704. doi: 10.1096/fj.07-8173com. [DOI] [PubMed] [Google Scholar]
- 17.Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing pro-inflammatory cytokines in macrophages through a JNK-and TLR4-dependent and retinol-independent mechanism. Mol Cell Biol. 2012;32(10):2010–2019. doi: 10.1128/MCB.06193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D, Dobrowolska G, Aicher LD, Chen M, Wright JH, Drueckes P, et al. Expression of the casein kinase 2 subunits in Chinese hamster ovary and 3T3 L1 cells provides information on the role of the enzyme in cell proliferation and the cell cycle. J Biol Chem. 1999;274(46):32988–32996. doi: 10.1074/jbc.274.46.32988. [DOI] [PubMed] [Google Scholar]
- 19.Eckel R, Fujimoto W, Brunzell J. Effect of in-vitro lifespan of 3T3-L1 cells on hormonal responsiveness of lipoprotein lipase activity. Int J Obes. 1981;5(6):571–577. [PubMed] [Google Scholar]
- 20.Tafuri SR. Troglitazone enhances differentiation, basal glucose uptake, and Glut1 protein levels in 3T3-L1 adipocytes. Endocrinology. 1996;137(11):4706–4712. doi: 10.1210/endo.137.11.8895337. [DOI] [PubMed] [Google Scholar]
- 21.Shahrestanaki MK, Aghaei M. A3 receptor agonist, Cl-IBMECA, potentiate glucose-induced insulin secretion from MIN6 insulinoma cells possibly through transient Ca2+ entry. Res Pharm Sci. 2019;14(2):107–114. doi: 10.4103/1735-5362.253357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshy S, Alizadeh P, Timchenko LT, Beeton C. Quantitative measurement of GLUT4 translocation to the plasma membrane by flow cytometry. J Vis Exp. 2010;45 doi: 10.3791/2429. pii: 2429. DOI: 10.3791/2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ataei N, Aghaei M, Panjehpour M. Cadmium induces progesterone receptor gene expression via activation of estrogen receptor in human ovarian cancer cells. Res Pharm Sci. 2018;13(6):493–499. doi: 10.4103/1735-5362.245961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahrestanaki MK, Arasi FP, Aghaei M. Adenosine protects pancreatic beta cells against apoptosis induced by endoplasmic reticulum stress. J Cell Biochem. 2018;120(5):7759–7770. doi: 10.1002/jcb.28050. [DOI] [PubMed] [Google Scholar]
- 25.Shahrestanaki MK, Arasi FP, Aghaei M. IPP-1 controls Akt/CREB phosphorylation extension in A2a adenosine receptor signaling cascade in MIN6 pancreatic β-cell line. Eur J Pharmacol. 2019;850:88–96. doi: 10.1016/j.ejphar.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Jaldin-Fincati JR, Pavarotti M, Frendo-Cumbo S, Bilan PJ, Klip A. Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol Metab. 2017;28(8):597–611. doi: 10.1016/j.tem.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Ma J, Nakagawa Y, Kojima I, Shibata H. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J Biol Chem. 2014;289(1):133–142. doi: 10.1074/jbc.M113.533240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, et al. Kinome screen identifies PFKFB3 and glucose metabolism as important regulators of the insulin/IGF-1 signalling pathway. J Biol Chem. 2015;290(43):25834–25846. doi: 10.1074/jbc.M115.658815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter and the insulin receptor challenge assumptions about insulin action and glucose homeostasis. J Biol Chem. 2003;278(36):33609–33612. doi: 10.1074/jbc.R300019200. [DOI] [PubMed] [Google Scholar]
- 30.Lorenzi O, Veyrat-Durebex C, Wollheim CB, Villemin P, Rohner-Jeanrenaud F, Zanchi A, et al. Evidence against a direct role of klotho in insulin resistance. Pflugers Arch. 2010;459(3):465–473. doi: 10.1007/s00424-009-0735-2. [DOI] [PubMed] [Google Scholar]
- 31.Chihara Y, Rakugi H, Ishikawa K, Ikushima M, Maekawa Y, Ohta J, et al. Klotho protein promotes adipocyte differentiation. Endocrinology. 2006;147(8):3835–3842. doi: 10.1210/en.2005-1529. [DOI] [PubMed] [Google Scholar]
- 32.Fan J, Sun Z. The antiaging gene klotho regulates proliferation and differentiation of adipose‐derived stem cells. Stem Cells. 2016;34(6):1615–1625. doi: 10.1002/stem.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samms RJ, Cheng CC, Kharitonenkov A, Gimeno RE, Adams AC. Overexpression of β-klotho in adipose tissue sensitizes male mice to endogenous FGF21 and provides protection from diet-induced obesity. Endocrinology. 2016;157(4):1467–1480. doi: 10.1210/en.2015-1722. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, et al. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19(6):4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]