

Abstract
Green fluorescent protein (GFP) has played an important role in biochemistry and cell biology as a reporter gene. It has been used to assess the potency of promoters for recombinant protein production. This investigation reveals evidences suggesting that the gfp GFP gene (EGFP) could be expressed without the promoter. In a study, a pLenti-F/GFP vector was constructed with the purpose to allow GFP expression in transduced cells but not in packaging cells; however, after transfecting the HEK293T cell line, GFP gene was expressed, compared to pLOX/CWgfp-transfected cells showed expression lag, lower levels and reduced percentage of GFP expression in the cells. This unexpected result could be due to auto transduction in packaging cell, possible retrotransposon activity in the cell line, possible contamination of pLenti-F/GFP with the pLOX/CWgfp and possible presence of a promoter within backbone of the vector. All the possibilities were ruled out. To exclude the possibility that a sequence within the region might act as a promoter, the fragment to be transfected was minimized to a region containing “from the start of the GFP gene to 5’LTR R”. The GFP gene was again expressed. Therefore, our findings suggest the EGFP does not need promoter for expression. This should appeal to the researchers designing GFP based assays to evaluate the potency of promoters, since possible aberrant expression may have a potential to influence on the results of a planned experiment.
Keywords: Green fluorescent proteins, Viral packaging, Promoterless
INTRODUCTION
Fluorescent proteins (FPs) are a homologous class of proteins containing a chromophore, which is composed of three amino acids, within their polypeptide which produce a visible wavelength upon emission. As a common research procedure, genes encoding engineered FPs are introduced into living cells to trace the location of the gene product and its dynamics using fluorescence microscopy (1). For a successful experiment involving FPs, several criteria should be met: the FP should be expressed efficiently while causing no toxicity for the cell, the signal should also be strong enough well above the level of auto fluorescence, it should have adequate photo stability necessary for the duration of the experiment and finally not to be influenced by environmental effects.
In case the FP is to be used as a fusion protein, it should not be oligomerized (2). Green FP (GFP) is one of the FPs first discovered by Shimomura et al. (3) as a companion protein to aequorin, a chemiluminescent protein produced in Aequorea jellyfish. During the last 20 years, GFP has promoted from a largely unknown protein to a commonly used tool in molecular biology, biochemistry, and pharmaceutical biotechnology. Its remarkable ability to create an efficiently emitting internal fluorophore is highly valuable (4). The GFP gene was first sequenced in 1992 (5). GFP is an 11-stranded β-barrel threaded by an α-helix which makes the axis of the cylinder. The chromophore is connected to the α-helix and is buried nearly completely in the center of the cylinder, called a β-can (6). Interestingly, GFP is resistant to heat, alkaline pH, organic and chaotropic salts, detergents, photo bleaching, and numerous proteases (7). Its mutants optimized for codon usage have been created for mammals (8), plants (9), yeast (10), and fungi (11). The stronger the promoters/enhancers, the higher are the emission intensity of the GFP (12). GFP can be expressed with sufficient efficiency in a cell-free in vitro translation system confirming the fact that the protein is capable of folding autonomously (13). Today, GFP has found its broad use in many organisms. Widely used as a real-time molecular and cellular analysis tool in bacteria, transgenic plants, fungi, and mammals. GFP is now considered to be a vital marker (14,15). As a general approach, the GFP gene is fused in frame with the gene of interest forming a chimera which upon expression can be detected using fluorescence microscopy. GFP is believed to rarely affect the protein activity or mobility, and is not generally toxic except in rare cases involving high GFP concentrations, for example, in retroviral packaging cells (16). GFP has been successfully used to indicate protease action, dimerization of transcription factors, calcium sensitivity, and quantitative monitoring of gene expression in organisms (1). However, so far, several pitfalls have been found with GFPs: the signal might be confounded by background fluorescence effect especially if it is not densely localized or highly expressed, and the chromophore formation occurs slowly post-translationally and requires oxygen (5). Thus far, there has been no report of a cryptic promoter within the GFP gene. Cryptic promoters differ from regular promoters in that they do not have a well-defined promoter initiation site sequence like TATAA box. They are independently regulated and the initiation mechanisms are not well-characterized. The existence of a cryptic promoter has been proven for some genes (17,18,19). In previous studies, as we were involved in designing and constructing a viral transfer vector which was supposed to express GFP in transduced cell line but not in packaging HEK293T cells, after transfection of the cells, we unexpectedly noticed that the promoterless GFP cassette was expressed (20,21). Therefore, the present study was launched to investigate the reason behind the observation. Our data demonstrate that the GFP gene can be expressed without any promoter.
MATERIALS AND METHODS
The HEK293T cell line
The HEK293T cell line was provided by Pasteur Institute of Iran, Tehran, I.R. Iran. The cells were maintained at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, Missouri state, USA) containing 100 U/mL of penicillin and 100 mg/mL streptomycin, and supplemented with 10% fetal calf serum (Sigma-Aldrich, Missouri state, USA).
Vector constructs
pLenti-final/GFP (pLenti-F/GFP)
We designed a new lenti transfer vector. The back bone of this vector was pLenti4-GW/H1/TO-laminshRNA (Invitrogen, California, USA). The vector was modified for the purpose of expressing GFP in transduced cells with no expression in packaging cells. In this vector, the CMV promoter (PCMV) was positioned between R and U5 in 5′LTR on the opposite strand. The position of the GFP gene was between 5’ and 3’ LTR and next to the 3’ LTR on the opposite strand and with the same direction as the promoter (Fig. 1A).
Fig. 1.
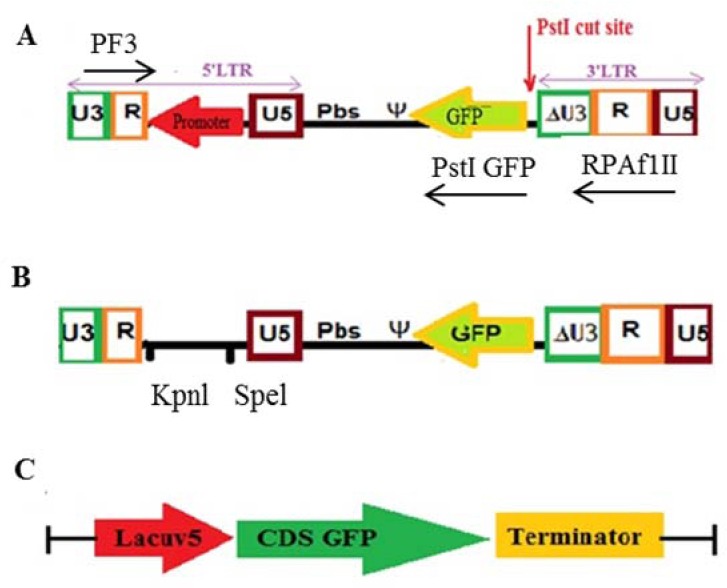
Vector constructs. (A) pLenti-F/GFP, (B) pLenti-pl/GFP, and (C) p-pro opti/GFP. GFP, Green florescent protein.
pLenti-promoterless/GFP (pLenti-pl/GFP)
During the construction of the pLenti-final/GFP, the promoter fragment was inserted in the final stage. Therefore, a stage before the completion of construction, the vector had no promoter (Fig. 1B).
p-Prokaryotic optimized/GFP (p-pro opti/GFP)
The GFP gene sequence was optimized according to E. coli expression system codon preferences, and placed under the control of a lacUV.5 promoter on pUC57 vector (GeneCust Luxembourg) (Fig. 1C).
Production of lentivirus in the HEK293T cell line
The HEK293T cell line was transfected with polyfect (Qiagen, Germany), following manufacturer’s instructions. In the first step, the HEK293T cell line was co-transfected with the PMD2G (envelope plasmid), psPAX2 plasmid (packaging plasmid), and pLenti-F/GFP (transfer plasmid). In parallel, the HEK293T cell line was co-transfected with the PMD2G, psPAX2, and pLOX/CWgfp plasmids as the positive control. After overnight transfection, the cells were analyzed for the expression of GFP using fluorescence microscopy (Nikon Inverted Microscope, Japan) and flow cytometry (FACS, Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA) and the medium was replaced with fresh DMEM + 10% FBS + penicillin/streptomycin. The cells were incubated at 37 °C, 5% CO2. After 24, 48, and 72 hrs, culture media were harvested and pooled and then fresh media added. The pooled media now contains the lentiviral particles.
Vector digestion and self-ligation to rule out contamination with pLOX/CWgfp
The maps of pLOX/CWgfp, pLenti-F/GFP, and pLenti-promoterless/GFP (pLenti-pl/GFP) were evaluated. Two restriction enzymes EcoRI and PstI were selected producing different digestion patterns in the vectors. The pLenti-F/GFP, pLenti-pl/GFP, and pLOX/CWgfp were digested separately with EcoRI and PstI.
The products of pLenti-F/GFP and pLenti-pl/GFP digestion with EcoRI were run on 1% agarose gel. The fragments of both plasmids were extracted from gel by DNA extraction kit according to the company’s instructions (QIAGEN, Germany). The extracted products were self-ligated by T4 DNA ligase (fermentas, Walthman, Massachusetts, USA) and transformed into E.coli strain TOP10F competented with CaCl2, and then cultured in LB agar medium containing ampicillin. After overnight culture, some colonies were picked up and cultured in LB broth medium containing ampicillin. After 16 h, plasmids were extracted (Bioneer, Korea) and used to transfect the HEK293T cell line.
Vector linearization for separation of the CDS of green florescent protein from plasmid backbone
The pLenti-F/GFP and pLenti-pl/GFP plasmids were linearized at the start of GFP gene using PstI (Fig. 1A).The linearized plasmids were then treated by calf intestinal alkaline phosphatase (CIAP), following the company’s instruction (Invitrogen, California, USA), in order to prevent self-ligation. The CIAP treated products were analyzed by agarose gel (1%) electrophoresis followed by gel extraction (gel extraction kit, QIAGEN, Hilden, Germany).
Amplification of the regions from 5’LTR R to 3’LTR R and from the GFP gene to 5’LTR R was conducted. A fragment spanning 5’LTR R to 3’LTR R containing the GFP gene was amplified from each of the pLenti-F/GFP and pLenti-pl/GFP vectors. Forward and reverse primers (forward 3 and reverse AflII) are shown in Table 1. Another fragment encompassing from GFP to 5’LTR was amplified from both vectors using forward 3 and reverse PstI-GFP primers (Table 1 and Fig. 1A). Agarose gel (1%) electrophoresis was carried out to analyze these polymerase chain reaction (PCR) products and gel extraction was performed.
Table 1.
The primer sequences used in this study.
| Primers | Primer sequences |
|---|---|
| F3 (Forward and revers primers) | 5´ATA CTT AAG CCT CAA TAA AGC TTG CCT TGA GTG CTT CAG GTA CC 3´ |
| RAflII (Forward and reverse primers) | 5´TGA GGC TTA AGC AGT GGG TTC C 3´ |
| RPstI GFP (Forward and reverse primers) | 5´ATT CTG CAG GTC GCC ACC ATG GTG AGC 3´ |
Transfection of the HEK293T cells by different fragments and constructs
The HEK293T cells was transfected with the pLenti-F/GFP, pLenti-pl/GFP, linearized pLenti-F/GFP, linearized pLenti-pl/GFP, newly extracted pLenti-F/GFP (plasmid without any contamination with pLOX/CWgfp), newly extracted pLenti-pl/GFP (plasmid without any contamination with pLOX/CWgfp), 5’LTR R to 3’LTR R fragment, the gfp gene to 5’LTR R fragment, p-prokaryotic optimized/GFP (p-pro opti/GFP), and pLOX/CWgfp in separate experiments. After overnight transfection, the cells were analyzed for the expression of GFP using fluorescence microscopy and flowcytometry.
RESULTS
Production of lentivirus in the HEK293T cell line
In the process of lentivirus production, in the HEK293T cell line transfected with the three plasmids including psPAX2, pLenti-F/GFP, and PMD2G, the GFP gene was found to be expressed although with delay (after 20 h) and reduced intensity compared to the HEK293T cells transfected with pLOX/CWgfp, psPAX2, and PMD2G (test cells) which became green after 8 h (Fig. 2). Furthermore, the percentage of the cells expressing GFP was 55% for the former vs. 90% for the latter (Fig. 3).
Fig. 2.
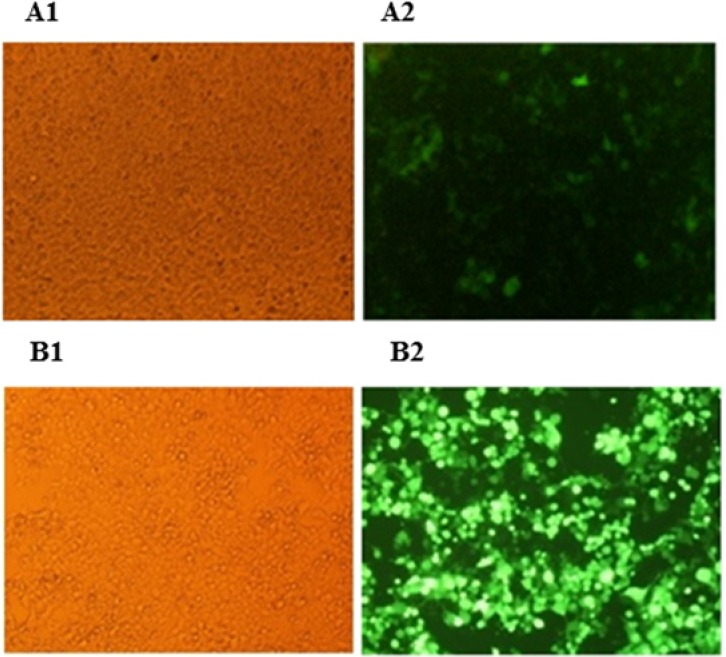
Green florescent protein expression in the HEK293T cells under fluorescence microscopy. (A1 and A2) The HEK293T cells were co-transfected with PMD2G, psPAX2 and pLenti-F/GFP; (B1 and B2) the HEK293T cells were co-transfected with PMD2G, psPAX2 and pLOX/CWgfp. GFP, Green florescent protein.
Fig. 3.
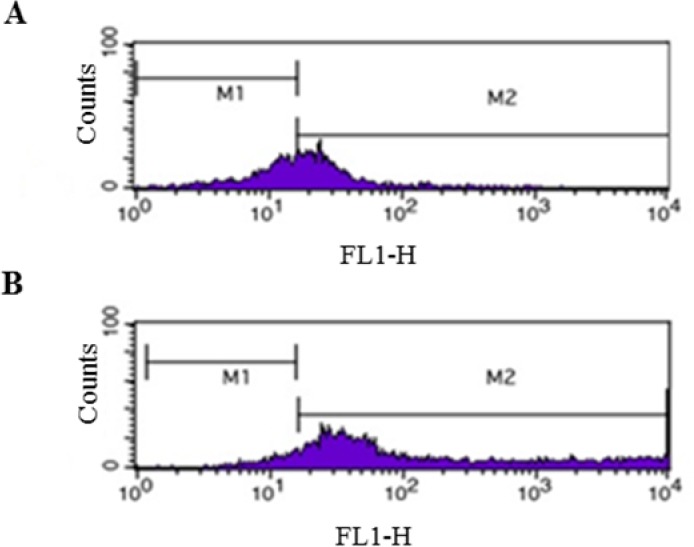
The flow cytometry of GFP expression in HEK293T cells. (A) The HEK293T cells were co-transfected with PMD2G, psPAX2 and pLenti-F/GFP; (B) The HEK293T cells were co-transfected with PMD2G, psPAX2 and pLOX/CWgfp (control). The horizontal axis shows the fluorescence intensity of GFP. GFP, Green florescent protein.
Transfection of HEK293T cell line with pLenti-F/GFP plasmid and pLenti-pl/GFP
To assess the GFP expression pattern, HEK293T cells were transfected with pLenti-F/GFP and pLenti-pl/GFP, separately. After 20 h, the cells became green, with the similar pattern of expression described above under fluorescence microscopy.
Vector digestion and self-ligation to rule out contamination with pLOX/CWgfp
PLenti-F/GFP and pLenti-pl/GFP have no restriction site for EcoRI but one for PstI, pLOX/CWgfp has four restriction sites for EcoRI and three for PstI. Following digestion of pLenti-F/GFP and pLenti-pl/GFP by EcoRI and ruling out the possible existence of pLOX/CWgfp within them, the HEK293T cell line was transfected with the resultant purified plasmid which yielded the same mentioned expression pattern.
Transfection of HEK293T cell line with p-pro opti/GFP
After 24 h of transfection of HEK293T cells with p-pro opti/GFP, GFP was not expressed.
Vector linearization for separation the CDS of GFP from plasmid backbone
After separation the CDS of GFP from plasmid backbone, HEK293T cells were transfected with GFP CDS. After 20 h, the cells became green, with the similar pattern of expression described above under fluorescence microscopy.
DISCUSSION
Fluorescent labeling is an important tool in biological studies nowadays. While a number of chemical dyes are available to label biological specimens, they have to be added exogenously and are generally incompatible with living systems. However, genetically encoded fluorophore such as members of the GFP family provide a solution for the purpose (22).
We demonstrated here evidence suggesting that the GFP gene (EGFP) can be expressed in promoterless cassettes. During an investigation, we designed and constructed a retroviral transfer vector for the purpose of expressing GFP in transduced cells and with no expression in packaging cells (20). In the production of lentivirus, we expected the GFP not to be expressed in the packaging cells due to the absence of any promoter in the upstream of GFP. However, after 20 h, in the HEK293T cell line transfected with the three plasmids including psPAX2, pLenti-F/GFP, and PMD2G, the GFP gene was found to be expressed although with delay and reduced intensity compared to the HEK293T cells transfected with pLOX/CWgfp, psPAX2, and PMD2G which became green after 8 h. Furthermore, the percentage of the cells expressing GFP was 55% for the former vs. 90% for the latter. To ensure the validity of the results, all the experiments were repeated and the same findings were obtained. Given the odd observation, the process of viral packaging was ceased and our investigations were focused on understanding the underlying reason.
To explain the result, we hypothesized either of several reasons could be the case:
-
a)
Auto transduction of the cells with the viruses produced during the process of packaging. In this view, the viral RNA could be reverse-transcribed, imported into the nucleus and stably integrated into the host genome. Since the promoter will now be positioned upstream of the GFP gene, it can be expressed (23,24). This might also explain the observed delayed expression of the gene after 20 h. To rule out the hypothesis, the cells were transfected with the pLenti-F/GFP plasmid alone to ensure the virus not to be produced. We found that GFP was expressed again, with the same pattern of delayed, decreased intensity and the same percentage of the expressing cells.
-
b)
Retrotransposase re-activation. In cell lines especially those derived from cancer cells, possible epigenetic modifications that inactivate retrotransposases can be removed leading to retrotransposase activity. If retrotransposons are active in the cell line, the promoter can be placed upstream of the GFP gene and results in its expression. To investigate the notion, the HEK293T cell line was transfected with the pLenti-pl/GFP vector, the promoterless version of our vector. Again, under fluorescence microscopy the cells became green, with the similar pattern of expression described above. Therefore, we conclude retrotransposition plays no role in the expression of the GFP.
-
c)
Random contamination of our plasmids with the pLOX/CWgfp vector. To rule out this possibility, all the vectors, pLenti-F/GFP, pLenti-pl/GFP, and pLOX/CWgfp, were digested separately with two restriction enzymes, EcoRI and PstI, which generated plasmid-specific digestion patterns as expected. Following digestion of pLenti-F/GFP and pLenti-pl/GFP by EcoRI and ruling out the possible existence of pLOX/CWgfp within them, the HEK293T cell line was transfected with the resultant purified plasmid which yielded the same mentioned expression pattern. From this point on, all the tests were performed with either the newly extracted pLenti-F/GFP or pLenti-pl/GFP plasmids.
-
d)
Possibility of a promoter sequence within the transfer vector backbone. For ruling out the likelihood, the pLenti-F/GFP and pLenti-pl/GFP were digested with PstI resulting in vector linearization at the adjacent of the GFP gene to separate the GFP gene from plasmid backbone at its upstream (Fig. 1A). After transfection of the HEK293T cell line with the linearized vector, the same GFP expression pattern was achieved.
Having ruled out all the logical possibilities mentioned above, we concluded that the GFP gene could be expressed without promoter. To confirm the idea, a shortened fragment of the vector spanning 5’LTR R to 3’LTR R fragment was amplified by PCR and transfected into the HEK293T cell line. Even under the circumstances, the GFP gene was found to be expressed. We repeated the experiment by producing a fragment from start codon (ATG) of the GFP gene to 5’LTR R fragment by PCR which led to the same finding.
In a parallel experiment, the HEK293T cells were transfected with p-pro opti/GFP. Unlike our expectations, GFP was not expressed. This could largely be due to the fact that the GFP gene on this vector had been optimized for prokaryotic codon usage. Although its sequence was different from the native one, its open reading frame was the same. The disruption in the native GFP sequence can turn off GFP expression. Therefore, there must be a eukaryotic cryptic promoter sequence in CDS of GFP that is interrupted.
Cryptic promoters have homology to the promoter sequences. They might be distributed randomly throughout the genome and are capable of driving transcription (25).
However, no significant attention had been paid to cryptic promoters from reporter genes until 2008 when Vopalensky et al. reported a cryptic promoter activity in the firefly luciferase gene (19). Recently, several researchers have revised their published results after finding cryptic promoters or cryptic splicing sites in the genomic fragments that were claimed to bear an internal ribosome entry sites (26,27,29).
The results of our experiment strongly suggest the presence of a cryptic promoter within the GFP gene and advocate the notion that this reporter gene might not be ideal for some experiments.
In a study in order to prove the cryptic promoter activity of a firefly luciferase (FLuc) gene, the investigators removed the CMV IE promoter, positioned upstream of FLuc and GFP genes, from the pFG vector creating a promoterless variant pFG(-P) in which FLuc was followed by GFP. Based on the finding that a significant amount of GFP had been produced in cells transfected with promoterless pFG(-P) vector, they suggested that it was a strong evidence for the cryptic promoter activity within the FLuc gene (19). In the light of our new finding, their conclusion might not be completely valid. Interestingly, results of the studies launched to investigate whether gene editing tools had been acting based on GFP, as the reporter gene, might not be valid, either. For instance, in a research, a reporter plasmid was designed in which the PCMV had been positioned upstream of a monomeric RFP (mRFF), as a reporter gene, a programmable nuclease target site and the enhanced GFP gene. mRFP was constitutively expressed from the PCMV, whereas the functional GFP was not expressed because of its out-of-frame position under normal conditions. Thus, cells transfected with this reporter plasmid expressed mRFP but not GFP. However, in case of programmable nuclease activity, a double-strand break was made in the target sequence, which upon its repair by non-homologous end-joining mechanism, could occasionally lead to frameshift mutations that position GFP in frame (30). Such experiments relying on the expression of GFP may give rise to false positive results.
Although the data obtained in this study are valid enough, further investigation is required to determine the transcription initiation site of the cryptic promoter using such techniques as 3’RACE. To find the exact sequence of the cryptic promoter, transfection of the truncated GFP gene cassettes with poly A signal could address the issue.
CONCLUSION
Finding a cryptic promoter within the GFP gene should appeal to the researchers working in the field and encourage them, when possible, to investigate the possible presence of cryptic promoters in plasmid backbones as well as other reporter genes. Failing to take the fact into consideration could potentially lead to confounded results of the planned experiments due to aberrant expression induced by these cryptic promoter elements. In summary, we could demonstrate that GFP does not need promoter for expression and this fact can negatively affect its role as a reporter gene to evaluate the power of promoters.
ACKNOWLEDGMENTS
The content of this paper was extracted from the MSc thesis submitted by Zahra Mohammdi which was financially supported (Grant No. 293249) by the Research council of Isfahan University of Medical Sciences, Isfahan, I.R. Iran.
REFERENCES
- 1.Arnold E, Jacobo-Molina A, Nanni RG, Williams RL, Lu X, Ding J, et al. Structure of HIV-1 reverse transcriptase/DNA complex at 7 Å resolution showing active site locations. Nature. 1992;357:85–89. doi: 10.1038/357085a0. [DOI] [PubMed] [Google Scholar]
- 2.Baird SD, Turcotte M, Korneluk RG, Holcik M. Searching for IRES. 2006;12(10):1755–1785. doi: 10.1261/rna.157806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu W, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J. Engineered GFP as a vital reporter in plants. Current Biol. 1996;6(3):325–330. doi: 10.1016/s0960-9822(02)00483-9. [DOI] [PubMed] [Google Scholar]
- 4.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90(3):1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 5.Chudakov DM, Lukyanov S, Lukyanov KA. Fluorescent proteins as a toolkit for in vivo imaging. Trends in biotechnology. 2005;23(12):605–613. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Cormack BP, Bertram G, Egerton M, Gow NA, Falkow S, Brown AJ. Yeast-enhanced green fluorescent protein (yEGFP): a reporter of gene expression in Candida albicans. Microbiology. 1997;143(Pt 2):303–311. doi: 10.1099/00221287-143-2-303. [DOI] [PubMed] [Google Scholar]
- 7.Ehrmann MA, Scheyhing CH, Vogel RF. In vitro stability and expression of green fluorescent protein under high pressure conditions. Lett Appl Microbio. 2001;32(4):230–234. doi: 10.1046/j.1472-765x.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- 8.Guo Y, German TL, Schultz RD. A cryptic promoter in potato virus X vector interrupted plasmid construction. BMC Mol Bio. 2007;8:17–22. doi: 10.1186/1471-2199-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanazono Y, Yu JM, Dunbar CE, Emmons RV. Green fluorescent protein retroviral vectors: low titer and high recombination frequency suggest a selective disadvantage. Hum Gene Ther. 1997;8(11):1313–1319. doi: 10.1089/hum.1997.8.11-1313. [DOI] [PubMed] [Google Scholar]
- 10.Haseloff J, Siemering KR, Prasher DC, Hodge S. Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci U S A. 1997;94(6):2122–2127. doi: 10.1073/pnas.94.6.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikawa M, Yamada S, Nakanishi T, Okabe M. Green fluorescent protein (GFP) as a vital marker in mammals. Curr Top Dev Biol. 1999;44:1–20. doi: 10.1016/s0070-2153(08)60465-2. [DOI] [PubMed] [Google Scholar]
- 12.Jakab G, Droz E, Brigneti G, Baulcombe D, Malnoe P. Infectious in vivo and in vitro transcripts from a full-length cDNA clone of PVY-N605, a Swiss necrotic isolate of potato virus Y. J Gen Virol. 1997;78(Pt 12):3141–3145. doi: 10.1099/0022-1317-78-12-3141. [DOI] [PubMed] [Google Scholar]
- 13.Kahn TW, Beachy RN, Falk MM. Cell-free expression of a GFP fusion protein allows quantitation in vitro and in vivo. Curr Biol. 1997;7(4):R207–R208. doi: 10.1016/s0960-9822(06)00100-x. [DOI] [PubMed] [Google Scholar]
- 14.Kim H, Um E, Cho SR, Jung C, Kim H, Kim JS. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods. 2011;8(11):941–943. doi: 10.1038/nmeth.1733. [DOI] [PubMed] [Google Scholar]
- 15.Kozak M. Alternative ways to think about mRNA sequences and proteins that appear to promote internal initiation of translation. Gene. 2003;318:1–23. doi: 10.1016/s0378-1119(03)00774-1. [DOI] [PubMed] [Google Scholar]
- 16.Kozak M. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res. 2005;33(20):6593–6602. doi: 10.1093/nar/gki958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorang JM, Tuori RP, Martinez JP, Sawyer TL, Redman RS, Rollins JA, et al. Green fluorescent protein is lighting up fungal biology. Appl Environ Microbiol. 2001;67(5):1987–1994. doi: 10.1128/AEM.67.5.1987-1994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mancuso G, Rugarli EI. A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Bio. 2008;6:31–44. doi: 10.1186/1741-7007-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masek T, Vopalensky V, Horvath O, Vortelova L, Feketova Z, Pospisek M. Hepatitis C virus internal ribosome entry site initiates protein synthesis at the authentic initiation codon in yeast. J Gen Virol. 2007;88(Pt 7):1992–2002. doi: 10.1099/vir.0.82782-0. [DOI] [PubMed] [Google Scholar]
- 20.Mohammadi Z, Shariati L, Khanahmad H, Kolahdouz M, Kianpoor F, Ghanbari JA, et al. A lentiviral vector expressing desired gene only in transduced cells: an approach for suicide gene therapy. Mol Biotechnol. 2015;57(9):793–800. doi: 10.1007/s12033-015-9872-3. [DOI] [PubMed] [Google Scholar]
- 21.Pourzadegan F, Shariati L, Taghizadeh R, Khanahmad H, Mohammadi Z, Tabatabaiefar MA. Using intron splicing trick for preferential gene expression in transduced cells: an approach for suicide gene therapy. Cancer Gene Ther. 2016;23(1):7–12. doi: 10.1038/cgt.2015.57. [DOI] [PubMed] [Google Scholar]
- 22.Phillips GN., Jr Structure and dynamics of green fluorescent protein. Curr Opin Struct Biol. 1997;7(6):821–827. doi: 10.1016/s0959-440x(97)80153-4. [DOI] [PubMed] [Google Scholar]
- 23.Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- 24.Telesnitsky A, Goff SP. Reverse Transcriptase and the Generation of Retroviral DNA. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 25.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 26.Vopalensky V, Masek T, Horvath O, Vicenova B, Mokrejs M, Pospisek M. Firefly luciferase gene contains a cryptic promoter. RNA. 2008;14(9):1720–1729. doi: 10.1261/rna.831808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wahlfors J, Loimas S, Pasanen T, Hakkarainen T. Green fluorescent protein (GFP) fusion constructs in gene therapy research. Histochem Cell Biol. 2001;115(1):59–65. doi: 10.1007/s004180000219. [DOI] [PubMed] [Google Scholar]
- 28.Yang TT, Sinai P, Green G, Kitts PA, Chen YT, Lybarger L, et al. Improved fluorescence and dual color detection with enhanced blue and green variants of the green fluorescent protein. J Biol Chem. 1998;273(14):8212–8216. doi: 10.1074/jbc.273.14.8212. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3(12):906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 30.Zimmer M. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem Rev. 2002;102(3):759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]