

Abstract
Heat shock proteins (HSPs) are induced after haemorrhagic stroke, which includes subarachnoid haemorrhage (SAH) and intracerebral haemorrhage (ICH). Most of these proteins function as neuroprotective molecules to protect cerebral neurons from haemorrhagic stroke and as markers to indicate cellular stress or damage. The most widely studied HSPs in SAH are HSP70, haeme oxygenase‐1 (HO‐1), HSP20 and HSP27. The subsequent pathophysiological changes following SAH can be divided into two stages: early brain injury and delayed cerebral ischaemia, both of which determine the outcome for patients. Because the mechanisms of HSPs in SAH are being revealed and experimental models in animals are continually maturing, new agents targeting HSPs with limited side effects have been suggested to provide therapeutic potential. For instance, some pharmaceutical agents can block neuronal apoptosis signals or dilate cerebral vessels by modulating HSPs. HO‐1 and HSP70 are also critical topics for ICH research, which can be attributed to their involvement in pathophysiological mechanisms and therapeutic potential. However, the process of HO‐1 metabolism can be toxic owing to iron overload and the activation of succedent pathways, for example, the Fenton reaction and oxidative damage; the overall effect of HO‐1 in SAH and ICH tends to be protective and harmful, respectively, given the different pathophysiological changes in these two types of haemorrhagic stroke. In the present study, we focus on the current understanding of the role and therapeutic potential of HSPs involved in haemorrhagic stroke. Therefore, HSPs may be potential therapeutic targets, and new agents targeting HSPs are warranted.
Keywords: heat shock proteins, intracerebral haemorrhage, review, stroke, subarachnoid haemorrhage, therapeutic target
1. INTRODUCTION
Stroke, which is the second leading cause of fatality worldwide and the primary cause of permanent disability among adults, includes ischaemic stroke and haemorrhagic stroke.1, 2 The latter accounts for approximately 10%‐20% of all strokes yet, have a higher mortality rate vs the former.3, 4, 5, 6 Furthermore, subarachnoid haemorrhage (SAH) and intracerebral haemorrhage (ICH) constitute haemorrhagic strokes, with worldwide incidences of 9.1 and 24.6 per 100,000 person years respectively.4, 7, 8 Despite increasing attention on the prevention of stroke and new methods for treating intracranial haemorrhage, incidences of SAH and ICH and the case fatality rate of ICH have not decreased over time.7, 8, 9 Additionally, even if patients survive, more than half of haemorrhagic stroke survivors never recover enough brain function to live on their own, which is attributed to the pathophysiological processes following haemorrhage.1, 10, 11 Thus, it is necessary to explore the pathophysiological mechanisms of haemorrhagic stroke to seek potential treatments.
Heat shock proteins (HSPs), a family of evolutionarily conserved molecular chaperones that includes HSP90, HSP70, HSP60, HSP40 and small heat shock proteins (sHSPs), which have various functions in proteostasis, are defined by the presence of heat shock elements in their promoters.12, 13 These proteins participate in pathophysiological neurological mechanisms by regulating stress responses, mitigating apoptotic signals, stabilizing the cytoskeleton and shuttling damaged proteins for degradation via the ubiquitin‐proteasome system or autophagy (Table 1).12, 14, 15 Therefore, the functional roles of HSPs may be potential targets for haemorrhagic stroke therapeutics (Figure 1). To explore the explicit mechanisms of HSPs in haemorrhagic stroke, several experimental models in animals have been established over time.
Table 1.
HSPs involved in haemorrhagic stroke
| HSPs | Pathophysiological processes | Potential mechanisms | The induction location | Effect and roles | Related agents | Reference | |
|---|---|---|---|---|---|---|---|
| Subarachnoid haemorrhage | |||||||
| HSP70 (including GRP78) | Cerebral vasospasm, neural cell apoptosis, immunoreaction and inflammation | Refold protein, degrade damaged proteins, inhibit apoptosis, mediate BBB disruption and cell death via aberrant proteolysis | Bilateral neocortex, hippocampus, thalamus, septum, hypothalamus, caudoputamen and basal forebrain | Neuroprotective molecule, significant marker for cellular stress or damage; crucial predictor of poor prognosis; blood biomarker for the early differential diagnosis of haemorrhagic stroke and ischemic stroke | Geranylgeranylacetone, modified HSP70 proteins (eg TAT‐Hsp70), valproic acid, atorvastatin, | 12, 14, 15, 16, 17, 22, 24, 25, 28, 31, 33, 34, 35, 36, 37, 38, 40, 41, 50, 51, 52, 53, 54, 55, 56, 57, 58, 96, 97, 98 | |
| HO‐1 | Cerebral vasospasm, lipid peroxidation | Metabolize haeme, remove haeme and iron, diastolic vascular smooth musclea | Microglia and cerebral blood vessels | Possible neuroprotective moleculeb; marker of cellular stress and damage in infarcted regions | Nicaraven, argon, carnosol, ebselen and CGS26393, HO‐1 protein combined with protein transduction domains | 14, 39, 40, 61, 62, 63, 64, 65, 66, 70, 71, 99, 100, 101 | |
| HSP20 and HSP27 | Cerebral vasoconstriction, apoptosis pathway | Solubilize misfolded proteins and hinder their aggregation, suppress cell death signaling and protect neurons against ischemic injury | Astrocytes in the ischaemic zone and the ischaemic penumbra | Important molecules in cerebral vasoconstriction without ATP necessarily involved in the function | AZX100 | 14, 28, 40, 73, 74, 75, 76, 77, 78 | |
| HSP90 | Apoptosis, inflammation, and BBB destruction | Stabilize and promote the function of P2X7 receptor, which is abundant in the nervous system and is associated with the pathophysiological process of inflammation and oxidative stress in EBI | Microglia and neurons of the hippocampus | Neurotoxic factor in the development of EBI | 17‐allylamino‐17‐demethoxygeldanamycin, A438079 | 79, 80, 81, 82, 83, 84, 85 | |
| Intracerebral haemorrhage | |||||||
| HO‐1 | Inflammation, oxidative stress and cytotoxicity | Increase oxidative stress, accelerate the accumulation of iron overload, promote inflammation and increase secondary injury | Microglia and cerebral blood vessels | Possible harmful moleculeb | Haemin, nicotinamide mononucleotides, minocycline | 14, 20, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 134, 144, 145, 146 | |
| HSP70 | GRP75 | Neural cell apoptosis, immunoreaction | Inhibit inflammation and neuronal apoptosis | Mainly located in mitochondria, reduced after intracerebral haemorrhage | Neuroprotective molecule | Minocycline, geranylgeranylacetone, geldanamycin, Di Dang Tang | 36, 135, 136, 137, 138, 139, 140, 147, 148, 149, 150 |
| GRP78 | Neural cell apoptosis | Inhibit neuronal apoptosis | Mainly located in endoplasmic reticulum | Neuroprotective molecule | |||
Abbreviations: BBB, blood‐brain barrier; EBI, early brain injury; GRP75, glucose‐regulated protein 75; GRP78, glucose‐regulated protein 78; HO‐1, haeme oxygenase‐1; HSP, heat shock protein; TAT, N‐terminal transactivator of transcription.
Carbon monoxide, one of the haeme metabolite, can up‐regulate soluble guanylyl cyclase, which contributes to cyclic guanosine monophosphate accumulation and subsequently leads to vascular smooth muscle relaxation.
The role of HO‐1 is still controversial, the overall effect of HO‐1 in SAH and ICH tends to be protective and harmful respectively.
Figure 1.
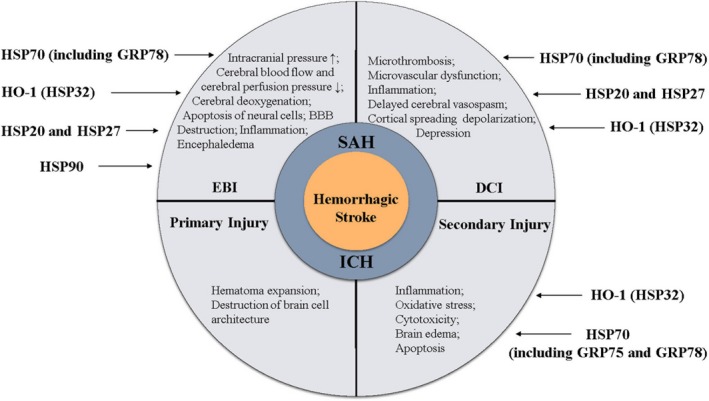
Schematic representation of the relationship between heat shock proteins and haemorrhagic stroke
Among all the SAH models, endovascular perforation in small animals is optimal, as the subsequent responses including changes in haemodynamics and the sudden elevation of intracranial pressure parallel human aneurysmal SAH better than autologous blood injection.16, 17 However, some studies have demonstrated that the double haemorrhage model, in which SAH is induced a second time 48 hours after the first autologous blood injection, directly corresponds with the time course of cerebral vasospasm in humans.18, 19 For ICH, disease models made by collagenase or autologous blood injection are both options, and each has its own pros and cons.20 With the gradual improvement of animal models, we have discovered more HSP functions in SAH and ICH. In the present review, we will summarize and integrate research findings on the role of HSPs in haemorrhagic stroke and their therapeutic potential.
2. THE PATHOPHYSIOLOGY OF SAH
Subarachnoid haemorrhage constitutes 5%‐10% of all strokes and is mainly (85%‐98.9%) caused by ruptured aneurysms.21, 22, 23, 24, 25, 26 Typically, the basic pathophysiological changes during SAH can be divided into two periods: early brain injury (EBI) and delayed cerebral ischaemia (DCI).27 The former takes place within the first 3 days of SAH, while the latter usually appears 3‐4 days after the aneurysm rupture and usually resolves after 12‐14 days.22, 28 The concept of DCI, which consists of delayed cerebral vasospasm and subsequent microvascular dysfunction, microthrombosis (especially in the cortical vessels), inflammation, cortical spreading depolarization, depression and ischaemia, was proposed over 150 years ago.22 Cerebral vasospasm was once considered to be a prerequisite for DCI; however, DCI can develop without angiographic vasospasms, and vasospasms may also end with no ischaemic lesions.29, 30 The EBI stage was proposed more recently and involves elevated intracranial pressure, decreased cerebral blood flow and cerebral perfusion pressure, cerebral deoxygenation, apoptosis of neural cells, destruction of the blood‐brain barrier (BBB), inflammation and encephaledema.16, 22, 24, 31, 32 The pathophysiological changes occurring during EBI might induce the development of DCI, but this has not yet been confirmed.22, 33, 34
2.1. The role of HSP70 plays in SAH
Present in all cells, constitutively active HSP70 is usually called HSC70 or HSP73, while inducible HSP70 protein, commonly called HSP72 or HSP70, is barely detectable in normal brain tissues but becomes the abundant protein when heat shock, ischaemia or other injuries occur.14 As the most widely studied HSP, HSP70 is known to be involved in protein refolding and the degradation of damaged proteins.24, 35, 36 HSP70 is involved in the development of SAH by participating in EBI and DCI, and acts as a critical molecule in cerebral vasospasm, neural cell apoptosis, immunoreaction and inflammation.14, 16, 17, 28, 36, 37, 38 HSP70 might be a potential target for SAH treatment and management, especially because no strategies explicitly targeting SAH have been developed yet.39 Additionally, HSP70 is a significant marker for cellular stress or damage and is also a crucial predictor of poor prognosis as it indicates the severity and extent of brain injury during SAH.16, 17, 31, 36, 38, 40 Moreover, because it is expressed at lower levels in haemorrhagic stroke than in ischaemic stroke, HSP70 can be used as a blood biomarker for the early differential diagnosis of haemorrhagic stroke and ischaemic stroke.36, 41
2.1.1. HSP70 and early brain injury
Although many efforts have been made to explore the therapeutic targets of delayed cerebral vasospasm, all of the new drugs found in recent years have failed to improve the clinical outcome of patients with SAH.22, 37 As discussed previously, the development of DCI is not caused solely by delayed cerebral vasospasm, and there might be causality between EBI and DCI; thus, agents directed at treating DCI would not eliminate the root causes of DCI, and this may explain the frustrating results from new drugs to treat SAH.22, 33 The alleviation of delayed vasospasm does not improve the clinical outcome in patients with SAH, which leads to a reasonable doubt as to whether delayed cerebral vasospasm affects the prognosis of SAH.22, 24, 31, 40, 42, 43, 44 Therefore, an increasing number of scholars have begun to support the theory that EBI is the chief cause of mortality and disability following SAH.24, 45, 46
Brain stress and acute ischaemia produced by elevated intracranial pressure, decreased cerebral blood flow and cerebral perfusion pressure can induce the expression of HSP70 4 hours after haemorrhage.16, 36 As the most common mechanism in EBI, apoptosis increases the permeability of the BBB and encephaloedema and decreases cerebral blood flow.31 It is well known that HSP70 is involved in many apoptotic mechanisms; however, related studies involving SAH are scant.14, 29 The results from literature concerning EBI after SAH showed that HSP70 could inhibit apoptosis via the regulation of the phosphorylation of anti‐apoptotic Akt kinase, which plays a critical role in EBI.24 Moreover, HSP70 can decrease the activity of matrix metalloproteinase‐9, which is a member of the zinc endopeptidase family, and is able to mediate BBB disruption and cell death via aberrant proteolysis.24, 47, 48 HSP70 contributes to the decrease of inflammation and cerebral oedema during EBI.14, 36
Endoplasmic reticulum (ER) stress is believed to exert important roles in EBI.46, 49, 50 Glucose‐regulated protein 78 (GRP78), which belongs to HSP70 family, is mainly localized in the ER51 Acting as a chaperone that folds and transports proteins, GRP78 can bind to three ER sensors, protein kinase RNA‐like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol‐requiring protein 1α (IRE1).31 The complexes are stable under normal cell conditions while decomposed under ER stress31. Unfolded proteins accumulate in the ER when the protein folding or processing reaction is impaired during EBI pathology, triggering the unfolded protein response (UPR) to alleviate ER stress.31, 49 The UPR signal is transmitted to cytoplasm and nucleus by decomposed GRP78 and subsequently gets initiated; PERK, ATF6 and IRE1 simultaneously dissociate from GRP78 and get activated to modulate UPR.49, 52, 53 Activated ATF6 subsequently increases GRP78 expression and up‐regulated GRP78 can in turn bind to and stabilize free PERK, ATF6 and IRE1 and alleviates ER stress by refolding damaged proteins and preventing them from aggregating.31, 49, 54 However, once UPR fails to control the extent of ER stress, downstream effectors such as C/EBP homologous protein (CHOP) and caspase‐12 can be activated by PERK, ATF6 and IRE1 pathways, and the apoptotic response is triggered (Figure 2).55, 56
Figure 2.
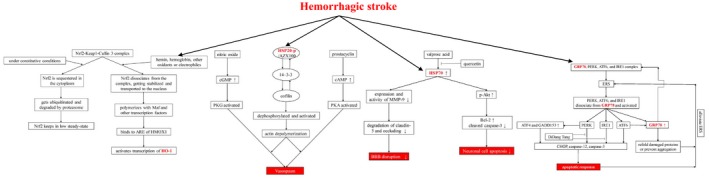
The potential mechanism of heat shock proteins involved in haemorrhagic stroke. PERK, protein kinase RNA‐like ER kinase; AFT6, activating transcription factor 6; IRE1, inositol‐requiring protein 1α; ERS, endoplasmic reticulum stress; AFT4, activating transcription factor 4; GADD153, growth arrest and DNA‐damage‐inducible gene 153; CHOP, C/EBP homologous protein; PKG, cGMP‐dependent protein kinase; PKA, cAMP‐dependent protein kinase; HSP20‐p, HSP20 phosphopeptide; BBB, blood‐brain barrier; MMP‐9, matrix metalloproteinases 9; p‐Akt, phosphorylated RAC‐alpha serine/threonine‐protein kinase; Bcl‐2, B‐cell lymphoma 2; Nrf2, nuclear factor erythroid‐2 related factor 2; Keap1, actin‐bound Kelch like‐ECH‐associated protein 1; ARE, antioxidant response elements; HMOX1, haeme oxygenase (decycling) 1
2.1.2. HSP70 and delayed cerebral vasospasm
Although it does not always occur and can be asymptomatic, delayed cerebral vasospasm plays a vital role in the outcome and prognosis of SAH.29, 30, 37, 57, 58, 59 Of patients with delayed cerebral vasospasm, 26%‐38% patients suffer sequelae and die despite undergoing satisfactory surgery to treat aneurysm and perioperative treatment.37, 60 Cerebral vasospasm was once considered the leading preventable cause of mortality and morbidity in patients with aneurysmal SAH.29 However, HSPs are likely to protect neural cells.14 Thus, many experimental studies have explored the mechanism and potential therapeutic value of HSPs in the treatment of delayed cerebral vasospasm.22, 37 HSP70 induction can be seen in the bilateral neocortex, hippocampus, thalamus, septum, hypothalamus,caudoputamen and basal forebrain 5 days after SAH.16, 17 It is known that HSP70 is highly sensitive to vasospasm, and immunostaining with HSP70 is more sensitive than histological examination for the detection of ischaemia and injury after SAH; it is even possible to build a quantifiable model of nerve damage in rats based on HSP70 sensitivity to vasospasm.16, 17, 37 However, the definite pathways involved in HSP70 induction caused by delayed cerebral vasospasm have remained unclear until now.17, 38
2.2. The roles that Haeme oxygenase‐1 (HO‐1), sHSPs and HSP90 play in SAH
2.2.1. The role HO‐1 plays in SAH
haeme oxygenase‐1, also known as HSP32, can be induced in microglia and cerebral blood vessels by the presence of haeme itself after brain haemorrhage and metabolizes haeme to ferrous iron, biliverdin and carbon monoxide (CO).14, 61, 62, 63 After aneurysm rupture, lysed red blood cells can trigger pathophysiological cascades including cerebral vasospasm and lipid peroxidation that can be attenuated by HO‐1 via the enhancement of haeme clearance, which intensifies iron sequestration and increases the levels of the antioxidant bilirubin.40, 64, 65, 66In addition, CO can up‐regulate soluble guanylyl cyclase, which contributes to cyclic guanosine monophosphate (cGMP) accumulation and subsequently leads to vascular smooth muscle relaxation.64, 65 However, bilirubin oxidation products have been implicated in vasospasm and clinical complications following SAH.67, 68 Thus, HO‐1 may simultaneously play a protective and harmful role in SAH. However, the overall effect of HO‐1 has been shown to be neuroprotective.14, 39, 63, 64, 65, 66, 69 Additionally, the overexpression of HO‐1 has been demonstrated to be a marker of cellular stress and damage in infarcted regions.70, 71
2.2.2. The roles that HSP20 and HSP27 play in SAH
Small heat shock proteins, including HSP20 and HSP27, are a family of low‐molecular‐weight HSPs. HSP27, also known as HSPB1, is seldom expressed in normal brain tissues and is induced mainly in astrocytes in the ischaemic zone and the ischaemic penumbra.14, 40 In contrast to HSP70, adenosine triphosphate (ATP) is not necessarily involved in the function of HSP27; moreover, HSP27 appears to solubilize misfolded proteins and hinder their aggregation rather than refold them.40 HSP20 was originally discovered as a byproduct of HSP27 purification, and HSP20 and HSP27 coexist in macromolecular aggregates.72 They are highly homologous in molecular structure, and both show high constitutive expression in smooth muscle and play a role in cerebral vasoconstriction after SAH.73, 74 Notably, phosphorylated HSP27 inhibits nucleotide‐dependent vasodilatation, while phosphorylated HSP20 promotes it.75, 76, 77 However, stress merely induces the expression and phosphorylation of HSP27, which accounts for impaired vasodilatation and cerebral vasospasm following SAH.74, 77 In contrast, Stetler et al demonstrated that the phosphorylation of HSP27 is essential to suppress cell death signalling and protect neurons against ischaemic injury.78 The ATP‐independent neuroprotective potential of HSP27 has been suggested to be critical in treating ischaemic brain injury.14 In addition, HSP27 and HSP20 are involved in the apoptosis pathway in numerous non‐neural systems, while their explicit roles in neural systems require further study.14, 73 A possible mechanism by which HSP20 mediates vasodilatation is shown in Figure 2.28
2.2.3. The role of HSP90 plays in SAH
HSP90 participates in the folding, maturation and homoeostasis of cellular proteins.79, 80 Studies have shown the facilitation of HSP90 in the development of apoptosis, inflammation and BBB destruction after ischaemic stroke81, 82, 83; and being essential for the stabilization and function of the ligand‐gated non‐selective cation channel, the P2X7 receptor, which is abundant in nervous system and associated with the pathophysiological process of inflammation and oxidative stress underlying EBI.84, 85 A recent study showed HSP90 played a similar role in SAH and was up‐regulated 2.5 times in both the microglia and neurons in the hippocampus 48 hours post‐SAH.79
2.3. HSPs as potential therapeutic targets for SAH
Anti‐vasospasm agents have always been a critical research focus because delayed cerebral vasospasm has long been considered an essential prognostic factor, while protection against the pathophysiology processes of EBI has also become a research focus in recent years. Nimodipine, an oral calcium channel blocker first introduced in 1985, was the drug typically used to treat SAH and was suggested to effectively prevent neuroischaemic events after SAH.22, 23, 28, 86 A recent randomized trial showed that the intrathecal application of nimodipine not only reduces DCI, but also improves clinical outcome and that this approach is safe.87, 88 Other vasodilators, such as fasudil, clazosentan and nicardipine, have also been shown to have some potential value for the treatment of vasoconstriction, although they may induce systemic hypotension and cerebral perfusion pressure.22, 28
Atorvastatin was suggested to ameliorate cellular apoptosis in EBIin a rat model, which might because of the inhibition of caspase‐3 and ER stress‐related proteins such as GRP78.46 Although the clinical value of atorvastatin targeting EBI remains unknown, the clinical application of statin in ameliorating DCI was widely researched. A retrospective study and a prospective cohort study have shown there is a correlation between statin treatment and the decrease of vasospasm incidence after SAH.89, 90 Moreover, randomized controlled phase II studies have demonstrated that acute administration of statin directly after SAH decreased the incidence of radiological vasospasm, and reduced the clinical signs of delayed ischaemic neurological deficits and mortality.91, 92 However, some experimental and clinical studies demonstrated the different or even contradictory results.93, 94, 95 Thus, further well‐designed multicentre randomized controlled trials are required.
Numerous emerging agents from experimental studies also show the therapeutic potential in SAH. The oral administration of geranylgeranylacetone is reportedly capable of inducing HSP70 expression and ameliorating DCI, although its actual clinical value remains unclear.37 Experimental studies have also shown that, when administered intravenously, modified HSP70 proteins, for example, HSP70 attached to an antibody or terminal transactivator of transcription (TAT) motif, are capable of crossing the BBB and subsequently lead to a decrease in focal cerebral ischaemia and a better outcome.96, 97, 98 In addition, HO‐1 protein combined with protein transduction domains significantly attenuated cerebral vasospasm when injected into the cerebral arteries of rats.64 The therapeutic potential of HO‐1 inducers, including nicaraven, argon, carnosol, ebselen and CGS26393, has also been revealed.39, 65, 71, 99, 100, 101 Additionally, nicaraven is known to cause minute side effects during treatment.65The intravenous administration of AZX100, a molecule constructed by combining phosphopeptide mimetics of HSP20 with a cell‐permeant peptide, was proven to successfully cross the BBB and prevent and reverse the decrease in cerebral perfusion without systemic hypotension even though the dose administered was 15‐fold higher than the minimally effective dose.28
Valproic acid reportedly exerts neuroprotective effects in EBI.24 By increasing HSP70 induction, valproic acid reduces the activity and expression of MMP‐9 and prevents the degradation of claudin‐5 and occludin, thus attenuating BBB disruption and brain oedema. On the other hand, valproic acid can inhibit neuronal cell apoptosis through phosphorylated anti‐apoptotic Akt kinase, the phosphorylation of which is regulated by HSP70 (Figure 2).24 Furthermore, because the HSP90 or the P2X7 receptor inhibition exerts neuroprotective effects, both the 17‐allylamino‐17‐demethoxygeldanamycin and A438079, which are specific inhibitors of HSP90 and P2X7, respectively, showed the therapeutic potential in SAH mice.79
Unfortunately, nearly all the new drugs developed in the last few decades to alleviate vasospasm led to no better outcome, and, still worse, no clinical treatment found significant amelioration in EBI.22, 39 Additionally, Leclerc et al suggested that all the currently available models of SAH might lead to considerable variation in results and make experimental findings difficult to convert into clinical outcome owing to the lack of spontaneous aneurysm rupture.59 Thus, more studies targeting cerebral vasospasm or EBI and experimental models more comparable to human SAH are warranted.
3. THE PATHOPHYSIOLOGY OF ICH
ICH leads to destructive outcome with high mortality and morbidity.6, 102 However, there are fewer therapeutic strategies for ICH than for other types of craniocerebral vascular disorders, such as ischaemic stroke and SAH.103, 104 Some studies have demonstrated that the mortality rate of ICH has not decreased for several decades.8, 105 Therefore, investigating the pathophysiology of ICH and exploring potential targets for ICH therapy are crucial in improving the outcome of ICH.
ICH‐associated cerebral injury includes primary injury and secondary injury. Primary injury occurs immediately after bleeding and is mainly because of haematoma expansion and the physical disruption of the brain's cellular architecture caused by haematoma.106 Based on this mechanism, several studies have investigated the benefit of haematoma removal and surgical decompression for ICH. However, the effect of surgical treatment for ICH is still controversial. Some studies have shown that the timely removal of haematomas is important to inhibit further damage.107 A clinical trial showed that surgery early in the progression of ICH might confer a small but clinically relevant survival advantage for ICH, excluding intraventricular haemorrhage.108 However, other studies have shown no benefit of clot removal surgery in ICH.109
Secondary injury during ICH follows primary injury and includes the inflammatory response to haematoma, the release and accumulation of haematoma components, oxidative stress and cytotoxicity.10, 105, 107 Among the pathophysiology of secondary injury, inflammation is considered to be a critical part of the whole brain injury process.105, 110, 111, 112 The presence of blood components in the parenchyma of the brain induces and triggers the inflammatory response, including the release of inflammatory mediators, enzyme activation, inflammatory cell migration and the breakdown and repair of brain tissue, which aggravates ICH damage through complicated pathways and leads to irreversible effects such as brain barrier disruption, brain oedema and brain cell death.10, 110, 113, 114 As the treatment for primary injury is time‐limited and still controversial, understanding the mechanism of secondary injury, especially inflammation, in ICH and exploring potential therapeutic targets is a method anticipated to improve the outcome of ICH.
3.1. The role of HO‐1 in ICH
As mentioned previously, HO‐1 is an isoenzyme of haeme oxygenase that catalyzes the rate‐limiting step in the metabolism of haeme and has been proven to play a neuroprotective role in SAH. Also, HO‐1 levels have already been proven to be associated with ICH.115 Unfortunately, although the role of HO‐1 in ICH has been researched extensively, the effect of HO‐1 in ICH is still controversial. Most scholars believe that the overall effect of HO‐1 in ICH tends to be harmful.14 HO‐1 metabolizes haeme to ferrous iron, biliverdin and CO. After ICH induction, the pro‐oxidant hydroxyl radical induced by the reaction of ferrous iron and hydrogen peroxide increases oxidative stress, and HO‐1 accelerate the accumulation of iron overload in the brain, both of which are involved in the HO‐1 induced damage.14, 116, 117 Moreover, a study proving the harmful role of HO‐1 demonstrated a marked reduction of ICH‐induced leucocyte infiltration and microglia/macrophage activation in HO‐1 knockout mice, which implied a function of HO‐1 in promoting inflammation and increasing secondary injury.116
Further studies revealed that HO‐1 levels fluctuate after ICH, and the effect of HO‐1 after ICH depends on the time‐point of the ICH process.118 In the early stage after ICH, HO‐1 is mainly expressed in microglia, the major phagocytes in the brain, and the activation of microglia contributes to neurological impairment.119 Furthermore, the activation of microglia leads to the development of M1‐like or M2‐like phenotypes in the microglia, which are in dynamic flux after ICH.120, 121 Between these two phenotypes, M1 microglia produce pro‐inflammatory factors that then contribute to brain damage, while M2 microglia induce anti‐inflammatory factors and are neuroprotective.122 Moreover, HO‐1 is also expressed in astrocytes in the late stages of ICH, and many studies have proven that HO‐1 overexpression in astrocytes provides neuroprotection and reduces brain barrier disruption, peri‐haematoma cell injury and mortality after ICH (Figure 3).123, 124, 125, 126 One of these studies revealed that HO‐1 induction could protect cortical astrocytes from haemoglobin toxicity,126 which may be one of the mechanisms of the neuroprotective function of HO‐1. The mechanisms described above may explain the different effects of HO‐1 at different time‐points of ICH. In contrast, another study showed that HO‐1 was up‐regulated in the early stage after ICH and exerted a protective effect against oxidative stress; however, in the late stage of ICH, the expression of HO‐1 may result in neurological dysfunction, and HO‐1 may be toxic.118
Figure 3.
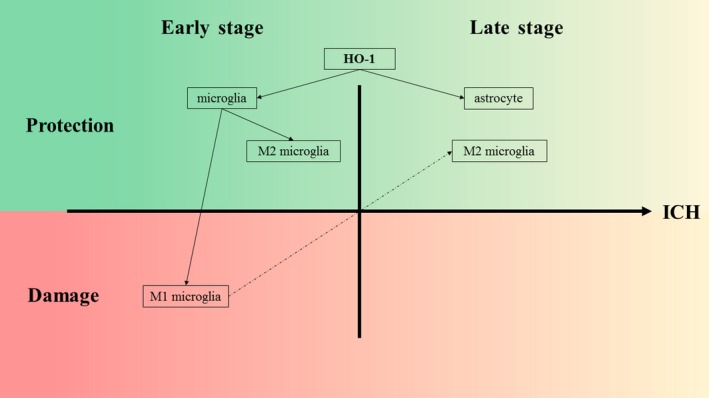
The expression of HO‐1 after ICH. In the early stage after ICH, HO‐1 is mainly expressed in microglia, and the activation of microglia leads to the development of M1‐like or M2‐like phenotypes. The former contributes to neurological impairments, while the latter is neuroprotective. Furthermore, the two subtypes of microglia are in dynamic flux after ICH. In the late stages of ICH, the overexpression of HO‐1 in astrocytes is neuroprotective and reduces blood‐brain barrier disruption, perihaematomal cell injury and mortality following ICH. M1 microglia, M1‐like phenotype of microglia; M2 microglia, M2‐like phenotype of microglia
Besides microglia, there are other types of tissue‐resident macrophages that may participate in different stages of ICH. Border‐associated macrophages (BAMs) in brain contain cells locating in the perivascular spaces of brain vessels, the leptomeningeal spaces and the choroid plexus, which have scavenger function and the ability to present antigen to lymphocytes.127 Although a recent study has shown BAMs attract granulocytes and promote vascular leakage after ischaemic stroke, it is still little known about the function of these macrophages under the ICH conditions.128 But researchers believe that perivascular macrophages and meningeal macrophages have been identified by the expression of CD163 in both humans and rats.129, 130 CD163 is up‐regulated and participates in haemoglobin clearance after ICH and may be a biomarker of functional outcome in ICH.131, 132, 133 A previous study has proved that CD163/HO‐1 pathway could regulate inflammation in haematoma surrounding tissues after ICH; specifically, about 24 hours after ICH onset, CD163 and HO‐1 expressions would be gradually enhanced and reach a peak after 72 hours, exhibiting anti‐inflammatory characteristics.134
3.2. The role of HSP70 in ICH
As indicated in the paragraphs above, HSP70 is a common and extensively studied HSP. The relationship between HSP70 and ICH has also been clarified. Studies demonstrated that HSP70 levels changed in patients with intracranial haemorrhage and were negatively associated with Glasgow Coma Scale scores and positively associated with bleeding volume,36, 135 and HSP70 is proven to ameliorate the neurological deficits in the delayed but not acute phase of ICH.136 The function of glucose‐regulated protein 75 (GRP75) and GRP78 in ICH have also been examined in many studies.
3.2.1. GRP75 and ICH
Glucose‐regulated protein 75 is a member of the HSP70 family and is mainly located in mitochondria.137 As GRP75 showed a potential protective effect in central nervous system disorders,138, 139 its role in ICH was also studied. In a rat ICH model, GRP75 expression was reduced upon ICH, and further research demonstrated that GRP75 overexpression in brain tissues with ICH inhibited inflammatory factors such as tumour necrosis factor‐α and interleukin‐1β and inhibited the expression of neuronal apoptosis markers (such as active caspase‐3 and B cell lymphoma 2 apoptosis regulator‐associated X apoptosis regulator).140 These results showed the protective role of GRP75 in ICH, which includes the inhibition of inflammation and neuronal apoptosis.
3.2.2. GRP78 and ICH
Glucose‐regulated protein 78 was also reported to be associated with ICH. A study of the role of a 45‐kD Ca2+‐binding protein demonstrated that the interaction of the Ca2+‐binding protein and GRP78 led to the inhibition of neuronal apoptosis.141 In addition, ER stress pathway is also involved tightly in cell apoptosis of secondary injury after ICH.142, 143 Thus, similar to GRP75, the role of GRP78 in ICH may be protective for brain tissues. However, additional studies on GRP78 and ICH are required.
3.3. Therapeutic strategies targeting HSPs in ICH
According to the role of HSPs in ICH, some therapeutic strategies targeting HSPs are potential methods to improve ICH outcome. For HO‐1, which is a key enzyme of haeme metabolism, the HO‐1 inducer haemin was proven to improve BBB function and neurological outcome in ICH models.20 Furthermore, nuclear factor erythroid‐2 related factor 2 (Nrf2), which can regulate the expression of HO‐1 and many other proteins with antioxidant and anti‐inflammatory effects, was shown to be a target for therapy in ICH (Figure 2).144 Therefore, Nrf2 activators targeting the Nrf2/HO‐1 axis are neuroprotective in ICH and may be potential drugs for ICH treatment. For instance, a study focusing on nicotinamide mononucleotides demonstrated that nicotinamide mononucleotides activated the Nrf2/HO‐1 pathway to attenuate brain injury after ICH.145 In addition, minocycline was proven to significantly reduce iron overload and iron handling proteins induced by HO‐1 and also reduce brain swelling, inflammation and neuronal loss, leading to a better outcome for ICH in a rat model.146
Similarly, a high level of HSP70 was detected in minocycline‐treated rats with ICH, which showed that minocycline exerted a neuroprotective effect by mediating HSP70.147, 148 Another study demonstrated that geranylgeranylacetone, which is known to be an HSP70 inducer, could reduce brain oedema and exerts neuroprotective effects in an ICH model.149 Administration of another HSP inducer geldanamycin, also resulted in amelioration of inflammation, neurobehavioral deficits and BBB destruction by the up‐regulation of HSP70.136 Moreover, mild hypothermia exerted a protective effect on rat neuronal injury after ICH by reducing ER stress, in which GRP78 plays a vital role.142, 143 Similarly, a kind of Chinese traditional medicine, Di Dang Tang, which involves four traditional Chinese medicinal substances, was proven to block the GRP78‐IRE1/PERK pathway post‐ICH and results in reduced ER stress‐mediated apoptosis (Figure 2).150
4. CONCLUSIONS
In the present study, we focus on the current understanding of the role and therapeutic potential of HSPs involved in haemorrhagic stroke. HSP70 acts as a critical neuroprotective molecule and participates in the processes of cerebral vasospasm, neural cell apoptosis, immunoreaction and inflammation during haemorrhagic stroke. Additionally, HSP70 is a significant marker for cellular stress or damage and can be used for outcome prediction and differential diagnosis. HO‐1 is capable of enhancing haeme clearance, intensifying iron sequestration and increasing the antioxidant bilirubin after haemorrhage; however, the metabolic process can be toxic because of iron overload and the succedent pathway, for example, the Fenton reaction and oxidative damage, and the overall effect of HO‐1 in SAH and ICH tends to be protective or harmful, respectively, given their different pathophysiological mechanisms. In addition, the role of HO‐1 in ICH was proven to vary at different stages during the ICH process, and further research is needed to explore the effects of HO‐1 in ICH. HSP20 and HSP27 mainly play a role in SAH. Despite being highly homologous in molecular structure, the phosphorylation of HSP20 and HSP27 mediates opposite impacts on SAH, as the former promotes nucleotide‐dependent vasodilatation, and the latter inhibits it. Even so, the ATP‐independent neuroprotective potential of HSP27 is suggested to be critical in ischaemic brain injury. Pathophysiological processes following initial haemorrhage present a high risk of disability and mortality among survivors, and new drugs developed over the years have failed to improve the clinical outcome of patients. HSPs represent potential therapeutic targets, and new agents targeting HSPs are warranted.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTION
AWS, YXZ and YHY wrote the paper and made the original figures. WHZ, JMZ and YCD critically revised the texts and figures. All authors read and approved the final manuscript.
ACKNOWLEDGEMENTS
This work was funded by China Postdoctoral Science Foundation (2017M612010) and National Natural Science Foundation of China (81701144, 81870916).
Shao A, Zhou Y, Yao Y, Zhang W, Zhang J, Deng Y. The role and therapeutic potential of heat shock proteins in haemorrhagic stroke. J Cell Mol Med. 2019;23:5846–5858. 10.1111/jcmm.14479
Anwen Shao, Yunxiang Zhou, and Yihan Yao contributed equally to this manuscript.
Contributor Information
Anwen Shao, Email: 21118116@zju.edu.cn, Email: anwenshao@sina.com.
Yongchuan Deng, Email: dyc001@zju.edu.cn.
REFERENCES
- 1. Rothwell PM, Coull AJ, Giles MF, et al. Change in stroke incidence, mortality, case‐fatality, severity, and risk factors in Oxfordshire, UK from 1981 to 2004 (Oxford Vascular Study). Lancet. 2004;363:1925‐1933. [DOI] [PubMed] [Google Scholar]
- 2. Polivka J, Polivka J, Pesta M, et al. Risks associated with the stroke predisposition at young age: facts and hypotheses in light of individualized predictive and preventive approach. EPMA Journal. 2019;10:81‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sudlow CL, Warlow CP. Comparable studies of the incidence of stroke and its pathological types: results from an international collaboration. International Stroke Incidence Collaboration. Stroke. 1997;28:491‐499. [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez‐Perez A, Gaist D, Wallander MA, McFeat G, Garcia‐Rodriguez LA. Mortality after hemorrhagic stroke: data from general practice (The Health Improvement Network). Neurology. 2013;81:559‐565. [DOI] [PubMed] [Google Scholar]
- 5. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics‐2017 update: a report from the American Heart Association. Circulation. 2017;135:e146‐e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics‐2018 update: a Report From the American Heart Association. Circulation. 2018;137:e67‐e492. [DOI] [PubMed] [Google Scholar]
- 7. Nieuwkamp DJ, Setz LE, Algra A, Linn FH, de Rooij NK, Rinkel GJ. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta‐analysis. Lancet Neurol. 2009;8:635‐642. [DOI] [PubMed] [Google Scholar]
- 8. van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta‐analysis. Lancet Neurol. 2010;9:167‐176. [DOI] [PubMed] [Google Scholar]
- 9. Jolink WM, Klijn CJ, Brouwers PJ, Kappelle LJ, Vaartjes I. Time trends in incidence, case fatality, and mortality of intracerebral hemorrhage. Neurology. 2015;85:1318‐1324. [DOI] [PubMed] [Google Scholar]
- 10. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurogibol. 2014;115:25‐44. [DOI] [PubMed] [Google Scholar]
- 11. Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109:315‐329. [DOI] [PubMed] [Google Scholar]
- 12. San Gil R, Ooi L, Yerbury JJ, Ecroyd H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol Neurodegener. 2017;12:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stetler RA, Gan Y, Zhang W, et al. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurogibol. 2010;92:184‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain: role of Hsp70, Hsp27, and HO‐1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4:685‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ouyang YB, Giffard RG. MicroRNAs regulate the chaperone network in cerebral ischemia. Transl Stroke Res. 2013;4:693‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matz PG, Sundaresan S, Sharp FR, Weinstein PR. Induction of HSP70 in rat brain following subarachnoid hemorrhage produced by endovascular perforation. J Neurosurg. 1996;85:138‐145. [DOI] [PubMed] [Google Scholar]
- 17. Harada S, Kamiya K, Masago A, Iwata A, Yamada K. Subarachnoid hemorrhage induces c‐fos, c‐jun and hsp70 mRNA expression in rat brain. NeuroReport. 1997;8:3399‐3404. [DOI] [PubMed] [Google Scholar]
- 18. Gules I, Satoh M, Clower BR, Nanda A, Zhang JH. Comparison of three rat models of cerebral vasospasm. Am J Physiol Heart Circ Physiol. 2002;283:H2551‐H2559. [DOI] [PubMed] [Google Scholar]
- 19. Guresir E, Schuss P, Borger V, Vatter H. Experimental subarachnoid hemorrhage: double cisterna magna injection rat model–assessment of delayed pathological effects of cerebral vasospasm. Transl Stroke Res. 2015;6:242‐251. [DOI] [PubMed] [Google Scholar]
- 20. Lu X, Chen‐Roetling J, Regan RF. Systemic hemin therapy attenuates blood‐brain barrier disruption after intracerebral hemorrhage. Neurobiol Dis. 2014;70:245‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Korja M, Lehto H, Juvela S, Kaprio J. Incidence of subarachnoid hemorrhage is decreasing together with decreasing smoking rates. Neurology. 2016;87:1118‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Lieshout JH, Dibue‐Adjei M, Cornelius JF, et al. An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg Rev. 2018;41:917‐930. [DOI] [PubMed] [Google Scholar]
- 23. van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet. 2007;369:306‐318. [DOI] [PubMed] [Google Scholar]
- 24. Ying GY, Jing CH, Li JR, et al. Neuroprotective effects of valproic acid on blood‐brain barrier disruption and apoptosis‐related early brain injury in rats subjected to subarachnoid hemorrhage are modulated by heat shock protein 70/matrix metalloproteinases and heat shock protein 70/AKT pathways. Neurosurgery. 2016;79:286‐295. [DOI] [PubMed] [Google Scholar]
- 25. Turan N, Heider RA, Zaharieva D, Ahmad FU, Barrow DL, Pradilla G. Sex differences in the formation of intracranial aneurysms and incidence and outcome of subarachnoid hemorrhage: review of experimental and human studies. Transl Stroke Res. 2016;7:12‐19. [DOI] [PubMed] [Google Scholar]
- 26. Nicholson P, O'Hare A, Power S, et al. Decreasing incidence of subarachnoid hemorrhage. J Neurointerv Surg. 2019;11:320‐322. [DOI] [PubMed] [Google Scholar]
- 27. Kamp MA, Lieshout J, Dibue‐Adjei M, et al. A Systematic and meta‐analysis of mortality in experimental mouse models analyzing delayed cerebral ischemia after subarachnoid hemorrhage. Transl Stroke Res. 2017;8:206‐219. [DOI] [PubMed] [Google Scholar]
- 28. Furnish EJ, Brophy CM, Harris VA, et al. Treatment with transducible phosphopeptide analogues of the small heat shock‐related protein, HSP20, after experimental subarachnoid hemorrhage: prevention and reversal of delayed decreases in cerebral perfusion. J Neurosurg. 2010;112:631‐639. [DOI] [PubMed] [Google Scholar]
- 29. Crowley RW, Medel R, Dumont AS, et al. Angiographic vasospasm is strongly correlated with cerebral infarction after subarachnoid hemorrhage. Stroke. 2011;42:919‐923. [DOI] [PubMed] [Google Scholar]
- 30. Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6:1‐9. [DOI] [PubMed] [Google Scholar]
- 31. Liu Q, Zhao D, Ji YX, et al. Role of glucose‐regulated protein 78 in early brain injury after experimental subarachnoid hemorrhage in rats. J Huazhong Univ Sci Technolog Med Sci. 2016;36:168‐173. [DOI] [PubMed] [Google Scholar]
- 32. Conzen C, Becker K, Albanna W, et al. The acute phase of experimental subarachnoid hemorrhage: intracranial pressure dynamics and their effect on cerebral blood flow and autoregulation. Transl Stroke Res. 2018. 10.1007/s12975-018-0674-3 [DOI] [PubMed] [Google Scholar]
- 33. Topkoru B, Egemen E, Solaroglu I, Zhang JH. Early brain injury or vasospasm? an overview of common mechanisms. Curr Drug Targets. 2017;18:1424‐1429. [DOI] [PubMed] [Google Scholar]
- 34. Nishikawa H, Nakatsuka Y, Shiba M, Kawakita F, Fujimoto M, Suzuki H. Increased plasma Galectin‐3 preceding the development of delayed cerebral infarction and eventual poor outcome in non‐severe aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2018;9:110‐119. [DOI] [PubMed] [Google Scholar]
- 35. Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. ScientificWorldJournal. 2010;10:1543‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alatas OD, Gurger M, Atescelik M, et al. Neuron‐specific enolase, S100 calcium‐binding protein B, and heat shock protein 70 levels in patients with intracranial hemorrhage. Medicine (Baltimore). 2015;94:e2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikaido H, Tsunoda H, Nishimura Y, Kirino T, Tanaka T. Potential role for heat shock protein 72 in antagonizing cerebral vasospasm after rat subarachnoid hemorrhage. Circulation. 2004;110:1839‐1846. [DOI] [PubMed] [Google Scholar]
- 38. Matz P, Weinstein P, States B, Honkaniemi J, Sharp FR. Subarachnoid injections of lysed blood induce the hsp70 stress gene and produce DNA fragmentation in focal areas of the rat brain. Stroke. 1996;27:504‐512; discussion 13. [DOI] [PubMed] [Google Scholar]
- 39. Hollig A, Weinandy A, Liu J, Clusmann H, Rossaint R, Coburn M. Beneficial properties of argon after experimental subarachnoid hemorrhage: early treatment reduces mortality and influences hippocampal protein expression. Crit Care Med. 2016;44:e520‐e529. [DOI] [PubMed] [Google Scholar]
- 40. Dietrich HH, Dacey RG Jr. Molecular keys to the problems of cerebral vasospasm. Neurosurgery. 2000;46:517‐530. [DOI] [PubMed] [Google Scholar]
- 41. Bustamante A, Lopez‐Cancio E, Pich S, et al. Blood biomarkers for the early diagnosis of stroke: the stroke‐chip study. Stroke. 2017;48:2419‐2425. [DOI] [PubMed] [Google Scholar]
- 42. Etminan N, Vergouwen MD, Ilodigwe D, Macdonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta‐analysis. J Cereb Blood Flow Metab. 2011;31:1443‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV‐034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double‐blind, placebo‐controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9‐17. [DOI] [PubMed] [Google Scholar]
- 44. Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS‐1): randomized, double‐blind, placebo‐controlled phase 2 dose‐finding trial. Stroke. 2008;39:3015‐3021. [DOI] [PubMed] [Google Scholar]
- 45. Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res. 2013;4:432‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qi W, Cao D, Li Y, et al. Atorvastatin ameliorates early brain injury through inhibition of apoptosis and ER stress in a rat model of subarachnoid hemorrhage. Biosci Rep. 2018;38:BSR20171035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82‐89. [DOI] [PubMed] [Google Scholar]
- 48. Kim N, Kim JY, Yenari MA. Anti‐inflammatory properties and pharmacological induction of Hsp70 after brain injury. Inflammopharmacology. 2012;20:177‐185. [DOI] [PubMed] [Google Scholar]
- 49. Zhang H, He X, Wang Y, et al. Neuritin attenuates early brain injury in rats after experimental subarachnoid hemorrhage. Int J Neurosci. 2017;127:1087‐1095. [DOI] [PubMed] [Google Scholar]
- 50. Li H, Yu JS, Zhang HS, et al. Increased expression of caspase‐12 after experimental subarachnoid hemorrhage. Neurochem Res. 2016;41:3407‐3416. [DOI] [PubMed] [Google Scholar]
- 51. Cao Y, Hao Y, Li H, et al. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33:809‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu F, Ma R, Zhang G, et al. Estrogen and propofol combination therapy inhibits endoplasmic reticulum stress and remarkably attenuates cerebral ischemia‐reperfusion injury and OGD injury in hippocampus. Biomed Pharmacother. 2018;108:1596‐1606. [DOI] [PubMed] [Google Scholar]
- 53. Lebeau P, Platko K, Al‐Hashimi AA, et al. Loss‐of‐function PCSK9 mutants evade the unfolded protein response sensor GRP78 and fail to induce endoplasmic reticulum stress when retained. J Biol Chem. 2018;293:7329‐7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Menzie‐Suderam JM, Mohammad‐Gharibani P, Modi J, et al. Granulocyte‐colony stimulating factor protects against endoplasmic reticulum stress in an experimental model of stroke. Brain Res. 2018;1682:1‐13. [DOI] [PubMed] [Google Scholar]
- 55. Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105‐118. [DOI] [PubMed] [Google Scholar]
- 56. Liu L, Zhang Y, Wang Y, Peng W, Zhang N, Ye Y. Progesterone inhibited endoplasmic reticulum stress associated apoptosis induced by interleukin‐1beta via the GRP78/PERK/CHOP pathway in BeWo cells. J Obstet Gynaecol Res. 2018;44:463‐473. [DOI] [PubMed] [Google Scholar]
- 57. Mortimer AM, Steinfort B, Faulder K, et al. The detrimental clinical impact of severe angiographic vasospasm may be diminished by maximal medical therapy and intensive endovascular treatment. J Neurointerv Surg. 2015;7:881‐887. [DOI] [PubMed] [Google Scholar]
- 58. Dorsch N. A clinical review of cerebral vasospasm and delayed ischaemia following aneurysm rupture. Acta Neurochir Suppl. 2011;110:5‐6. [DOI] [PubMed] [Google Scholar]
- 59. Leclerc JL, Garcia JM, Diller MA, et al. A comparison of pathophysiology in humans and rodent models of subarachnoid hemorrhage. Front Mol Neurosci. 2018;11:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dorsch NW. Cerebral arterial spasm–a clinical review. Br J Neurosurg. 1995;9:403‐412. [DOI] [PubMed] [Google Scholar]
- 61. Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629‐652. [DOI] [PubMed] [Google Scholar]
- 62. Kuroki M, Kanamaru K, Suzuki H, Waga S, Semba R. Effect of vasospasm on heme oxygenases in a rat model of subarachnoid hemorrhage. Stroke. 1998;29:683‐689; discussion 8‐9. [DOI] [PubMed] [Google Scholar]
- 63. Matz P, Turner C, Weinstein PR, Massa SM, Panter SS, Sharp FR. Heme‐oxygenase‐1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996;713:211‐222. [DOI] [PubMed] [Google Scholar]
- 64. Ogawa T, Hanggi D, Wu Y, et al. Protein therapy using heme‐oxygenase‐1 fused to a polyarginine transduction domain attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:2231‐2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shimada Y, Tsunoda H, Zang L, Hirano M, Oka T, Tanaka T. Synergistic induction of heme oxygenase‐1 by nicaraven after subarachnoid hemorrhage to prevent delayed cerebral vasospasm. Eur J Pharmacol. 2009;620:16‐20. [DOI] [PubMed] [Google Scholar]
- 66. Ono S, Komuro T, Macdonald RL. Heme oxygenase‐1 gene therapy for prevention of vasospasm in rats. J Neurosurg. 2002;96:1094‐1102. [DOI] [PubMed] [Google Scholar]
- 67. Clark JF, Sharp FR. Bilirubin oxidation products (BOXes) and their role in cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1223‐1233. [DOI] [PubMed] [Google Scholar]
- 68. Clark JF, Harm A, Saffire A, Biehle SJ, Lu A, Pyne‐Geithman GJ. Bilirubin oxidation products seen post subarachnoid hemorrhage have greater effects on aged rat brain compared to young. Acta Neurochir Suppl. 2011;110:157‐162. [DOI] [PubMed] [Google Scholar]
- 69. Suzuki H, Kanamaru K, Tsunoda H, et al. Heme oxygenase‐1 gene induction as an intrinsic regulation against delayed cerebral vasospasm in rats. J Clin Invest. 1999;104:59‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matz PG, Massa SM, Weinstein PR, Turner C, Panter SS, Sharp FR. Focal hyperexpression of hemeoxygenase‐1 protein and messenger RNA in rat brain caused by cellular stress following subarachnoid injections of lysed blood. J Neurosurg. 1996;85:892‐900. [DOI] [PubMed] [Google Scholar]
- 71. Ulbrich F, Goebel U. The molecular pathway of argon‐mediated neuroprotection. Int J Mol Sci. 2016;17:1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kato K, Goto S, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20‐kDa protein that is highly homologous to alpha B crystallin. J Biol Chem. 1994;269:15302‐15309. [PubMed] [Google Scholar]
- 73. Li F, Xie Y, Wu Y, et al. HSP20 exerts a protective effect on preeclampsia by regulating function of trophoblast cells via Akt pathways. Reprod Sci. 2018. https://doi.org/10.1933719118802057 [DOI] [PubMed] [Google Scholar]
- 74. Macomson SD, Brophy CM, Miller W, Harris VA, Shaver EG. Heat shock protein expression in cerebral vessels after subarachnoid hemorrhage. Neurosurgery. 2002;51:204‐211; discussion 10‐1. [DOI] [PubMed] [Google Scholar]
- 75. Woodrum DA, Brophy CM, Wingard CJ, Beall A, Rasmussen H. Phosphorylation events associated with cyclic nucleotide‐dependent inhibition of smooth muscle contraction. Am J Physiol. 1999;277:H931‐H939. [DOI] [PubMed] [Google Scholar]
- 76. Brophy CM, Beall A, Mannes K, et al. Heat shock protein expression in umbilical artery smooth muscle. J Reprod Fertil. 1998;114:351‐355. [DOI] [PubMed] [Google Scholar]
- 77. Knoepp L, Beall A, Woodrum D, et al. Cellular stress inhibits vascular smooth muscle relaxation. J Vasc Surg. 2000;31:343‐353. [DOI] [PubMed] [Google Scholar]
- 78. Stetler RA, Gao Y, Zhang L, et al. Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J Neurosci. 2012;32:2667‐2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zuo Y, Wang J, Liao F, et al. Inhibition of heat shock protein 90 by 17‐AAG reduces inflammation via P2X7 receptor/NLRP3 inflammasome pathway and increases neurogenesis after subarachnoid hemorrhage in mice. Front Mol Neurosci. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lei W, Mullen N, McCarthy S, et al. Heat‐shock protein 90 (Hsp90) promotes opioid‐induced anti‐nociception by an ERK mitogen‐activated protein kinase (MAPK) mechanism in mouse brain. J Biol Chem. 2017;292:10414‐10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Z, Guo LM, Wang Y, et al. Inhibition of HSP90alpha protects cultured neurons from oxygen‐glucose deprivation induced necroptosis by decreasing RIP3 expression. J Cell Physiol. 2018;233:4864‐4884. [DOI] [PubMed] [Google Scholar]
- 82. Qi J, Liu Y, Yang P, et al. Heat shock protein 90 inhibition by 17‐Dimethylaminoethylamino‐17‐demethoxygeldanamycin protects blood‐brain barrier integrity in cerebral ischemic stroke. Am J Transl Res. 2015;7:1826‐1837. [PMC free article] [PubMed] [Google Scholar]
- 83. Qi J, Han X, Liu HT, et al. 17‐Dimethylaminoethylamino‐17‐demethoxygeldanamycin attenuates inflammatory responses in experimental stroke. Biol Pharm Bull. 2014;37:1713‐1718. [DOI] [PubMed] [Google Scholar]
- 84. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47:15‐31. [DOI] [PubMed] [Google Scholar]
- 85. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29:3781‐3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hanggi D, Perrin J, Eicker S, et al. Local delivery of nimodipine by prolonged‐release microparticles‐feasibility, effectiveness and dose‐finding in experimental subarachnoid hemorrhage. PLoS ONE. 2012;7:e42597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hanggi D, Etminan N, Aldrich F, et al. Randomized, open‐label, phase 1/2a study to determine the maximum tolerated dose of intraventricular sustained release nimodipine for subarachnoid hemorrhage (NEWTON [Nimodipine Microparticles to Enhance Recovery While Reducing Toxicity After Subarachnoid Hemorrhage]). Stroke. 2017;48:145‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hanggi D, Etminan N, Macdonald RL, et al. NEWTON: nimodipine microparticles to enhance recovery while reducing toxicity after subarachnoid hemorrhage. Neurocrit Care. 2015;23:274‐284. [DOI] [PubMed] [Google Scholar]
- 89. Moskowitz SI, Ahrens C, Provencio JJ, Chow M, Rasmussen PA. Prehemorrhage statin use and the risk of vasospasm after aneurysmal subarachnoid hemorrhage. Surg Neurol. 2009;71:311‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Woo SW, Kim JH, Kang HI, Kim DR, Moon BG, Kim JS. High‐dose simvastatin is effective in preventing cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a prospective cohort study in Korean patients. J Korean Neurosurg Soc. 2015;58:328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tseng M‐Y, Hutchinson Peter J, Czosnyka M, Richards H, Pickard John D, Kirkpatrick PJ. Effects of acute pravastatin treatment on intensity of rescue therapy, length of inpatient stay, and 6‐month outcome in patients after aneurysmal subarachnoid hemorrhage. Stroke. 2007;38:1545‐1550. [DOI] [PubMed] [Google Scholar]
- 92. Lynch John R, Wang H, McGirt Matthew J, et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2005;36:2024‐2026. [DOI] [PubMed] [Google Scholar]
- 93. Naraoka M, Matsuda N, Shimamura N, et al. Long‐acting statin for aneurysmal subarachnoid hemorrhage: A randomized, double‐blind, placebo‐controlled trial. J Cereb Blood Flow Metab. 2018;38:1190‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lizza BD, Kosteva A, Maas MB, et al. Preadmission statin use does not improve functional outcomes or prevent delayed ischemic events in patients with spontaneous subarachnoid hemorrhage. Pharmacotherapy. 2014;34:811‐817. [DOI] [PubMed] [Google Scholar]
- 95. Choi KS, Kim JM, Yi HJ, et al. Dose‐related effect of statins in patients with endovascular coiling or microsurgical clipping for aneurysmal subarachnoid hemorrhage: updated study‐level meta‐analysis. Eur J Clin Pharmacol. 2017;73:1071‐1081. [DOI] [PubMed] [Google Scholar]
- 96. Doeppner TR, Nagel F, Dietz GP, et al. TAT‐Hsp70‐mediated neuroprotection and increased survival of neuronal precursor cells after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2009;29:1187‐1196. [DOI] [PubMed] [Google Scholar]
- 97. Zhan X, Ander BP, Liao IH, et al. Recombinant Fv‐Hsp70 protein mediates neuroprotection after focal cerebral ischemia in rats. Stroke. 2010;41:538‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Doeppner TR, Ewert TA, Tonges L, et al. Transduction of neural precursor cells with TAT‐heat shock protein 70 chaperone: therapeutic potential against ischemic stroke after intrastriatal and systemic transplantation. Stem Cells. 2012;30:1297‐1310. [DOI] [PubMed] [Google Scholar]
- 99. Satoh T, Ishige K, Sagara Y. Protective effects on neuronal cells of mouse afforded by ebselen against oxidative stress at multiple steps. Neurosci Lett. 2004;371:1‐5. [DOI] [PubMed] [Google Scholar]
- 100. Chou AK, Chen TI, Winardi W, et al. Functional neuroprotective effect of CGS 26303, a dual ECE inhibitor, on ischemic‐reperfusion spinal cord injury in rats. Exp Biol Med (Maywood). 2007;232:214‐218. [PubMed] [Google Scholar]
- 101. Martin D, Rojo AI, Salinas M, et al. Regulation of heme oxygenase‐1 expression through the phosphatidylinositol 3‐kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919‐8929. [DOI] [PubMed] [Google Scholar]
- 102. Weimar C, Kleine‐Borgmann J. Epidemiology, prognosis and prevention of non‐traumatic intracerebral hemorrhage. Curr Pharm Des. 2017;23:2193‐2196. [DOI] [PubMed] [Google Scholar]
- 103. Liao KH, Sung CW, Huang YN, Li WJ, Yu PC, Wang JY. Therapeutic potential of drugs targeting pathophysiology of intracerebral hemorrhage: from animal models to clinical applications. Curr Pharm Des. 2017;23:2212‐2225. [DOI] [PubMed] [Google Scholar]
- 104. Behrouz R. Re‐exploring tumor necrosis factor alpha as a target for therapy in intracerebral hemorrhage. Transl Stroke Res. 2016;7:93‐96. [DOI] [PubMed] [Google Scholar]
- 105. Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Schlunk F, Greenberg SM. The pathophysiology of intracerebral hemorrhage formation and expansion. Transl Stroke Res. 2015;6:257‐263. [DOI] [PubMed] [Google Scholar]
- 107. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781‐1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mendelow AD, Gregson BA, Rowan EN, Murray GD, Gholkar A, Mitchell PM. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet (London, England). 2013;382:397‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Morgenstern LB, Hemphill JC 3rd, Anderson C, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41:2108‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894‐908. [DOI] [PubMed] [Google Scholar]
- 111. Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurogibol. 2010;92:463‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53‐63. [DOI] [PubMed] [Google Scholar]
- 113. Wilkinson DA, Pandey AS, Thompson BG, Keep RF, Hua Y, Xi G. Injury mechanisms in acute intracerebral hemorrhage. Neuropharmacology. 2018;134:240‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jiang B, Li L, Chen Q, et al. Role of glibenclamide in brain injury after intracerebral hemorrhage. Transl Stroke Res. 2017;8:183‐193. [DOI] [PubMed] [Google Scholar]
- 115. Matz PG, Weinstein PR, Sharp FR. Heme oxygenase‐1 and heat shock protein 70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery. 1997;40:152‐162; discussion 60‐2. [DOI] [PubMed] [Google Scholar]
- 116. Wang J, Dore S. Heme oxygenase‐1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wu J, Hua Y, Keep Richard F, Nakamura T, Hoff Julian T, Xi G. Iron and iron‐handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964‐2969. [DOI] [PubMed] [Google Scholar]
- 118. Wang G, Yang Q, Li G, et al. Time course of heme oxygenase‐1 and oxidative stress after experimental intracerebral hemorrhage. Acta Neurochir. 2011;153:319‐325. [DOI] [PubMed] [Google Scholar]
- 119. Zhang Z, Song Y, Zhang Z, et al. Distinct role of heme oxygenase‐1 in early‐ and late‐stage intracerebral hemorrhage in 12‐month‐old mice. J Cereb Blood Flow Metab. 2017;37:25‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mazar J, Rosado A, Shelley J, Marchica J, Westmoreland TJ. The long non‐coding RNA GAS5 differentially regulates cell cycle arrest and apoptosis through activation of BRCA1 and p53 in human neuroblastoma. Oncotarget. 2017;8:6589‐6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Taylor RA, Chang CF, Goods BA, et al. TGF‐beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Investig. 2017;127:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. 2017;54:1874‐1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen‐Roetling J, Song W, Schipper HM, Regan CS, Regan RF. Astrocyte overexpression of heme oxygenase‐1 improves outcome after intracerebral hemorrhage. Stroke. 2015;46:1093‐1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chen‐Roetling J, Kamalapathy P, Cao Y, Song W, Schipper HM, Regan RF. Astrocyte heme oxygenase‐1 reduces mortality and improves outcome after collagenase‐induced intracerebral hemorrhage. Neurobiol Dis. 2017;102:140‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Benvenisti‐Zarom L, Regan RF. Astrocyte‐specific heme oxygenase‐1 hyperexpression attenuates heme‐mediated oxidative injury. Neurobiol Dis. 2007;26:688‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Regan RF, Guo Y, Kumar N. Heme oxygenase‐1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1‐4. [DOI] [PubMed] [Google Scholar]
- 127. Coles JA, Myburgh E, Brewer JM, McMenamin PG. Where are we? The anatomy of the murine cortical meninges revisited for intravital imaging, immunology, and clearance of waste from the brain. Prog Neurogibol. 2017;156:107‐148. [DOI] [PubMed] [Google Scholar]
- 128. Pedragosa J, Salas‐Perdomo A, Gallizioli M, et al. CNS‐border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol Commun. 2018;6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kim WK, Alvarez X, Fisher J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol. 2006;168:822‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mato M, Ookawara S, Sakamoto A, et al. Involvement of specific macrophage‐lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc Natl Acad Sci USA. 1996;93:3269‐3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Liu R, Cao S, Hua Y, Keep RF, Huang Y, Xi G. CD163 expression in neurons after experimental intracerebral hemorrhage. Stroke. 2017;48:1369‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Garton A, Gupta VP, Christophe BR, Connolly ES Jr. Biomarkers of functional outcome in intracerebral hemorrhage: interplay between clinical metrics, CD163, and ferritin. J Stroke Cerebrovasc Dis. 2017;26:1712‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Leclerc JL, Lampert AS, Loyola Amador C, et al. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J Cereb Blood Flow Metab. 2018;38:262‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Liu B, Hu B, Shao S, et al. CD163/hemoglobin oxygenase‐1 pathway regulates inflammation in hematoma surrounding tissues after intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2015;24:2800‐2809. [DOI] [PubMed] [Google Scholar]
- 135. Fang HY, Ko WJ, Lin CY. Inducible heat shock protein 70, interleukin‐18, and tumor necrosis factor alpha correlate with outcomes in spontaneous intracerebral hemorrhage. J Clin Neurosci. 2007;14:435‐441. [DOI] [PubMed] [Google Scholar]
- 136. Manaenko A, Fathali N, Chen H, et al. Heat shock protein 70 upregulation by geldanamycin reduces brain injury in a mouse model of intracerebral hemorrhage. Neurochem Int. 2010;57:844‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ran Q, Wadhwa R, Kawai R, et al. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res Comm. 2000;275:174‐179. [DOI] [PubMed] [Google Scholar]
- 138. Zhang G, Han M, Wang X, Xiao A. GRP75 involves in retinal ganglion cell apoptosis after rat optic nerve crush. Journal of molecular neuroscience : MN. 2015;56:422‐430. [DOI] [PubMed] [Google Scholar]
- 139. Yang L, Liu X, Hao J, et al. Glucose‐regulated protein 75 suppresses apoptosis induced by glucose deprivation in PC12 cells through inhibition of Bax conformational change. Acta Biochim Biophys Sin. 2008;40:339‐348. [DOI] [PubMed] [Google Scholar]
- 140. Lv LJ, Li J, Qiao HB, et al. Overexpression of GRP75 inhibits inflammation in a rat model of intracerebral hemorrhage. Molecular medicine reports. 2017;15:1368‐1372. [DOI] [PubMed] [Google Scholar]
- 141. Shen J, Zhou T, Li H, et al. Cab45s inhibits neuronal apoptosis following intracerebral hemorrhage in adult rats. Brain Res Bull. 2018;143:36‐44. [DOI] [PubMed] [Google Scholar]
- 142. Guo C, Geng Y, Song F, et al. Mild hypothermia protects rat neuronal injury after intracerebral hemorrhage via attenuating endoplasmic reticulum response induced neuron apoptosis. Neurosci Lett. 2016;635:17‐23. [DOI] [PubMed] [Google Scholar]
- 143. Zhuo F, Qiu G, Xu J, et al. Both endoplasmic reticulum and mitochondrial pathways are involved in oligodendrocyte apoptosis induced by capsular hemorrhage. Mol Cell Neurosci. 2016;72:64‐71. [DOI] [PubMed] [Google Scholar]
- 144. Chen‐Roetling J, Regan RF. Targeting the Nrf2‐heme oxygenase‐1 axis after intracerebral hemorrhage. Curr Pharm Des. 2017;23:2226‐2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wei CC, Kong YY, Li GQ, Guan YF, Wang P, Miao CY. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO‐1 signaling pathway. Sci Rep. 2017;7:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Dai S, Hua Y, Keep RF, Novakovic N, Fei Z, Xi G. Minocycline attenuates brain injury and iron overload after intracerebral hemorrhage in aged female rats. Neurobiol Dis. 2018. [DOI] [PubMed] [Google Scholar]
- 147. Pu J, Shi W, Wang Z, et al. Effects of minocycline on the expression of NGF and HSP70 and its neuroprotection role following intracerebral hemorrhage in rats. J Biomed Res. 2011;25:292‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shi W, Wang Z, Pu J, et al. Changes of blood‐brain barrier permeability following intracerebral hemorrhage and the therapeutic effect of minocycline in rats. Acta Neurochir Suppl. 2011;110:61‐67. [DOI] [PubMed] [Google Scholar]
- 149. Sinn DI, Chu K, Lee ST, et al. Pharmacological induction of heat shock protein exerts neuroprotective effects in experimental intracerebral hemorrhage. Brain Res. 2007;1135:167‐176. [DOI] [PubMed] [Google Scholar]
- 150. Huang Q, Lan T, Lu J, et al. DiDang tang inhibits endoplasmic reticulum stress‐mediated apoptosis induced by oxygen glucose deprivation and intracerebral hemorrhage through blockade of the GRP78‐IRE1/PERK pathways. Front Pharmacol. 2018;9:1423. [DOI] [PMC free article] [PubMed] [Google Scholar]