

Abstract
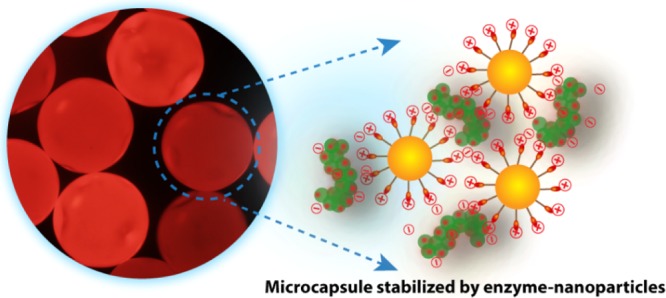
Enzyme immobilization is an essential prerequisite for biocatalysis. In this context, emulsion provides an excellent template for assembling enzymes at the oil–water interface. A microfluidic approach has been adopted to produce oil-in-water-type emulsions stabilized by gold nanoparticle–catalase conjugates. In situ ring-opening polymerization of the oil phase produces solid core enzyme-immobilized microcapsules (MCs). These resultant MCs exhibited a Km value of 42 mM and shows 1.1-fold higher activity compared to free enzymes. Finally, the robust MCs showed excellent recyclability, which can meet the demand of industrial biotechnological applications.
1. Introduction
Exploration for suitable catalysts has been expanded significantly from the last few decades because of the increasing demand of environmental friendly production in the industry. In this quest, biocatalysts have much to offer because of their ease of production, substrate specificity, and green chemistry. Progress in biotechnology has paved the way for the widespread application of biocatalysis in industrial organic synthesis.1−3 An example includes the production of high-fructose corn syrup by xylose isomerase that catalyzes the isomerization of d-glucose to d-fructose.4 In another instance, peroxidases are used to catalyze the synthesis of phenolic resins and replaced the use of conventional phenol formaldehydes.5 Despite the great potential of enzymes, their industrial applications have been restricted because of long-term stability; for example, denaturation or inactivation of enzymes by heat, proteolysis, or action of organic solvents. Furthermore, the recovery of enzymes and difficulty in reusability limit their application in the industry.6
To overcome these limitations, enzymes have been immobilized on various solid supports. Immobilization provides certain benefits—(i) storage and operational stability, (ii) easy and safer handling of enzymes, (iii) easy recovery, and (iv) reusability. In many instances, immobilization of enzymes also improves activity, selectivity, specificity, and resistance to inhibitors.7−11 Various methods have been developed in past decades to immobilize enzymes and can be summarized into following categories—support binding,12,13 cross-linking,14 and entrapment.15 In the case of carrier binding, it is important to have optimum interactions of enzymes with the support materials. Strong binding prevents enzyme leaching from carrier surfaces, but often times, it irreversibly deactivates the enzymes. On the other hand, weak binding cannot prevent enzyme leaching from the support materials during the operational process, thus hampering the reusability.16 An optimum binding can be achieved by self-assembly between enzymes and carriers through noncovalent interactions.17 To date, a variety of synthetic scaffolds have been used for enzyme immobilization through noncovalent interactions including nanoparticles,18 gels,19 macromolecules,20,21 and nanoreactors.22 In this context, emulsion is an attractive candidate for enzyme immobilization because of its ease of preparation and large-scale production. Recent studies by Rotello et al. have shown that emulsions can be used as a template for enzyme immobilization at the oil–water interface.23 A thin layer of enzyme–nanoparticle conjugates was assembled on the emulsion droplets, and the resulting microcapsules (MCs) showed high enzymatic activity. In their next approach, solidification of the core was achieved by cross-linking the oil phase to attain reusability.24 In both cases, synthesis of polydisperse emulsions limits their practical applications because many important properties such as enzyme kinetics, rheology, interparticle interactions, and shelf life largely depend on the size of the emulsions.
We address the limitation using a microfluidic device which can produce monodisperse emulsion in a simple way with high frequency. Herein, we report one-step fabrication of enzyme-immobilized polymeric MCs on a microfluidic platform. The micron-size oil-in-water emulsions were stabilized by gold nanoparticle–catalase conjugates, and simultaneously, the solidification of the oil core was achieved by the polymerization reaction using the Grubbs catalyst and dicyclopentadiene (DCPD) monomer. Later, we have demonstrated that the solidified scaffold retained high enzymatic activity as well as provided excellent recyclability. To the best of our knowledge, this is the first microfluidic synthesis of enzyme–nanoparticle-stabilized solid core MCs.
2. Experimental Section
2.1. Materials
Gold(III) chloride trihydrate (≥99.9% trace metal basis), catalase from bovine liver (lyophilized powder, 2000–5000 units/mg protein), Grubbs catalyst, DCPD, and 1,2,4-trichlorobenzene (TCB) were purchased from Sigma-Aldrich. Hydrogen peroxide (H2O2, 30%) was purchased from RANKEM. Millipore water (18.2 MΩ·cm at 25 °C) was used in all experiments. SU-8 2035 photoresist and developer solution were purchased from MicroChem for fabricating the silica master with designed microchannels. The polydimethylsiloxane (PDMS; SYLGARD 184) package was purchased from Dow corning corporation and used for fabricating the microfluidic chip.
2.2. Synthesis of Trimethylammonium Tetraethylene Glycol-Functionalized Au Nanoparticles
Cationic Au nanoparticles were prepared through a two-step process reported earlier.25 In brief, 100 mg of thiol ligand, that is, HS-C11-tetra(ethylene glycol)lyated trimethylammonium bromide, in 3 mL of dichloromethane was added to 20 mg of hydrophobic Au nanoparticles dispersed in 15 mL of dichloromethane. The resulting mixture was stirred in an oil bath at 35 °C for 2 days forming black precipitation. The black precipitate was then washed three times with dichloromethane and finally dissolved in Millipore water.
2.3. Microfluidic Device Fabrication
Microfluidic devices were fabricated using soft lithography.26 First, the master template was designed using AutoCAD software. Then, a negative photoresist SU-8 2035 was spin-coated onto a 4 × 4 silicon wafer, and the design was transferred on the wafers using maskless photolithography (X-pert SF100, Intelligent Instruments). The next step involves dipping the surface in the propylene glycol monomethyl ether acetate developer and rinsing by 2-propanol. Finally, the patterned surface was baked for 2 min to stabilize the structure. Figure S1a represents the structure of the master on silicon wafers. Next, PDMS 184 SYLGARD with a curing agent in a ratio of 10:1(w/w) was poured onto the wafer and degassed in a vacuum desiccator until the trapped air bubbles disappeared. The unit was placed in an oven at 60 °C for 1 h. Once cured, the microchannels imprinted on the PDMS mold were peeled from the wafer and punched with a biopsy punch with a diameter of 2.5 mm. PDMS was then plasma-bonded to a glass slide with oxygen plasma treatment in a radio frequency plasma cleaner system for 60 s. Finally, the device was placed in a 70 °C oven for 5 h and cooled to room temperature for further use.
2.4. Preparation of Polymerized Core Catalytic MCs
MCs were produced in a flow focusing microchannel device with one horizontal-Y inlet and one vertical-Y inlet channel. The channel dimension was 100 μm in depth and 200 μm in width with an aqueous solution flow rate of 200 μL/min and an oil-phase flow rate of 50 μL/min, respectively. Figure S1b represents a typical microfluidic channel used for this experiment. The aqueous phases consisted of the solution of cationic Au NPs (2 μM) and catalase (2 μM) in phosphate-buffered saline (PBS) buffer (pH 7.4). The oil phase comprised a Grubbs catalyst in toluene (1 mg/mL) and DCPD in TCB (2:3 v/v). The flow was controlled by syringe infusion pumps (Harvard Apparatus, catalog no. 703009) and was connected to the device by silicone tubing (inner diameter, 0.8 mm). Figure S2 represents the size distribution of enzyme-immobilized MCs.
2.5. Activity Assay
The enzymatic activity of the MCs was determined spectrophotometrically by using the hydrogen peroxide decomposition assay. In brief, 900 μL of hydrogen peroxide (10 mM) was added to solid core MCs, and decomposition was monitored for 80 s at 240 nm. The same experiment was also performed with the free enzyme. The assays were implemented in triplicates, and averages were reported.
3. Results and Discussion
The enzyme-immobilized MCs were consisted of enzymes, nanoparticles, and a polymerized oil core. Catalase from bovine liver was used as an enzyme component with a negative zeta potential of −6.1 ± 0.24 at pH 7.4 (pI = 5.4). The trimethylammonium tetraethyleneglycol-functionalized Au nanoparticles (Figure 1a) of approx. 4 nm of diameter with a positive zeta potential of 50.86 ± 3.20 were synthesized in order to bind with enzymes as well as to minimize the denaturation of enzymes upon binding.27 The solidified oil core was prepared via ring-opening metathesis polymerization by mixing the DCPD monomer with the first-generation Grubbs catalyst in TCB oil. To stabilize the oil-in-water emulsions, formation of low-valent enzyme–nanoparticle conjugates is highly desired.23 The reduction in interfacial tension by low-valent conjugates was evaluated using pendant drop tensiometry, with TCB as the major phase and water as the minor phase (Figure 2a). In a typical experiment, a drop of aqueous phase was introduced in TCB solution using a J-shaped needle and the interfacial tension between two fluids was measured for 1200 s.28 The interfacial tension of the water–TCB biphasic system was 49 mN/m. A small reduction in interfacial tension was observed when Au NPs (39 mN/m) were used as a surfactant to stabilize the emulsions. The lowest interfacial tension (12.4 mN/m) was observed for enzymes because of their low surface charges and their ability to form a viscoelastic film around the emulsion, providing stability to the droplets. Unlike NPs, the enzyme–NP conjugates exhibited a very low interfacial tension of 16.7 mN/m because of the formation of low-valent conjugates.
Figure 1.

(a) Chemical structure of (i) trimethylammonium tetraethyleneglycol-functionalized Au nanoparticles, (ii) catalase from bovine liver, (iii) first-generation Grubbs catalyst, and (iv) DCPD monomer. (b) Schematic illustration for one-step synthesis of enzyme–nanoparticle-immobilized MCs using a microfluidic device. (c) Cross-sectional view of polymer core oil-in-water emulsions stabilized by enzyme–nanoparticle conjugates at the interface.
Figure 2.

(a) Pendant drop tensiometry measurement of interfacial tension of the aqueous droplet in TCB. The interfacial assembly of Au NPs, enzymes, and enzyme–NP conjugates reduced the surface tension. Microscopy images of emulsions. (b) OM image of enzyme–NP-stabilized oil-in-water emulsion, (c) fluorescence microscopy image of Nile dye-encapsulated oil-in-water emulsion, (d) low-resolution TEM image of the emulsion, and (e) high-resolution TEM image of the emulsion shell.
In the current study, the enzyme–nanoparticle complex was formed inside the microchannel of a microfluidic device. The double Y-shaped microfluidic channel was fabricated in PDMS following a traditional soft lithography technique. The width and depth of the microchannel are 200 and 100 μm, respectively. It consisted of a horizontal-Y-shaped inlet, a vertical-Y-shaped inlet, and an outlet channel. The aqueous solution of Au NPs and enzyme (PBS buffer, pH 7.4) was passed through two arms of the horizontal-Y inlet channel with a flow rate of 200 μL/min. Simultaneously, TCB oil was passed through both arms of the vertical-Y inlet channel with a flow rate of 50 μL/min. We hypothesized that upon injection of nanoparticles and the enzyme through the horizontal-Y channel, low-valent complexes were formed through electrostatic interaction.29 As the oil phase sheared off the aqueous phase, the oil droplets were formed and concomitantly the low-valent complexes migrated to the oil–water interface, providing stabilization to the MCs.
The freshly prepared MCs exhibited a high level of monodispersity when examined under an optical microscope), as indicated by the narrow size distribution with a mean diameter of 742 ± 3.0 μm (Figure 2b). No MCs were formed when only NPs were passed through the horizontal Y-inlet channel. It proves that the formation of the low-valent enzyme–nanoparticle complex is the key for stabilization of emulsions. Figure 2c shows Nile red (oil soluble)-encapsulated fluorescent MCs which confirm the formation of oil-in-water-type emulsions. To investigate the nanoscopic structure, the emulsions were drop-cast on a transmission electron microscopy (TEM) grid, and the low-resolution TEM image (Figure 2d) showed the formation of a wrinkle upon drying of MCs. It revealed a membrane-like structure which was possibly formed because of extended cross-linking of nanoparticle–enzyme conjugates at the oil–water interface. The high-resolution TEM image in Figure 2e confirmed that the membrane is composed of closely packed Au nanoparticles.
After establishing the protocol, the next step was to synthesize the oil-in-water emulsions with a polymerized oil core. It was achieved by a co-flowing Grubbs catalyst and DCPD monomer in two different arms of the vertical-Y-shaped channel simultaneously. Upon mixing, polymerization occurred within 5 min, and polymerized MCs were collected in the Eppendorf tube for further characterization. The optical microscopy (OM) image in Figure 3a represented the formation of monodisperse MCs with an average diameter of 580 ± 10.0 μm (see the Supporting Information, Figure S2). It is clearly evident that the size of the polymerized MCs was reduced upon polymerization of the liquid core compared to nonpolymerized MCs. The fluorescence microscopy image (Figure 3b) showed the encapsulation of Nile red dye inside the solid core. The polymerized MCs were also examined under a scanning electron microscope, and the representative image in Figure 3c demonstrated the uniform size particles with an average diameter of 562 ± 12 μm. When a single MC was sectioned into pieces and examined under a scanning electron microscope, it clearly revealed the formation of a solid core upon polymerization (Figure 3d). The polymerization was further confirmed by IR spectroscopy as shown in Figure 4. The peak at 1570 and 1613 cm–1 is attributed to the C=C stretching frequency of the monomer, but a new broad peak appeared at 1700 cm–1 corresponded to the C=C stretching frequency of the ring-opened product. Similarly, the =C–H stretching frequency shifted from 939 to 971 cm–1 in the polymerized product.
Figure 3.

(a) OM image of the MCs with a polymerized core. (b) Fluorescence microscopy image of MCs. (c) Scanning electron microscopy (SEM) image of dried polymerized core MCs. (d) Cross-sectional SEM image of polymerized MCs.
Figure 4.
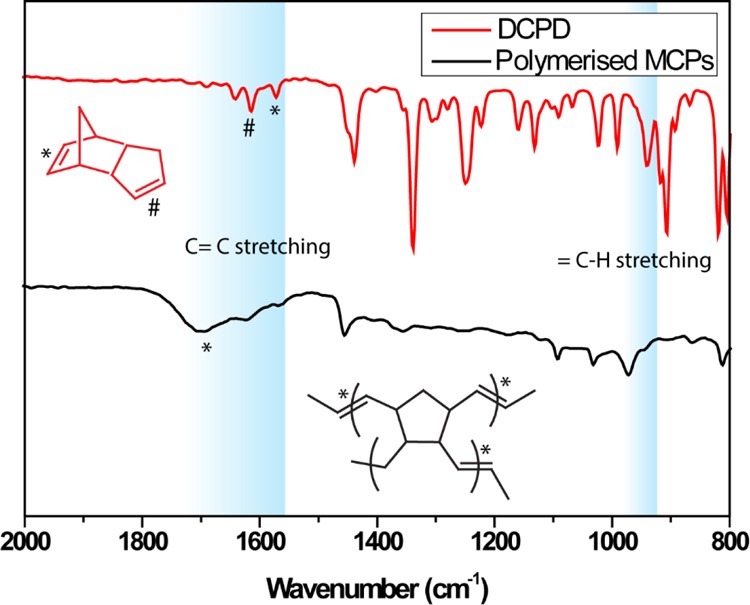
IR spectrum of DCPD and enzyme-immobilized polymerized MCs.
Next, the assessment of catalytic activity of enzyme-immobilized polymerized MCs was carried out using a standard protocol. The immobilization of catalase on MCs was quantified by the Coomassie (Bradford) protein assay, and the amount of residual enzymes in the supernatant solution was measured after the formation of MCs. The catalytic performance of catalase immobilized on MCs was determined by the decomposition of hydrogen peroxide in an aqueous medium.30 The activity of immobilized catalase was 1.1-fold higher compared to free enzymes in solution, with no detectable autodegradation of hydrogen peroxide (Figure 5a,b). This observation clearly demonstrated that the polymerization of the oil core has no adverse effect on catalytic activity. The slight enhancement in enzymatic activity might be due to the presence of hydrophobic environment.31 We have further evaluated the decomposition kinetics of H2O2 in the presence of polymerized MCs. Initial rates of H2O2 decomposition (see the Supporting Information, Figure S3) were obtained for various concentrations of H2O2 solution for MCs. Km, that is, affinity of the enzymes toward the substrate, was found to be 42.39 mM from the Lineweaver–Burk plot (Figure 5c) for the enzyme-immobilized solid core MCs prepared by the microfluidic device.
Figure 5.

(a) Activity assay of enzyme-immobilized MCs and free enzymes in hydrogen peroxide (10 mM). (b) Relative activity of free enzymes vs immobilized enzymes. (c) Lineweaver–Burk plot of the enzyme-immobilized MC rate as a function of hydrogen peroxide concentration.
The important feature of any heterogeneous catalyst is its ability to perform catalysis in multiple cycles without much compromising the activity. The recyclability of the MCs was examined through repeated identical enzymatic activity assays. As shown in Figure 6, the microparticles exhibited >95% retention of activity even after eight reaction cycles. The polymerization of the oil core trapped by the enzyme–nanoparticle conjugates at the emulsion interface and prevents the enzyme leaching during repetitive cycles.
Figure 6.
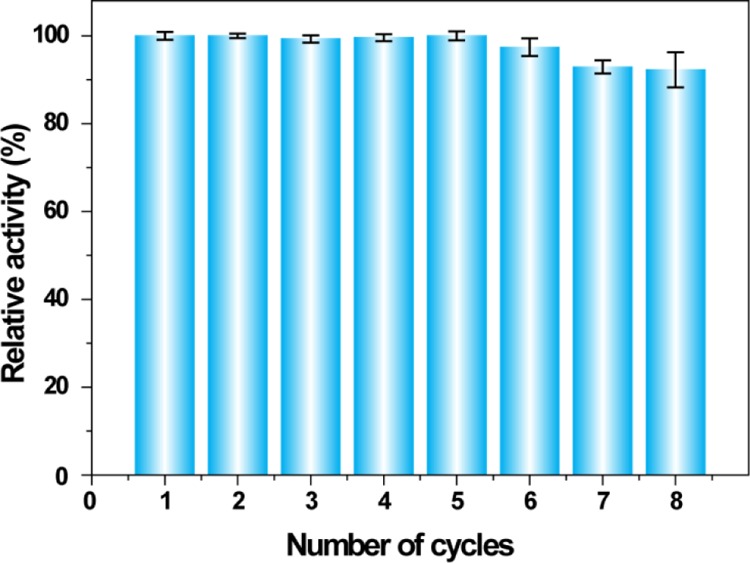
Reusability test of the MCs. The activity remains similar after eight catalytic cycles.
4. Conclusions
In summary, we have presented a microfluidic approach that enables one-step fabrication of enzyme-immobilized MCs through core polymerization of enzyme-nanoparticle-stabilized emulsions. The resulting monodisperse MCs retained their activity after multiple catalytic cycles. The extension of this approach for various biocatalytic applications is currently being explored.
Supporting Informationback
Digital image of master on a silicon wafer and a double Y-shaped microfluidic chip, droplet size distribution of polymerized MCs, and characterization details (PDF)
Acknowledgments
This research was supported by SERB-DST (ECR/2017/000442).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01321.
(PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Faber K.; Faber K.. Biotransformations in Organic Chemistry; Springer, 1992; Vol. 4. [Google Scholar]
- Tramper J. Chemical versus biochemical conversion: When and how to use biocatalysts. Biotechnol. Bioeng. 2000, 52, 290–295. . [DOI] [PubMed] [Google Scholar]
- Wandrey C.; Liese A.; Kihumbu D. Industrial biocatalysis: past, present, and future. Org. Process Res. Dev. 2000, 4, 286–290. 10.1021/op990101l. [DOI] [Google Scholar]
- Jensen V. J.; Rugh S.. [33] Industrial-scale production and application of immobilized glucose isomerase. Methods in Enzymology; Academic Press, 1987; Vol. 136, pp 356–370. [Google Scholar]
- Pokora A. R.; Cyrus W. L. Jr. Phenolic developer resins, US Patent 4,647,952, 1987.
- Homaei A. A.; Sariri R.; Vianello F.; Stevanato R. Enzyme immobilization: an update. J. Chem. Biol. 2013, 6, 185–205. 10.1007/s12154-013-0102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos J. C. S. d.; Barbosa O.; Ortiz C.; Berenguer-Murcia A.; Rodrigues R. C.; Fernandez-Lafuente R. Importance of the Support Properties for Immobilization or Purification of Enzymes. ChemCatChem 2015, 7, 2413–2432. 10.1002/cctc.201500310. [DOI] [Google Scholar]
- Rodrigues R. C.; Ortiz C.; Berenguer-Murcia Á.; Torres R.; Fernández-Lafuente R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. 10.1039/c2cs35231a. [DOI] [PubMed] [Google Scholar]
- Bilal M.; Asgher M.; Cheng H.; Yan Y.; Iqbal H. M. N. Multi-point enzyme immobilization, surface chemistry, and novel platforms: a paradigm shift in biocatalyst design. Crit. Rev. Biotechnol. 2019, 39, 202–219. 10.1080/07388551.2018.1531822. [DOI] [PubMed] [Google Scholar]
- Klibanov A. M. Enzyme stabilization by immobilization. Anal. Biochem. 1979, 93, 1–25. 10.1016/s0003-2697(79)80110-4. [DOI] [PubMed] [Google Scholar]
- Hernandez K.; Berenguer-Murcia A.; Rodrigues R.; Fernandez-Lafuente R. Hydrogen peroxide in biocatalysis. A dangerous liaison. Curr. Org. Chem. 2012, 16, 2652–2672. 10.2174/138527212804004526. [DOI] [Google Scholar]
- Jesionowski T.; Zdarta J.; Krajewska B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. 10.1007/s10450-014-9623-y. [DOI] [Google Scholar]
- D’souza S. Immobilized enzymes in bioprocess. Curr. Sci. 1999, 77, 69–79. [Google Scholar]
- Lee D. H.; Park C. H.; Yeo J. M.; Kim S. W. Lipase immobilization on silica gel using a cross-linking method. J. Ind. Eng. Chem. 2006, 12, 777–782. [Google Scholar]
- Betigeri S. S.; Neau S. H. Immobilization of lipase using hydrophilic polymers in the form of hydrogel beads. Biomaterials 2002, 23, 3627–3636. 10.1016/s0142-9612(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; van Pelt S. Enzyme immobilisation in biocatalysis: why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. 10.1039/c3cs60075k. [DOI] [PubMed] [Google Scholar]
- Keighron J. D.; Keating C. D. Enzyme: nanoparticle bioconjugates with two sequential enzymes: stoichiometry and activity of malate dehydrogenase and citrate synthase on Au nanoparticles. Langmuir 2010, 26, 18992–19000. 10.1021/la1040882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolatti E. P.; Valério A.; Henriques R. O.; Moritz D. E.; Ninow J. L.; Freire D. M. G.; Manoel E. A.; Fernandez-Lafuente R.; de Oliveira D. Nanomaterials for biocatalyst immobilization—state of the art and future trends. RSC Adv. 2016, 6, 104675–104692. 10.1039/c6ra22047a. [DOI] [Google Scholar]
- Wang Q.; Yang Z.; Gao Y.; Ge W.; Wang L.; Xu B. Enzymatic hydrogelation to immobilize an enzyme for high activity and stability. Soft Matter 2008, 4, 550–553. 10.1039/b715439a. [DOI] [PubMed] [Google Scholar]
- Borchmann D. E.; Carberry T. P.; Weck M. “Bio”-Macromolecules: Polymer-Protein Conjugates as Emerging Scaffolds for Therapeutics. Macromol. Rapid Commun. 2014, 35, 27–43. 10.1002/marc.201300792. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Wu S.; Liu X.; Hegazy M.; Wu G.; Huang X. Multifunctional and Programmable Modulated Interface Reactions on Proteinosomes. ACS Appl. Mater. Interfaces 2018, 10, 38565–38573. 10.1021/acsami.8b11216. [DOI] [PubMed] [Google Scholar]
- Peters R. J. R. W.; Louzao I.; van Hest J. C. M. From polymeric nanoreactors to artificial organelles. Chem. Sci. 2012, 3, 335–342. 10.1039/c2sc00803c. [DOI] [Google Scholar]
- Samanta B.; Yang X.-C.; Ofir Y.; Park M.-H.; Patra D.; Agasti S. S.; Miranda O. R.; Mo Z.-H.; Rotello V. M. Catalytic Microcapsules Assembled from Enzyme–Nanoparticle Conjugates at Oil–Water Interfaces. Angew. Chem., Int. Ed. 2009, 48, 5341–5344. 10.1002/anie.200901590. [DOI] [PubMed] [Google Scholar]
- Jeong Y.; Duncan B.; Park M.-H.; Kim C.; Rotello V. M. Reusable biocatalytic crosslinked microparticles self-assembled from enzyme-nanoparticle complexes. Chem. Commun. 2011, 47, 12077–12079. 10.1039/c1cc14448k. [DOI] [PubMed] [Google Scholar]
- You C.-C.; Miranda O. R.; Gider B.; Ghosh P. S.; Kim I.-B.; Erdogan B.; Krovi S. A.; Bunz U. H. F.; Rotello V. M. Detection and identification of proteins using nanoparticle–fluorescent polymer “chemical nose”sensors. Nat. Nanotechnol. 2007, 2, 318. 10.1038/nnano.2007.99. [DOI] [PubMed] [Google Scholar]
- Wan J. Microfluidic-based synthesis of hydrogel particles for cell microencapsulation and cell-based drug delivery. Polymers 2012, 4, 1084–1108. 10.3390/polym4021084. [DOI] [Google Scholar]
- Hong R.; Fischer N. O.; Verma A.; Goodman C. M.; Emrick T.; Rotello V. M. Control of Protein Structure and Function through Surface Recognition by Tailored Nanoparticle Scaffolds. J. Am. Chem. Soc. 2004, 126, 739–743. 10.1021/ja037470o. [DOI] [PubMed] [Google Scholar]
- Glogowski E.; Tangirala R.; He J.; Russell T. P.; Emrick T. Microcapsules of pegylated gold nanoparticles prepared by fluid– fluid interfacial assembly. Nano Lett. 2007, 7, 389–393. 10.1021/nl062581h. [DOI] [PubMed] [Google Scholar]
- Böker A.; He J.; Emrick T.; Russell T. P. Self-assembly of nanoparticles at interfaces. Soft Matter 2007, 3, 1231–1248. 10.1039/b706609k. [DOI] [PubMed] [Google Scholar]
- Sengupta S.; Dey K. K.; Muddana H. S.; Tabouillot T.; Ibele M. E.; Butler P. J.; Sen A. Enzyme Molecules as Nanomotors. J. Am. Chem. Soc. 2013, 135, 1406–1414. 10.1021/ja3091615. [DOI] [PubMed] [Google Scholar]
- Manoel E. A.; dos Santos J. C. S.; Freire D. M. G.; Rueda N.; Fernandez-Lafuente R. Immobilization of lipases on hydrophobic supports involves the open form of the enzyme. Enzyme Microb. Technol. 2015, 71, 53–57. 10.1016/j.enzmictec.2015.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.