

Abstract
There is an increasing interest in determining the role of ribosomal proteins (RPs) in the regulation of MDM2-p53 pathway in coordinating cellular response to stress. Herein, we report a novel regulatory role of ribosomal protein S25 (RPS25) in MDM2-mediated p53 degradation and a feedback regulation of S25 by p53. We demonstrated that S25 interacted with MDM2 and inhibited its E3 ligase activity, resulting in the reduction of MDM2-mediated p53 ubiquitination and the stabilization and activation of p53. S25, MDM2 and p53 formed a ternary complex following ribosomal stress. The nucleolar localization and MDM2-binding domains of S25 were critical for its role in MDM2-mediated p53 regulation. Knockdown of S25 by siRNA attenuated the induction and activation of p53 following ribosomal stress. S25 stabilized and cooperated with MDMX to regulate MDM2 E3 ligase activity. Furthermore, S25 was identified to be a transcriptional target of p53;p53 directly bound to S25 promoter and suppressed S25 expression. Our results suggest that there is a S25–MDM2–p53 regulatory feedback loop, which may have an important role in cancer development and progression.
Keywords: RPS25, p53, MDM2, ribosomal stress
INTRODUCTION
The tumor suppressor p53 protects cells from transformation and tumorigenesis by activating the transcriptional expression of many genes whose protein products induce cell growth arrest, apoptosis or senescence in response to stress signals.1 MDM2 negatively regulates p53 by inhibiting its transcriptional activity and promoting its ubiquitination and degradation.2–4 Mdm2 itself is a transcriptional target of p53 and deletion of p53 gene completely rescues the lethality of mdm2 knockout mice.5,6 Thus, MDM2 forms an autoregulatory feedback loop with p53 to regulate cellular homeostasis.7 Cytotoxic and genotoxic stressors induce modifications of both p53 and MDM2 proteins, which uncouple the MDM2–p53 interaction to stabilize and activate p53.8,9 Moreover, MDM2 is able to interact with other proteins independent of p53, thereby contributing to cellular responses to different stimuli.10
Disturbing ribosomal biogenesis triggers ribosomal stress (also called as nucleolar stress). Emerging evidence have revealed that p53 also has a crucial role in mediating the cellular response to ribosomal stress. Ribosomal proteins (RPs) are believed to have extraribosomal functions and participate in a surveillance network for cell growth, division and apoptosis.11 Studies over the past decade have established a critical role for RPs in mediating p53 signaling in response to ribosomal stress. A group of RPs has been revealed as the regulator of p53 in response to ribosomal stress.12–22 Disruption of ribosomal RNA synthesis, processing and RP imbalance activates p53 by increasing the interactions between RPs and MDM2.19,23,24 Knockdown of certain RPs including L5, L11 and S7 attenuates p53 signaling after ribosomal stress, indicating that these RPs have a non-redundant role in ribosomal stress-induced p53 activation. Ribosomal protein L26 has previously been demonstrated to increase the translation of p53 mRNA by binding to its 5’ untranslated region.25 Further studies reveal that L26 also inhibits the MDM2-mediated ubiquitination of p53, and the MDM2-mediated ubiquitination and degradation of L26 decreased p53 translation,26 suggesting that RPs employ similar but not the same mechanisms to regulate p53. Several RPs including S7,22 S27L21 and S27a20 have been demonstrated as the substrate of MDM2, indicating the mutual regulation between RPs and MDM2. In addition, it has been demonstrated that cancer-associated mutation in the MDM2 zinc finger domain (MDM2C305F) disrupts the interaction between ribosomal protein L11 and MDM2, resulting in the escape from L11-mediated negative regulation.27 Further study in transgenic mice has revealed that disruption of the RP–MDM2 interaction attenuates the p53-mediated response to ribosomal stress, and accelerates c-MYC-induced lympho-magenesis.28
The role of RPs in p53 regulation has not been completely understood. Zhang and Lu29 suggest there may be a common RPs–MDM2–p53 pathway involved in coordinating the inhibition of cell growth with cell-cycle arrest and apoptosis. Several RPs (L5, L11, L23, L26, S7, S27a and S27L) have been reported to regulate the MDM2–p53 pathway, but some RPs (L29, L30 and S12) do not bind to MDM2 and do not inhibit MDM2-mediated p53 degradation.20 We have been interested in studying the mechanism of MDM2–p53 interaction and have identified several novel MDM2-interacting proteins, including ribosomal protein S7,13 PA28γ30 and RYBP (RING1- and YY1-binding protein).31 In a yeast two-hybrid screening for novel MDM2-interacting proteins, we identified that human ribosomal protein S25 (RPS25) interacted with MDM2. The function of S25 has not been fully elucidated. It may be involved in viral replication of Dicistroviridae and hepatitis C viruses.32,33 S25 is overexpressed in human leukemia cells exhibiting adriamycin resistance.34 It may have a role in p53-mediated apoptosis35 and cell-cycle arrest.36 However, the mechanisms underlying these biological phenomena remain largely unknown.
In this study, we attempted to address the following questions: (1) Is the MDM2-mediated p53 degradation regulated by S25? (2) Does S25 affect p53 functions? (3) Is MDMX involved in the S25-MDM2-p53 interaction? and (4) Does p53 also regulate S25 expression? We anticipated that these studies would provide a basis for the existence of the S25–MDM2–p53 regulatory loop and shed lights on the role of S25 in human cancer development and progression.
RESULTS
S25 interacts with MDM2
Following identification of S25 as a novel MDM2-interacting protein in our yeast two-hybrid screening, we conducted several experiments to confirm the interaction between S25 and MDM2 in mammalian cells. First, ectopically expressed S25 bound to MDM2 in COS7 cells, as shown by co-immunoprecipitation (co-IP) and immunoblotting (IB) assays (Figure 1a). The binding between endogenous S25 and MDM2 was demonstrated in A549 cells (Figure 1b). We further revealed that the binding between S25 and MDM2 was direct by using pull-down assays (Figure 1c). To determine the sites responsible for the binding between S25 and MDM2, different deletion mutants of both S25 and MDM2 were cloned, and co-IP assays were performed. Our results indicated that an S25 fragment (amino acids 42–93) was able to bind to MDM2 (Figure 1d1), and that the acidic domain of MDM2 (amino acids 180–298) interacted with S25 (Figure 1d2; Supplementary Figure S1). S25 did not directly bind to p53 but p53 was co-immunoprecipitated with S25 when MDM2 was co-expressed (Figure 1e), indicating that S25 formed a ternary complex with p53 through MDM2. We further examined the binding of S25, MDM2 and p53 after actinomycin D (Act D) treatment and found that Act D increased the interactions of S25 with MDM2 and p53 in HCT116 p53+/+ cells (Figure 1f, left panel). We also demonstrated the enhanced binding between MDM2 and S25 in HCT116 p53−/− cells after Act D treatment (Figure 1f, right panel). Taken together, these results suggest that S25 directly interacts with MDM2 and forms a complex with p53 in response to ribosomal stress.
Figure 1.
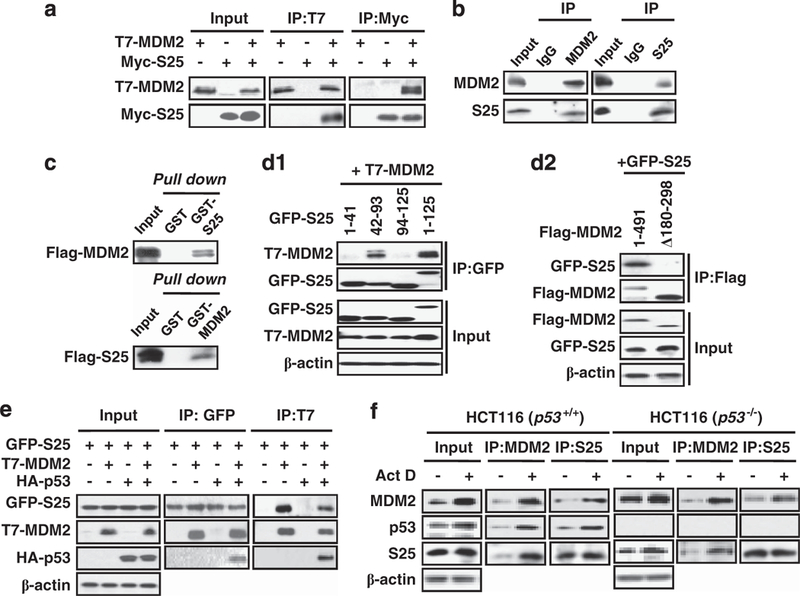
S25 interacts with MDM2. (a) The interaction between exogenous MDM2 and S25. COS7 cells were transfected with T7-MDM2, Myc-S25 or both for 24 h. Cell lysates were then either immunoprecipitated with Myc and T7 antibodies or immunoblotted with Myc and T7 antibodies. (b) The binding between endogenous S25 and MDM2 in A549 cells. The cell lysates were immunoprecipitated with mouse IgG or an antibody against MDM2 or S25, then the immunoprecipitates were further separated by SDS–PAGE and the target proteins were detected with antibodies against S25 and MDM2. (c) S25 binds to MDM2 in vitro. GST, GST-MDM2 or GST-S25 proteins were incubated with in vitro-translated Flag-S25 or Flag-MDM2 at 4 °C overnight. The bound proteins were separated using GST beads and detected by IB with a Flag antibody. (d) Mapping the binding domains in S25 and MDM2. COS7 cells were transfected with combinations of plasmids as indicated in the figure. Whole cell lysates were subjected to IP with a GFP (d1) or Flag (d2) antibody followed by IB with T7 and GFP. (e) S25 forms a ternary complex with p53 through MDM2. COS7 cells were transfected with combinations of plasmids as indicated in the figure. Whole cell lysates were subjected to IP with GFP or T7 antibodies followed by IB with T7, HA and GFP. (f) Ribosomal stress enhances the S25–MDM2 interaction. HCT116 p53+/+ and p53−/− cells were exposed to Act D (5 nM) for 6h. Whole cell lysates were then immunoprecipitated with an MDM2 or S25 antibody. Target proteins in the immunoprecipitates were separated by SDS-PAGE and detected by IB.
S25 stabilizes and activates p53
Considering that MDM2 is a negative regulator of p53, we hypothesized that S25 would regulate p53 stability and function through MDM2-mediated mechanism. As shown in Figure 2a, ectopic overexpression of S25 upregulated the level of endogenous p53 in several cancer cell lines. The MDM2 levels were also increased, independent of p53, as ectopic expression of S25 increased MDM2 protein level in p53-negative HCT116 cells (Figure 2a). To determine the effect of S25 on p53 localization, GFP-S25 was transfected into U2OS cells, followed by immunofluorescence staining with an anti-p53 antibody. As shown in Figure 2b, S25 mainly resided in the nucleolus and overexpression of S25 increased nuclear accumulation of p53, while GFP resided in both the nuclear and cytoplasm and its overexpression had minimal effects on p53 nuclear import. To determine whether S25 acts on p53 and MDM2 at the post-translational level, S25 was overexpressed in HCT116 p53+/+ and p53−/− cells, respectively, followed by exposure to the protein synthesis inhibitor cycloheximide (CHX, 10 μg/ml) for various times. The p53 and MDM2 proteins were stabilized in cells transfected with S25, compared with the cells transfected with the control vector (Figures 2c1 and c2). To determine whether ectopic S25 affects MDM2 transcription, we transfected HCT116 p53+/+ and HCT116 p53−/− cells with S25 vector and detected the expression levels of MDM2 mRNA. As shown in Figure 2d, S25 overexpression upregulated MDM2 mRNA level in HCT116 p53+/+ but not in HCT116 p53−/− cells, indicating that increased MDM2 mRNA is due to p53 activation and that S25 does not have direct effect on MDM2 mRNA level.
Figure 2.
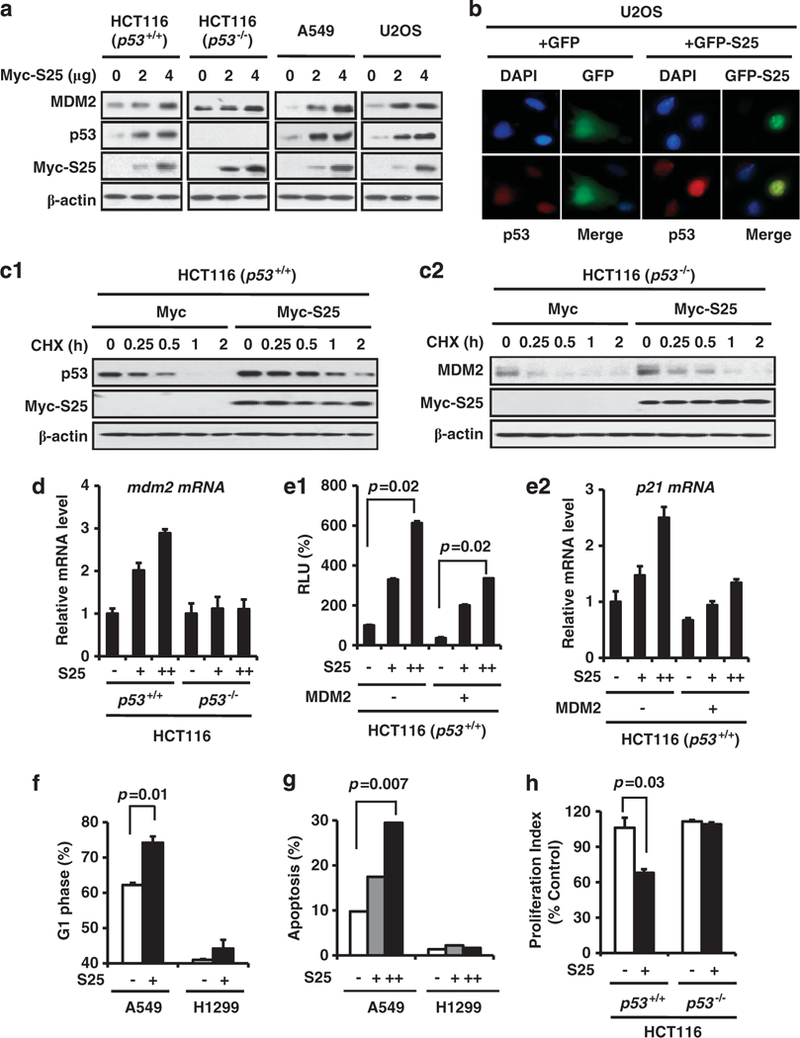
S25 stabilizes and activates p53. (a) Ectopic expression of S25 stabilizes endogenous p53. HCT116 (p53+/+), HCT116 (p53−/−), A549 and U2OS cells were transfected with a Myc-S25 plasmid for 24 h. The protein levels of MDM2 and p53 were assessed by IB. (b) S25 enhances the nuclear accumulation of p53. U2OS cells were transfected with GFP or GFP-S25 plasmid for 24 h. The cells were fixed and stained with p53 antibody, followed by staining with a secondary goat anti-mouse IgG AlexaFluor-594 antibody. (c) S25 prolongs p53 and MDM2 protein half-life. (c1) HCT116 p53+/+ and (c2) HCT116 p53−/− cells were transfected with Myc-S25 or control vectors, followed by exposure to the protein synthesis inhibitor CHX (10 μg/ml) for various times. Target proteins were detected by IB. The relative p53 and MDM2 levels at different time points were plotted. (d) HCT116 p53+/+ and HCT116 p53−/− cells were transfected with Myc-S25 plasmid. The MDM2 mRNA levels were determined by real-time PCR analyses. (e) S25 increases the transactivational activity of p53. A549 cells were co-transfected with Myc-S25 and p21waf1 luciferase reporter plasmids in the presence or absence of ectopically expressed MDM2 (e1). The luciferase assay was performed 24 h later. Relative luciferase units are the mean values ± s.d. of duplicates. In a separate experiment, the levels of p21 mRNA were determined by real-time PCR (e2). (f) A549 and H1299 cells were transfected with a GFP or GFP-S25 vector for 24 h and then the GFP-positive populations were examined for their DNA content. (g) A549 and H1299 cells were transfected with various amounts of Myc-S25 for 24 h. The apoptotic cells were detected by Annexin V/PI staining and flow cytometry. (h) HCT116 (p53+/+) and HCT116 (p53−/−) cells were incubated with a Myc-S25 vector. Cell proliferation was determined by the BrdUrd incorporation assay.
To examine the consequences of p53 stabilization by S25, we transiently transfected hCt116 p53+/+ cells with S25 and the p21waf1 promoter luciferase reporter (an indicator of p53 activity), and the data showed that S25 increased the p21 luciferase activity in a dose-dependent manner (Figure 2e1). The activity of p21 luciferase was inhibited by overexpressed MDM2, which was partially reversed by S25 co-transfection (Figure 2e1). We further confirmed that S25 overexpression increased p21 mRNA level and reversed MDM2-mediated downregulation of p21 mRNA level (Figure 2e2). To determine the biological significance of S25-mediated p53 transactivation, we transfected A549 (p53 wt) and H1299 (p53 null) cells with S25 and examined the cell-cycle distribution and apoptosis in the transfected cells. As shown in Figures 2f and g, the overexpression of S25 induced G1-phase arrest and increased apoptosis in A549 cells, but not in H1299 cells. Ectopic expression of S25 inhibited cell proliferation in HCT116 p53+/+ but not in HCT116 p53−/− cells (Figure 2h).
Nucleolar localization and MDM2-binding domains of S25 are critical for p53 regulation
We identified that the nucleolar location signal sequence was in the N terminus of S25 using a set of S25 deletion mutants (Figure 3a). With the deletion of amino-acid residues 1–41, the GFP fusion protein was not shown in the nucleus (Figure 3b). To determine the role of S25 nucleolar location in stabilizing p53, Mdm2−/−p53−/− mouse embryonic fibroblast (MEF) cells were transfected with p53, MDM2 and S25 deletion mutants. As shown in Figure 3c1, the S25 deletion mutant (amino acids 1–93), which includes both the nucleolar location signal sequence and MDM2-binding domain, stabilized p53 as efficiently as full-length S25. We further demonstrated that the S25 deletion mutant containing amino acids 1–93, but not other S25 deletion mutants, was able to reverse the MDM2-mediated inhibition of p21 luciferase activity and mRNA as efficiently as full-length S25 (Figures 3c2 and c3), which indicates that MDM2 interaction and nucleolar location are critical for S25-mediated stabilization and activation of p53.
Figure 3.
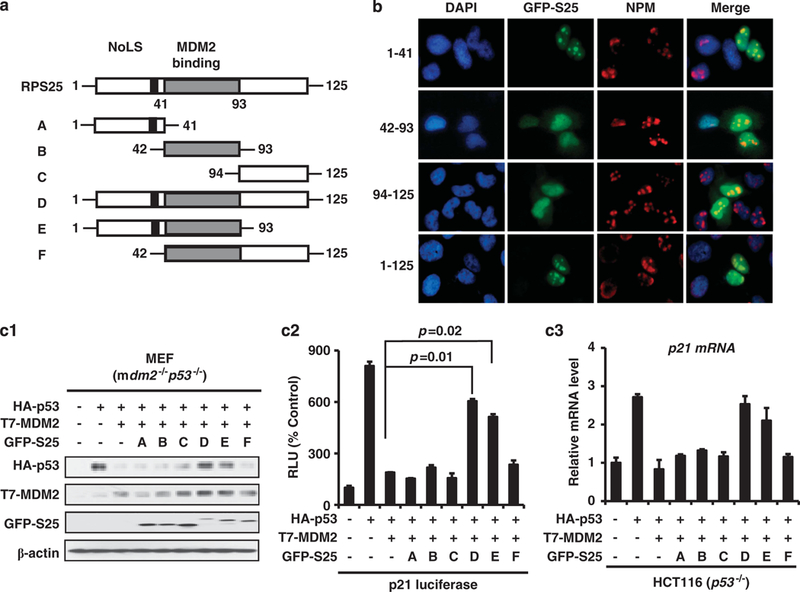
Nucleolar localization and MDM2-binding domains of S25 are critical for p53 regulation. (a) S25 mutant deletions. (b) The nucleolar localization signal is in the N terminus of S25. U2OS cells were transfected with GFP-S25 or mutant deletions of S25 for 24 h. The cells were fixed and stained with mouse anti-human nucleophosmin (NPM) antibody, followed by staining with a secondary goat anti-mouse IgG AlexaFluor-594 antibody, and the GFP signal was detected by fluorescence microscopy. (c) Nucleolar localization and MDM2-binding domains of S25 are critical for p53 stabilization and activation. (c1) Mdm2−/− p53−/− MEF cells were transfected with HA-p53, T7-MDM2 and GFP-S25 mutant deletions, then were collected and lysed. The whole cell lysates were subjected to IB with the indicated antibodies. (c2) Mdm2−/− p53−/− MEF cells were transfected with HA-p53, T7-MDM2, GFP-S25 mutant deletions and p21waf1 luciferase reporter plasmids as indicated. The luciferase assay was performed 24 h later. The relative luciferase units are the mean values ± s.d. of duplicates. (c3) HCT116 p53−/− cells were transfected with HA-p53, T7-MDM2 and GFP-S25 mutant deletions, the changes in the expression of p21 mRNA were determined by quantitative PCR analyses.
S25 inhibits the MDM2-mediated ubiquitination of p53
We next determine the mechanism by which S25 stabilizes p53. S25-MDM2 interaction may abolish the effect of MDM2 on p53 ubiquitination. To test this, HCT116 p53−/− cells were co-transfected with p53, MDM2 and S25, with or without ubiquitin. MDM2 transfection resulted in an increase in ubiquitinated p53, while S25 co-transfection decreased the ubiquitination of p53 (Figure 4a). This in-vivo effect of S25 on p53 ubiquitination was also validated in mdm2−/− p53−/− MEF cells by an immunoprecipitation assay (Figure 4b). We further examined the inhibitory effect of S25 on the ubiquitination of endogenous p53 in HCT116 cells. As shown in Figure 4c, the presence of exogenous S25 decreased the MDM2-associated p53 ubiquitination.
Figure 4.
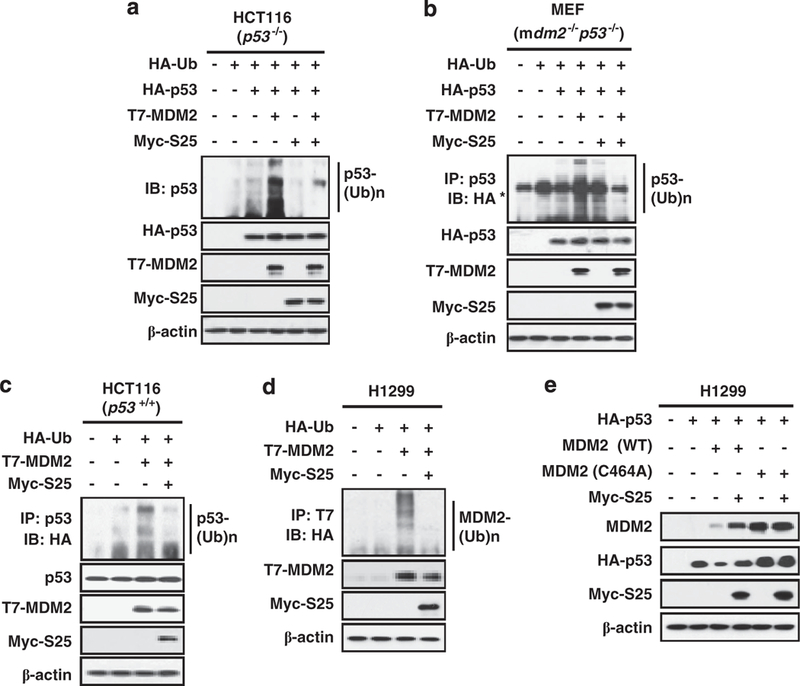
S25 innibits the MDM2-mediated ubiquitination of p53. (a) HCT116 p53−/− cells were transfected with combinations of HA-Ub, HA-p53 and T7-MDM2, with or without Myc-S25 plasmids, as indicated for 24 h. The cells were treated with MG132 (20 μM) for 2h before harvesting. The cell lysates were subjected to IB with the antibody against p53. (b) S25 inhibits the ubiquitination of exogenous p53. Mdm2−/−p53−/− MEF cells were transfected with combinations of HA-Ub, HA-p53 and T7-MDM2 with or without Myc-S25 plasmids, as indicated, for 24 h. The cells were treated with MG132 (20 μM) for 2 h before harvesting. The cell lysates were subjected to IP with an anti-p53 antibody, followed by IB with an anti-HA antibody. Note (*) indicates a non-specific band. (c) S25 inhibits the ubiquitination of endogenous p53. HCT116 cells were transfected with HA-Ub, T7-MDM2 and Myc-S25 plasmids as indicated in the figure. The cells were treated with MG132 (20μM) for 2 h before harvesting. Whole cell lysates were subjected to IP with an anti-p53 antibody, followed by IB with an anti-HA antibody to detect ubiquitinated p53. (d) S25 inhibits the ubiquitination of MDM2. H1299 cells were transfected with HA-Ub, T7-MDM2 and Myc-S25 as indicated in the figure for 24 h. The cells were treated with MG132 (20 μM) for 2 h before harvesting. Whole cell lysates were subjected to IP with an anti-T7 antibody, followed by IB with an anti-HA antibody to detect ubiquitinated MDM2. (e) S25 inhibits the E3 ligase activity of MDM2. H1299 cells were transfected with HA-p53 and GFP-S25 in the presence of wild-type MDM2 or a mutant MDM2 without E3 ligase activity (C464A). The cells were harvested 24 h after transfection, and the cell lysates were subjected to IB with the indicated antibodies.
To determine whether the effects of S25 on p53 ubiquitination are associated with the inhibition of the E3 ligase activity of MDM2, S25 and MDM2 were co-transfected into H1299 cells. As shown in Figure 4d, the autoubiquitination of MDM2 was markedly decreased in the presence of S25. We confirmed this effect by using wild-type or mutant MDM2 without E3 ligase activity (464A). Transfection with S25 prevented the decrease of p53 in cells transfected with wild-type MDM2, whereas no apparent change in the expression of p53 was observed in cells transfected with the mutant MDM2 (Figure 4e).
Knockdown of S25 attenuates p53 induction and activation
To demonstrate that S25 is essential for the regulation of p53 in response to ribosomal stress, S25 was knocked down by siRNA. As shown in Figure 5a, exposure to Act D triggered ribosomal stress and subsequent activation of p53 in A549 cells. The introduction of S25 siRNA into A549 cells attenuated the Act D-induced p53 stabilization and significantly decreased the level of MDM2. The similar effect of S25 knockdown on p53 induction by 5-fluorouracil (5-FU) was also observed (Figure 5b). We further determined whether S25 is needed for p53 stability under genotoxic stress. As shown in Figure 5c, ablation of S25 resulted in a reduction of p53 after exposure to etoposide (Eto).
Figure 5.
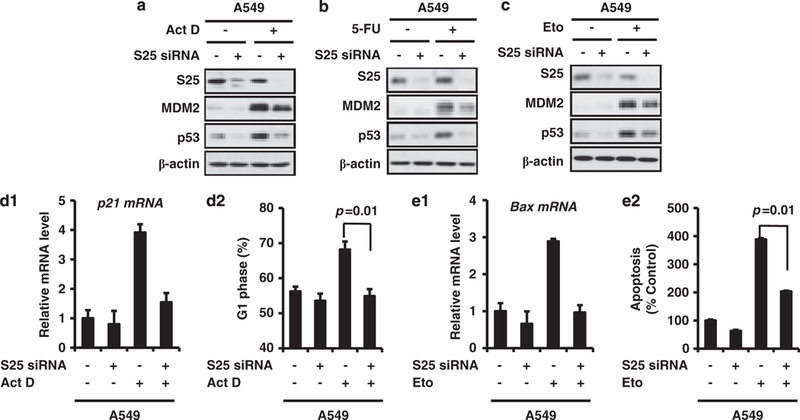
S25 knockdown attenuates the stabilization and activation of p53. (a) A549 cells were transfected with 50 nM control siRNA or S25 siRNA for 48 h. Then, cells were treated with Act D (5 nM) for 6 h or with 5-FU (100 μM) (b) or etoposide (Eto, 50 μM) (c) for 24 h before harvesting, and cell lysates were subjected to IB with the indicated antibodies. (d) S25 ablation inhibits Act D-induced p21 upregulation and G1 arrest. A549 cells were transfected with 50 nM control siRNA or S25 siRNA for 24 h. The cells were exposed to Act D (5 nM) for 24 h before harvesting. Cells were then stained with PI and examined for their DNA content (d2). In a separate experiment, p21 mRNA levels were determined by quantitative PCR analyses (d1). (e) S25 ablation inhibits etoposide-induced Bax expression and apoptosis. A549 cells were transiently transfected with 50 nM of a control or S25 siRNA pool for 24 h, followed by treatment with etoposide (50 μM) for 24 h. The apoptotic cells were detected by flow cytometry (e2). In a separate experiment, Bax mRNA levels were determined by quantitative PCR analyses (e1).
To determine if S25 is vital for the function of p53 under ribosomal stress, A549 cells were transfected with S25 siRNA and then treated with Act D or etoposide. The results from quantitative PCR and cell-cycle analysis indicated that Act D induced an increase in p21 mRNA level and G1 phase arrest in A549 cells, which was abrogated by S25 siRNA, but not by control siRNA (Figures 5d1 and d2). S25 siRNA also inhibited etoposide-induced Bax expression and apoptosis in A549 cells (Figures 5e1 and e2).
MDMX facilitates S25–MDM2 interaction to regulate MDM2 E3 ligase activity
The MDM2 homolog, MDMX, is an important regulator of p53 by cooperating with MDM2 to promote p53 poly-ubiquitination.37 MDMX is also a substrate of MDM2-mediated ubiquitination and subsequent proteasomal degradation.38 To test our hypothesis that the inhibition of MDM2 E3 ligase activity by S25 will stabilize MDMX, we overexpressed S25 in U2OS and HCT116 cells, and determined the level of MDMX protein. Ectopic expression of S25 stabilized MDMX in both U2OS and HCT116 cells (Figure 6a). To determine the role of MDMX in S25-mediated inhibition of MDM2 E3 ligase activity, mdm2−/− p53−/− MEF cells were transfected S25, MDMX and MDM2. Our IB results revealed that MDMX and S25 act synergistically to stabilize MDM2 (Figure 6b). To further demonstrate the importance of MDMX in S25-mediated inhibition of MDM2 E3 ligase activity, we transfected both mdm2−/− p53−/− and mdmx−/− p53−/− MEF cells with exogenous MDM2 and S25. As shown in Figure 6c, ectopic expression of S25 stabilized MDM2 in both cell lines, indicating that S25–MDM2 interaction does not rely on MDMX.
Figure 6.
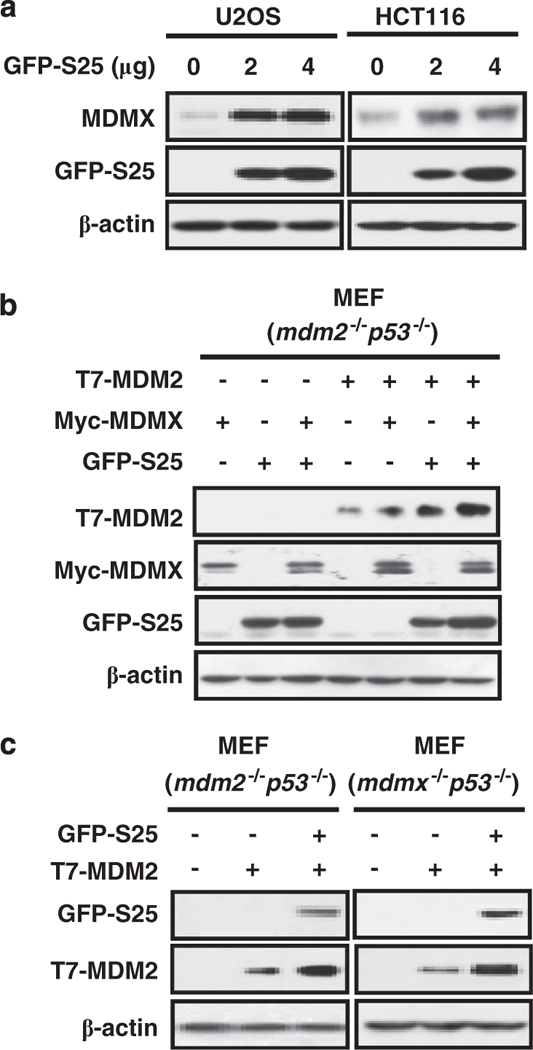
MDMX is involved in S25-mediated regulation of MDM2 E3 ligase activity. (a) S25 stabilizes MDMX. U2OS and HCT116 cells were transfected with a GFP-S25 plasmid for 24 h. The protein levels of MDMX were assessed by IB. (b) Mdm2−/− p53−/− MEF cells were transfected with combinations of T7-MDM2, GFP-S25 and Myc-MDMX plasmids as indicated. Cells were harvested 24 h after transfection, and cell lysates were subjected to IB with the indicated antibodies. (c) Mdm2−/−p53−/− and mdmx−/−p53−/− MEF cells were transfected with T7-MDM2 and GFP-S25 plasmids as indicated. Cells were harvested 24 h after transfection, and cell lysates were subjected to IB with the indicated antibodies.
p53 negatively regulates S25 transcription
We treated HCT116 (p53+/+ and p53−/−) cells with Act D for various times and found that Act D dramatically decreased the expression of S25 in a time-dependent manner in p53-positive HCT116 cells, but not in p53-negative HCT116 cells (Figure 7a). The inhibitory effect of Act D on S25 protein level was also confirmed in U2OS (p53 wt) cells (Supplementary Figure S2a). We further demonstrated that Act D-associated S25 inhibition was not related to its degradation, as treating the cells with Act D and MG132 together, could not restore S25 expression (Figure 7b).
Figure 7.
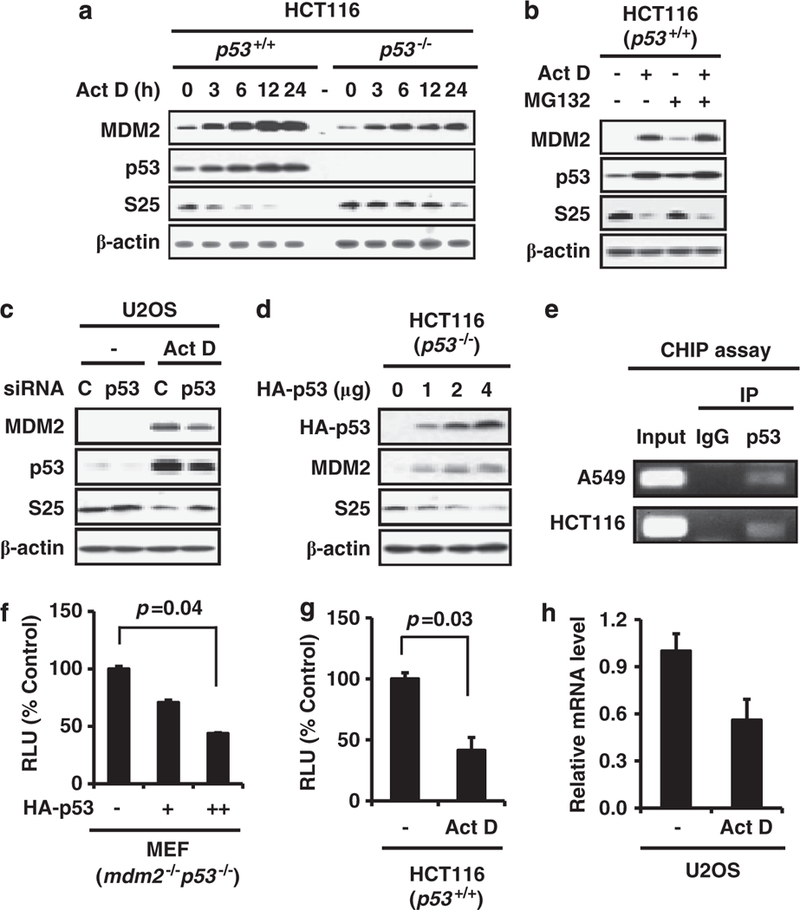
p53 suppresses the transcription of S25. (a) HCT116 (p53 +/+ and p53 –/–) cells were seeded and allowed to attach overnight, then treated with Act D (5 nM) for different time points, as indicated. The cells were harvested, and the cell lysates were subjected to IB with specific antibodies. (b) HCT116 cells were treated with Act D (5nM) for 24 h, then MG132 (20 μm) was added for another 6h. The cells were then harvested, and the whole cell lysates were subjected to IB. (c) U2OS cells were transfected with control (C) or p53 siRNA (50 nM) for 24 h, followed by treatment with Act D (5nM) for another 24 h. The cells were harvested, and the whole cell lysates were subjected to IB with specific antibodies. (d) HCT116 (p53–/–) cells were transfected with increasing amounts of p53. The level of S25 protein was determined 24 h later. (e) HCT116 and A549 cell lysates were subjected to IP with a p53 antibody or IgG. The immunoprecipitated DNA was eluted and subjected to PCR assays with primers specific for the S25 promoter fragment spanning the p53 binding site. (f) Mdm2–/– p53–/– MEF cells were transfected with increasing amounts of p53 and S25 luciferase reporter plasmids as indicated. The luciferase assay was performed 24 h later. (g) HCT116 cells were transfected with the S25 luciferase reporter plasmid for 24 h, and treated with Act D (5 nM) for another 24 h. The cells were then harvested, and the luciferase assay was performed. The relative luciferase units are the mean values ±s.d. of duplicates. (h) U2OS cells were treated with Act D (5nM) for 24 h. The cells were harvested, and the total RNA was isolated, followed by reverse transcription to cDNA. The relative level of S25 mRNA was measured by real-time PCR, and was normalized to that of GAPDH.
To confirm that the observed decrease in S25 after Act D treatment was due to p53 activation, U2OS cells were treated with p53 siRNA, followed by Act D treatment. As shown in Figure 7c, p53 knockdown abrogated the decrease in S25 by Act D. In addition, we overexpressed p53 in HCT116 p53−/− cells and found that p53 decreased S25 protein level in a dose-dependent manner (Figure 7d). To elucidate the inhibitory effect of p53 on S25 expression, we performed a computational prediction of the potential p53 binding site in the S25 promoter. A consensus p53 binding site with several mismatches was found in the S25 promoter region, which located between the −893 and −861 bases of the S25 gene (Supplementary Figure S2b). We confirmed the binding of p53 to S25 promoter by chromatin immunoprecipitation assay in both HCT116 and A549 cells (Figure 7e). S25 promoter was cloned and constructed in a luciferase reporter plasmid system. Over-expression of p53 in mdm2−/−p53−/− MEF cells decreased S25 luciferase activity (Figure 7f). We also examined the effect of Act D on S25 luciferase activity. Consistent with the overexpression study, p53 activation triggered by Act D, also decreased S25 luciferase activity (Figure 7g). We further demonstrated that Act D decreased S25 mRNA level in U2OS cells (Figure 7h), which indicates p53 activation suppresses S25 gene transcription.
DISCUSSION
In this study, we identified and validated RPS25 as a novel MDM2-interacting protein. The interaction between S25 and MDM2 resulted in p53 protein stabilization and functional activation. We provided at least five new discoveries in this study. First, S25 directly bound to the central domain of MDM2 and formed a ternary complex with p53. Second, S25 inhibited MDM2-mediated p53 ubiquitination by suppressing the E3 ligase activity of MDM2. Third, MDMX was involved in the S25–MDM2–p53 interaction. Fourth, S25 was a transcriptional target of p53 and there was an S25-MDM2-p53 feedback regulatory loop. Finally, S25-mediated p53 activation had an important role in cellular response to ribosomal stress. These results may help us better understand the role of RPs in p53 regulation and cancer development and progression in general. Considering that MDM2 is highly expressed in various human cancers and can be used as an ideal target for anticancer drugs. The identification of S25–MDM2 interaction also provides a potential target for anticancer drug development.
Accumulating evidence suggests that p53, together with RPs, takes part in a surveillance mechanism for genome integrity and protein translation, and is activated by ribosomal stress.29 S25 positively regulates p53, acting as a tumor suppressor. S25 exerts its function by regulating the MDM2–p53 interaction. The MDM2 central domain is critical for p53 ubiquitination and degradation.39,40 S25 binding to this domain may abolish MDM2-mediated ubiquitination and subsequent degradation of p53, resulting in p53 activation and induction of p53-mediated cell-cycle arrest and apoptosis. Thus, our findings provide mechanistic insight into the role of S25 in regulating p53 function. In the current study, although we demonstrated that S25 alone was sufficient to interact with MDM2 and inhibit its E3 ligase activity, it needs to be further investigated whether any other cofactors are also involved in this process (as observed in L5 and L11).41
It has been shown that amino-acid deprivation increased S25 mRNA in a p53-dependent manner, and the enhanced S25 mRNA was maintained in the nucleus by p53.35,42 Amino-acid starvation also decreased S25 protein level in p53 wild-type but not in p53 null cells, indicating that p53 induces S25 transcription but inhibits its translation upon amino-acid deprivation. In opposition to their results, we found p53 suppressed S25 transcription. The levels of RPs are generally regulated by both their efficiency of translation and their rate of turnover. We excluded the possibility of proteasome-mediated degradation of S25, as blocking this process could not reverse the S25 inhibition observed in our study. We found that overexpression or induction of p53 decreased S25 mRNA level by p53 direct binding to S25 promoter and suppressing its transcription. The differences between our study and the others may be partially explained by the cell models used and conditions for cell stress. Increasing evidence suggests that p53 may have a critical role in regulating ribosomal biogenesis.29,43,44 In this study, we demonstrated that in response to ribosomal stress, p53 activation directly inhibited the transcription of RPS25, which added additional evidence for the importance of p53 in coordinating ribosomal biogenesis and cell-cycle control. Of note, p53 was already activated at 3 h after ribosomal stress and S25 protein level began to decrease at 3–6 h after ribosomal stress, which indicates that at the early phase of ribosomal stress, RP(s)-mediated activation of p53, in turn, inhibits the production of RP, to delay ribosomal biogenesis and also prevent the overactivation of p53 (Figure 8).
Figure 8.
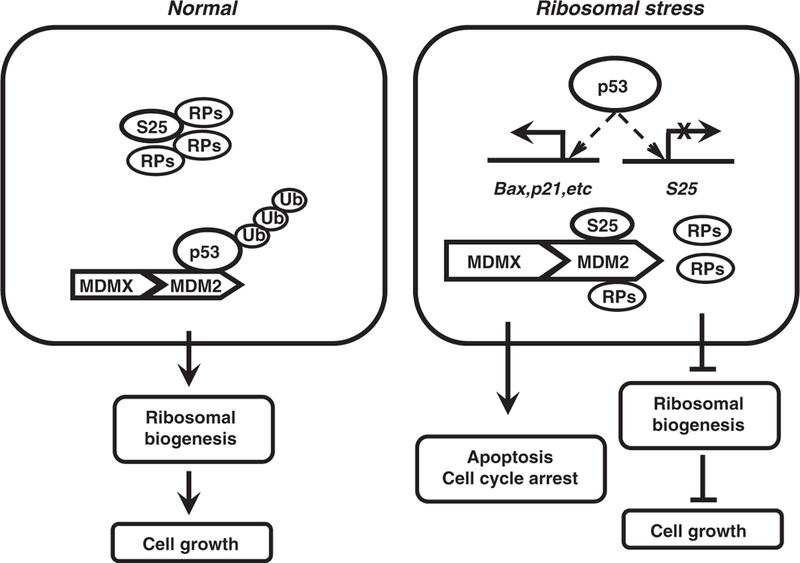
Proposed mode of action for S25–MDM2–p53 interaction. Under normal conditions, p53 is strictly maintained at low level by MDM2 (with the assistance of MDMX) and RPs majorly participate in the assembly of translation machinery (left panel). Under ribosomal stress, the free RPs (S25 and other counterparts) interact with MDM2 to inhibit its E3 ligase activity, stabilizing and activating p53 (right panel). The activated p53 induces the expression of cell cycle and apoptosis regulators (p21, Bax, and so on) and inhibits the production of the ribosomal biogenesis components (ribosomal RNA, RP and other factors), coordinating cell growth, cell-cycle control and apoptosis.
There is an interest in studying the relation between MDMX, MDM2 and p53.45 MDMX enhances MDM2-mediated p53 ubiquitination and degradation by forming hetero-oligomers with MDM2.46,47 However, MDMX may stabilize and activate p53 through other mechanisms.48–50 MDMX regulates p53 in response to ribosomal stress.51 Exogenous L11 activates the MDM2-mediated ubiquitination of MDMX, while L11 ablation reduces the downregulation of MDMX after Act D treatment. Zhu et al.22 report that MDMX facilitates the inhibition of the MDM2 E3 ligase activity by S7. In our study, we observed that S25 stabilized MDMX and that MDMX cooperated with S25 to inhibit MDM2 E3 ligase activity, which was different from the effects observed with L11 but consistent with that observed with S7, indicating that RPs may employ various mechanisms regarding the involvement of MDMX in regulation of p53. Based on the existing reports and our results, we speculate that in normal cells, MDMX that associates with MDM2 is ubiquitinated and subjected to subsequent proteasomal degradation by MDM2. Disruption of ribosome biogenesis triggers the release of S25 from the nucleolus to the nucleoplasm, where it interacts with MDM2, stabilizing MDMX. The stabilized MDMX then acts together with S25 to inhibit the E3 ligase activity of MDM2, leading to p53 stabilization and activation. When the stress is removed, S25 disassociates from MDM2, restoring the E3 ligase activity of MDM2, leading to the turnover of MDMX and p53, restricting their expression to the basal level.
Ribosomal dysfunction appears to lead to an increased predisposition to cancer and differential expression of RPs has been detected in human cancers.52,53 Considering that p53 is frequently mutated in human cancers and p53 mutations increase the expression of some RPs at the transcriptional level,54,55 it is possible that the mutant p53 in human cancer loses its ability to transcriptionally regulate S25, leading to overactivation of RP synthesis and cell proliferation.
In conclusion, our data demonstrate that S25 is a novel regulator of the MDM2–p53 circuit in ribosomal stress. Our results not only shed lights on the biological consequences of RP–MDM2–p53 interplay, but also reveal the existence of a novel S25-p53 feedback regulatory loop. The physiological role of S25 in cancer development and progression and the response to normal and pathological stimuli need to be investigated in future studies.
MATERIALS AND METHODS
Antibodies and plasmids
Rabbit polyclonal antiserum against human S25 was generated by ProteinTech (Chicago, IL, USA) using the full-length human S25 protein as the antigen. The T7-MDM2, HA-p53 and p21waf1 luciferase vectors were kindly provided by Dr X Chen (University of California, Davis). The HA-Ub vector was provided by Dr Y Shi (Harvard University). The Myc-MDMX vector was a kind gift from Dr J Chen (H Lee Moffitt Cancer Center and Research Institute). The constructs expressing full-length and deletions of the MDM2 and S25 proteins were generated by proof-reading PCR.
Cell culture and transfection
COS7 and A549 cells were purchased from the ATCC (Manassas, VA, USA). U2OS and HCT116 cells with or without p53 were provided by Dr X Chen (University of California, Davis) and Dr B Vogelstein (Johns Hopkins University), respectively. The mdmx−/− p53−/− and mdm2−/− p53−/− MEF cell lines were kindly provided by Dr G Lozano (University of Texas, MD Anderson Cancer Center). All cells were cultured under standard conditions described by the ATCC. Plasmids were transfected with Lipofectin (Invitrogen, Carlsbad, CA, USA) into cells as indicated.
IP and IB
Cells were collected and lysed in NP-40 lysis buffer (137 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Nonidet P-40, 10mM Tris, pH 7.5, and 5% (v/v) protease inhibitor mixture (Sigma-Aldrich, St Louis, MO, USA). Equal amounts of clear cell lysates were incubated at 4 °C overnight with appropriate antibodies. IP was conducted using the antibodies indicated in the figures, as reported previously.30 The immunocomplex was captured by protein G-Sepharose beads (Amersham Biosciences, Chandler, AZ, USA) and the bound proteins were eluted, resolved on SDS–PAGE, and detected by IB.
In-vivo ubiquitination assay
Cells were transfected with various vectors as indicated in the figures. After 24 h, cell lysates were immunoprecipitated with corresponding antibodies, and the bound proteins were purified with protein G-Sepharose beads, resolved on SDS–PAGE, and detected by an HA antibody.
Luciferase assay
Cells were co-transfected with promoter luciferase vectors with the Renilla luciferase reporter (as an internal control; Promega, Madison, WI, USA) for 24 h. The luciferase activity of the promoter reporter was determined using the Dual-Luciferase Reporter Assay System (Promega) according to the provided protocol and was normalized to the corresponding Renilla luciferase reporter activity.
siRNA treatment
The siRNA against human RPS25 was purchased from Dharmacon (Lafayette, CO, USA) and the transfection was performed according to manufacturer’s protocol. For IB assays using the siRNA pool, cells were incubated with 50 nM of each siRNA for 48 h, then treated with actinomycin D (Act D, 5 nM) for 6h, or with 5-fluorouracil (5-FU, 100 μM), etoposide (Eto, 50 mM) for 24 h. For cell-cycle assays, 50 nM of S25 or control siRNA was incubated with cells for 24 h, then Act D (5 nM) or etoposide (50 mM) was added for another 24 h before harvesting cells.
Immunofluorescence staining
U2OS cells grown on coverslips were transfected with GFP-S25 and deletions for 24 h. Cells were then fixed, and immunofluorescent staining was carried out as described previously.22 The mouse monoclonal anti-p53 and anti-nucleophosmin antibodies and a secondary goat anti-mouse IgG AlexaFluor-594 conjugated antibody (Molecular Probes, Eugene, OR, USA) were used to detect p53 and nucleophosmin. Immunofluorescence images were captured by confocal microscopy.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation assay was performed according to manufacturer’s protocol (Millipore, Billerica, MA, USA). The primer sequences used for the S25 chromatin immunoprecipitation assay were p53 RE1: 5′-TGCCATTCACGTCCATGCCATT-3′ and p53 RE2: 5′-TGCTGTA-GAAGTGGGCTGGTTGTA-3′, which generated a 158-bp fragment. The p53 antibody DO-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used for the IP. Normal mouse IgG was used as a negative control. One-tenth of the original DNA lysates were used as the input.
Statistical analysis
The means of the relative densitometry of the p53 bands, survival indices, and apoptotic indices between different treatment groups were analyzed using two-tailed paired t-tests. P<0.05 was considered to be statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr J Chen for the kind gift of the MDMX plasmid; Dr G Lozano for the mdmx−/− p53−/− and mdm2−/− p53−/− MEF cell lines; and Drs G Jin, D Chen, Z Zhang, L Ao, X Yang, J Qin and S Voruganti for helpful discussions and excellent technical support. This work was supported in part by NIH/NCI grants R01 CA112029, R01 CA121211 and a Susan G Komen Foundation grant BCTR0707731 (to RZ). MW was supported by NIH grant R01 CA91980. HW was supported by the One Hundred Talents Program, Chinese Academy of Sciences, grants from National Nature Science Foundation (30870513, 31070680, 91029715 and 81025017) and Ministry of Science and Technology of China (2007CB947100), the Science and Technology Commission of Shanghai Municipality (08391910800, 10391902100), National Science and Technology Major Project ‘Key New Drug Creation and Manufacturing Program’ (2009ZX09102-114, 2009ZX09301-011).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–299. [DOI] [PubMed] [Google Scholar]
- 3.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993; 362: 857–860. [DOI] [PubMed] [Google Scholar]
- 4.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387: 299–303. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev 1993; 7: 1126–1132. [DOI] [PubMed] [Google Scholar]
- 6.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378: 206–208. [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–310. [DOI] [PubMed] [Google Scholar]
- 8.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- 10.Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 2010; 17: 93–102. [DOI] [PubMed] [Google Scholar]
- 11.Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell 2009; 34: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J 2004; 23: 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen D, Zhang Z, Li M, Wang W, Li Y, Rayburn ER et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene 2007; 26: 5029–5037. [DOI] [PubMed] [Google Scholar]
- 14.Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 2004; 279: 44475–44482. [DOI] [PubMed] [Google Scholar]
- 15.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 2004; 24: 7654–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol 2004; 24: 7669–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai MS, Shi D, Jin Y, Sun XX, Zhang Y, Grossman SR et al. Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J Biol Chem 2006; 281 : 24304–24313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003; 3: 577–587. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent riboso-mal-stress checkpoint pathway. Mol Cell Biol 2003; 23: 8902–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun XX, DeVine T, Challagundla KB, Dai MS. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J Biol Chem 2011; 286: 22730–22741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong X, Zhao Y, He H, Sun Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 2011; 30: 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Poyurovsky MV, Li Y, Biderman L, Stahl J, Jacq X et al. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell 2009; 35: 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun XX, Dai MS, Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribo-somal protein interaction. J Biol Chem 2007; 282: 8052–8059. [DOI] [PubMed] [Google Scholar]
- 24.Sun XX, Dai MS, Lu H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem 2008; 283: 12387–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005; 123: 49–63. [DOI] [PubMed] [Google Scholar]
- 26.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell 2008; 32: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol 2007; 27: 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 2010; 18: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009; 16: 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Zhang R. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J 2008; 27: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen D, Zhang J, Li M, Rayburn ER, Wang H, Zhang R. RYBP stabilizes p53 by modulating MDM2. EMBO Rep 2009; 10: 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishiyama T, Yamamoto H, Uchiumi T, Nakashima N. Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res 2007; 35: 1514–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 2009; 23: 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Center MS. Regulation of ribosomal protein S25 in HL60 cells isolated for resistance to adriamycin. FEBS Lett 1992; 298: 142–144. [DOI] [PubMed] [Google Scholar]
- 35.Adilakshmi T, Laine RO. Ribosomal protein S25 mRNA partners with MTF-1 and La to provide a p53-mediated mechanism for survival or death. J Biol Chem 2002; 277: 4147–4151. [DOI] [PubMed] [Google Scholar]
- 36.Castro ME, Leal JF, Lleonart ME, Ramon YCS, Carnero A. Loss-of-function genetic screening identifies a cluster of ribosomal proteins regulating p53 function. Carcinogenesis 2008; 29: 1343–1350. [DOI] [PubMed] [Google Scholar]
- 37.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 2001; 29: 92–95. [DOI] [PubMed] [Google Scholar]
- 38.Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 2003; 23: 5113–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meulmeester E, Frenk R, Stad R, de Graaf P, Marine JC, Vousden KH et al. Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol 2003; 23: 4929–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Argentini M, Barboule N, Wasylyk B. The contribution of the acidic domain of MDM2 to p53 and MDM2 stability. Oncogene 2001; 20: 1267–1275. [DOI] [PubMed] [Google Scholar]
- 41.Horn HF, Vousden KH. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 2008; 27: 5774–5784. [DOI] [PubMed] [Google Scholar]
- 42.Laine RO, Shay NF, Kilberg MS. Nuclear retention of the induced mRNA following amino acid-dependent transcriptional regulation of mammalian ribosomal proteins L17 and S25. J Biol Chem 1994; 269: 9693–9697. [PubMed] [Google Scholar]
- 43.Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K et al. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell 2012; 45: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Budde A, Grummt I. p53 represses ribosomal gene transcription. Oncogene 1999; 18: 1119–1124. [DOI] [PubMed] [Google Scholar]
- 45.Mancini F, Di Conza G, Monti O, Macchiarulo A, Pellicciari R, Pontecorvi A et al. Puzzling over MDM4-p53 network. Int J Biochem Cell Biol 2010; 42: 1080–1083. [DOI] [PubMed] [Google Scholar]
- 46.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation ofp53. Proc Natl Acad Sci USA 2003; 100: 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci 2007; 120(Part 3): 371–378. [DOI] [PubMed] [Google Scholar]
- 48.Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol 2000; 20: 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP et al. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep 2001; 2:1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, Giglio S et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J 2009; 28: 1926–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. EMBO J 2006; 25: 5614–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer 2003; 3: 179–192. [DOI] [PubMed] [Google Scholar]
- 53.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood 2010; 115: 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loging WT, Reisman D. Elevated expression of ribosomal protein genes L37, RPP-1, and S2 in the presence of mutant p53. Cancer Epidemiol Biomarkers Prev 1999; 8: 1011–1016. [PubMed] [Google Scholar]
- 55.Artero-Castro A, Castellvi J, Garcia A, Hernandez J, Ramon y Cajal S, Lleonart ME. Expression of the ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors. Hum Pathol 2011; 42: 194–203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.