

Abstract
Recent work on hippocampal LTP has focused on gene expression induced with high-frequency stimulation, as well as the signal transduction cascades responsible for the induction of these genes. Many scenarios for LTP lasting for greater than 5 hours include some or all of the following processes: 1) tagging of potentiated synapses, possibly by phosphorylation; 2) signaling to the nucleus; 3) kinase cascades and transcription factors in the nucleus;, 4) expression of immediate-early genes and/or synaptic proteins; and, finally, 5) targeting of newly synthesized proteins (or RNAs) to the potentiated synapses (and not to the unpotentiated synapses). Unfortunately, most scenarios proposed for the late-phase expression of LTP are still highly speculative at this time. A critical review of the literature relating to the role of gene expression in hippocampal LTP and a discussion of recent work on the subject will be presented.
Keywords: LTP, Gene expression, CREB, Dendrites, Synaptic tagging
In a field characterized by enormous complexity and persistent controversy, research on learning and memory maintains as fundamental the distinction between short- and long-term memory (STM and LTM, respectively). This enduring concept has been validated in a wide range of paradigms in research performed on simple systems to human beings. In humans, these two forms of memory can be dissociated by a number of behavioral and clinical conditions, including disease (Alzheimer’s, multiple sclerosis, epilepsy, encephalitis), neurotoxic agents (hypoxia, alcohol, heavy metals, anesthetics), neuropsychological conditions (schizophrenia, depression), and various forms of trauma, temporal lobe dysfunction, and medications. The biochemical conversion from STM to LTM may be disrupted by blocking RNA or protein synthesis using a number of chemically distinct antibiotics. As demonstrated in early work by Agranoff et al. (1) on the conditioned avoidance learning in goldfish, the conversion between STM and LTM is only sensitive to agents that disrupt gene expression when they are administered during a surprisingly narrow time window relative to training. Protein synthesis inhibitors have no affect on STM or on established LTM nor are they effective when administered immediately before training; the inhibitors must be applied within the first hour after training to prevent the conversion to LTM. Although these facts are well established, the interpretation remains ambiguous. Is there a distinct gene or class of genes that are required to render a transient memory permanent, or does disrupting protein synthesis impair the growth or maintenance of synaptic connections indirectly? If specific genes are required, what are they? How does synaptic activity stimulate synthesis of RNA and protein that is essential for LTM? How does transcription, which takes place in the nucleus, or translation, which takes place on ribosomes, lead to the strengthening of only the appropriate synaptic connection on that neuron? Studies on LTP in the hippocampus are providing new information and interpretations on many of these questions.
Role of Protein Synthesis in LTP
Evidence supporting the idea that hippocampal LTP requires new protein synthesis has accumulated in the form of pharmacological experiments using hippocampal slices (CA1) and in vivo preparations (dentate gyrus). From experiments using inhibitors of protein synthesis, including emetine, cycloheximide, puromycin, and anisomycin (2-4), it was concluded that the maintenance of long-term potentiation required the production of new proteins. Stanton and Sarvey (3) found that emetine could irreversibly block protein synthesis and LTP, as could cycloheximide reversibly. Furthermore, emetine was found to have no deleterious affects on various membrane properties measured intracellularly. The field at that point (mid-1980s), however, was hardly sold on the idea—anisomycin, the only drug tested that did not block LTP in the slice study was found to have a significant effect in vivo in the freely moving rat (2) and the largest effect of all the drugs tested in the anesthetized rat (5). By far the largest criticism, however, now seems to be that many of these drugs seem to block LTP induction (versus late-phase LTP or maintenance of LTP), which is more likely to be related to nonspecific actions of the drugs than to the protein synthesisblocking actions. This possibility is illustrated by the data shown by Otani et al. (5), in which all of the aforementioned drugs showed at least some effect on the earliest time point plotted (probably post-tetanic potentiation and/or short-term potentiation) when the excitatory postsynaptic potential slopes were examined. Interestingly, cycloheximide, emetine, and puromycin were all found to dramatically inhibit calcium influxes evoked by depolarizing pulses in Purkinje neurons (6). This property, if true in hippocampal neurons, could have profound effects on LTP induction but have little to do with inhibition of protein synthesis and late-phase LTP expression or stabilization (note that researchers in ref. 3 were using 1.5 μM emetine, but those in ref. 6 used 50 μM emetine).
Later, it became clear that anisomycin, which seemed to have little or no effect on the immediate induction of LTP, was able to block the late consolidation of LTP in a manner only apparent 3–4 hours after LTP induction. Indeed, this is what was reported initially by Krug et al. (2). Interestingly, these results are consistent with behavioral studies showing that protein synthesis inhibitors can block long-term memory but leave short-term memory relatively intact (7). Perhaps the more convincing part of the anisomycin story is that it was found to be effective in blocking late-phase LTP, even when applied or injected after tetanization (5, 8). By applying the drug after the tetanization, the studies avoided possible criticism that the effects of the drug were caused by an artifactual inhibition of LTP induction processes. Remarkably, Otani et al. (5) found an 89% inhibition of protein synthesis measured within the 15 minutes after anisomycin injection, indicating a very rapid onset of the drug’s action. This makes it even more exciting that anisomycin injected immediately after tetanus, but not 15 minutes after the tetanus, inhibited LTP consolidation. These data suggest that the crucial protein synthesis event(s) take place within the first 15~30 minutes after the tetanic stimulation.
Role of mRNA Synthesis in LTP
The picture becomes more complicated when the data regarding new RNA synthesis are examined. In the dentate gyrus in vivo, Otani et al. (5) found the RNA synthesis inhibitor actinomycin D to be quite ineffective in blocking LTP (in fact, a slight enhancement of LTP was observed). This result is far different than the data shown by Nguyen et al. (9) recording from CA1 in slices in vitro, where it was found that actinomycin D and another RNA synthesis inhibitor, 5,6-di-chloro-1-β-D-ribofuranosyl-benzimidazole, blocked late-phase LTP. The differences between these two studies may have been clarified somewhat by the study of Frey et al. (10), which showed data from CA1 in vitro and dentate gyrus in vivo. The study by Frey et al. saw significant decay of LTP in the treated animals only after 5 hours post-LTP-induction; the Otani study followed the LTP out to only 3 hours. Therefore, it is possible that the effect of RNA synthesis inhibitors on late-phase LTP was missed in the earlier study. The results are far from conclusive, however; here, significant effects of actinomycin D on LTP measured by excitatory postsynaptic potential slopes were not observed until after 7 hours and by population spikes not until after 5 hours after induction (versus less than 2 hours in the study by Nguyen et al. [9]). These discrepancies should not be taken as evidence against a role for RNA synthesis in LTP (indeed, maybe only the very late phases of LTP require new RNA synthesis); rather, they indicate that further investigation on the topic is warranted.
Unlike protein synthesis inhibitors, which can be injected or applied immediately after tetanic stimulation and still effectively block LTP, the RNA synthesis inhibitors must be in place in the preparation at the time of LTP induction to be effective. Actinomycin D or 5,6-dichloro-1-β-D-ribofuranosyl-benzimidazole has no effect on LTP if delivered immediately after tetanization (10). Although this could be taken as argument that these drugs can influence the LTP induction process, these drugs do have in their favor that post-tetanic potentiation and early LTP out to 2 hours are relatively intact (5, 9, 10) and the drugs seem to be free of side effects in Purkinje neurons (6). One may therefore conclude that if RNA synthesis were involved in late-phase expression of LTP, it would have to take place very early in the LTP consolidation process.
Signal Transduction Mechanisms in LTP-Related Gene Expression
In contrast to the diverse pharmacological interventions that can disrupt induction of LTP, present evidence indicates a surprisingly conserved molecular mechanism in the protein-synthesis-dependent phase of learning. Protein kinase A has been implicated in late-LTP (11, 12) and in long-term facilitation in Aplysia sp. (13). In both cases, phosphorylation of the transcription factor CREB has been shown to be an important downstream event. Phosphorylation of CREB induces transcription of genes containing CRE enhancer elements in the promoter region (see ref. 14) for review). In Aplysia sp., microinjection of CRE-containing oligonucleotides into cultured neurons blocks long-term facilitation but leaves short-term facilitation unchanged (13). In Drosophila melanogaster, genetically induced expression of CREB repressor isoform blocks LTM (15), and expression of an activator isoform enhances LTM (16). In the mouse, targeted mutation of the CREB gene disrupts LTM, but STM is normal (17); when oligonucleotides directed against CREB mRNA are infused into the dorsal hippocampus of rats, learning in a water maze test is impaired 48 hours later (LTM), but STM is not affected (18).
Several other signaling pathways can lead to phosphorylation of CREB, including Ca2+/calmodulin-dependent protein (CaM) kinases, mitogen-activated protein kinase, ribosomal S6 kinase 2, and protein kinase C (14, 19), which suggests a number of possible routes for conveying a signal from an activated synapse to the nucleus. Determination of those kinases involved specifically in LTP-related gene expression, however, is an area of intense investigation, because many of the kinase inhibitors also interfere with the LTP induction processes. In response to synaptic activation, appropriate kinases could translocate from the synapse to the nucleus to activate CREB-dependent gene expression (20), or influx of calcium ions could diffuse to the nucleus to activate appropriate intranuclear kinases (21). Other synapse-to-nucleus signaling molecules have been suggested, including nuclear factor κB, and neurotrophin receptors (22). Pharmacological evidence (23) and imaging methods (24) in hippocampal neurons support the hypothesis that Ca2+/CaM translocates from the subsynaptic membrane to the nucleus in response to synaptic activation to initiate CREB-dependent gene expression.
Multiple factors, including kinases, phosphatases, and timing of action potentials, are sure to interact as a complex system to regulate gene expression (25). Indeed, phosphorylation of CREB, although probably necessary, is not sufficient for CRE-mediated gene expression associated with LTP; in slices of hippocampus, CRE-mediated gene expression was found to correlate with the induction of late LTP, but not early LTP, whereas CREB was phosphorylated at Ser133 in both early and late LTP induction protocols (26). Interestingly, the CRE-mediated gene expression associated with late LTP was dependent on activation of L-type calcium channels. Thus it remains unclear which additional factors are required for induction of late LTP-associated genes.
Problems in Targeting New Proteins to Potentiated Synapses
LTP has among its features synapse specificity. That is, only the synapses that are coactive with postsynaptic depolarization (or firing) are potentiated (27, 28; but see 29). As outlined by Lisman (30), this presents a problem for many hypotheses involving gene expression; new proteins synthesized in the nucleus must find their way to the few synapses (of up to 10,000) that may have been potentiated. Thus, some kind of tag of the potentiated synapses is a theoretical must. Despite this apparent necessity, the idea of synaptic tagging has enjoyed little in terms of experimental support—until very recently, that is. Frey and Morris (31) showed, in two different experimental paradigms, that decremental LTP (lasting 4–6 hours, early LTP) could be rescued by late LTP (lasting at least 8 hours) induced at another input (early LTP was induced with either an abbreviated stimulation protocol, or with a standard protocol in the presence of a protein synthesis inhibitor) (Box 1) These results also carry with them the strong implication of a postsynaptic locus for the maintenance of LTP, because the convergence onto the postsynaptic neuron is necessary for the rescue.
Box 1 and Figure 1: Synaptic Tagging during LTP.
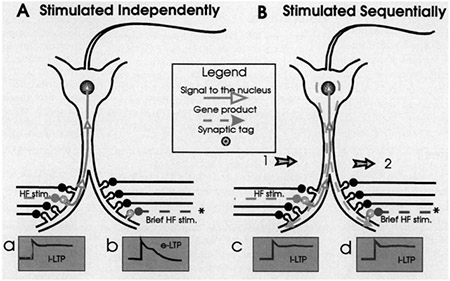
Independent pathways can be stimulated to produce either early LTP (4–6 hours, eLTP) or late LTP (more than 8 hours, ILTP) In late LTP (light blue), high-frequency stimulation presumably creates a synaptic tag (gray) and possibly a signal to the nucleus to induce new protein synthesis necessary for the late phase (a). In early LTP (blue), very brief high-frequency stimulation can induce the tag but not the gene product, potentiation fades by 6 hours (b) *, Early-LTP can also be induced with full high-frequency stimulation in the presence of protein synthesis inhibitors. Frey and Morris (31) showed that if the brief high-frequency (brief HF, blue) stimulation is preceded by (or followed by) a late-LTP-inducing (HF, light blue) stimulation to a converging input (c), the (expected) early LTP stays up— it has been “rescued” by the other input (d). A possible explanation for these results is that new proteins (or mRNAs) induced by the HF stimulation can be hijacked by all tagged synapses Note that this explanation places the site of new protein or mRNA synthesis in the postsynaptic neuron. Arrows 1 and 2 represent the sequential stimulation of S1 and S2 inputs, respectively The order, however, is not crucial for the rescue effect
If it were necessary to perpetually maintain a post-translational modification for a tag (such as phosphorylation, for example), why shouldn’t the same type of modification be maintained as the expression of late-phase LTP? After all, there is evidence of the phosphorylation of AMPA receptors as the expression of LTP (32). The Frey and Morris study not only provided evidence against the idea of a “perpetual tag” by demonstrating that the tag lasts for a limited period of time, but also suggested that new proteins must be utilized by the potentiated synapse in somewhat under 3 hours The identity and nature of the tag, and gene product, remain undetermined
Dendritic mRNA as a Solution?
The observation of polyribosomes in close proximity to dendritic spines (33) has led to the suggestion that local protein synthesis within dendrites would be an ideal way that new proteins could be synthesized on demand in response to the induction of LTP. Evidence of a possible dendritic synthesis of CaM kinase II has been reported (34), and the fact that the transcript for an RNA polymerase III (35) has been found in dendrites suggests that such a process is feasible. That such a scenario alone is responsible for the long-term maintenance of LTP, however, is unlikely in light of the results of experiments in CA1 dendrites separated from their cell bodies (36); in this preparation, LTP could be induced, but it did not last more than 3 hours. Thus, de novo protein synthesis from existing mRNA in the dendrites was not sufficient for the late-phase maintenance of LTP. The results from LTP in isolated dendrites suggest instead that protein or mRNA is synthesized at the soma and subsequently transported to the dendrites. Of the mRNAs that have been found to localize in the dendrites, several have been determined to increase with LTP, including the microtubule-associated protein MAP2, Cam kinase IIα (37), and the spectrinlike molecule Arc. Interestingly, Arc mRNA has even been shown to localize selectively to stimulated regions of the dendritic tree (38). Thus, a possible scenario for late-phase expression of LTP involves mRNA synthesis at the nucleus and transport to activated regions of the dendrite Signals for the translation of new proteins from the new mRNA would therefore have to persist from the time of LTP induction until the mRNA reaches the subsynaptic site (perhaps the “synaptic tag” of Frey and Morris is just this). Alternatively, the dendritic mRNA induced with LTP-inducing stimulation may not be directly involved with late-phase expression of LTP. This leaves us with the current challenge to prove a direct involvement of these mRNAs and their proteins in LTP maintenance (versus induction).
What Are the Genes?
With the rapid development of techniques in molecular and cell biology accessible to neurobiologists, tremendous progress has been made in determining the identity of genes turned on by LTP-inducing stimulation. A comprehensive picture of which gene products make it to the synapse to participate in the expression of LTP, however, has not yet emerged. This is largely because of the general lack of agreement in the field about how LTP is actually expressed in hippocampal neurons. Disagreements notwithstanding, there has been no lack of candidate genes with expression patterns that correlate with the induction of LTP. Although the identity and nature of a large number of these genes remain undetermined (39), some have been identified as synaptic proteins (such as synaptophysin [40] and GAP-43 [41], kinases (such as CaM kinase II [34] and PKCζ [42]), glutamate receptors (12), a metabotropic glutamate receptor-associated protein (homer [43]), and immediate-early genes (such as zif268 [44] and krox-20 [45]). Other immediate early genes not classified as transcription factors that are induced with LTP are a protease (tissue plasminogen activator [46]), brain-derived neurotrophic factor (47) and its receptor (48), a cytoskeletal protein (arc [38]), and a Ras-like protein (rheb [49]). Only as more is known about the expression of LTP, be it structural changes in spines, modification or insertion of ion channels, or increases in neurotransmitter release, will the picture become clear as to which genes are important in the stabilization of LTP, and which are compensatory in response to increased whole-cell activity from increases in synaptic effectiveness (relating to the phenomenon recently coined as “meta-plasticity” [50]). On the other hand, by working backward from the nucleus and looking at proteins that localize to synapses, we may begin to understand more about how late-phase LTP is expressed. We look forward to rapid progress on this front.
References
- 1.Agranoff BW, Davis RE, Brink JJ. Memory fixation in the goldfish. Proc Natl Acad Sci U S A 1965;54:788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krug M, Lossner B, Ott T. Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull 1984;13:39–42. [DOI] [PubMed] [Google Scholar]
- 3.Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci 1984;4:3080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deadwyler SA, Dunwiddie T, Lynch G. A critical level of protein synthesis is required for long-term potentiation. Synapse 1987;1:90–5. [DOI] [PubMed] [Google Scholar]
- 5.Otam S, Marshall CJ, Tate WP, Goddard GV, Abraham WC. Maintenance of long-term potentiation in rat dentate gyrus requires protein synthesis but not messenger RNA synthesis immediately post-tetanization. Neurosci 1989;28:519–26. [DOI] [PubMed] [Google Scholar]
- 6.Linden DJ. A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron 1996;17:483–90. [DOI] [PubMed] [Google Scholar]
- 7.Squire LR, Barondes SH. Variable decay of memory and its recovery in cycloheximide-treated mice. Proc Natl Acad Sci U S A 1972;69:1416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 1988;452:57–65. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science 1994,265:1104–7. [DOI] [PubMed] [Google Scholar]
- 10.Frey U, Frey S, Schollmeier F, Krug M. Influence of actinomycin D, an RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J Physiol 1996;490:703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abel T, Nguyen PV, Barad M, Duel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 1997;88: 615–26. [DOI] [PubMed] [Google Scholar]
- 12.Nayak A, Zastrow DJ, Lickteig R, Zahniser NR, Browning MD. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature 1998,394:680–3. [DOI] [PubMed] [Google Scholar]
- 13.Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature 1990; 345:718–21. [DOI] [PubMed] [Google Scholar]
- 14.Fields RD. Signaling from neural impulses to genes. THE NEUROSCIENTIST 1996;2:315–25. [PMC free article] [PubMed] [Google Scholar]
- 15.Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, et al. Induction of a dominant-negative CREB transgene blocks long-term memory in Drosophila. Cell 1994;79:49–58. [DOI] [PubMed] [Google Scholar]
- 16.Yin JC, Del Vecchio M, Zhou H, Tully T. CREB as a memory modulator, induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell 1995;81:107–15. [DOI] [PubMed] [Google Scholar]
- 17.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994;79:59–68. [DOI] [PubMed] [Google Scholar]
- 18.Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci U S A 1997;94:2693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 1998;21:869–83. [DOI] [PubMed] [Google Scholar]
- 20.Adams SR, Harootuman AT, Buechler YJ, Taylor SS, Tsien RY. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991;349:694–7. [DOI] [PubMed] [Google Scholar]
- 21.Chawla S, Hardmgham GE, Quinn DR, Badmg H. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaMK IV. Science 1998;281:1505–9. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T Messengers from the synapse to the nucleus (MSNs) that establish late phase of long-term potentiation (LTP) and long-term memory. Neurosci Res 1996; 25:1–6. [DOI] [PubMed] [Google Scholar]
- 23.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 1996;16:89–101. [DOI] [PubMed] [Google Scholar]
- 24.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 1998;392: 198–202. [DOI] [PubMed] [Google Scholar]
- 25.Fields RD, Eshete F, Stevens B, Itoh K. Action potential-dependent regulation of gene expression: temporal specificity in Ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci 1997; 17: 7252–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of hippocampus. Neuron 1996;16:973–82. [DOI] [PubMed] [Google Scholar]
- 27.Levy WB, Steward O. Synapses as associative memory elements in the hippocampal formation. Brain Res 1979;175: 233–45. [DOI] [PubMed] [Google Scholar]
- 28.Abraham WC, Goddard GV. Asymmetric relationships between homosynaptic long-term potentiation and heterosynaptic long-term depression. Nature 1983;305:717–19. [DOI] [PubMed] [Google Scholar]
- 29.Engert F, Bonhoeffer T. Synapse specificity of long-term potentiation breaks down at short distances. Nature 1997;388:279–84. [DOI] [PubMed] [Google Scholar]
- 30.Lisman J What does the nucleus know about memories? J NIH Res 1995;7:43–6. [Google Scholar]
- 31.Frey U, Morris RGM. Synaptic tagging and long-term potentiation. Nature 1997; 385:533–6. [DOI] [PubMed] [Google Scholar]
- 32.Barria A, Muller D, Derkach V, Griffith LC, Soderhng TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 1997;276:2042–5. [DOI] [PubMed] [Google Scholar]
- 33.Steward O, Levy WB. Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci 1982;2:284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouyang Y, Kantor D, Hams KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci 1997;17:5416–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiedge H, Fremeau RT, Weinstock PH, Arancio O, Brosius J. Dendritic localization of neural BC1 RNA Proc Natl Acad Sci U S A 1991;88:2093–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frey U, Krug M, Brodeman R, Reymann K, Matthies H. Long-term potentiation induced in dendrites separated from rat’s CA1 pyramidal somata does not establish a late phase. Neurosci Lett 1989;97:135–9. [DOI] [PubMed] [Google Scholar]
- 37.Roberts LA, Large CH, Higgins MJ, Stone TW, O’Shaughnessy CT, Moms BJ. Increase expression of dendritic mRNA following the induction of long-term potentiation. Brain Res Mol Brain Res 1998;56:38–44. [DOI] [PubMed] [Google Scholar]
- 38.Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 1998;21:741–51. [DOI] [PubMed] [Google Scholar]
- 39.Nedivi E, Hevroni D, Naot D, Israeli D, Citn Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 1993;363:718–22. [DOI] [PubMed] [Google Scholar]
- 40.Mullany P, Lynch MA. Changes in protein synthesis of the synaptic vesicle protein, synaptophysin, in entorhinal cortex following induction of long-term potentiation in dentate gyrus: an age-related study in the rat. Neuropharmacol 1997;36:973–80. [DOI] [PubMed] [Google Scholar]
- 41.Namgung U, Matsuyama S, Routtenberg A. Long-term potentiation activates the GAP-43 promoter: selective participation of the hippocampal mossy cells. Proc Natl Acad Sci U S A 1997;94:11675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osten P, Valsamis L, Hams A, Sacktor TC. Protein synthesis-dependent formation of protein kinase Mzeta in long-term potentiation. J Neurosci 1996;16:2444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 1997;386:284–8. [DOI] [PubMed] [Google Scholar]
- 44.Worley PF, Bhat RV, Baraban JM, Erickson CA, McNaughton BL, Barnes CA. Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long-term enhancement. J Neurosci 1993;13:4776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inokuchi K, Murayama A, Ozawa F. mRNA differential display reveals Krox-20 as a neural plasticity-regulated gene in the rat hippocampus. Biochem Biophys Res Commun 1996;221:430–6. [DOI] [PubMed] [Google Scholar]
- 46.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 1993;361:453–7. [DOI] [PubMed] [Google Scholar]
- 47.Patterson SL, Grover LM, Schwartzkrom PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron 1992;9:1081–8. [DOI] [PubMed] [Google Scholar]
- 48.Bramham CR, Southard T, Sarvey JM, Herkenham M, Brady LS. Unilateral LTP taggers bilateral increase in hippocampal neurotrophin and trk receptor mRNA expression in behaving rats: evidence for interhemispheric communication. J Comp Neurol 1996;368:371–82. [DOI] [PubMed] [Google Scholar]
- 49.Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, Worley PF. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 1994;269:16333–9. [PubMed] [Google Scholar]
- 50.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 1996;19:126–30. [DOI] [PubMed] [Google Scholar]