

Abstract
Acquired von Willebrand syndrome (AVWS) associated with severe aortic stenosis (AS) has been frequently subclassified into a subtype 2A based on the deficiency of high-molecular-weight (HMW) multimers as it is seen in inherited von Willebrand disease (VWD) type 2A. However, the multimeric phenotype of VWD type 2A does not only include an HMW deficiency but also a decrease in intermediate-molecular-weight (IMW) multimers and an abnormal inner triplet band pattern. These additional characteristics have not been evaluated in AVWS associated with severe AS. Therefore, we recruited N = 31 consecutive patients with severe AS and performed a high-resolution Western blot with densitometrical band quantification to characterize the von Willebrand factor (VWF) multimeric structure and reevaluate the AVWS subtype classification. Study patients showed an isolated HMW VWF multimer deficiency without additional abnormalities of the IMW portions and the inner triplet structure in 65%. In conclusion, the multimeric pattern of AVWS associated with severe AS does neither resemble that seen in AVWS type 2A nor that seen in inherited VWD type 2A. Therefore, a subclassification into a type 2A should not be used.
Keywords: von Willebrand factor, aortic stenosis, multimer analysis
Background
Acquired von Willebrand syndrome (AVWS) type 2 has been frequently found in patients with severe aortic stenosis (AS) with a prevalence up to 80%.1 In these patients, AVWS is characterized by proteolytic loss of high-molecular-weight (HMW) multimers of von Willebrand factor (VWF) due to a high shear force.2,3 Passage through a stenosed aortic valve induces a structural molecular change in VWF, which makes the molecule sensitive to proteolytic cleavage by the metalloprotease ADAMTS13 with subsequent degradation of HMW multimers.4,5 High-molecular-weight multimers represent the hemostatically most active form of VWF, and thus, its deficiency results in bleeding complications ranging from epistaxis to gastrointestinal hemorrhage. Acquired von Willebrand syndrome and associated primary hemostasis abnormalities are corrected by surgical6,7 or interventional8 valve replacement that supports this mechanistic concept.
It has been suggested that the abnormal VWF multimeric structure in the setting of AS resembles that of an inherited type 2A von Willebrand disease (VWD),2 which has led to the frequent subclassification into AVWS type 2A in various studies.9–14 Type 2A VWD is characterized by the deficiency of HMW multimers, the reduction in intermediate molecular weight (IMW) multimers and an abnormal triplet band pattern of the lower molecular weight multimers.15–18 Neither IMW multimers nor the multimeric triplet configuration has been evaluated in patients with AVWS associated with AS, which could have led to a misclassification in previous studies.
Therefore, in our present project, we aimed to analyze the complete VWF multimeric structure in patients with severe AS including the low- and intermediate-molecular-weight multimer portions and the triplet structure.
Materials and Methods
Laboratory Analysis
Blood sampling was performed after echocardiographic diagnosis of severe AS as described below. von Willebrand factor antigen (VWF:Ag), VWF activity (VWF:GPIbM), and factor VIII coagulation activity (FVIII:C) were determined using photo-optical measurements (Healthcare Diagnostics, Siemens, Erlangen, Germany) and were abbreviated based on the approved nomenclature.19 von Willebrand factor multimeric structure analysis was assessed using a Western blot according to the protocol of Ott et al.15 Briefly, VWF multimeric bands were electrophoretically separated using a low-resolving (1%) and a high-resolving (2%) agarose gel. For immunolocalization, we used a polyclonal rabbit anti-VWF (DAKO, Glostrup, Denmark) as the primary antibody and a Cy5-labeled ECL Plex goat anti-rabbit IgG (GE Healthcare Life Sciences, Marlborough, Massachusetts) as the secondary antibody. von Willebrand factor multimeric bands were densitometrically quantified using CCD camera and Image Quant LAS 4000 software (GE Healthcare Life Sciences). A normal VWF multimeric pattern consists of low-molecular-weight (LMW, bands 1-3), intermediate-molecular-weight (IMW, bands 4-13), and high-molecular-weight (HMW, bands >13) bands. Acquired von Willebrand syndrome type 2 was defined by the loss of HMW bands compared to normal plasma.
A decrease in HMW and IMW VWF multimers accompanied by an abnormal triplet band pattern was regarded indicative of an AVWS “type 2A.” The resulting VWF multimer structure was compared to the multimer pattern of previously diagnosed AVWS type 2A and inherited type 2A VWD of our institutional database.
von Willebrand factor multimeric band pattern was analyzed by experienced laboratory physicians blinded to echocardiographic and baseline patient characteristics. Our institution is an experienced center for VWF multimer analysis with more than 1400 VWF Western blots every year.
Study Patients
Consecutive patients with the diagnosis of severe AS (N = 31) were prospectively enrolled after providing written informed consent in accordance with the Declaration of Helsinki under a protocol reviewed by the local Ethics Committee. Patients with a history of surgical or interventional valve replacement and patients with other significant valve diseases, acute decompensated heart failure, or liver cirrhosis were excluded. Severe AS was defined as an echocardiographic peak aortic jet velocity ≥4m/s, a mean transvalvular pressure gradient ≥40 mm Hg, and an aortic valve area (AVA; calculated by continuity equation) <1 cm2. The stenosis-induced shear stress that impacts on vWF was calculated as 4μ ×V m/r (μ = blood viscosity [estimated as 0.035 poise]; V m = mean transvalvular blood velocity; r = radius of the stenosis, which is calculated: [AVA/π]1/2). Bleeding symptoms in the previous 12 months were evaluated using a standardized screening questionnaire adapted from those by Rapaport.20
Statistics
The study was designed as a monocentric hypothesis-generating study. All P values are descriptive and presented to 3 decimal values. Categorical data are presented using counts and percentages, and Fisher exact test (2 × 2 tables) was calculated. Continuous measurements are presented using arithmetic mean and standard deviation (SD). For the comparison of 2 independent groups, if normality was assumed, the 2-sample t test was used. If normality was not assumed, the exact Mann-Whitney U test was used.
Results
Patient baseline characteristics and echocardiographic findings are shown in Table 1. Study patients had a mean age of 79 ± 8 years and a mean body mass index (kg/m2) of 26 ± 6, and 68% were male gender. The most frequent clinical symptoms were chest pain (51.6%) and dyspnea (New York Heart Association [NYHA] I [no shortness of breath in ordinary physical activity]:4/31 [12.9%], NYHA II [mild shortness of breath]: 14/31 [45.2%], NYHA III [moderate shortness of breath]: 10/31 [32.2%], NYHA IV [shortness of breath at rest]: 3/31 [9.7%]). Bleeding episodes in the previous 12 months were reported in 12 (38.7%) out of 31 patients (gingivorrhagia/epistaxis: 9.7%, spontaneous hematoma: 19.4%, prolonged bleeding after surgery: 0%, gastro-intestinal hemorrhage: 25.8%, cerebral hemorrhage: 0%, menorrhagia: 0%, and hematuria: 9.7% of all patients). A total of 54.8% of all patients were on antiaggregatory and 25.8% on anticoagulant treatment at the time of bleeding symptom evaluation. All patients were diagnosed with severe AS with a peak aortic jet velocity of 4.7 ± 0.5 m/s and a mean transvalvular gradient of 56 ± 10 mm Hg in the presence of normal systolic ejection fraction. Considering hemodynamic findings, comorbidities and clinical status patient management ranged from conservative treatment (5/31; 16.1%) to surgical valve replacement (11/31; 35.5%) or TAVR (transcatheter aortic valve replacement (14/31; 45.2%). One patient died before projected surgical intervention.
Table 1.
Baseline patient characteristics including clinical, echocardiographic and laboratory data.
| N = 31 | ||
|---|---|---|
| Baseline patient characteristics | ||
| Age, mean ± SD, years | 79 ± 8 | |
| Male gender (%) | 21/31 (68%) | |
| BMI, mean kg/m2 | 26 ± 6 | |
| Height, mean ± SD, cm | 166 ± 9 | |
| Weight, mean ± SD, kg | 72.6 ± 14.9 | |
| Current smoking (%) | 19.4 | |
| Diabetes (%) | 29.0 | |
| Coronary artery disease (%) | 51.6 | |
| Echocardiographic data | ||
| Peak aortic jet velocity, mean ± SD, m/s | 4.7 ± 0.5 | |
| Max./Mean aortic gradient, mean ± SD, mm Hg | 90 ± 17/ 56 ± 10 | |
| AVA, mean ± SD, cm2 | 0.64 ± 0.14 | |
| Systolic ejection fraction (%) | 60 ± 9 | |
| Intraventricular septum thickness, mean ± SD, mm | 1.7 ± 0.2 | |
| LVEDD, mean ± SD, cm | 4.9 ± 0.7 | |
| LVESD, mean ± SD, cm | 3.2 ± 0.6 | |
| Shear stress, mean ± SD, dyn/cm2 | 107 ± 12 | |
| Bleeding history (previous 12 months) | ||
| GI bleeding (%) | 8/31 (25.8%) | |
| Gingivorrhagia and/or epistaxis (%) | 3/31 (9.7%) | |
| Spontaneous hematoma (%) | 6/31 (19.4%) | |
| Hematuria (%) | 3/31 (9.7%) | |
| Laboratory data | ||
| Hemoglobin, mean ± SD, g/dL | 11.7 ± 2.0 | |
| Thrombocyte count, mean ± SD, G/L | 228 ± 71 | |
| aPTT, mean ± SD, seconds | 28.8 ± 7.8 | |
| C-reactive protein, mean ± SD, mg/dL | 1.2 ± 0.9 | |
| proBNP, mean ± SD, pg/mL | 3427 ± 5112 | |
| VWF:Ag (%) | 198 ± 92 | |
| VWF:GPIbM (%) | 164 ± 66 | |
| FVIII:C (%) | 177 ± 52 | |
| HMW multimer deficiency (%) | 20/31 (65%) | |
| IMW multimer deficiency (%) | 0/31 (0%) | |
| LMW multimer deficiency (%) | 0/31 (0%) | |
| Abnormal triplet structure (%) | 0/31 (0%) | |
Abbreviations: aPTT, activated partial thromboplastin time; AVA, aortic valve area; FVIII:C, factor VIII coagulation activity; HMW, high molecular weight; IMW, intermediate molecular weight; LMW, low molecular weight; LVEDD, left ventricular end diastolic diameter; LVESD, left ventricular end systolic diameter; pro-BNP, pro-brain natriuretic peptide; SD, standard deviation; VWF:Ag, von Willebrand factor antigen; VWF:GPIbM, von Willebrand factor activity.
Laboratory findings including results of VWF:Ag, VWF:GPIbM, FVIII:C, and VWF multimer analyses are shown in Tables 1 and 2. Patients had a mean hemoglobin level of 11.7 ± 2.0 g/dL, mean thrombocyte count of 228 ± 71 G/L, and a mean activated partial thromboplastin time (aPTT) of 28.8 ± 7.8 seconds. Low-resolution gel electrophoresis revealed an HMW multimer deficiency without an additional decrease in LMW or IMW portions in 65% of all patients (Table 1 and Figure 1). High-resolution gel analysis showed a normal inner multimeric triplet structure in all study patients (Figure 2). Among patients with reported bleeding episodes, 91.7% had an abnormal multimeric pattern, whereas only 8.3% had a normal multimeric structure. Noteworthily, none of the blood samples showed a multimeric pattern identical to AVWS type 2A or VWD type 2A (Figures 1 and 2). Results of assays for VWF:Ag and VWF:GPIbM were in the range of normal in most patients (83.9%), and VWF:GPIbM/VWF:Ag ratio <0.7 was detectable in only 20.0% of all patients with HMW multimer deficiency, and thus, only the multimer analysis yielded the correct diagnosis. Differences in patient characteristics, echocardiographic, and laboratory findings between patients with and without AVWS multimeric pattern are shown in Table 2.
Table 2.
Comparison of baseline characteristics between patients with versus without abnormal VWF multimer structure.
| Abnormal Multimeric Structure (n = 20) | Normal Multimeric Structure (n = 11) | P Value | |
|---|---|---|---|
| Patient characteristics | |||
| Age, mean ± SD, years | 81 ± 5 | 76 ± 11 | .238 |
| Male gender (%) | 14/20 (70%) | 7/11 (64%) | 1.000 |
| BMI, mean ± SD, kg/m2 | 27 ± 6 | 26 ± 4 | .645 |
| Echocardiographic data | |||
| Peak aortic jet velocity, mean ± SD, m/s | 4.8 ± 0.5 | 4.6 ± 0.4 | .274 |
| Peak aortic gradient, mean ± SD, mm Hg | 93 ± 18 | 86 ± 15 | .301 |
| Mean aortic gradient, mean ± SD, mmHg | 57 ± 11 | 54 ± 9 | .574 |
| AVA, mean ± SD, cm2 | 0.65 ± 0.13 | 0.69 ± 0.18 | .521 |
| Systolic ejection fraction (%) | 59 ± 10 | 61 ± 4 | .910 |
| Shear stress, mean ± SD, dyn/cm2 | 107 ± 13 | 106 ± 14 | .961 |
| Laboratory characteristics | |||
| Hemoglobin, mean ± SD, g/dL | 11.3 ± 2.2 | 12.5 ± 1.5 | .098 |
| Thrombocyte count, mean ± SD, G/L | 233 ± 83 | 219 ± 42 | .620 |
| aPTT, mean ± SD, sec | 25.4 ± 1.4 | 30.2 ± 8.9 | .068 |
| C-reactive protein, mean ± SD, mg/dL | 1.2 ± 1.0 | 1.2 ± 0.8 | .782 |
| proBNP, mean ± SD, pg/mL | 4500 ± 5930 | 1477 ± 2259 | .043 |
| VWF:Ag (%) | 176 ± 74 | 210 ± 100 | .470 |
| VWF:GPIbM (%) | 160 ± 59 | 166 ± 72 | .813 |
| FVIII:C (%) | 187 ± 50 | 157 ± 52 | .119 |
| VWF:GPIbM/VWF:Ag ratio | 0.81 ± 0.12 | 1.01 ± 0.20 | .001 |
| Bleeding history | |||
| GI bleeding (%) | 7/20 (35%) | 1/11 (9.1%) | .202 |
| Gingivorrhagia and epistaxis (%) | 3/20 (15%) | 0/11 (0%) | .535 |
| Spontaneous hematoma (%) | 6/20 (30%) | 0/11 (0%) | .065 |
| Hematuria (%) | 2/20 (10%) | 1/11 (9.1%) | 1.000 |
Abbreviations: aPTT, activated partial thromboplastin time; AVA, aortic valve area; BMI, body mass index; FVIII:C, factor VIII coagulation activity; GI, gastro-intestinal; proBNP, pro-brain natriuretic peptide; SD, standard deviation; VWF:Ag, von Willebrand factor antigen; VWF:GPIbM, von Willebrand factor activity.
Note: The bold numbers are below the significance level p < 0.05.
Figure 1.

Illustration of VWF multimer low-resolution gel electrophoresis (left side of the panel) and densitometrical band quantification (right side of the panel) of 4 individuals in comparison with a reference standard. Top row (patient A with severe AS) shows a normal VWF multimer electrophoresis band pattern with a normal densitometrical quantification of low- (bands 1-3), intermediate- (bands 4-13), and high- (bands >13) molecular-weight VWF multimers. The middle top row (patient B with severe AS) depicts an isolated deficiency of high-molecular-weight multimers both in the electrophoresis and in the densitometrical quantification in comparison with a reference standard. The middle bottom row (patient C with myeloproliferative syndrome) illustrates the electrophoresis and densitometrical pattern of a previously diagnosed typical AVWS type 2A characterized by a decrease in the high- and intermediate-molecular-weight multimers. The bottom row (patient D) shows intermediate- and high-molecular-weight VWF multimer deficiency of a young child with previously diagnosed VWD type 2A. The VWF multimer structure of patient B does not resemble the multimer pattern as it is seen in patient C and D. AS denotes aortic stenosis; AVWS, acquired von Willebrand syndrome; VWF, von Willebrand factor.
Figure 2.
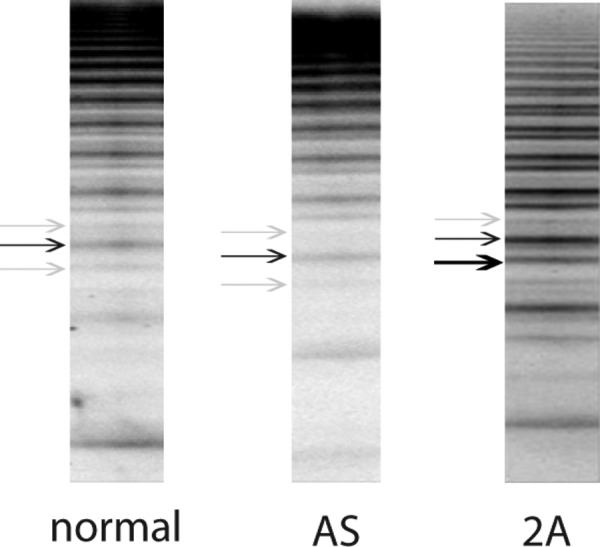
Illustration of the inner triplet sub-band structure of VWF multimers. Plasma samples of a healthy person (left panel), a patient with severe AS with isolated HMW VWF deficiency (middle panel) and a patient with previously diagnosed typical AVWS type 2A associated with myeloproliferative syndrome (right panel) were electrophoretically separated on an SDS high-resolution (2%) agarose gel. The characteristic triplet subband structure consisting of 2 satellite (grey arrows) and 1 main band (black arrow) can be seen in the left and the middle panel, whereas the right panel shows an abnormal subband composition (black bold arrow) typical of AVWS type 2A. AVWS denotes acquired von Willebrand syndrome; SDS, sodium dodecyl sulfate; VWF, von Willebrand factor.
Discussion
von Willebrand disease is the most common inherited bleeding disorder and is categorized into a partial quantitative (type 1), a qualitative (type 2) and a total quantitative (virtually complete deficiency; type 3) subtype according to the revised classification by the VWF subcommittee of the ISTH published in 2006.21
Although laboratory tests (such as the measurement of plasma concentration of VWF antigen, VWF binding activity, FVIII:C, and VWF:GPIbM/VWF:Ag ratio) represent the diagnostic cornerstone of VWD, these assays are frequently unspecific in AVWS. Therefore, VWF multimer analysis using protein electrophoresis remains the gold standard for AVWS diagnosis. Although, historically, AVWS has not been further subclassified, the similarity of the multimeric band pattern that is found in VWD type 2 and AVWS in severe AS has led to an analogous subclassification. von Willebrand disease type 2 is further subcategorized into type 2A, 2B, 2N, and 2M. von Willebrand disease type 2A and type 2B are the only subtypes to feature HMW multimer deficiency and can be distinguished using multimeric structure and a ristocetin-induced platelet aggregation (RIPA) assay. Although VWD type 2A is characterized by a reduction in both HMW and IMW multimers, an abnormal triplet structure, and a hypoactive RIPA, VWD type 2B shows an isolated HMW multimer deficiency, preserved triplet structure, and hyperactive RIPA. The RIPA assay is frequently not reliable in AVWS, thus VWF multimer structure analysis remains the gold standard to diagnose AVWS.
Acquired von Willebrand syndrome in the setting of AS has become an extensively studied topic in recent years and has been frequently subcategorized into type 2A, according to the VWF multimeric band phenotype. Our literature research revealed that neither IMW multimers nor the multimer triplet structure has been evaluated in the setting of AS. Hence, we supposed that AVWS could have been previously misclassified.
Consequently, we recruited 31 consecutive patients with severe AS, evaluated bleeding symptoms via questionnaire, and analyzed the VWF multimeric structure. The prevalence of VWF multimer abnormalities was high, whereas a bleeding history was present in only the minority of patients, indicating a “hemostatic rescue mechanism,” which has been recently proposed.1 Besides a slight difference in pro-brain natriuretic peptide levels and VWF:GPIbM/VWF:Ag ratio, all other laboratory parameters were comparable between patients with and without HMW VWF multimer deficiency (Table 2).
Results of VWF:Ag, VWF:GPIbM, (VWF:GPIbM/VWF:Ag ratio), and FVIII:C were unspecific in patients with an abnormal multimeric phenotype, which underscores the importance of gel electrophoresis for the diagnosis of AVWS. Multimeric band pattern analysis revealed a preserved triplet structure and normal band distribution and density in the IMW portions in all patients. A deficiency in the HMW portions accounted exclusively for the abnormal band pattern and, therefore, did neither resemble the multimer band pattern seen in AVWS type 2A nor that seen in VWD type 2A.
Noteworthily, in our study, the bleeding phenotype in patients with VWF multimer abnormalities appeared to be less common than expected, which is in accordance with previous findings.1 We suppose that the hemostatic capacity is still sufficient in patients with HMW VWF multimer deficiency under steady state conditions, whereas external influences (ie, trauma, surgery, and infection) might result in bleeding complications. It is unclear whether an additional decrease of IMW multimers in the setting of AS would have resulted in an increased bleeding prevalence compared to the present isolated HMW multimer deficiency.
There is a demand for a standardization of VWF multimer Western blot results in order to improve the diagnostic value, comparability, and reproducibility of the test and to avoid misclassifications. To achieve a test harmonization, a required minimum number of multimer bands on agarose gels and a clear definition for multimer subcategorization into the LMW, IMW, and HMW subset is necessary. In our present experiments, VWF multimer gel electrophoresis contained at least 19 bands, displayed the multimer triplet structure in the high-resolution gel, and bands 4 to 13 were defined as IMW whereas bands >13 were defined as HMW multimers.
Conclusion
Our present study reveals that multimeric pattern of AVWS in severe AS is not identical to that seen in AVWS type 2A or VWD type 2A and should not be subclassified as type 2A anymore. A lack of electrophoresis standardizations might have contributed to previous misclassifications, and thus, there is a demand for standardized diagnostic guidelines to avoid future AVWS misclassifications in clinical practice and research.
Footnotes
Authors’ Contributions: Joerg Kellermair and Helmut W. Ott contributed equally.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Natorska J, Mazur P, Undas A. Increased bleeding risk in patients with aortic valvular stenosis: from new mechanisms to new therapies. Thromb Res. 2016;139:85–89. [DOI] [PubMed] [Google Scholar]
- 2. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349(4):343–349. [DOI] [PubMed] [Google Scholar]
- 3. Casonato A, Sponga S, Pontara E, et al. von Willebrand factor abnormalities in aortic valve stenosis: pathophysiology and impact on bleeding. Thromb Haemost. 2011;106(1):58–66. [DOI] [PubMed] [Google Scholar]
- 4. Tsai HM, Sussman II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83(8):2171–2179. [PubMed] [Google Scholar]
- 5. Panzer S, Badr Eslam R, Schneller A, et al. Loss of high-molecular-weight von Willebrand factor multimers mainly affects platelet aggregation in patients with aortic stenosis. Thromb Haemost. 2010;103(2):408–414. [DOI] [PubMed] [Google Scholar]
- 6. Frank RD, Lanzmich R, Haager PK, Budde U. Severe Aortic Valve Stenosis: sustained cure of acquired von Willebrand syndrome after surgical valve replacement. Clin Appl Thromb Hemost. 2017; 23(3):229–234. doi: 10.1177/1076029616660759 [DOI] [PubMed] [Google Scholar]
- 7. Bander J, Elmariah S, Aledort LM, et al. Changes in von Willebrand factor-cleaving protease (ADAMTS-13) in patients with aortic stenosis undergoing valve replacement or balloon valvuloplasty. Thromb Haemost. 2012;108(1):86–93. [DOI] [PubMed] [Google Scholar]
- 8. Spangenberg T, Budde U, Schewel D, et al. Treatment of acquired von Willebrand syndrome in aortic stenosis with transcatheter aortic valve replacement. JACC. Cardiovasc Interv. 2015;8(5):692–700. [DOI] [PubMed] [Google Scholar]
- 9. Caspar T, Jesel L, Desprez D, et al. Effects of transcutaneous aortic valve implantation on aortic valve disease-related hemostatic disorders involving von Willebrand factor. Can J Cardiol. 2015;31(6):738–743. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi N, Tanabe K, Yoshitomi H, et al. Impairment of platelet retention rate in patients with severe aortic valve stenosis. J Cardiol. 2013;62(3):171–175. [DOI] [PubMed] [Google Scholar]
- 11. Warkentin TE, Moore JC, Anand SS, Lonn EM, Morgan DG. Gastrointestinal bleeding, angiodysplasia, cardiovascular disease, and acquired von Willebrand syndrome. Transfus Med Rev. 2003;17(4):272–286. [DOI] [PubMed] [Google Scholar]
- 12. Hillegass WB, Limdi NA. Valvular heart disease and acquired type 2A von Willebrand syndrome: the “hemostatic” waring blender syndrome. JAMA Cardiol. 2016;1(2):205–206. [DOI] [PubMed] [Google Scholar]
- 13. Solomon C, Budde U, Schneppenheim S, et al. Acquired type 2A von Willebrand syndrome caused by aortic valve disease corrects during valve surgery. Br J Anaesth. 2011;106(4):494–500. [DOI] [PubMed] [Google Scholar]
- 14. Rauch A, Caron C, Vincent F, et al. A novel ELISA-based diagnosis of acquired von Willebrand disease with increased VWF proteolysis. Thromb Haemost. 2016;115(5):950–959. [DOI] [PubMed] [Google Scholar]
- 15. Ott HW, Griesmacher A, Schnapka-Koepf M, et al. Analysis of von Willebrand factor multimers by simultaneous high- and low-resolution vertical SDS-agarose gel electrophoresis and Cy5-labeled antibody high-sensitivity fluorescence detection. Am J Clin Pathol. 2010;133(2):322–330. [DOI] [PubMed] [Google Scholar]
- 16. Ruggeri ZM. Structure and function of von Willebrand factor. Thromb Haemost. 1999;82(2):576–584. [PubMed] [Google Scholar]
- 17. Enayat MS, Hill FG. Analysis of the complexity of the multimeric structure of factor VIII related antigen/von Willebrand protein using a modified electrophoretic technique. J Clin Pathol. 1983;36(8):915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gralnick HR, Williams SB, McKeown LP, et al. In vitro correction of the abnormal multimeric structure of von Willebrand factor in type IIa von Willebrand’s disease. Proc Natl Acad Sci U S A. 1985;82(17):5968–5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bodo I, Eikenboom J, Montgomery R, et al. Platelet-dependent von Willebrand factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13(7):1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rapaport SI. Preoperative hemostatic evaluation: which tests, if any? Blood. 1983;61(2):229–231. [PubMed] [Google Scholar]
- 21. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–2114. [DOI] [PubMed] [Google Scholar]