

Abstract
Objective:
We aimed to investigate whether soluble CD40 ligand (CD40L) levels are higher in patients with isolated coronary artery ectasia (CAE) compared to patients with angiographically normal coronary arteries and those with stable coronary artery disease (CAD).
Materials and Methods:
In all, 55 patients with isolated CAE without stenosis, 55 with stable CAD, and 55 control participants with angiographically normal coronary arteries were included. The CAE severity was determined according to the Markis classification. Plasma levels of soluble CD40 ligand were measured by enzyme-linked immunosorbent assay.
Results:
The baseline characteristics of the 3 groups were similar. Plasma levels of soluble CD40 ligand were significantly higher in patients with CAE and CAD than in controls (2.6 ± 3.1 ng/mL and 2.0 ± 3.1 ng/mL vs 1.8 ± 2.1 ng/mL, P = .004). No difference was found between the CAE and CAD groups. Soluble CD40 ligand level was significantly higher in the type 1 Markis subgroup than that in the type 3 or type 4 subgroups (P = .01). A receiver operating characteristic curve analysis revealed that soluble CD40 ligand level >1.2 ng/mL identified patients with isolated CAE.
Conclusion:
Significantly higher levels of soluble CD40 ligand were detected in patients with CAE than that in control participants with normal coronary arteries, suggesting that soluble CD40 ligand may be involved in the pathogenesis of CAE. The CD40–CD40 ligand system likely plays a role in the pathogenesis of CAE.
Keywords: inflammation, isolated coronary artery ectasia, soluble CD40 ligand
Introduction
Coronary artery ectasia (CAE) is characterized by the dilation of the coronary arteries, particularly localized or diffuse dilation with luminal dilation over the normal adjacent segment or a vessel diameter 1.5-fold wider than that of a normal vessel.1 The prevalence of CAE is 0.3% to 5% among patients who undergo coronary angiography.2 The isolated form of CAE, which has been defined as CAE without important coronary artery stenosis, constitutes a small portion of the total number of CAE cases with a rate of 0.1 % to 0.8%.1,3,4
Coronary artery ectasia clinically predisposes patients without coronary artery disease (CAD) to adverse coronary events, such as vasospasm, thrombosis, myocardial ischemia, and dissection.4,5 Therefore, determining the factors associated with the presence and severity of CAE may be beneficial for managing those patients. The mechanism of CAE remains unclear. Some congenital disorders, such as polycystic kidney disease and Ehlers-Danlos syndrome, are associated with CAE.6,7 In addition, CAE can be diagnosed in patients with several types of vasculitis or connective tissue disorders.8,9 However, most patients with CAE do not have a congenital disorder or connective tissue disease.
Previous studies have shown that inflammation and atherosclerosis play major roles in the development of CAE, although the underlying reasons for ectasia formation are not fully understood.10 The CAE may be a variant of CAD, as CAE is associated with inflammation and frequently accompanies CAD. More severe inflammation can be involved in the pathogenesis of CAE.11
The CD40 ligand (CD40L/CD154) is a transmembrane glycoprotein retaining structural homology with tumor necrosis factor-α (TNF-α), and it was initially found in activated T lymphocytes.12 Subsequently, it was detected in platelets and it is expressed rapidly after platelets are activated.13 The CD40L receptor CD40 is found in immune, endothelial, and smooth muscle cells (SMCs), as well as macrophages and fibroblasts.14 The CD40 has gained considerable interest due to evidence linking the CD40/CD40L dyad with atherosclerosis, inflammation, and thrombosis.15
The physiological role and pathophysiological importance of soluble CD40 (sCD40) in patients with CAE are unclear. As CAE seems to be a form of atherosclerosis, and inflammation plays a major role in atherosclerotic vascular processes, sCD40-mediated inflammation may also be involved in CAE. Therefore, we aimed to investigate whether sCD40L levels are higher in patients with isolated CAE compared to patients with angiographically normal coronary arteries and those with stable CAD.
Materials and Methods
The present study was an observational, case-control, comparative study. The relationship between sCD40L levels and the presence of CAE was investigated. Approximately 3000 participants who had undergone elective diagnostic coronary angiography at our institution were scanned to find patients with apparent CAE. The indications for coronary angiography were either the presence of typical angina or positive or equivocal results of noninvasive screening tests for myocardial ischemia. We selected 55 (26 males and 29 females; mean age, 57.0 ± 10.3 years) consecutive patients diagnosed with isolated CAE without any atherosclerotic lesions (CAE group) and 55 (27 males and 28 females; mean age, 56.5 ± 9.1 years) consecutive patients with completely normal coronary arteries (control group).
Finally, the CAD group consisted of 55 (34 males and 21 females; mean age, 59.8 ± 9.9 years) consecutive participants who were selected from catheterized patients during the same study period and who had ≥50% stenotic lesions on a coronary angiogram.
The following patients were excluded—those who developed secondary coronary aneurysms following balloon angioplasty, received a coronary stent, underwent brachytherapy or atherectomy, who were diagnosed with vascular ectasia associated with Kawasaki disease or any known collagen vascular disease, who developed myocardial infarction or had a coronary artery bypass grafting surgery, those with significant organic valvular heart disease, peripheral arterial disease, congestive heart failure, congenital heart disease, atrial fibrillation, hypothyroidism, hyperthyroidism, thrombocytopenia, hemolytic failure, respiratory tract disease (chronic obstructive pulmonary disease, chronic bronchitis, or pulmonary embolism), pulmonary hypertension, any autoimmune or neoplastic disease, chronic kidney (creatinine > 1.5 mg/dL) or hepatic failure (aspartate transaminase or alanine transaminase more than third the normal value), or actively infectious diseases, and those receiving active treatment with antiaggregants, anticoagulants, or steroids.
Study Protocol
A detailed medical history was obtained from all patients and a complete physical examination was performed. A 12-lead electrocardiogram (ECG) was taken, and 2 experienced specialists performed a detailed transthoracic ECG. The diagnosis of hypertension was established if systolic blood pressure was ≥140 mm Hg or diastolic blood pressure was ≥90 mm Hg, measured at least twice separately, or if a patient was taking antihypertensive medication. The diagnosis of diabetes mellitus was established by a fasting blood glucose ≥126 mg/dL or if a patient was taking antidiabetic medication. Fasting blood glucose was defined as impaired at 100 to 126 mg/dL. Hyperlipidemia was defined as a total cholesterol level ≥200 mg/dL or a history of statin use except in the last 3 months. Patients who were smoking before hospitalization were accepted as smokers.
Informed consent was obtained from all patients prior to the study. The local ethics committee approved the study protocol, and all patients signed written informed consent. This study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and International Conference on Harmonization guidelines.
Coronary Angiography
Coronary angiography was performed using the Judkin technique without nitroglycerin, and 6F right and left heart catheters were employed through femoral or radial arteries. Two experienced interventional cardiologists who were blinded to the laboratory measurements and clinical status of the participants analyzed the coronary angiograms. The CAE was diagnosed when a segment of a coronary artery was dilated at least 1.5 times more than the adjacent segment.1 The CAE was classified according to the system proposed by Markis et al.3 Severity of CAE was graded according to the following criteria—type 1, diffuse ectasia of 2 or 3 vessels; type 2, diffuse ectasia in 1 vessel and localized ectasia in another vessel; type 3, diffuse ectasia in 1 vessel only; and type 4, localized or segmental ectasia.1 The location of ectatic coronary segment, number of coronary vessels with ectasia, and Markis classifications were recorded.
Patients with coronary lesions with a stenosis diameter ≥50% in ≥1.5-mm vessels were diagnosed with CAD. Complexity of CAD was evaluated using the Synergy between percutaneous coronary intervention with Taxus and cardiac surgery (SYNTAX) score. Coronary lesions with ≥50% diameter stenosis in ≥1.5-mm vessels were scored separately and added together to provide the cumulative SYNTAX score, which was prospectively calculated using the SYNTAX score algorithm on the baseline diagnostic angiogram.16,17 Two experienced interventional cardiologists analyzed the SYNTAX score, and the final judgment was made by consensus in cases of disagreement. The final score was calculated from the individual lesion scores by analysts who were blinded to the procedural data and clinical outcomes.
Routine Laboratory Measurements
Blood was taken from the antecubital vein of the study participants between 08:00 and 10:00 am after 12 hours of fasting . Serum was separated by centrifugation (3000g, 10 minutes, 4°C) and stored at −80°C. Biochemical blood tests were assayed photometrically with an Architect c1600 Analyzer (Abbott Diagnostics, Chicago, Illinois). C-reactive protein (CRP) levels were measured using the nephelometric method and the IMMAGE 800 Immunochemistry system (Beckman Coulter, Brea, California). Complete blood counts were obtained using the LH500 Automatic Hematology Analyzer (Beckman Coulter). Fasting blood glucose levels were determined using the hexokinase method. Total cholesterol levels were determined using enzymatic methods. High-density lipoprotein concentrations were obtained using the accelerator selective detergent method. Triglyceride levels were determined by employing the glycerol-3-phosphate oxidase method. Low-density lipoprotein concentrations were determined using the Friedewald formula.
Soluble CD40L Measurement
Blood was drawn from the femoral artery or radial artery before angiography after inserting a sheath. Pressure cuffs were not used within 30 minutes of blood sampling. The blood was placed immediately in evacuated tubes containing 3.8% sodium citrate (BD Biosciences, San Jose, California) and centrifuged immediately for 20 minutes at 2000 rpm and 4°C. Plasma samples were stored at −80°C until use. Plasma concentrations of sCD40L were measured using a commercial enzyme-linked immunosorbent assay kit (eBioscience, San Diego, California). The minimum limit of detection for was 0.06 ng/mL, and the interassay and intra-assay coefficients of variation were <6.8% and <4%, respectively. The assay is highly specific for human sCD40L, and the manufacturer reports <0.5% cross-reactivity with available related molecules.
Statistical Analyses
Statistical analyses were performed using SPSS software version 20 (SPSS Inc, Chicago, Illinois). The variables were investigated using visual (histograms and probability plots) and analytical methods (Shapiro-Wilk test) to determine whether they were normally distributed. Descriptive analyses are presented using means and standard deviations for normally distributed variables. Mean, standard deviation, lowest median value, highest frequency value, and ratio value were used. The Kolmogorov-Smirnov test was used to assess the distribution of the data. Analysis of variance followed by Tukey post hoc method, the Kruskal-Wallis, and Mann-Whitney U tests was used to analyze the quantitative data. The χ2 test was used to analyze the qualitative data. A P value <.05 was considered significant. The ability to predict the presence of CAE based on sCD40L levels was analyzed using a receiver operating characteristic (ROC) curve analysis. Sensitivity and specificity values were determined when a significant cutoff value was observed. A 5% type 1 error level was significantly predictive of the test variables when evaluating the area under the curve.
Results
The demographic, clinical, and laboratory data of the patients are summarized in Table 1. No differences in age, sex, or body mass index were observed among the 3 groups (P > .05 for all). No differences in cardiovascular risk factors, such as hyperlipidemia, hypertension, diabetes mellitus, smoking, or family history, were detected between the groups (all Ps > .05).
Table 1.
Demographic, Clinical, and Angiographic Characteristics of the Study Population.
| Variables | NCA (n = 55) | CAE (n = 55) | CAD (n = 55) | P Value |
|---|---|---|---|---|
| Age (years) | 56.5 ± 9.1 | 57.0 ± 10.3 | 59.8 ± 9.9 | .217 (NS) |
| Gender | ||||
| Male | 27 (49.0%) | 26 (47.2%) | 34 (61.8%) | .261 (NS) |
| Female | 28 (50.9%) | 29 (52.7%) | 21 (38.1%) | |
| BMI (kg/m2) | 28.4 ± 4.0 | 29.3 ± 4.2 | 27.4 ± 4.0 | .148 (NS) |
| Diabetes mellitus | 15 (27.2%) | 15 (27.2%) | 20 (36.3%) | .440 (NS) |
| Hypertension | 21 (38.1%) | 38 (69%) | 35 (63.6%) | .114 (NS) |
| Hypercholesterolemia | 9 (16.3%) | 13 (23.6%) | 12 (21.8%) | .966 (NS) |
| Family history of CAD | 7 (12.7%) | 13 (23.6%) | 11 (20.0%) | .702 (NS) |
| Smoker | 8 (14.5%) | 16 (29.0%) | 13 (23.63%) | .051 (NS) |
| Marcis classification | ||||
| Type 1 | 17 (30.9%) | |||
| Type 2 | 12 (21.8%) | |||
| Type 3 | 14 (25.4%) | |||
| Type 4 | 12 (21.8%) | |||
| CAE distribution | ||||
| Left main | 4 (3.0%) | |||
| Left anterior descending | 46 (40.7%) | |||
| Left circumflex | 20 (17.6%) | |||
| Right | 43 (38.0%) | |||
| SYNTAX score | 13.2 ± 9.1 | |||
Abbreviations: BMI, body mass index; CAD, coronary artery disease; CAE, coronary artery ectasia; NCA, normal coronary artery, NS, not significant.
Patients’ angiographic characteristics are shown in Table 1. Ectasia involved the left main coronary artery in 4 (3.0%), the left anterior descending artery in 46 (40.7%), the left circumflex artery in 20 (17.6%), and the right coronary artery in 43 (38.0%) patients. The types of CAE according to the Markis classification of coronary ectasia were as follows in decreasing order of severity: 17 (30.9%) patients with type 1 CAE, 12 (21.8%) with type 2 CAE, 14 (25.4%) with type 3 CAE, and 12 (21.8%) with type 4 CAE. Complexity of CAD was evaluated with the SYNTAX score, and the mean SYNTAX score was 13.2 ± 9.1 in the CAD group. No differences in any of the laboratory parameters were observed among the 3 groups (all Ps > .05; Table 2).
Table 2.
Laboratory Findings of the Study Population, Mean Value ± Standard Deviation.
| NCA (n = 55) | CAE (n = 55) | CAD (n = 55) | P Value | |
|---|---|---|---|---|
| WBC (103/μL) | 6.7 ± 1.6 | 6.3 ± 1.8 | 6.7 ± 1.7 | .510 (NS) |
| Hemoglobin (103/μL) | 13.6 ± 1.7 | 13.6 ± 1.5 | 13.4 ± 1.7 | .423 (NS) |
| Platelet (103/μL) | 281 ± 72 | 272 ± 67 | 261 ± 64 | .321 (NS) |
| AST (U/L) | 21.7 ± 8.4 | 23.1 ± 6.1 | 23.5 ± 12.4 | .189 (NS) |
| ALT (U/L) | 22.4 ± 8.2 | 25.3 ± 10.8 | 23.3 ± 21.6 | .966 (NS) |
| TSH (mIU/L) | 1.4 ± 0.9 | 1.7 ± 1.6 | 1.8 ± 1.8 | .915 (NS) |
| FBG (mg/dL) | 111.9 ± 41.9 | 109.6 ± 42.6 | 111.7 ± 40.5 | .819 (NS) |
| HbA1c (%) | 6.1 ± 0.9 | 6.0 ± 0.9 | 6.4 ± 1.7 | .756 (NS) |
| Urea (mg/dL) | 30.0 ± 10.3 | 33.8 ± 17.7 | 33.9 ± 12.9 | .489 (NS) |
| Creatinine (mg/dL) | 0.7 ± 0.1 | 0.8 ± 0.3 | 0.8 ± 0.4 | .476 (NS) |
| Total cholesterol (mg/dL) | 196 ± 44.3 | 199.6 ± 37.8 | 203 ± 45.9 | .672 (NS) |
| LDL-C (mg/dL) | 122.1 ± 34.4 | 123.1 ± 32.9 | 133.5 ± 39.6 | .055 (NS) |
| HDL-C (mg/dL) | 47.6 ± 13.3 | 42.6 ± 8.4 | 44.4 ± 9.8 | .142 (NS) |
| Triglyceride (mg/dL) | 151.7 ± 69.2 | 169.8 ± 84.5 | 161.1 ± 86.4 | .159 (NS) |
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; CAD, coronary artery disease; CAE, coronary artery ectasia; FBG, fasting blood glucose; HbA1c, hemoglobin A1c; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; NCA, normal coronary artery; NS, not significant; TSH, thyroid-stimulating hormone; WBC, white blood cell.
The plasma levels of sCD40L are summarized in Table 3. As shown in Figure 1, patients with isolated CAE had significantly higher levels than in the control group (P < .05). However, no differences were detected between the CAE and CAD groups. Levels of sCD40L were significantly associated with the Markis classification for CAE (Table 4 and Figure 2). Relatively high levels of type 1 sCD40L, type 3 SCD40L, and type 4 sCD40L were identified as the root cause behind the differences (means: 4.61, 1.55, and 0.85 ng/mL respectively; P < .05).
Table 3.
Plasma sCD40L Levels of the Study Population, Mean Value ± Standard Deviation.
| NCA (n = 55) | CAE (n = 55) | CAD (n = 55) | P Value | |
|---|---|---|---|---|
| sCD40L | 1.8 ± 2.1 | 2.6 ± 3.1 | 2.0 ± 3.1 | .004 |
Abbreviations: CAD, coronary artery disease; CAE, coronary artery ectasia; NCA, normal coronary artery; sCD40L, soluble CD40 ligand.
Figure 1.
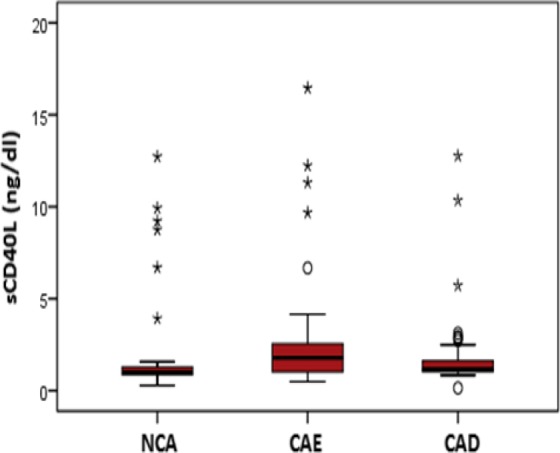
The comparison of soluble CD40 ligand levels between 3 groups, 2.6 ± 3.1 ng/mL in coronary artery ectasia group, 2.0 ± 3.1 ng/mL in coronary artery disease group, and 1.8 ± 2.1 ng/mL in normal coronary artery group.
Table 4.
Correlations Between CAE Severity and sCD40L Levels.
| Markis Classification | CAE (n = 55) | Median Value | Mean Value ± Standard Deviation | Kruskall-Wallis, χ2 | P Value | Post Hoc |
|---|---|---|---|---|---|---|
| Type 1 | 17 | 3.33 | 4.61 ± 4.20 | 18.39 | 0.01a | Type 1 > type 3a, Type 1 > type 4a |
| Type 2 | 12 | 1.29 | 2.50 ± 3.15 | |||
| Type 3 | 14 | 1.55 | 1.40 ± 0.63 | |||
| Type 4 | 12 | 0.85 | 1.19 ± 0.61 |
Abbreviations: CAE, coronary artery ectasia; sCD40L, soluble CD40 ligand. aStatistically significant.
Figure 2.
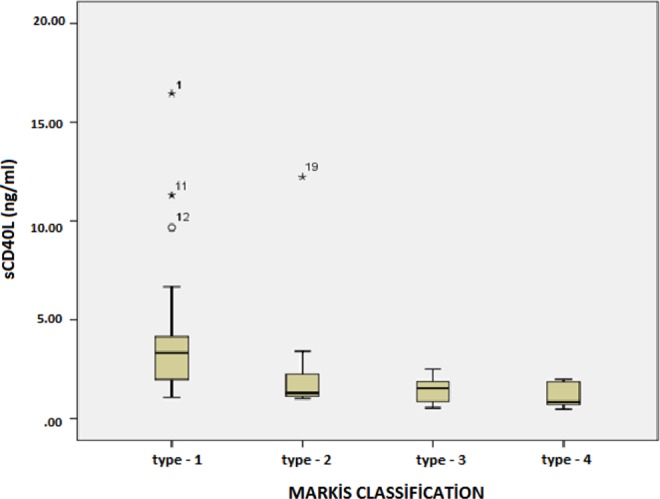
The soluble CD40 ligand is highest in patients with the most severe type of coronary artery ectasia and lowest in patients with the least severe type of coronary artery ectasia.
As shown in Figure 3, an ROC curve analysis was performed in the isolated CAE and control groups to detect the sCD40L cutoff value for predicting patients with isolated CAE. The ROC curve for the sCD40L revealed that a cutoff value >1.2 ng/L was correlated with CAE (75% sensitivity, 65.5% specificity, 65.5% positive predictive value, and 71.4% negative predictive value; area under the curve: 0.684, 95% confidence interval: 0.576-0.792; P < .002).
Figure 3.
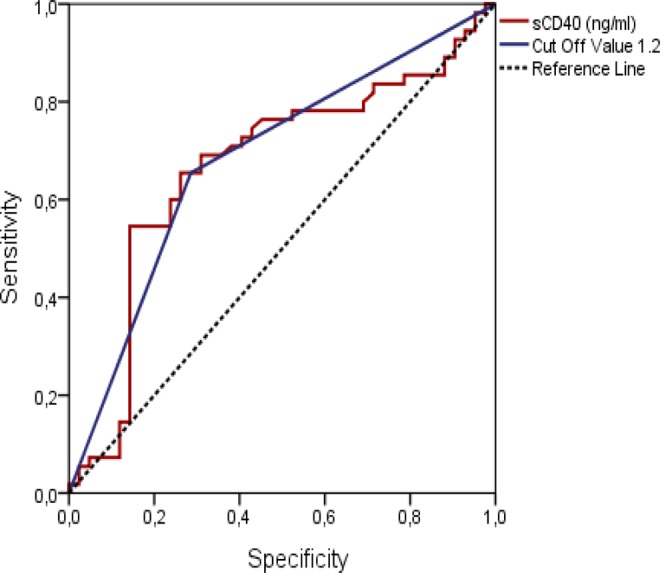
The ROC curve for soluble CD40 ligand revealed that cutoff values over 1.2 ng/L were correlated with coronary artery ectasia (75% sensitivity, 65.5% specificity, positive predictive value: 65.5%, and negative predictive value: 71.4%; area under the curve: 0.684, 95% CI: 0.576-0.792, P < .002). 95% CI indicates 95% confidence interval; ROC, receiver operating characteristic.
Discussion
Patients with CAE had significantly higher sCD40 levels than patients with angiographically normal coronary arteries. This is the first study demonstrating a relationship between CAE and sCD40. We also found that sCD40L level was significantly related with the Markis classification of CAE severity. In addition, plasma levels of sCD40L were similar between patients with CAE and those with CAD.
Although a definitive link between atherosclerosis and ectasia has not been confirmed, it has been suggested that CAE is a variant of CAD, and atherosclerosis is thought to be the major cause of CAE.1,3,18 Markis et al3 reported that histological changes in patients with CAE are the same as those observed in atherosclerotic plaques, such as diffuse intimal and medial degeneration and hyalinization. The most common mechanism for CAE seems to be inflammation-induced injury of the vascular tunica media.16,18 Prior pathological examinations and studies that have demonstrated chronic inflammatory cell infiltration, destruction of the arterial media, and increases in the levels of inflammatory markers, such as the plasma-soluble adhesion molecules, interleukin (IL)-1, TNF-α, and IL-10, support the hypothesis of inflammation-induced injury to the tunica media in patients with CAE.16,17,19,20 Atheromatous ulcerations found in ectatic segments have suggested that atherosclerosis is the most common cause of CAE.18,21 A similar mechanism is the turbulent blood flow in ectatic segments, because a loss of laminar flow causes an increase in red blood cell aggregation and thrombogenicity.5,18,21
The prevalence of CAE varies from 0.3% to 5% in angiographic series.2–22 It may appear as a relatively innocent clinical entity; however, it can cause cardiac events such as stable or unstable angina pectoris, myocardial infarction, and cardiac death.23–25 Several studies have shown that patients with isolated CAE have an increased risk of mortality, equivalent to that of patients with obstructive CAD.3–5 In the current study, plasma sCD40L level is similar within CAE and CAD patient groups. Furthermore, normal coronary artery control group plasma sCD40L level is less than CAE and CAD patient groups. These results that align with the previous studies suggest that CAE may be a variant of CAD. At least, it suggests that they have similar pathophysiological and clinical process.
Evidence suggests that both CD40 and CD40L are expressed by endothelial cells, SMCs, and macrophages in atherosclerotic lesions, and that they are prevalent within advanced, rupture-prone lesions.14,26 The physiological interaction between CD40L and CD40 causes a variety of reactions, including increased expression of adhesion molecules and secretion of IL-1, IL-6, and TNF-α, all of which recruit more inflammatory cells that aggravate local microinflammation.14,27 Furthermore, CD40/CD40L enhances the expression of tissue factor and matrix metalloproteinases, thereby maintaining a prothrombotic milieu and decreased plaque stability.26,28,29 The CD40 signaling also results in increased production of reactive oxygen species that antagonize endothelial nitric oxide activity, leading to endothelial dysfunction.30 All of these actions are explained by the important role of the CD40/CD40L dyad in endothelial dysfunction, inflammation, atherosclerosis, and CAE.
The CD40/CD40L expression is upregulated in atheroma-associated cells, but the exact regulatory mechanisms remain largely unknown.31,32 In vitro studies have shown that CD40/CD40L interactions at the endothelial cell surface result in activation of the endothelium and SMC and subsequent expression of adhesion molecules, a step that initiates atherogenesis.31,32 In addition, recent evidence suggests that CD40/CD40L has a role in dendritic cells and T-lymphocyte interactions inside the vascular wall.33 Furthermore, sCD40L has an unfavorable effect on the vascular redox state and endothelium-dependent relaxation via the activation of specific redox-sensitive intracellular pathways.34 Cell and animal studies have demonstrated that CD40L destabilizes endothelial nitric oxide synthase messenger ribonucleic acid and increases the production of vascular oxygen.34 Therefore, pro-inflammatory cytokines, reduced bioavailability of nitric oxide, and overexpression of adhesion molecules promote leukocyte recruitment and migration into the tunica media, participating in the formation of atheroma. All of these actions are important in the relationship between CD40/CD40L and CAE.
Platelets are the main source of sCD40L, being responsible for >95% of the circulating levels.35 In addition to atheroma formation and plaque weakening, CD40/CD40L interactions have an eminent role in thrombotic events after plaque rupture. These interactions induce the expression of tissue factor on macrophages and the expression of thrombomodulin, favoring local procoagulant and prothrombotic status.36
Coronary ectasia may promote thrombosis via abnormal flow conditions.36,37 Studies that have used the “thrombolysis in myocardial infarction frame count method” have shown that coronary blood flow is impaired in patients with isolated CAE.37 Within the ectatic segment itself, the flow is relatively slow or static, which promotes the activation of platelets and formation of thrombi. Thrombosis of CAE is also mediated both by platelet- and endothelial-derived prothrombogenic, pro-inflammatory mechanisms and may be further propagated in the presence of chronic thrombus.37,38 Finally, the presence of thrombi increases platelet activity in patients with isolated CAE and may contribute to the development of more intracoronary thrombi and greater pro-inflammatory activation within the ectatic segment and to more distal microembolization.36–38 As per our hypothesis, platelet activation is important and is probably the main factor in the pro-inflammatory and prothrombotic process triggered by CAE. Moreover, we suggest that sCD40L is the main biomarker of CAE platelet activation and plays an active role in each step of its pathophysiological process. Consequently, sCD40L may cause a vicious prothrombotic/pro-inflammatory circle. Consistent with these data, we found close correlations between sCD40L and CAE.
The CD40/CD40L interaction participates in the development and progression of atherosclerosis and plays a role in oxidative stress and inflammation.39–41 Antoniades et al42 reported higher sCD40L levels in patients with hypercholesterolemia, unstable angina, or acute myocardial infarction. Tousoulis et al43 showed that plasma levels of sCD40L are higher in patients with stable angina than in healthy participants but lower than in patients with acute coronary syndrome. Durakoğlugil et al44 revealed a significant increase in serum levels of sCD40 in patients with slow coronary flow. In the current study, plasma levels of sCD40L were similar in the CAE and CAD patient groups. Furthermore, the levels were lower in the control group than in the CAE and CAD groups. These results agree with those of previous studies suggesting that CAE may be a variant of CAD or has a similar pathophysiological process.
We cannot speculate whether sCD40L itself is the only factor involved in the pathophysiology of CAE. However, it seems to be important for prothrombotic and pro-inflammatory status in patients with CAE. A second major finding of this study is that there were differences between lesion number and distribution in the Markis classification, suggesting a relationship between plasma levels of sCD40L and CAE severity. Although the subgroup samples were relatively small, the mean sCD40L levels in some subgroups were nearly the same, suggesting that a larger sample would improve the reliability of the results. These findings indicate that sCD40L may have a role in the pathogenesis of CAE. As mentioned above, the relationship between sCD40L level and CAE may be causative, or it could be that sCD40L increased as a result of CAE. We cannot clarify this in the current study.
Study Limitations
Our study had some limitations, including the small number of patients and the fact that they were all Caucasians. None of the patients underwent intravascular ultrasonography to detect atherosclerotic remodeling in ectatic arteries. Hence, the coexistence of nonobstructive CAD (<40%) in patients with “isolated” CAE cannot be absolutely established. Nevertheless, patients with isolated CAE do not routinely undergo intravascular ultrasound in clinical practice, and CAE is usually diagnosed by coronary angiography. A number of drugs affect plasma levels of sCD40.45 Although patients using antiaggregant, anticoagulant, and anti-inflammatory drugs were excluded from the study, other drugs such as statins can affect sCD40 levels. Furthermore, markers that play a role in aggregation and inflammation, such as E-selectin, P-selectin, vascular cell adhesion molecule 1, intercellular adhesion molecule 1, and hs-CRP, were not evaluated in relation to sCD40L levels.
Conclusion
A number of studies have shown a relationship between sCD40L and cardiovascular diseases. Our study is the first to investigate the relationship between sCD40L and CAE. Detailed studies are needed to evaluate the exact pathophysiological role of sCD40L, which has autocrine, paracrine, and endocrine effects and causes endothelial dysfunction and atherothrombosis through its pro-inflammatory, procoagulant, and proatherogenic properties in patients with CAE.
Footnotes
Authors’ Note: The English in this document has been checked by at least 2 professional editors, both native speakers of English. For a certificate, please see http://www.textcheck.com/certificate/LydgVi.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Swaye PS, Fisher LD, Litwin P, et al. Aneurysmal coronary artery disease. Circulation. 1983;67(1):134–138. [DOI] [PubMed] [Google Scholar]
- 2. Şarlı B, Baktır AO, Sağlam H, et al. Neutrophil-to-lymphocyte ratio is associated with severity of coronary artery ectasia. Angiology. 2014:65(2):147–151. [DOI] [PubMed] [Google Scholar]
- 3. Markis JE, Joffe CD, Cohn PF, Feen DJ, Herman MV, Gorlin R. Clinical significance of coronary arterial ectasia. Am J Cardiol. 1976;37(2):217–222. [DOI] [PubMed] [Google Scholar]
- 4. Al-Harthi SS, Nouh MS, Arafa M, Al-Nozha M. Aneurysmal dilatation of the coronary arteries: diagnostic patterns and clinical significance. Int J Cardiol. 1991;30(2):191–194. [DOI] [PubMed] [Google Scholar]
- 5. Krüger D, Stierle U, Herrmann G, Simon R, Sheikhzadeh A. Exercise induced myocardial ischemia in isolated coronary artery ectasias and aneurysms (“dilated coronaropathy”). J Am Coll Cardiol. 1999;34(5):1461–1470. [DOI] [PubMed] [Google Scholar]
- 6. Dieter RS, Murtaugh T, Black J, Russell DC. Coronary arteriomegaly in a patient with Ehlers-Danlos syndrome and multiple aneurysms—a case report. Angiology. 2003;54(6):733–736. [DOI] [PubMed] [Google Scholar]
- 7. Pourafkari L, Ghaffari S, Zamani B, Tolui M. Coronary artery ectasia in a patient with polycystic kidney disease. J Cardiovasc Thorac Res. 2011;3(3):101–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mavrogeni S, Manoussakis MN, Karagiorga TC, et al. Detection of coronary artery lesions and myocardial necrosis by magnetic resonance in systemic necrotizing vasculitides. Arthritis Rheum. 2009;61(8):1121–1129. [DOI] [PubMed] [Google Scholar]
- 9. Mueller F, Knirsch W, Harpes P, Prêtre R, Valsangiacomo Buechel E, Kretschmar O. Long-term follow-up of acute changes in coronary artery diameter caused by Kawasaki disease: risk factors for development of stenotic lesions. Clin Res Cardiol. 2009;98(8):501–507. [DOI] [PubMed] [Google Scholar]
- 10. Mavrogeni S. Coronary artery ectasia: from diagnosis to treatment. Hell J Cardiol. 2010;51(2):158–163. [PubMed] [Google Scholar]
- 11. Turhan H, Erbay AR, Yaşar AS, Balcı M, Biçer A, Yetkin E. Comparison of C-reactive protein levels in patients with coronary artery ectasia versus patients with obstructive coronary artery disease. Am J Cardiol. 2004;94(10):1303–1306. [DOI] [PubMed] [Google Scholar]
- 12. Armitage RJ, Fanslow WC, Strockbine L, et al. Molecular and biological characterization of a murine ligand for CD40. Nature. 1992;357(6373):80–82. [DOI] [PubMed] [Google Scholar]
- 13. Henn V, Slupsky JR, Grafe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391(6667):591–594. [DOI] [PubMed] [Google Scholar]
- 14. Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. 2001;58(1):4–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santilli F, Basili S, Ferroni P, Davi G. CD40/CD40L system and vascular disease. Intern Emerg Med. 2007;2(4):256–268. [DOI] [PubMed] [Google Scholar]
- 16. Turhan H, Erbay AR, Yaşar AS, et al. Plasma soluble adhesion molecules; intercellular adhesion molecule-1, vascular cell adhesion molecule-1 and E-selectin levels in patients with isolated coronary artery ectasia. Coron Artery Dis. 2005;16(1):45–50. [DOI] [PubMed] [Google Scholar]
- 17. Soto ME, Reyes-Villatoro MA, Márquez R, Cardoso G, Posadas-Sánchez R, Juárez-Orozco LE. Evaluation and analysis of plasma soluble adhesion molecules in patients with coronary ectasia and atherosclerotic coronary artery disease. Arch Med Res. 2014;45(6):478–483. [DOI] [PubMed] [Google Scholar]
- 18. Antoniadis AP, Chatzizisis YS, Giannoglou GD. Pathogenetic mechanisms of coronary ectasia. Int J Cardiol. 2008;130(3):335–343. [DOI] [PubMed] [Google Scholar]
- 19. Brunetti ND, Salvemini G, Cuculo A, et al. Coronary artery ectasia is related to coronary slow flow and inflammatory activation. Atherosclerosis. 2014;233(2):636–640. [DOI] [PubMed] [Google Scholar]
- 20. Tokgözoğlu L, Ergene O, Kınay O, Nazlı C, Hasçelik G, Hoşcan Y. Plasma interleukin-6 levels are increased in coronary artery ectasia. Acta Cardiol. 2004;59(5):515–519. [DOI] [PubMed] [Google Scholar]
- 21. Aboeata AS, Sontineni SP, Alla VM, Esterbrooks DJ. Coronary artery ectasia: current concepts and interventions. Front Biosci (Elite Ed). 2012;4:300–310. [DOI] [PubMed] [Google Scholar]
- 22. Hartnell GG, Parnell BM, Pridie RB. Coronary artery ectasia: its prevalence and clinical significance in 4993 patients. Br Heart J. 1985;54(4):392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Demopoulos VP, Olympios CD, Fakiolas CN, et al. The natural history of aneurysmal coronary artery disease. Heart. 1997;78(2):136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baman TS, Cole JH, Devireddy CM, et al. Risk factors and outcomes in patients with coronary artery aneurysms. Am J Cardiol. 2004;93(12):1549–1551. [DOI] [PubMed] [Google Scholar]
- 25. Dogan A, Tunc T, Ergene O, et al. Evaluation of overall fibrinolytic activity in patients with coronary artery ectasia: global fibrinolytic capacity. Int J Cardiovasc Imaging. 2003;19(6):465–471. [DOI] [PubMed] [Google Scholar]
- 26. Lutgens E, Daemen MJ. CD40-CD40L interactions in atherosclerosis. Trends Cardiovasc Med. 2002;12(1):27–32. [DOI] [PubMed] [Google Scholar]
- 27. Kiener PA, Moran-Davis P, Rankin BM, Wahl AF, Aruffo A, Hollenbaugh D. Stimulation of CD40 with purified soluble gp39 induces proinflammatory responses in human monocytes. J Immunol. 1995;155(10):4917–4925. [PubMed] [Google Scholar]
- 28. Malik N, Greenfield BW, Wahl AF, Kiener PA. Activation of human monocytes through CD40 induces matrix metalloproteinases. J Immunol. 1996;156(10):3952–3960. [PubMed] [Google Scholar]
- 29. Zhou L, Stordeur P, de Lavareille A, et al. CD40 engagement on endothelial cells promotes tissue factor dependent procoagulant activity. Thromb Haemost. 1998;79(5):1025–1028. [PubMed] [Google Scholar]
- 30. Urbich C, Dernbach E, Aicher A, Zeiher AM, Dimmeler S. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. 2002;106(8):981–986. [DOI] [PubMed] [Google Scholar]
- 31. Schonbeck U, Gerdes N, Varo N, et al. Oxidized low-density lipoprotein augments and 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors limit CD40 and CD40L expression in human vascular cells. Circulation. 2002;106(23):2888–2893. [DOI] [PubMed] [Google Scholar]
- 32. Schonbeck U, Libby P. CD40 signaling and plaque instability. Circ Res. 2001;89(12):1092–1103. [DOI] [PubMed] [Google Scholar]
- 33. Pryshchep O, Ma-Krupa W, Younge BR, et al. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118(12):1276–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen C, Chai H, Wang X, et al. Soluble CD40 ligand induces endothelial dysfunction in human and porcine coronary artery endothelial cells. Blood. 2008;112(8):3205–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heeschen C, Dimmeler S, Hamm CW. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. 2003;348(12):1104–1111. [DOI] [PubMed] [Google Scholar]
- 36. Aukrust P, Damas JK, Solum NO. Soluble CD40 ligand and platelets: self-perpetuating pathogenic loop in thrombosis and inflammation? J Am Coll Cardiol. 2004;43(12):2326–2328. [DOI] [PubMed] [Google Scholar]
- 37. Papadakis MC, Manginas A, Cotileas P, et al. Documentation of slow coronary flow by the TIMI frame count in patients with coronary ectasia. Am J Cardiol. 2001;88(9):1030–1032. [DOI] [PubMed] [Google Scholar]
- 38. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lutgens E, Lievens D, Beckers L, Donners M, Daemen M. CD40 and its ligand in atherosclerosis. Trends Cardiovasc Med. 2007;17(4):118–123. [DOI] [PubMed] [Google Scholar]
- 40. Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol. 2005;25(11):2428–2434. [DOI] [PubMed] [Google Scholar]
- 41. Rizvi M, Pathak D, Freedman JE, Chakrabarti S. CD40-CD40 ligand interactions in oxidative stress, inflammation and vascular disease. Trends Mol Med. 2008;14(12):530–538. [DOI] [PubMed] [Google Scholar]
- 42. Antoniades C, Bakogiannis C, Tousoulis D, Antonopoulos AS, Stefanadis C. The CD40/ CD40 ligand system: linking inflammation with atherothrombosis. J Am Coll Cardiol. 2009;54(8):669–677. [DOI] [PubMed] [Google Scholar]
- 43. Tousoulis D, Antoniades C, Nikolopoulou A, et al. Interaction between cytokines and sCD40 L in patients with stable and unstable coronary syndromes. Eur J Clin Invest. 2007;37(8):623–628. [DOI] [PubMed] [Google Scholar]
- 44. Durakoğlugil ME, Kocaman SA, Çetin M, et al. Increased circulating soluble CD40 levels in patients with slow coronary flow phenomenon: an observational study. Anadolu Kardiyol Derg. 2013;13(1):39–44. [DOI] [PubMed] [Google Scholar]
- 45. Han SH, Koh KK, Quon MJ, Lee Y, Shin EK. The effects of simvastatin, losartan, and combined therapy on soluble CD40 ligand in hypercholesterolemic, hypertensive patients. Atherosclerosis. 2007;190(1):205–211. [DOI] [PubMed] [Google Scholar]