

ZAP is an IFN-induced host factor that can inhibit a wide range of viruses, and there is great interest in fully characterizing its antiviral mechanism. This is the first study that defines the antiviral capacities of individual ZAP isoforms in the absence of endogenous ZAP expression and, hence, cross talk with other isoforms. Our data demonstrate that ZAP is expressed as four different forms: ZAPS, ZAPM, ZAPL, and ZAPXL. The longer ZAP isoforms better inhibit alphaviruses and HBV, while all isoforms equally inhibit Ebola virus transcription and replication. In addition, there is no difference in the abilities of ZAP isoforms to enhance the induction of type I IFN expression. Our results show that the full spectrum of ZAP activities can change depending on the virus target and the relative levels of basal expression and induction by IFN or infection.
KEYWORDS: Ebola virus, PARP13, ZAP, alphavirus, alternative splice variants, hepatitis B virus, interferon
ABSTRACT
Given the unprecedented scale of the recent Ebola and Zika viral epidemics, it is crucial to understand the biology of host factors with broad antiviral action in order to develop novel therapeutic approaches. Here, we look into one such factor: zinc finger antiviral protein (ZAP) inhibits a variety of RNA and DNA viruses. Alternative splicing results in two isoforms that differ at their C termini: ZAPL (long) encodes a poly(ADP-ribose) polymerase (PARP)-like domain that is missing in ZAPS (short). Previously, it has been shown that ZAPL is more antiviral than ZAPS, while the latter is more induced by interferon (IFN). In this study, we discovered and confirmed the expression of two additional splice variants of human ZAP: ZAPXL (extralong) and ZAPM (medium). We also found two haplotypes of human ZAP. Since ZAPL and ZAPS have differential activities, we hypothesize that all four ZAP isoforms have evolved to mediate distinct antiviral and/or cellular functions. By taking a gene-knockout-and-reconstitution approach, we have characterized the antiviral, translational inhibition, and IFN activation activities of individual ZAP isoforms. Our work demonstrates that ZAPL and ZAPXL are more active against alphaviruses and hepatitis B virus (HBV) than ZAPS and ZAPM and elucidates the effects of splice variants on the action of a broad-spectrum antiviral factor.
IMPORTANCE ZAP is an IFN-induced host factor that can inhibit a wide range of viruses, and there is great interest in fully characterizing its antiviral mechanism. This is the first study that defines the antiviral capacities of individual ZAP isoforms in the absence of endogenous ZAP expression and, hence, cross talk with other isoforms. Our data demonstrate that ZAP is expressed as four different forms: ZAPS, ZAPM, ZAPL, and ZAPXL. The longer ZAP isoforms better inhibit alphaviruses and HBV, while all isoforms equally inhibit Ebola virus transcription and replication. In addition, there is no difference in the abilities of ZAP isoforms to enhance the induction of type I IFN expression. Our results show that the full spectrum of ZAP activities can change depending on the virus target and the relative levels of basal expression and induction by IFN or infection.
INTRODUCTION
A recent outbreak of chikungunya virus (CHIKV) (family Togaviridae, genus Alphavirus) in India and the Indian Ocean islands (1–3) and its subsequent spread to the Caribbean Islands and Florida underscore the capacity of alphaviruses to cause significant morbidity and mortality. However, there is nothing other than insect control and general medical support available to combat their diseases. In the 2013–2016 outbreak in West Africa, Ebola virus (EBOV) (family Filoviridae, genus Ebolavirus), for which there was no approved therapy or vaccine, killed more than 10,000 people and reached and spread within the United States (4). Given the unprecedented scale of these recent viral epidemics, it is crucial to understand the biology of host factors with broad antiviral activity in order to develop novel approaches for vaccine and drug development.
The interferon (IFN) response is the first line of defense against viral infections. Type I IFNs (IFN-α/β) signal through a common receptor present on the cell surface and upregulate several hundred IFN-stimulated genes (ISGs) (5). One such ISG is the ZC3HAV1 gene-encoded zinc finger antiviral protein (ZAP), which exhibits broad-spectrum antiviral activity. ZAP was first discovered to inhibit the retrovirus Moloney murine leukemia virus (MMLV) (6). Since then, ZAP has been shown to block a variety of RNA and DNA viruses, including other retroviruses, alphaviruses, filoviruses, hepatitis B virus (HBV), coxsackievirus B3, Japanese encephalitis virus, influenza A virus, and Newcastle disease virus (7–15). However, some viruses are not inhibited by ZAP (7, 13), and it is not completely understood what determines the specificity of ZAP. Its direct and indirect antiviral mechanisms involve recruitment of both 5′-to-3′ and 3′-to-5′ mRNA decay machineries to degrade viral RNA, TRIM25-mediated inhibition of viral translation, Ago2-dependent regulation of host mRNAs, induction of IFN expression, and synergy with other ISGs (7, 9, 12, 16–22). Recently, ZAP has been shown to bind to regions in the human immunodeficiency virus (HIV) genome with elevated levels of CG dinucleotide motifs, leading to depletion of cytoplasmic unspliced viral RNA and inhibition of viral replication (23). The authors of that study speculate that suppressed CG dinucleotide content observed in viral genomes might be a mechanism of viral evasion of ZAP.
ZAP exists as two alternative splice variants that differ at their C termini (reviewed in reference 5). Isoform 1 (902 amino acids [aa]), designated ZAPL, encodes a C-terminal poly(ADP-ribose) polymerase (PARP)-like domain that is absent in isoform 2 (699 aa), designated ZAPS. The PARP-like domain found in ZAPL has been rapidly evolving (24), and ZAPL shows greater antiviral activity than ZAPS (24, 25). We previously identified a prenylation site at the C terminus of murine ZAPL that contributes to its enhanced antiviral activity (25). Interestingly, the PARP-like domain of ZAPL lacks the catalytically active triad motif (H-Y-E) and has no detectable auto-ADP-ribosylating activity (26), suggesting that the PARP-like domain might promote ZAP antiviral activity in a way that is independent of its catalytic activity. On the other hand, ZAPS, which lacks the PARP-like domain entirely, is induced to a greater degree than ZAPL upon IFN treatment or pathogen sensing (11, 12) and interacts with RIG-I to enhance IFN-β production (12). However, it is not clear what the specific activities of ZAPS and ZAPL are. Almost all published studies to date that have examined the antiviral function of ZAPS and ZAPL were conducted in cells capable of expressing endogenous ZAP isoforms, and the vast majority of studies have utilized overexpression of ZAPS or only the N-terminal one-third of ZAPS (NZAP). Since ZAP can multimerize with itself (27), the true antiviral spectrum of individual ZAP isoforms can be masked by synergistic interactions with or inhibition from other isoforms. In instances where ZAPS and ZAPL antiviral activities have been compared (9–11, 25), ZAPL is more antiviral than ZAPS against Sindbis virus (SINV) and HIV-1, whereas ZAPS is more antiviral than ZAPL against xenotropic murine leukemia virus-related virus (XMRV) and HBV, which is likely due to the lower protein expression level of ZAPL in those studies. Taken together, these data suggest that ZAP isoforms have differential antiviral activities.
In this study, we have discovered and confirmed the expression of two new ZAP isoforms, which we designate ZAPM (medium) and ZAPXL (extralong). For the first time, we have utilized an innovative ZAP replacement approach in ZAP knockout (KO) cells by a transposon-based inducible system. This allows us to begin dissecting the antiviral role of individual ZAP isoforms in the absence of endogenous ZAP protein expression. We have screened the ability of the individual ZAP isoforms to inhibit a panel of viruses to gain a global view of ZAP’s antiviral influence, providing mechanistic insight that may ultimately lead to the development of ZAP-based therapeutics.
RESULTS
Identification of novel alternatively spliced ZAP variants.
Genes coding for proteins homologous to ZAP are found across vertebrate species. A schematic of human ZAP isoforms based on UniProt domain data is shown in Fig. 1A. The amino-terminal region of ZAP possesses four CCCH-type zinc finger domains (aa 73 to 193), which are required for RNA binding (28). ZAP also encodes a tetrachlorodibenzo-rhodioxin-inducible PARP (TIPARP) homology domain (aa 512 to 563) (29) of unknown function and a WWE domain (aa 594 to 681), containing conserved tryptophan and glutamic acid residues and predicted to mediate protein-protein interactions in ubiquitin and ADP-ribose conjugation pathways (30). Alternative splicing results in two ZAP isoforms that differ at their C termini: isoform 1 is a 902-aa protein, designated ZAPL, while isoform 2, designated ZAPS, encodes a truncated (699-aa) form missing the C-terminal PARP-like domain (aa 716 to 902 of ZAPL).
FIG 1.

Discovery of novel ZAP isoforms. (A and B) Schematics of ZAPL and ZAPS (A) and the newly discovered ZAPXL and ZAPM (B). Light green shading indicates sequences shared by all four isoforms, whereas the hatched region is shared only between the XL and L isoforms. The XL and M isoforms of ZAP contain an extended exon 4 that is shaded dark green. The pink and blue lollipops represent nonsynonymous and synonymous single nucleotide polymorphisms found in the human ZAP gene in 293T cells, respectively. (C) Total RNA was isolated from 293T cells, cDNA was synthesized, and PCR was carried out with the indicated primers that target the shared extended exon 4 and the unique ZAPL/XL or ZAPS/M 3′ UTR. PCR was performed on duplicate 293T cDNA samples synthesized using oligo(dT)20 or a ZAP-specific 3′-UTR primer, and the results were consistent.
Recent data available on the NCBI, Ensembl, and UCSC genome browsers suggest the expression of a third isoform, annotated as ZAPXL (variant X1) (NCBI accession number XP_005250558.1; Gencode transcript ENST00000464606.5). ZAPXL is similar to ZAPL but contains an additional 121-aa sequence from an extended exon 4 (Fig. 1B). We found evidence for the newly annotated ZAPXL isoform in transcriptome sequencing (RNAseq) experiments performed in primary human fetal liver cells with or without type I IFN treatment (W. M. Schneider and C. M. Rice, unpublished data). Interestingly, this isoform, along with ZAPS, appears to be induced by IFN treatment. However, from the RNAseq data, we could not rule out the possibility that ZAPM (medium), a novel isoform similar to ZAPS, also exists. The putative ZAPM would be intermediate in size between ZAPS and ZAPL and identical to ZAPS except for the same extended exon 4 as seen in ZAPXL (Fig. 1B). Since ZAPS and ZAPM, and ZAPL and ZAPXL, would share the same 3′ untranslated regions (UTRs), respectively, we performed PCR on 293T cDNA using primers targeting the extended exon 4 (absent in ZAPS and ZAPL) and the 3′ UTRs of ZAPS/M and ZAPL/XL (see primer target sites in Fig. 1B). We detected the presence of the extended exon 4 in both ZAPXL (PCR 1, expected product size of about 1.6 kb) and ZAPM (PCR 2, expected product size of about 1 kb), therefore confirming the mRNA expression of the newly discovered isoforms (Fig. 1C). The data were consistent regardless of whether we synthesized the 293T cDNA using oligo(dT)20 (Fig. 1C) or ZAP-specific 3′-UTR primers (data not shown). Our sequencing analysis also discovered in 293T cells a synonymous variant in extended exon 4, 514V (Fig. 1B, blue lollipop), compared to the annotated sequence for ZAPXL (variant X1; NCBI accession number XP_005250558.1). In addition, we found that in 293T cells, each of the ZAP isoforms exists as two haplotypes (with numbers based on ZAPXL) (Fig. 1B, pink lollipops): 485R/687H (haplotype 1 for ZAPS and ZAPL) or 485R/543R/687H (haplotype 1 for ZAPM and ZAPXL), and 485K/687Q (haplotype 2 for ZAPS and ZAPL) or 485K/543C/687Q (haplotype 2 for ZAPM and ZAPXL).
We then asked whether the ZAPM and ZAPXL proteins are expressed. We isolated ZAP from 293T cells using a monoclonal antibody that recognizes the shared N-terminal end of all four isoforms and subjected the immunoprecipitate to mass spectrometric analysis. We found peptides that are unique to the extended exon 4 region of ZAPM and ZAPXL (labeled in red), suggesting that these protein isoforms are expressed (Fig. 2). To characterize the regulation of expression of the different ZAP isoforms, we performed Western blot analysis in 293T cells in the presence or absence of IFN-β treatment. The expected sizes for ZAP isoforms are 78 kDa (ZAPS), 91 kDa (ZAPM), 101 kDa (ZAPL), and 114 kDa (ZAPXL) (labeled with arrows in Fig. 3A). All four isoforms are expressed at baseline, with ZAPL being the predominant isoform (Fig. 3A, left). Upon IFN-β treatment, the levels of all forms increase, but the short isoform is upregulated the most. All four isoforms are absent from lysates harvested from zinc finger nuclease (ZFN)-mediated ZAP KO 293T cells (clones 32 and 89), demonstrating that they are bona fide ZAP isoforms (Fig. 3A, right). We quantified the relative protein levels of all four ZAP isoforms at baseline and upon IFN-β treatment by quantitative Western blotting (Fig. 3B). We found that ZAPL has the highest protein expression level at baseline (∼8-fold higher than that of ZAPS, ∼50-fold higher than that of ZAPXL, and ∼100-fold higher than that of ZAPM). ZAPS (3.5-fold) and ZAPM (2.2-fold) have the most dramatic increases in protein levels upon IFN-β treatment, while ZAPL and ZAPXL levels remain relatively the same. We also examined the expression of ZAP isoforms in cervical (HeLa), lung (A549), and liver (HepG2) cell lines. Similar to what we observed in 293T cells, ZAPL is the most abundantly expressed isoform at baseline in A549 and HepG2 cells, whereas ZAPS is induced the most following IFN-β treatment (Fig. 3C). ZAPM is also dramatically upregulated by IFN-β treatment in HeLa cells and less so in A549 cells and is not detectable in HepG2 cells (Fig. 3C). Together, the data show that these novel ZAP isoforms are indeed expressed as proteins and demonstrate differential baseline expression and type I IFN induction in various cell types.
FIG 2.

Mass spectrometric identification of ZAPM/XL-specific peptides. A denaturing ZAP immunoprecipitation was carried out, and the immunoprecipitates were digested with trypsin and subjected to mass spectrometric analysis. Identified peptides aligning to the shared region of ZAPXL (top), ZAPL (middle), and ZAPS (bottom) isoforms are highlighted in green. ZAPS/L- and ZAPM/XL-specific peptides are identified and highlighted in blue (VALVNDSLSDVTSTTSSR) and red (VALVNGK, TGATGFGLTMAVK, NLVPTTPGESTAPAQVSTLPQSPAALSSSNR, and KTTGSAQYSLSDVTSTTSSR), respectively. The experiment was performed once.
FIG 3.

Basal expression and IFN-β induction of ZAP isoforms in different cell lines. (A) Parental 293T cells and ZAP KO 293T cell clones (clones 32 and 89) were treated with 1 nM IFN-β for 6 and 24 h to induce ZAP expression. Lysates were harvested and quantified, and equal amounts of protein were loaded on the gel for probing with ZAP (top) and β-actin (bottom) antibodies. IB, immunoblotting. (B) Quantification of protein expression of the four ZAP isoforms at baseline and upon 1 nM IFN-β treatment. The mean normalized signals for ZAPS, ZAPM, ZAPL, and ZAPXL detected in 6 lysate samples from two independent experiments done in triplicate are plotted in the graph. (C) HeLa, A549, and HepG2 cells were treated with IFN-β for 6, 24, and 48 h to induce ZAP expression. Lysates were harvested and quantified, and equal amounts of protein were loaded on the gel for probing with ZAP (top) and β-actin (bottom) antibodies. The data are representative of results from 2 independent experiments.
Generation of ZAP KO 293T cell lines with inducible expression of individual ZAP isoforms.
In order to systemically study the activities of individual ZAP isoforms without endogenous ZAP isoform expression, we attempted to establish stable cell lines in ZFN-mediated ZAP KO 293T cells (12) reconstituted with single isoforms. As we discovered that there are two haplotypes for each ZAP isoform, we also overexpressed both of them to determine if they have functionally distinct activities. We chose a transposon-based delivery method and set up an inducible system to express and select for N-terminally red fluorescent protein (RFP)-fused ZAP isoforms in ZAP KO 293T cells. We tested these ZAP-inducible cell lines for their expression levels of RFP-ZAP upon treatment with doxycycline (Dox) via Western blotting (Fig. 4A) and fluorescence microscopy (Fig. 4B). We found that fluorescently tagged ZAP isoforms of both haplotypes are expressed at similar levels in all of the cells, providing us with a system to interrogate the antiviral activities of all four ZAP isoforms.
FIG 4.

Dox-inducible expression of ZAP isoforms in ZAP KO 293T cells. Stable cell lines with inducible expression of N-terminally RFP-tagged ZAP isoforms were generated in ZAP knockout 293T cells by use of the ePB transposon system. All four ZAP isoforms can be detected by immunoblotting (A) and fluorescence imaging (B). Expected sizes for ZAP isoforms are as follows: 106 kDa for RFP-ZAPS, 119 kDa for RFP-ZAPM, 130 kDa for RFP-ZAPL, and 142 kDa for RFP-ZAPXL. Dox titration was first carried out on one of the cell lines, and 1 μg/ml Dox was used to induce individual ZAP isoforms in all the cell lines. The data are representative of results from 2 independent experiments.
Differential inhibition of alphaviruses by ZAP.
First, we used the ZAP-inducible 293T cell lines to interrogate the sensitivity of alphaviruses to the antiviral activities of the different isoforms. We utilized green fluorescent protein (GFP)-expressing Old World alphaviruses (SINV, Ross River virus [RRV], and O’nyong-nyong virus [ONNV]) and a New World alphavirus (a Venezuelan equine encephalitis virus [VEEV] vaccine strain). Based on previous studies, we expected VEEV to be relatively less sensitive to ZAP (7). The numbers of infected cells are similar across uninduced cell lines (Fig. 5A). Upon Dox treatment and ZAP induction, ZAPL and ZAPXL are more antiviral than ZAPS and ZAPM against all alphaviruses tested compared to the RFP control (Fig. 5A). Although there are some exceptions (for example, ZAPL against SINV and RRV), ZAP isoforms of haplotype 2 are in general similarly active or less antiviral than those of haplotype 1 (Fig. 5A). We calculated fold inhibition of alphavirus replication for the individual ZAP isoforms and compared the fold inhibitions for ZAPM, ZAPL, and ZAPXL to that seen with ZAPS. We found that for the same ZAP haplotype, (i) fold inhibition by ZAPS is not significantly different from that by ZAPM and (ii) ZAPL and ZAPXL are significantly more inhibitory than ZAPS (Fig. 5B). Taken together, these data suggest that ZAPL and ZAPXL are the most inhibitory ZAP isoforms against all four alphaviruses tested.
FIG 5.

Inhibition of alphaviruses by the four ZAP isoforms. (A) ZAP KO 293T cell lines were treated with Dox to induce expression of the N-terminally RFP-tagged ZAP isoforms for 24 h prior to infection with GFP-expressing SINV (MOI = 10; 8 h p.i.), RRV (MOI = 10; 24 h p.i.), ONNV (MOI = 0.1; 18 h p.i.), and the VEEV vaccine strain (TC-83) (MOI = 1; 8 h p.i.). Cells were harvested and fixed, and their percent infection was determined by flow cytometry. The mean values for triplicate samples are plotted; error bars represent the standard deviations (SD). The data are representative of results from 2 independent experiments. (B) Fold inhibition of alphavirus replication by ZAP isoforms. Data from panel A are represented. Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001 [by one-way ANOVA and Dunnett’s multiple-comparison test]). ns, not significant. (C) ZAP KO 293T cell lines with inducible expression of ZAP isoforms were infected with SINV, RRV, and the CHIKV vaccine strain (181/clone 25) at an MOI of 0.1. Medium overlying the cells was harvested at 0, 12, 24, and 48 h p.i. from duplicate wells, and the viral titer was determined by infection of BHK-21 cells in standard plaque assays. The mean values for duplicate samples are plotted; error bars represent the SD. The data are representative of results from 2 independent experiments. (D) ZAP KO 293T cell lines with inducible expression of ZAP isoforms were treated with Dox and infected at 40°C (nonpermissive temperature) with a replication-defective SINV, Toto1101/Luc:ts6. Lysates were prepared, and luciferase activity was determined at the indicated times after infection. ZAP KO 293T cells expressing RFP alone served as a control. The mean values for quadruplicate samples are plotted; error bars represent the SD. Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ****, P < 0.0001 [by two-way ANOVA and Dunnett’s multiple-comparison test]). The data are representative of results from 2 independent experiments. RLU, relative light units.
Interestingly, we observed differences in alphavirus sensitivity to ZAP when we infected cells with GFP-expressing viruses. ONNV, like VEEV, is relatively insensitive to ZAP inhibition overall compared to SINV and RRV (Fig. 5A). Similarly, when we investigated the effects of ZAP on alphavirus production at different time points using wild-type (untagged) viruses, SINV and RRV were inhibited by ZAP isoforms (haplotype 1), whereas the vaccine strain (181/clone 25) of CHIKV, a close relative of ONNV, was highly resistant to all ZAP isoforms (haplotype 1) (Fig. 5C). However, the greater antiviral effects of ZAPL and ZAPXL that we observed with GFP-expressing alphaviruses are not apparent with wild-type (untagged) viruses.
We also investigated each isoform’s ability to inhibit viral translation, a function previously described for ZAP-mediated inhibition of SINV. We used a temperature-sensitive luciferase reporter SINV, Toto1101/Luc:ts6 (7), that is replication defective at a nonpermissive temperature (40°C), allowing us to measure translation of the incoming viral RNA genome only. At early time points, ZAPL and ZAPXL are most effective at blocking SINV translation (Fig. 5D). At 3 h postinfection (p.i.), all ZAP isoforms of both haplotypes are able to significantly inhibit viral translation (P < 0.0001 by two-way analysis of variance [ANOVA]), although ZAPM, haplotype 2, is the least efficacious (Fig. 5D).
Our observations in Fig. 5 prompted us to determine whether there are obvious sequence differences in the alphavirus genomes that are differentially targeted by ZAP. We quantified the number of CG dinucleotides across viral genes and GFP in the GFP reporter alphaviruses tested since ZAP specifically targets regions in the HIV genome that are dense in CG dinucleotides (23). We found that SINV, VEEV, ONNV, RRV, and CHIKV contain many CG dinucleotide motifs across their genomes, but CG dinucleotide densities of viral genes do not correlate with the extent of viral sensitivity to ZAP: RRV > SINV > VEEV > ONNV = CHIKV (Fig. 6). In addition, a previous study mapped the minimal ZAP-responsive fragment (ZRF) in SINV to the nsP4 region (nucleotides [nt] 6062 to 6715) of the pToto1101 infectious clone (28). We aligned the genomes of the four alphaviruses tested in this study and identified the homologous ZRF for calculation of CG dinucleotide frequency. We observed that SINV has the highest CG content in the ZRF compared to other alphaviruses (Fig. 6). It is likely that the ZRF is found in different regions among the alphavirus genomes. Finally, GFP is not the determinant for differential targeting by ZAP, as the GFP reporter in all the alphavirus constructs carries the same number of CG motifs (Fig. 6).
FIG 6.

Alphaviruses encode many CG dinucleotide motifs across their genomes. The number of CG dinucleotide motifs per kilobase of genome is calculated for the whole genome; for the nonstructural and structural polyprotein regions of the RRV, SINV, VEEV, ONNV, and CHIKV constructs tested in Fig. 5; and for the GFP reporter gene of the GFP-expressing alphaviruses tested in Fig. 5A. CG dinucleotide frequencies found in individual viral nonstructural (nsP1 to -4) and structural (capsid, E3, E2, 6K, and E1) genes are also shown. ZRF represents the minimal ZAP-responsive fragment identified in SINV previously (28). The viruses are arranged in the following order of susceptibility to ZAP: RRV > SINV > VEEV > ONNV = CHIKV.
Ebola virus replication is attenuated by ZAP isoforms.
Overexpressed ZAPS (in the context of endogenous ZAP isoforms) has been shown to inhibit Ebola virus (EBOV) replication through reduction of L polymerase gene mRNA levels; increased expression of the L gene can partially antagonize the inhibitory effects of ZAP (8). To study ZAP effects, we used an Ebola minigenome (31) that carries the firefly luciferase (Fluc) gene flanked by virus-specific gene start and end sequences and plasmids encoding the nucleocapsid proteins required for viral transcription and replication (Fig. 7A). We first determined whether ZAP inhibits Fluc expression, the reporter for the minigenome system. We transfected the ZAP-inducible cell lines with a construct where Fluc expression is driven by the simian virus 40 (SV40) promoter, independently of Ebola activities. We found that the longer ZAP isoforms have some inhibitory effects against Fluc expression (Fig. 7B). Next, we cotransfected the ZAP-inducible cell lines with the Ebola minigenome and plasmids required for viral transcription and replication. Luciferase activities were measured 2 days following transfection and were normalized to luciferase levels determined in the absence of Ebola minigenome replication (Fig. 7B), to determine the specific effects of ZAP isoforms on minigenome replication and expression (Fig. 7C). We showed that all ZAP isoforms in both haplotypes significantly inhibit minigenome expression compared to cells expressing the RFP control (Fig. 7C). We conclude that there are no differences in the abilities of different ZAP isoforms to inhibit Ebola virus transcription and replication.
FIG 7.
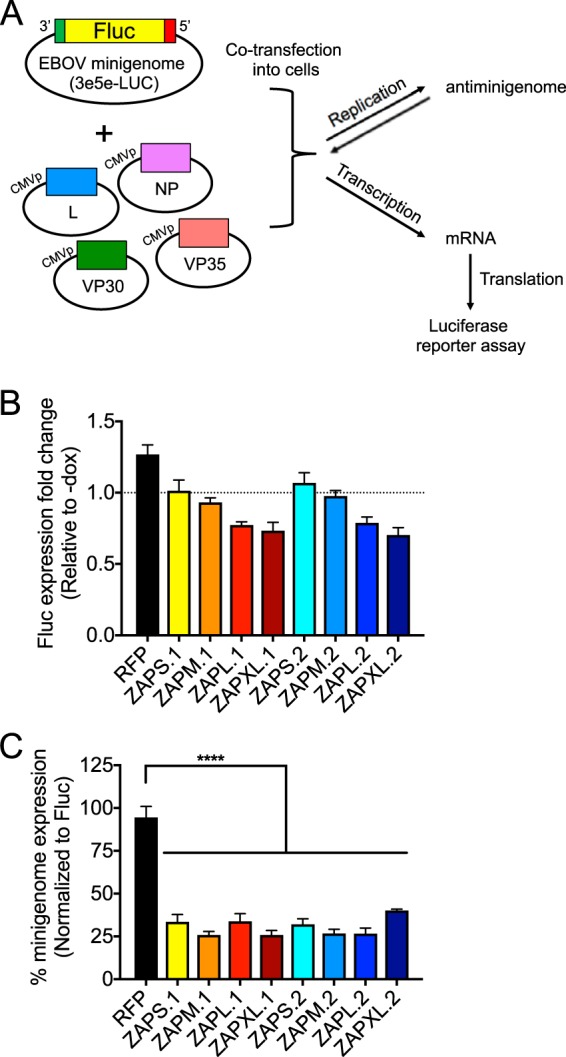
Inhibition of the Ebola minigenome system by ZAP isoforms. (A) The Ebola minigenome system (5 plasmids) consists of the viral nucleocapsid proteins (L, NP, VP30, and VP35) under the control of the CMV promoter (CMVp) and the minigenome, which carries the 3′ and 5′ ends of the Ebola viral genome, required for replication and transcription, flanking the Fluc gene. The minigenome is replicated and transcribed by the polymerase complex to generate mRNA of the Fluc reporter gene that can be translated and quantified in a luciferase reporter assay. (B) Stable cell lines with inducible expression of N-terminally RFP-tagged ZAP isoforms were transfected with a Fluc reporter construct (see Materials and Methods about the pGL3-Control vector) and treated with Dox to induce the expression of individual ZAP isoforms. Cell lysates were harvested at 1 day posttransfection for luciferase assays. The luciferase activity of Dox-treated cells was compared to the mean luciferase activity of uninduced cells to calculate the Fluc expression fold change for each ZAP-inducible cell line. Mean fold changes calculated from 2 independent experiments done in triplicate are plotted; error bars represent the SD. (C) Stable cell lines with inducible expression of N-terminally RFP-tagged ZAP isoforms were transfected with the Ebola minigenome system and treated with Dox to induce the expression of individual ZAP isoforms 1 day later. Cell lysates were harvested at 2 days posttransfection for luciferase assays. The luciferase activity of Dox-treated cells was compared to the mean luciferase activity of uninduced cells to calculate the minigenome expression fold change for each ZAP-inducible cell line. The values were then normalized by dividing the minigenome expression fold changes by the average Fluc expression fold changes and multiplying by 100. Mean percentages for triplicate wells are plotted; error bars represent the SD. Asterisks indicate statistically significant differences (****, P < 0.0001 [by one-way ANOVA and Dunnett’s multiple-comparison test]). The data are representative of results from 2 independent experiments.
The longer ZAP isoforms strongly inhibit HBV replication and protein expression.
Next, we determined the effects of ZAP isoforms on HBV replication. ZAPS has been shown to inhibit HBV by accelerating the decay of HBV pregenomic RNA (pgRNA) (11), which serves as the mRNA transcript for viral capsid and polymerase proteins as well as the template for reverse transcription (RT) for formation of the HBV DNA genome for packaging and production of progeny virus. Since HBV replicates exclusively in hepatocytes, we examined the anti-HBV effects of ZAP isoforms in HepG2 cells. We attempted to generate ZAP KO HepG2 clones, but CRISPR/Cas9-mediated gene editing of the first coding exon of ZAP led to incomplete KO and expression of a truncated ZAP protein (data not shown). We therefore cotransfected wild-type HepG2 cells, which express ZAP endogenously, with a replication-competent HBV DNA plasmid (pHBV1.3x) and plasmids expressing individual ZAP isoforms and measured secreted hepatitis B e antigen (HBeAg) and hepatitis B surface antigen (HBsAg) levels in the supernatant (Fig. 8). All four haplotype 1 ZAP isoforms exhibit strong inhibitory effects against HBV viral antigen production. For haplotype 2, while all four isoforms were significantly inhibitory, the magnitude of inhibition was greatest for ZAPL and ZAPXL. These data, together with the data in Fig. 5, show that, generally, ZAPL and ZAPXL can block both alphavirus and HBV replication more potently than the shorter isoforms. Interestingly, however, the shorter isoforms (ZAPS and ZAPM) of haplotype 1 display more potent activity against HBV than haplotype 2, with inhibitory effects nearing the levels of the longer isoforms for haplotype 1.
FIG 8.
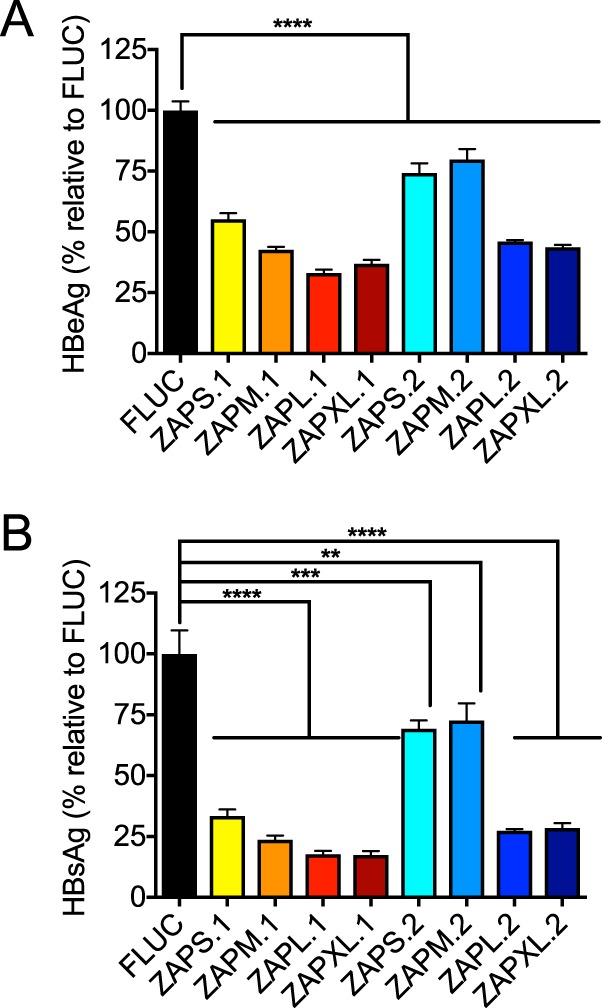
ZAP isoforms exhibit differential activities against HBV. HepG2 cells were cotransfected with ZAP-expressing constructs and pHBV1.3x, and secreted HBeAg (A) and HBsAg (B) levels in the supernatant were measured. Mean values for quadruplicate wells are plotted; error bars represent the SD. Asterisks indicate statistically significant differences (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [by one-way ANOVA and Dunnett’s multiple-comparison test]). The data are representative of results from 3 independent experiments.
Immune activation by ZAP isoforms.
In addition to directly inhibiting the replication of a range of viruses, ZAP can modulate the innate immune response by activating RIG-I and synergizing with other ISGs (12, 21). ZAP enhances IFN-β production upon RIG-I stimulation, with ZAPS being more active than ZAPL (12). However, whether ZAP requires RIG-I for its activity, and vice versa, still remains controversial (10, 11, 32). These comparisons in the past were done in the presence of the endogenous ZAP isoforms, so the relative contribution of the isoforms to enhancement of IFN-β production and whether they can function independently have not been carefully dissected. We utilized the individual ZAP isoform-inducible cell lines to examine the ability of each isoform to facilitate RIG-I-mediated IFN-β production. We challenged the cells with Sendai virus (SeV), a well-established RIG-I stimulus that upregulates IFN-β (33, 34). We focused on haplotype 1 here since haplotype 1 is equally as or more potently inhibitory against all the viruses tested. IFN-β upregulation was measured by Fluc activity driven by the IFN-β promoter in infected cells, and the effect of each isoform in the context of haplotype 1 was compared to that in ZAP KO cells reconstituted with the RFP control. We found that enhancement of IFN-β upregulation by ZAP isoforms was minimal yet significant when cells were stimulated with the higher dose of SeV (Fig. 9). All of the isoforms induce IFN-β upregulation, with ZAPXL being slightly more active (Fig. 9). We conclude that there is no major difference in the abilities of individual ZAP isoforms to enhance type I IFN production.
FIG 9.
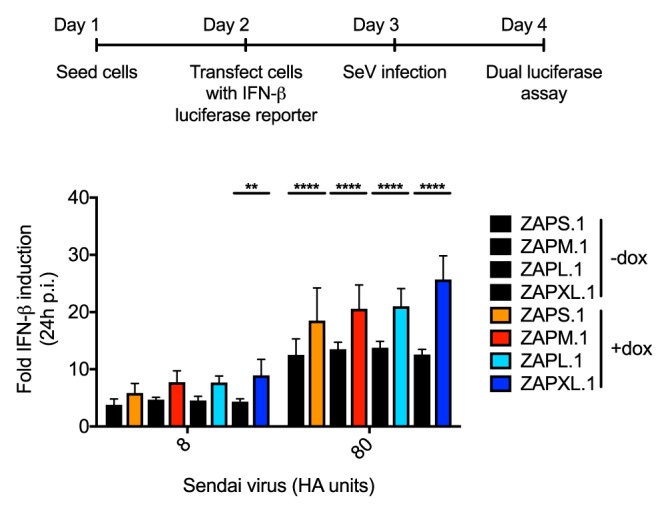
Type I IFN activation by ZAP isoforms. ZAP KO 293T cells with inducible ZAP isoform-specific expression were transfected with a construct expressing Fluc under the control of the IFN-β promoter. An Rluc luciferase construct was cotransfected for normalization purposes. The cells were treated with Dox to induce the expression of individual ZAP isoforms and infected with SeV (Cantell strain) at two different doses (8 and 80 hemagglutinating [HA] units/well) 1 day later to trigger RIG-I-mediated IFN-β production. Cell lysates were harvested at 24 h p.i. for dual-luciferase assays. The normalized Fluc values were set to 1 for each mock-infected cell line and used to calculate the fold IFN-β induction in 3 independent experiments. Asterisks indicate statistically significant differences (**, P < 0.01; ****, P < 0.0001 [by two-way ANOVA and Tukey’s multiple-comparison test]). Mean values for a total of 9 samples (3 independent experiments done with triplicate wells) are plotted; error bars represent the SD.
DISCUSSION
We are only beginning to dissect the relative contribution of splice isoforms and paralogs of ISGs to the innate immune response. How effectively the IFN response can thwart a viral infection depends on the activities and tissue-specific expression and induction levels of ISGs and their isoforms. For example, ADAR1 is an IFN-inducible adenosine deaminase that is alternatively spliced. The longer isoform (p150) of ADAR1 is IFN inducible, whereas the amino-terminally truncated isoform (p110) is constitutively expressed (35), similar to ZAPS and ZAPL, respectively. Due to differences in their subcellular localizations, it is speculated that the two ADAR isoforms differentially target cytoplasmic and nuclear viruses by A-to-I editing (35). In addition, OAS, another ISG, senses viral double-stranded RNA (dsRNA) and activates RNase L, leading to the cleavage of viral and host RNAs and blocking of viral replication. Three OAS genes encode catalytically active proteins, but a recent study found OAS3 to be the most critical for activation of the RNase L response against a panel of diverse viruses, suggesting different roles for the OAS paralogs in virus inhibition (36).
In this study, we found that the longer ZAP isoforms of both haplotypes can more effectively inhibit GFP-expressing alphaviruses (Fig. 5), while for HBV, there were haplotype differences in the patterns of sensitivity, with haplotype 2 shorter isoforms showing less potent, but still significant, inhibition, while differences in haplotype 1 were less pronounced (Fig. 8). However, all isoforms can similarly block Ebola virus gene expression (Fig. 7). These data suggest that the C-terminal ends of ZAPL and ZAPXL, which contain the PARP-like domain and prenylation site, are critical for full ZAP antiviral activity against alphaviruses and HBV, and this phenotype is haplotype dependent for HBV. The longer ZAP isoforms may utilize the enzymatically dead PARP-like domain to recruit binding partners or negatively regulate ADP-ribosylation of other proteins, resulting in enhanced antiviral activity. Localization to a membrane compartment due to prenylation may also position ZAPL and ZAPXL perfectly to block endocytic viruses. Given that all isoforms similarly enhanced IFN-β production (Fig. 9), it is possible that this mechanism, rather than direct antiviral activity, plays a greater role in Ebola inhibition. Surprisingly, we did not observe greater antiviral effects of ZAPL and ZAPXL with wild-type (untagged) alphaviruses (Fig. 5C). The reason(s) for this is unclear but may be attributable to the differences in the assay readouts (percentage of cells infected for GFP viruses and virion production for the untagged viruses). It is also possible that attenuation of the viruses due to insertion of the GFP marker results in amplification of the differences between the antiviral activities of the different ZAP isoforms.
Among the viruses that are known to be sensitive to ZAP, we did not identify any that are differentially targeted by the novel ZAP isoforms. Since it seems plausible that the isoforms with the extended exon 4 exist for biologically relevant reasons, these data suggest that the relevant viruses have not yet been tested or that ZAPM and ZAPXL play cellular roles independent of viral inhibition. In future studies, it would be informative to screen the ZAP isoforms against a broad panel of viruses to further characterize the antiviral spectrum of ZAP. This could reveal the specific viral targets of ZAPM and ZAPXL. By classifying more viruses as ZAP sensitive or insensitive, this would also identify common features shared by the ZAP-sensitive viruses (genome characteristics, life cycles, and cellular pathways usurped by the viruses) that might explain the specificity of ZAP. In addition, since ZAP’s antiviral activity depends on self-interaction (27), it is possible that some of the isoforms might not function optimally when expressed individually. Future experiments in which ZAP KO cells are reconstituted with various combinations of the ZAP isoforms might reveal synergistic interactions between multiple isoforms.
Alternatively, ZAPM and ZAPXL might modulate the antiviral state without directly targeting the virus. It is possible that the extended exon 4 sequence confers upon ZAP the ability to interact with novel cellular factors or pathways. The protein sequence encoded by the extended exon 4 contains regions with low homology (24% to 41%) to several components of the Wnt/β-catenin pathway, such as Wnt 8a (WNT8A), WD repeat-containing and planar cell polarity effector protein fritz homolog (WDPCP), and low-density lipoprotein receptor-related protein 11 precursor (LRP11). The Wnt/β-catenin pathway is important for stem cell pluripotency and cell fate decisions during development (37). In the absence of Wnt signaling, the transcriptional coregulator β-catenin is phosphorylated by casein kinase 1 (CK1) and the adenomatous polyposis coli (APC)/Axin/glycogen synthase kinase 3β (GSK3β) complex, leading to β-catenin ubiquitination and proteasomal degradation. In the presence of Wnt signaling, the coreceptor LRP5/6 complexes with Frizzled, stabilizing β-catenin, which translocates to the nucleus to upregulate Wnt target genes. This is particularly interesting since GSK3β also modulates ZAP’s antiviral activity by phosphorylation, leading to enhanced activity (38). Furthermore, β-catenin has been reported to negatively regulate the type I IFN response (39, 40). Further studies are required to determine whether ZAPM and ZAPXL are recruited and interact with the Wnt signaling pathway components to influence this important cellular signaling pathway.
Interestingly, among the Old World alphaviruses tested here, SINV and RRV are more sensitive to ZAP inhibition than ONNV and CHIKV (Fig. 5). It is possible that the sensitive alphaviruses encode a higher frequency of CG dinucleotide motifs, as this has been shown to be an important determinant for recognition of HIV mutants by ZAP (23). However, when we compared the distributions and densities of CG dinucleotide motifs present in these alphaviruses, we saw CGs distributed along the entire length of the viral genomes (Fig. 6). None of the viral genes reflect differences in CG content that are proportional to viral sensitivity to ZAP, such that RRV (most sensitive) has the most CGs, while ONNV and CHIKV (most resistant) have the least (Fig. 6). A previous study on ZAP recognition of CGs found that ZAP-sensitive viruses as a group clearly have a significantly higher ratio of observed CGs/expected CGs than the ZAP-insensitive viruses (23). However, several ZAP-sensitive viruses, such as HBV, MMLV, and Marburg virus, have similar ratios of observed CGs/expected CGs as ZAP-insensitive viruses such as vesicular stomatitis virus and poliovirus (∼0.5). It is not clear what additional molecular determinants confer ZAP sensitivity on viruses with similar overall CG dinucleotide contents. Moreover, there were CG peaks throughout the HIV genome, particularly near the 5′ and 3′ ends, although only the CG peaks that were engineered into the envelope region were significantly bound by ZAP (23), suggesting that the context of CG dinucleotides is also a critical factor. Until the mechanism of ZAP recognition of CG dinucleotide motifs is fully characterized, it is difficult to speculate on how many CG motifs over what length of the viral genome are sufficient for ZAP inhibition. Alternatively, the resistant viruses might actively evade or antagonize ZAP and hence are not blocked by the antiviral effects. Additional studies are required to determine whether specific viral gene segments or proteins are responsible for differences that we observed in alphavirus sensitivity to ZAP.
In conclusion, we have discovered that in addition to the two known ZAP isoforms, ZAPS and ZAPL, there are two additional isoforms containing an extended exon 4, ZAPM and ZAPXL. Thus, at least four different splice variants of human ZAP are expressed and contribute to its antiviral activity. We found that within a virus family, there can be differential susceptibility to ZAP. Moreover, some viruses display differential susceptibilities to the ZAP isoforms, with the longer isoforms exhibiting greater antiviral potential. For other viruses, the four isoforms exhibit similar antiviral activities. Our findings advance our understanding of innate immunity and help elucidate the mechanism and specificity of the broad-spectrum antiviral factor ZAP. Thus, the work is highly relevant, since an enhanced mechanistic understanding of such broad-spectrum antiviral factors can inform new strategies to fight a wide range of viral infections.
MATERIALS AND METHODS
Cells, plasmids, viruses, and infections.
ZFN-mediated ZAP KO 293T cells (clones 32 and 89) were obtained from Akinori Takaoka at Hokkaido University (12). ZAP KO 293T cells, ZAP KO 293T cells (clone 89) with inducible expression of RFP alone or RFP-tagged individual ZAP isoforms, HeLa cells, and A549 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Baby hamster kidney 21 (BHK-21) cells were cultured in minimum essential medium (MEM) supplemented with 7.5% FBS. HepG2 cells were cultured in DMEM supplemented with 10% FBS and 1% nonessential amino acids. As indicated, cultures were treated with 1 nM IFN-β (PBL Assay Science) in the culture medium.
Human ZAPM and ZAPXL were amplified and cloned from 293T cDNA as previously described (20). Total RNA was isolated from 293T cells by using an RNeasy minikit (Qiagen) and reverse transcribed using the SuperScript III first-strand synthesis system for RT-PCR (Invitrogen) and oligo(dT)20 or specific primers targeting the 3′ UTRs of ZAPS/M and ZAPL/XL (ZAPS/M 3′ UTR, 5′-ACTTGATGAGCCCAGGGCATG-3′; ZAPL/XL 3′ UTR, 5′-GTCTGCGGCAATTTAGTTCTG-3′). The presence of ZAPM and ZAPXL was confirmed by PCR amplification of the correctly sized fragments using primers targeting the 5′ end of the extended exon 4 (5′-GATGTTAACGGTAAATACAAAGGGAAGAC-3′) and the 3′ UTRs of ZAPS/M and ZAPL/XL (see the above-mentioned primer sequences). Full-length ZAPM and ZAPXL were amplified using the same forward primer that targets the 5′ end of all human ZAP isoforms (5′-GTTTTGTACAGCCACCATGGCGGACCCGGAGGTG-3′) and isoform-specific reverse primers (ZAPM, 5′-GGTAGCGGCCGCTTACTCTGGCCCTCTCTTCATC-3′; ZAPXL, 5′-GGTAGCGGCCGCCTAACTAATCACGCAGGCTTTG-3′). The resultant PCR products were digested and cloned into the BsrGI and NotI sites of a modified pTRIPZ construct (Open Biosystems) under the control of the cytomegalovirus (CMV) promoter to generate constructs expressing untagged ZAPM and ZAPXL (pTRIPZ-ZAPM and pTRIPZ-ZAPXL). The 3′ ends of ZAPM and ZAPXL, including the extended exon 4, were digested out of the pTRIPZ-ZAPM and pTRIPZ-ZAPXL plasmids using SmaI (present in the ZAP gene) and XhoI (present in the pTRIPZ vector downstream of ZAP). The ZAPM and ZAPXL fragments were ligated into the SmaI and XhoI sites of pTRIP-RFP-NZAP to generate pTRIP constructs expressing N-terminally RFP-fused ZAPM and ZAPXL (pTRIP-RFP-ZAPM and pTRIP-RFP-ZAPXL). pTRIP-RFP-ZAPS and pTRIP-RFP-ZAPL were generated previously (20).
Initial attempts at ZAP isoform reconstitution with a stable expression lentiviral cassette were unsuccessful and prompted us to take advantage of the enhanced PiggyBac (ePB) transposable element system provided by the Brivanlou laboratory at The Rockefeller University, New York, NY (41). The system consists of two components: the transposon and transposase plasmids. Cotransfection of both plasmids allows for gene transfer and stable inducible expression. The ePB transposon vector carries a puromycin resistance gene and delivers all of the machinery necessary for Dox-inducible expression of each ZAP isoform: a Tet-responsive element (TRE) driving RFP-ZAP expression and CMV promoter-driven TetR. The ePB transposase mediates gene transfer. We digested out the RFP-labeled ZAP isoforms from the respective pTRIP-RFP-ZAP constructs using BsiWI and NotI and transferred them into the BsrGI and NotI sites of the ePB transposon vector for facile detection of ZAP expression upon Dox induction.
SINV expressing enhanced GFP (EGFP) (TE/5′2J/GFP) or Fluc (Toto1101/Luc:ts6), O’nyong-nyong virus (ONNV) expressing EGFP (generously provided by Steve Higgs, Kansas State University), the Ross River virus (RRV) T48 strain (generously provided by Richard Kuhn, Purdue University), RRV expressing EGFP (generously provided by Mark Heise, University of North Carolina), and chikungunya virus (CHIKV) vaccine strain 181/clone 25 (generously provided by Scott Weaver, The University of Texas Medical Branch at Galveston) have been previously described (42–46). The Venezuelan equine encephalitis virus (VEEV) vaccine strain (TC-83) was generously provided by Ilya Frolov, University of Alabama at Birmingham. Stocks were generated in BHK-21 cells (ATCC) as previously described (7). Titers for multiplicity-of-infection (MOI) calculations and viral production by ZAP-inducible 293T cell lines were determined in BHK-21 cells. Viral infections were performed as previously described, and GFP-positive infected cells were analyzed on a BD LSRII flow cytometer (7).
The EBOV minigenome system was a kind gift of Elke Muhlberger at Boston University (31). pCAAGS expression constructs encode Zaire EBOV nucleoprotein (NP), the RNA polymerase L protein, VP30 protein, and VP35 protein under the control of the CMV promoter. The minigenome (3e5eLuc) contains a Fluc gene and is flanked by the virus-specific gene start and end sequences required for viral replication and transcription. The Fluc construct (pGL3-Control vector) used for normalization of ZAP activity against Ebola minigenome expression was purchased from Promega. A pHBV1.3x plasmid was obtained from Yosef Shaul at the Weizmann Institute of Science and was used in transfections as previously described (47). The SeV Cantell stock was propagated by inoculation into the allantoic cavity of 10-day-old embryonated chicken eggs. Following incubation at 37°C for 48 h, allantoic fluid was harvested, and titers were determined by hemagglutination of chicken red blood cells. SeV infection was carried out by incubating cells with the virus inoculum in Opti-MEM I reduced serum medium (Gibco) for 1 h at 37°C prior to the addition of fresh medium. The IFN-β-pGL3 Fluc reporter was previously reported (48). A Renilla luciferase (Rluc) construct (pRL-Rluc), where Rluc is driven by the CMV promoter, was generated by modifying the pRL-HL plasmid, a kind gift from Stanley Lemon (49). Specifically, the nucleotides between the NotI and ApaI sites which contained the hepatitis C virus (HCV) internal ribosome entry site (IRES) and the open reading frame encoding Fluc were removed.
Immunoprecipitation, proteomics methods, and data analysis.
Following ZAP immunoprecipitation as previously described (20), proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Regions of the gel were selected for excision and analysis based on Western blotting performed in parallel. Gel bands were reduced, alkylated (10 mM dithiothreitol [DTT], 30 mM iodoacetamide; Sigma), digested with trypsin (Promega) as described previously (50), and analyzed by reversed-phase nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Dionex 3000, Q-Exactive plus; Thermo Scientific). Peptides were separated using a gradient increasing from 5% solvent B–95% solvent A to 40% solvent B–60% solvent A (solvent A, 0.1% formic acid; solvent B, 0.1% formic acid–80% acetonitrile) over 35 min. MS and MS/MS data were recorded at resolutions of 35,000 and 17,500, respectively. Resolution is defined as the observed m/z value divided by the smallest difference, Δ(m/z), for two ions that can be separated: (m/z)/Δ(m/z).
Since contaminating proteins of nonhuman origin can be present, such as bovine proteins found in FBS, data were quantified and searched against a UniProt human database (January 2013) concatenated with common contaminants (51), using ProteomeDiscoverer v. 1.4.0.288 (Thermo Scientific) combined with Mascot v. 2.5 (Matrix Science). Oxidation of methionine, protein N-terminal acetylation, carbamidomethylation of lysine, and GlyGly modification of lysine were allowed as variable modifications, and all cysteines were treated as carbamidomethylated. Peptide matches were filtered using a Percolator (52)-calculated false discovery rate of 5%.
Immunoblotting.
Polypeptides were resolved by SDS-PAGE using 4% to 12% NuPAGE Bis-Tris gels (Invitrogen) and transferred to a nitrocellulose membrane (GE Healthcare). ZAP immunodetection was achieved with a 1:5,000 dilution of rabbit anti-ZAP (catalog number ab154680; Abcam), followed by incubation with a 1:20,000 dilution of goat anti-rabbit horseradish peroxidase (HRP) (catalog number 31462; Thermo Fisher Scientific). Actin was detected using a 1:50,000 dilution of mouse HRP-tagged anti-actin antibody (catalog number A3854; Sigma). The proteins were visualized by using ECL Prime Western blotting detection reagent (GE Healthcare). For quantification of ZAP isoforms, basal and IFN-β-induced ZAP expression was quantified with fluorescence detection on a Li-Cor CLX system. The membranes were blocked with Odyssey blocking buffer (phosphate-buffered saline [PBS], product number 927-40000; Li-Cor) and stained with a 1:5,000 dilution of rabbit anti-ZAP (catalog number ab154680; Abcam) and a 1:20,000 dilution of mouse anti-actin (catalog number A5441; Millipore Sigma) primary antibodies, followed by a 1:10,000 dilution of goat anti-rabbit (800CW, product number 926-32211; Li-Cor) and goat anti-mouse (680RD, product number 926-68070; Li-Cor) antibodies. Quantitation was performed using the Li-Cor Image Studio Lite software package and graphed using GraphPad Prism software.
Fluorescence microscopy.
ZAP KO 293T cells reconstituted with each RFP-ZAP isoform were seeded the day before Dox treatment and imaged 24 h after Dox treatment using a Nikon Eclipse TE300 inverted microscope. A ×20 magnification was used for the images in Fig. 4B, and both untreated and Dox-treated samples were imaged under a red fluorescence channel.
Generation of ZAP isoform-inducible cell lines.
ZAP KO 293T cells (clone 89) were transfected with a 1:1 ratio of the ePB transposon vector that encodes RFP only or N-terminally RFP-tagged human ZAP isoforms (S, M, L, and XL; haplotype 1 or 2) and the transposase plasmid. Transfection was performed using X-tremegene 9 (Roche) according to the manufacturer’s instructions. The transfected cells were allowed to rest for 2 days prior to selection with 1 μg/ml puromycin. Leaky expression (noted by RFP-labeled protein expression in the absence of Dox) was observable immediately after transfection but gradually diminished until it was negligible 1 week after transfection. Bulk resistant cells were expanded and treated with 1 μg/ml Dox to confirm inducible expression of ZAP isoforms by immunoblotting and fluorescence microscopy.
EBOV minigenome expression.
ZAP KO 293T cells stably expressing RFP-tagged ZAP isoforms or control RFP were transfected with a Fluc reporter construct or cotransfected with expression constructs (under the control of the CMV promoter) encoding Zaire EBOV NP, L protein, VP30, VP35, and a minigenome carrying Fluc. Cells were then treated with Dox to induce the expression of the individual ZAP isoforms. Cell lysates were harvested at 1 day (Fluc-transfected cells) or 2 days (minigenome-transfected cells) posttransfection for a luciferase assay (Promega) according to the manufacturer’s instructions. The Fluc expression fold change was calculated by dividing the relative luciferase units of cells treated with Dox (with ZAP isoform expression) by the average relative luciferase units of uninduced cells. The average Fluc expression fold change for each inducible cell line was used for normalization. The minigenome expression fold change was calculated by dividing the relative luciferase units of cells treated with Dox (with ZAP isoform expression) by the average relative luciferase units of uninduced cells. Normalized percent minigenome expression for each cell line was then determined by dividing the minigenome expression fold change by the average Fluc expression fold change and multiplying by 100.
HBeAg and HBsAg chemiluminescence immunoassays.
For quantitative analysis of secreted HBeAg and HBsAg, 50 μl of supernatants was loaded into 96-well plates of a chemiluminescence immunoassay (CLIA) kit according to the manufacturer’s instructions (Autobio Diagnostics Co., Zhengzhou, China). Plates were read using a FLUOstar Omega luminometer. Concentrations are expressed in national clinical units (NCU) per milliliter.
Measurement of IFN-β stimulation.
ZAP KO 293T cells stably expressing RFP-tagged ZAP isoforms or control RFP were transfected with a construct expressing Fluc under the control of the IFN-β promoter (IFN-β-pGL3). An Rluc construct (pRL-Rluc) was cotransfected for normalization purposes. The cells were treated with Dox to induce the expression of individual ZAP isoforms and infected with SeV (Cantell strain) 1 day later to trigger RIG-I-mediated IFN-β production. Cell lysates were harvested at 24 h p.i. for a dual-luciferase reporter assay (Promega) according to the manufacturer’s instructions. The Fluc values were normalized using the Rluc values and set to 1 for each mock-infected cell line. The normalized Fluc values for infected cell lines were then used to calculate fold IFN-β induction compared to the respective mock-infected cell lines.
Statistical analyses.
All statistical analyses were performed using GraphPad Prism.
Data availability.
The sequence for human ZAPM is available in GenBank under accession number MN030155.
ACKNOWLEDGMENTS
We thank Joseph Luna at The Rockefeller University for analyzing the RNAseq expression data, Ali Brivanlou at The Rockefeller University for the ePB transposon and transposase plasmids, Alison Ashbrook at The Rockefeller University and Lisa Miorin and Richard Cadagan at Icahn School of Medicine at Mount Sinai for generation of the SeV stock, Akinori Takaoka at Hokkaido University for the ZAP KO 293T cells, and Oliver Fregoso and Melanie Dimapasoc at the University of California, Los Angeles, for help with analysis of alphavirus CG content. We also thank Steve Higgs at Kansas State University, Richard Kuhn at Purdue University, Mark Heise at the University of North Carolina, Scott Weaver at The University of Texas Medical Branch at Galveston, and Ilya Frolov at the University of Alabama at Birmingham for the alphavirus constructs; Elke Muhlberger at Boston University for the Ebola minigenome constructs; and Yosef Shaul at the Weizmann Institute of Science for the pHBV1.3x plasmid. Finally, we thank the Proteomics Resource Center at The Rockefeller University for their help with mass spectrometric analysis.
The work was supported in part by a Rockefeller University Women & Science postdoctoral fellowship (to M.M.H.L.); National Institutes of Health (NIH) fellowships F31AI138409 (to E.G.A.), F32DK107164 (to E.M.), and F32DK095666 (to W.M.S.); NIH supplement fellowship R01AI091707 (to J.P.); NIH grants R01AI091707 and R01AI143295 (to C.M.R.); NIH grant R01AI114873 (to M.R.M.); a Robertson Foundation grant (to E.M.); and anonymous donors.
REFERENCES
- 1.Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M. 2007. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis 7:319–327. doi: 10.1016/S1473-3099(07)70107-X. [DOI] [PubMed] [Google Scholar]
- 2.Powers AM, Logue CH. 2007. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J Gen Virol 88:2363–2377. doi: 10.1099/vir.0.82858-0. [DOI] [PubMed] [Google Scholar]
- 3.Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel MP, Brehin AC, Cubito N, Despres P, Kunst F, Rey FA, Zeller H, Brisse S. 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med 3:e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coltart CE, Lindsey B, Ghinai I, Johnson AM, Heymann DL. 2017. The Ebola outbreak, 2013-2016: old lessons for new epidemics. Philos Trans R Soc Lond B Biol Sci 372:20160297. doi: 10.1098/rstb.2016.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li MM, MacDonald MR, Rice CM. 2015. To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol 25:320–329. doi: 10.1016/j.tcb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao G, Guo X, Goff SP. 2002. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297:1703–1706. doi: 10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 7.Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J Virol 77:11555–11562. doi: 10.1128/jvi.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller S, Möller P, Bick MJ, Wurr S, Becker S, Günther S, Kümmerer BM. 2007. Inhibition of filovirus replication by the zinc finger antiviral protein. J Virol 81:2391–2400. doi: 10.1128/JVI.01601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Chen G, Lv F, Wang X, Ji X, Xu Y, Sun J, Wu L, Zheng YT, Gao G. 2011. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc Natl Acad Sci U S A 108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Tu F, Zhu Y, Gao G. 2012. Zinc-finger antiviral protein inhibits XMRV infection. PLoS One 7:e39159. doi: 10.1371/journal.pone.0039159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, Cuconati A, Block TM, Guo JT, Guo H. 2013. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog 9:e1003494. doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayakawa S, Shiratori S, Yamato H, Kameyama T, Kitatsuji C, Kashigi F, Goto S, Kameoka S, Fujikura D, Yamada T, Mizutani T, Kazumata M, Sato M, Tanaka J, Asaka M, Ohba Y, Miyazaki T, Imamura M, Takaoka A. 2011. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat Immunol 12:37–44. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- 13.Chiu HP, Chiu H, Yang CF, Lee YL, Chiu FL, Kuo HC, Lin RJ, Lin YL. 2018. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog 14:e1007166. doi: 10.1371/journal.ppat.1007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CH, Zhou LG, Chen GF, Krug RM. 2015. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc Natl Acad Sci U S A 112:14048–14053. doi: 10.1073/pnas.1509745112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Yan K, Wei L, Yang J, Lu C, Xiong F, Zheng C, Xu W. 2015. Zinc finger antiviral protein inhibits coxsackievirus B3 virus replication and protects against viral myocarditis. Antiviral Res 123:50–61. doi: 10.1016/j.antiviral.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Guo X, Ma J, Sun J, Gao G. 2007. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci U S A 104:151–156. doi: 10.1073/pnas.0607063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y, Wang X, Goff SP, Gao G. 2012. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J 31:4236–4246. doi: 10.1038/emboj.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. 2011. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, Hsiang TY, Krug RM, Sullivan CS. 2013. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14:435–445. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li MM, Lau Z, Cheung P, Aguilar EG, Schneider WM, Bozzacco L, Molina H, Buehler E, Takaoka A, Rice CM, Felsenfeld DP, MacDonald MR. 2017. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (ZAP). PLoS Pathog 13:e1006145. doi: 10.1371/journal.ppat.1006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karki S, Li MM, Schoggins JW, Tian S, Rice CM, MacDonald MR. 2012. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLoS One 7:e37398. doi: 10.1371/journal.pone.0037398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng X, Wang X, Tu F, Wang Q, Fan Z, Gao G. 2017. TRIM25 is required for the antiviral activity of zinc finger antiviral protein. J Virol 91:e00088-17. doi: 10.1128/JVI.00088-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takata MA, Goncalves-Carneiro D, Zang TM, Soll SJ, York A, Blanco-Melo D, Bieniasz PD. 2017. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 550:124–127. doi: 10.1038/nature24039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerns JA, Emerman M, Malik HS. 2008. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet 4:e21. doi: 10.1371/journal.pgen.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charron G, Li MM, MacDonald MR, Hang HC. 2013. Prenylome profiling reveals S-farnesylation is crucial for membrane targeting and antiviral activity of ZAP long-isoform. Proc Natl Acad Sci U S A 110:11085–11090. doi: 10.1073/pnas.1302564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Luscher B. 2008. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol Cell 32:57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Law LM, Albin OR, Carroll JW, Jones CT, Rice CM, MacDonald MR. 2010. Identification of a dominant negative inhibitor of human zinc finger antiviral protein (ZAP) reveals a functional endogenous pool and critical homotypic interactions. J Virol 84:4504–4512. doi: 10.1128/JVI.02018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo X, Carroll JW, MacDonald MR, Goff SP, Gao G. 2004. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J Virol 78:12781–12787. doi: 10.1128/JVI.78.23.12781-12787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katoh M, Katoh M. 2003. Identification and characterization of human TIPARP gene within the CCNL amplicon at human chromosome 3q25.31. Int J Oncol 23:541–547. [PubMed] [Google Scholar]
- 30.Aravind L. 2001. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem Sci 26:273–275. doi: 10.1016/S0968-0004(01)01787-X. [DOI] [PubMed] [Google Scholar]
- 31.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H, Komano J, Saitoh Y, Yamaoka S, Kozaki T, Misawa T, Takahama M, Satoh T, Takeuchi O, Yamamoto N, Matsuura Y, Saitoh T, Akira S. 2013. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc Natl Acad Sci U S A 110:12379–12384. doi: 10.1073/pnas.1310604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111. doi: 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 34.Baum A, Sachidanandam R, García-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107:16303–16308. (Erratum, 108:3092, 2011.). doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuel CE. 2011. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YZ, Banerjee S, Wang YY, Goldstein SA, Dong BH, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A 113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klaus A, Birchmeier W. 2008. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 38.Sun L, Lv F, Guo X, Gao G. 2012. Glycogen synthase kinase 3beta (GSK3beta) modulates antiviral activity of zinc-finger antiviral protein (ZAP). J Biol Chem 287:22882–22888. doi: 10.1074/jbc.M111.306373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baril M, Es-Saad S, Chatel-Chaix L, Fink K, Pham T, Raymond VA, Audette K, Guenier AS, Duchaine J, Servant M, Bilodeau M, Cohen E, Grandvaux N, Lamarre D. 2013. Genome-wide RNAi screen reveals a new role of a WNT/CTNNB1 signaling pathway as negative regulator of virus-induced innate immune responses. PLoS Pathog 9:e1003416. doi: 10.1371/journal.ppat.1003416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith JL, Jeng S, McWeeney SK, Hirsch AJ. 2017. A microRNA screen identifies the Wnt signaling pathway as a regulator of the interferon response during flavivirus infection. J Virol 91:e02388-16. doi: 10.1128/JVI.02388-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacoste A, Berenshteyn F, Brivanlou AH. 2009. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 5:332–342. doi: 10.1016/j.stem.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Frolova EI, Fayzulin RZ, Cook SH, Griffin DE, Rice CM, Frolov I. 2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J Virol 76:11254–11264. doi: 10.1128/jvi.76.22.11254-11264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, Rossi SL, Partidos CD, Adams AP, Seymour RL, Weger J, Borland EM, Sherman MB, Powers AM, Osorio JE, Weaver SC. 2012. Attenuation of chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J Virol 86:6084–6096. doi: 10.1128/JVI.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brault AC, Foy BD, Myles KM, Kelly CL, Higgs S, Weaver SC, Olson KE, Miller BR, Powers AM. 2004. Infection patterns of O’nyong nyong virus in the malaria-transmitting mosquito, Anopheles gambiae. Insect Mol Biol 13:625–635. doi: 10.1111/j.0962-1075.2004.00521.x. [DOI] [PubMed] [Google Scholar]
- 45.Morrison TE, Whitmore AC, Shabman RS, Lidbury BA, Mahalingam S, Heise MT. 2006. Characterization of Ross River virus tropism and virus-induced inflammation in a mouse model of viral arthritis and myositis. J Virol 80:737–749. doi: 10.1128/JVI.80.2.737-749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuhn RJ, Niesters HG, Hong Z, Strauss JH. 1991. Infectious RNA transcripts from Ross River virus cDNA clones and the construction and characterization of defined chimeras with Sindbis virus. Virology 182:430–441. doi: 10.1016/0042-6822(91)90584-X. [DOI] [PubMed] [Google Scholar]
- 47.Doitsh G, Shaul Y. 2004. Enhancer I predominance in hepatitis B virus gene expression. Mol Cell Biol 24:1799–1808. doi: 10.1128/mcb.24.4.1799-1808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin R, Genin P, Mamane Y, Hiscott J. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol 20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honda M, Kaneko S, Matsushita E, Kobayashi K, Abell GA, Lemon SM. 2000. Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology 118:152–162. doi: 10.1016/S0016-5085(00)70424-0. [DOI] [PubMed] [Google Scholar]
- 50.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 51.Bunkenborg J, Garcia GE, Paz MI, Andersen JS, Molina H. 2010. The minotaur proteome: avoiding cross-species identifications deriving from bovine serum in cell culture models. Proteomics 10:3040–3044. doi: 10.1002/pmic.201000103. [DOI] [PubMed] [Google Scholar]
- 52.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequence for human ZAPM is available in GenBank under accession number MN030155.