

Signaling through the chemokine receptor CXCR2 in oligodendroglia is important for developmental myelination in rodents, while chemical inhibition or nonspecific genetic deletion of CXCR2 appears to augment myelin repair in animal models of the human demyelinating disease multiple sclerosis (MS). To better understand the biology of CXCR2 signaling on oligodendroglia, we generated transgenic mice in which Cxcr2 is selectively ablated in oligodendroglia upon treatment with tamoxifen. Using a viral model of neuroinflammation and demyelination, we demonstrate that genetic silencing of CXCR2 on oligodendroglia did not affect clinical disease, neuroinflammation, or demyelination, yet there was increased remyelination. These findings support and extend previous findings suggesting that targeting CXCR2 may offer a therapeutic avenue for enhancing remyelination in patients with demyelinating diseases.
KEYWORDS: chemokine receptors, demyelination, oligodendrocyte, remyelination, virus
ABSTRACT
CXCR2 is a chemokine receptor expressed on oligodendroglia that has been implicated in the pathogenesis of neuroinflammatory demyelinating diseases as well as enhancement of the migration, proliferation, and myelin production by oligodendroglia. Using an inducible proteolipid protein (Plp) promoter-driven Cre-loxP recombination system, we were able to assess how timed ablation of Cxcr2 in oligodendroglia affected disease following intracranial infection with the neurotropic JHM strain of mouse hepatitis virus (JHMV). Generation of Plp-Cre-ER(T)::Cxcr2flox/flox transgenic mice (termed Cxcr2-CKO mice) allows for Cxcr2 to be silenced in oligodendrocytes in adult mice following treatment with tamoxifen. Ablation of oligodendroglia Cxcr2 did not influence clinical severity in response to intracranial infection with JHMV. Infiltration of activated T cells or myeloid cells into the central nervous system (CNS) was not affected, nor was the ability to control viral infection. In addition, the severity of demyelination was similar between tamoxifen-treated mice and vehicle-treated controls. Notably, deletion of Cxcr2 resulted in increased remyelination, as assessed by g-ratio (the ratio of the inner axonal diameter to the total outer fiber diameter) calculation, compared to that in vehicle-treated control mice. Collectively, our findings argue that CXCR2 signaling in oligodendroglia is dispensable with regard to contributing to neuroinflammation, but its deletion enhances remyelination in a preclinical model of the human demyelinating disease multiple sclerosis (MS).
IMPORTANCE Signaling through the chemokine receptor CXCR2 in oligodendroglia is important for developmental myelination in rodents, while chemical inhibition or nonspecific genetic deletion of CXCR2 appears to augment myelin repair in animal models of the human demyelinating disease multiple sclerosis (MS). To better understand the biology of CXCR2 signaling on oligodendroglia, we generated transgenic mice in which Cxcr2 is selectively ablated in oligodendroglia upon treatment with tamoxifen. Using a viral model of neuroinflammation and demyelination, we demonstrate that genetic silencing of CXCR2 on oligodendroglia did not affect clinical disease, neuroinflammation, or demyelination, yet there was increased remyelination. These findings support and extend previous findings suggesting that targeting CXCR2 may offer a therapeutic avenue for enhancing remyelination in patients with demyelinating diseases.
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory neurodegenerative disease characterized by multifocal regions of central nervous system (CNS) neuroinflammation, demyelination, and axonal loss that ultimately results in extensive neurologic disability (1). Preclinical animal models of MS indicate that CNS infiltration of neutrophils, monocyte/macrophages, and inflammatory T cells, including those that are autoreactive to specific proteins embedded in the myelin sheath, is important in disease initiation and maintaining demyelination. Through the use of mouse models of MS, chemokines and chemokine receptors have been implicated as being important in attracting targeted populations of activated immune cells into the CNS and have been considered relevant targets for clinical intervention for MS patients (2–11). CXCR2 is a receptor for ELR-positive CXC chemokines, e.g., CXCL1 and CXCL2, and is expressed on polymorphonuclear neutrophils (PMN) as well as glia (12–16). CXCR2 signaling on neutrophils promotes demyelination in models of experimental autoimmune encephalomyelitis (EAE), cuprizone-induced demyelination, as well as virus-induced demyelination (8, 17–19). However, CXCR2 has additional roles that extend beyond influencing neutrophil activity, as it is expressed on immature oligodendrocyte progenitor cells (OPCs) as well as mature myelinating oligodendrocytes (20). In vitro studies have demonstrated that signaling through CXCR2 influences OPC proliferation and differentiation (21), whereas in vivo studies argue that CXCR2 controls the positional migration of OPCs within the spinal cord and regulates OPC numbers to ensure the structural integrity of the white matter during CNS development (22). Indeed, mice devoid of CXCR2 exhibit a paucity of OPCs and structural misalignments that persist into adulthood of the mouse, resulting in reduced numbers of mature oligodendrocytes and total myelin within the white matter (23). Moreover, either genetic deletion or antibody-mediated targeting of CXCR2 increases myelin synthesis in demyelinated cerebellum slice cultures (24). The concept of CXCR2 signaling on oligodendroglia regulating myelin synthesis is further supported by a recent report by Liu and colleagues (25) that demonstrated that targeted ablation of CXCR2 on oligodendroglia enhanced remyelination following toxin-induced demyelination. Collectively, these findings argue that CXCR2 signaling on oligodendroglia ultimately contributes to maintaining myelin integrity and axonal protection both under homeostatic conditions and following demyelination.
Intracranial (i.c.) inoculation of susceptible mice with the neurotropic JHM strain of mouse hepatitis virus (JHMV) results in an acute encephalomyelitis characterized by widespread viral replication in glial cells with relative sparing of neurons (26, 27). CNS-infiltrating CD4+ and CD8+ T cells control viral replication through the secretion of gamma interferon (IFN-γ) and cytolytic activity, yet sterile immunity is not acquired and virus persists in white matter tracts, resulting in an immune-mediated demyelinating disease with clinical and histologic similarities to the human demyelinating disease MS (28, 29). Previous studies employing the JHMV model have demonstrated important roles for select chemokines in participating in host defense as well as demyelination by attracting targeted populations of leukocytes into the CNS (30–36). More recently, we have shown that treatment of JHMV-infected mice with blocking antibody specific for CXCR2 led to an increase in clinical disease associated with more severe demyelination (37). Examination of spinal cords revealed that the increase in white matter damage in anti-CXCR2-treated mice was associated with oligodendroglia apoptosis, arguing for a protective role for CXCR2 signaling in a model of virus-induced demyelination (37). Nonetheless, other resident cells as well as inflammatory cells express CXCR2, making it difficult to assign a specific role for CXCR2 signaling on oligodendroglia in contributing to either protection or disease progression. The present study was undertaken to better understand how selective deletion of Cxcr2 within oligodendroglia lineage cells in adult mice influences host defense and disease in response to JHMV infection of the CNS.
RESULTS
Generation and characterization of Cxcr2-CKO mice.
We sought to ablate Cxcr2 signaling within oligodendrocytes and their progenitors to assess the impact on disease and repair in an animal model of neuroinflammation and demyelination. A Plp-Cre-ER(T)::Cxcr2flox/flox mouse line (referred to as Cxcr2-CKO) that utilizes Cre recombinase to ablate Cxcr2 in a selective and inducible manner was employed (17, 38–41). The proteolipid protein (PLP) regulatory element promotes expression of Cre-ER(T) (42) and has previously been shown to accurately reflect endogenous PLP expression spatially and temporally in oligodendroglia (42). To aid in visualizing cells actively expressing Cre recombinase, Cxcr2-CKO mice were crossed to a Cre-inducible Rosa26-tdTomato reporter line on the C57BL/6 mouse background (Fig. 1A). Ablation of Cxcr2 was confirmed within oligodendrocyte lineage cells following ex vivo culture of oligodendroglia generated from the brains of postnatal day 1 (P1) Cxcr2-CKO mice. Addition of (Z)-4-hydroxytamoxifen (4-OHT; 100 nM) induced Cre-mediated recombination at the Cxcr2 locus, as detected by PCR using primers that specifically generate an amplicon following excision of exon 2 of Cxcr2 (Fig. 1B). 4-OHT induced expression of tdTomato red in the majority of cultured cells from the brains of P1 Cxcr2-CKO mice at day 6 posttreatment, with the tdTomato red being distributed throughout the cell body and the cell morphology resembling oligodendrocytes (Fig. 1C). Immunocytochemical staining revealed >90% of cultured oligodendroglia expressing the late-stage O1 surface marker, in contrast to vehicle-treated cultures (Fig. 1D).
FIG 1.

Cre-mediated recombination is detected in vitro. (A) Schematic diagram showing the genetic strategy used to generate tamoxifen-inducible knockout of Cxcr2 within oligodendroglia. (B) Cre-mediated recombination at the Cxcr2 locus was detected by PCR from oligodendroglia-enriched cultures derived from postnatal day 1 (P1) Cxcr2-CKO mice following addition of 100 nM (Z)-4-hydroxytamoxifen (4-OHT). gDNA, genomic DNA. (C) Representative immunofluorescent staining for O1 (a marker for mature oligodendrocytes) from cultured oligodendroglia following 6 days of treatment with 4-OHT, showing a morphology characteristic of mature oligodendrocytes and colocalization of O1 (green) and tdTomato red. (D) Treatment of cultured oligodendroglia with 4-OHT resulted in expression of tdTomato in >90% of O1-positive oligodendrocytes (****, P < 0.0001; 91.4% ± 2.02%) derived from P1 Cxcr2-CKO mice, in contrast to vehicle treatment (3.92% ± 0.14%); data are presented as the average + SEM and represent those from 3 independent experiments.
Tamoxifen treatment results in enriched tdTomato red expression in spinal cord oligodendroglia.
To determine the cellular specificity of Cre activity in vivo, 4-week-old Cxcr2-CKO mice were treated with 1 mg tamoxifen by intraperitoneal (i.p.) administration twice daily for 5 days, and the targeting of Cxcr2 was subsequently determined (43, 44). Recombination at the Cxcr2 locus was detected by PCR in the brain and spinal cord of Cxcr2-CKO mice but not in the spleen, liver, or kidney (Fig. 2A). In response to tamoxifen treatment, there was a dramatic increase in tdTomato expression that was distributed throughout the cell body as well as processes. To identify cells expressing tdTomato within the CNS in response to tamoxifen treatment, immunohistochemical staining for defined cellular markers of the CNS was performed. In addition to oligodendroglia, we focused our attention on neurons (45, 46) and macrophages/microglia (45, 47), as these cells have been reported to express CXCR2. Expression of CXCR2 by astrocytes has not been reported, and we did not concentrate on this population of cells (48, 49). We found that the majority of tdTomato-positive cells in tamoxifen-treated Cxcr2-CKO mice were oligodendroglia, as defined by staining either oligodendrocyte transcription factor 2 (Olig2) (50, 51) or glutathione S-transferase π (GST-π), a marker of mature oligodendrocytes (52, 53). In tamoxifen-treated mice, ∼82% of tdTomato-positive cells (cells in which the tdTomato signal was distributed throughout the cell body) expressed nuclear-specific markers (Olig2, 42.1% ± 7.7%; GST-π, 39.9% ± 7.9%) (Fig. 2B, C, and F). MAP2-positive neurons accounted for <4% of tdTomato-positive cells (3.3% ± 0.8%) in tamoxifen-treated Cxcr2-CKO mice (Fig. 2D and F). Similarly, very few IBA1-positive macrophages/microglia expressed tdTomato (2.6% ± 0.3%) (Fig. 2E and F). We have previously demonstrated that neutrophils require CXCR2 to traffic and accumulate within the CNS in response to JHMV infection (54). Tamoxifen treatment did not modulate CXCR2 expression in neutrophils, as no differences were observed in surface expression of CXCR2 on splenic neutrophils obtained from tamoxifen-treated Cxcr2-CKO mice and vehicle-treated mice, as determined by flow cytometry (Fig. 2G). These results demonstrate that tamoxifen-induced targeting of Cxcr2 is highly specific to oligodendrocyte lineage cells. It is important to note that we did not perform CXCR2 immunohistochemical staining in experimental tissues, as we have previously reported that there are no validated CXCR2-specific antibodies, as we routinely detected nonspecific CXCR2 staining in experimental Cxcr2−/− mice (24). The remaining tdTomato-positive cells present within the spinal cords of tamoxifen-treated mice (∼12%) most likely represent oligodendroglia cellular processes extending from the cell bodies in which neither Olig2 or GST-π is detected.
FIG 2.

Cxcr2 is ablated in vivo following tamoxifen treatment. (A) Detection of Cre-mediated recombination at the Cxcr2 locus in the brain and spinal cord of Cxcr2-CKO mice 2 weeks following tamoxifen treatment. (B to E) Phenotyping of tdTomato-positive cells was performed via immunofluorescent staining for defined cell-specific markers. Representative staining of spinal cords from tamoxifen-treated (n = 5) Cxcr2-CKO mice for oligodendroglia via GST-π (B), Olig2 (C), neurons (MAP2) (D), and macrophage/microglia (IBA1) (E). Magnifications, ×20. Arrows in panels B and C represent dual-positive cells. (F) Quantification of dual-positive cells in the spinal cords of tamoxifen-treated mice; the data were derived from 2 independent experiments, with the data presented as the average ± SEM. (G) The surface expression of CXCR2 on neutrophils was not affected in tamoxifen-treated Cxcr2-CKO mice (n = 2), in contrast to the vehicle-treated controls (n = 2); the data are presented as the average ± SEM. MFI, mean fluorescence intensity.
No defects in myelin formation/synthesis following Cxcr2 ablation in oligodendroglia.
We next evaluated whether tamoxifen-mediated targeting of Cxcr2 in oligodendroglia in adult mice affected myelin integrity, as previous studies have indicated that adult Cxcr2−/− animals have deficient myelin formation (22, 23). Cxcr2-CKO mice were treated twice daily with either 1 mg tamoxifen or vehicle, followed by resting for 2 weeks, at which point the spinal cords were removed to evaluate spinal cord myelin. Similar to the findings of Liu et al. (25), we did not detect any differences in either the size or behavior in tamoxifen-treated Cxcr2-CKO mice compared to their vehicle-treated littermates. Assessment of spinal cord myelin via toluidine blue staining revealed no overt differences between tamoxifen-treated Cxcr2-CKO mice and vehicle-treated control Cxcr2-CKO mice (Fig. 3A and B). As an additional test of myelin integrity, electron microscopy (EM) analysis of spinal cord sections was performed. Assessment of the g ratio, the ratio of the inner axonal diameter to the total outer fiber diameter, is commonly employed as a structural index of myelin. We did not detect any changes in qualitative differences in myelin thickness between naive tamoxifen-treated Cxcr2-CKO mice and vehicle-treated Cxcr2-CKO mice (Fig. 3C and D); subsequent calculation of the g ratios between experimental groups confirmed that targeting of CXCR2 in oligodendroglia in adult mice did not alter myelin integrity (Fig. 3E and F). In all experimental groups, the g ratios averaged ∼0.8, indicating normal myelinated axons in the spinal cord (Fig. 3E and F) (55–57). These findings argue that inducible deletion of CXCR2 in oligodendroglia lineage cells in young (4-week-old) mice does not affect the myelin content in a nondisease state.
FIG 3.
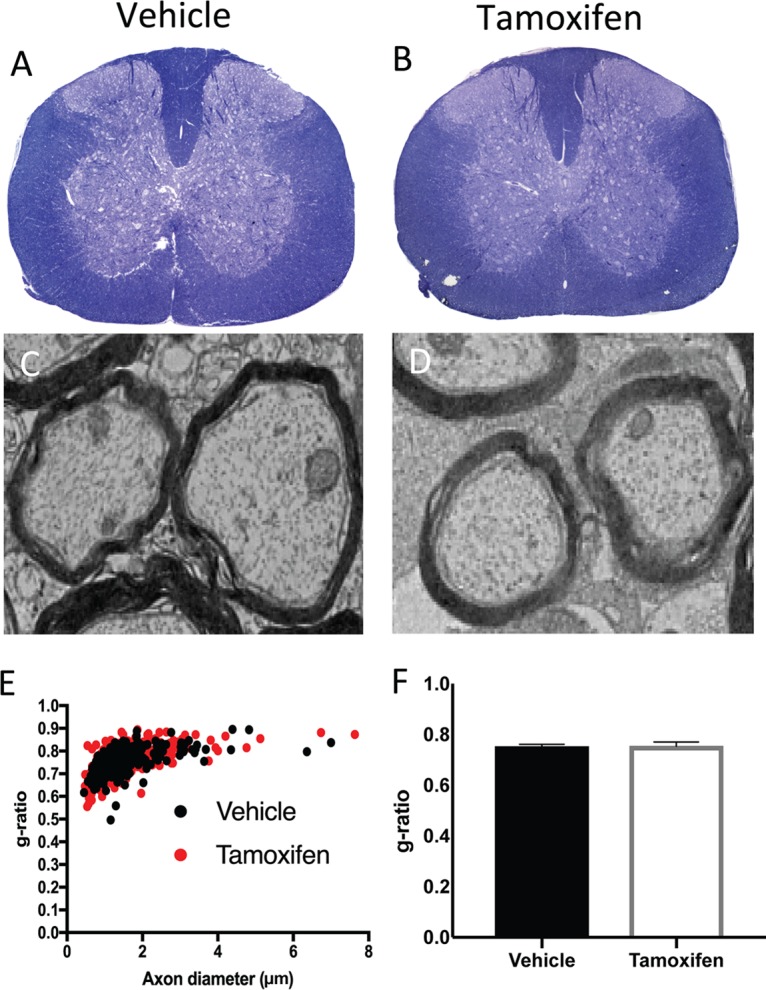
Myelin integrity and ultrastructure are not affected following tamoxifen-treatment of naive Cxcr2-CKO mice. Cxcr2-CKO mice were treated with either vehicle or tamoxifen for 1 week and sacrificed 2 weeks later to evaluate myelin integrity. (A and B) Representative spinal cord semithin sections stained with toluidine blue demonstrate no visible differences in myelin between vehicle-treated (A) and tamoxifen-treated (B) mice. (C and D) Representative EM images showing myelinated axons within the spinal cords of vehicle-treated (C) and tamoxifen-treated (D) Cxcr2-CKO mice. Analysis was performed in the ventral and lateral white matter tracts. Magnifications, ×1,200. (E) Scatter plot depicting the g ratios of individual axons as a function of the axonal diameter. (F) Calculation of the g ratios of vehicle-treated (n = 3, 0.75 ± 0.004, 222 axons) and tamoxifen-treated (n = 3, 0.75 ± 0.004, 298 axons) Cxcr2-CKO mice. The data were derived from two independent experiments with 2 to 4 mice per group and are presented as the mean ± SEM.
Cxcr2 ablation within oligodendrocytes does not impair antiviral responses.
Intracranial (i.c.) instillation of the neuroadapted JHM strain of mouse hepatitis virus (JHMV) results in an acute encephalomyelitis followed by a chronic demyelinating disease characterized by viral persistence in white matter tracts accompanied by immune-mediated demyelination (26). Tamoxifen- or vehicle-treated Cxcr2-CKO mice were infected i.c. with JHMV (250 PFU), and clinical disease was monitored. Vehicle-treated Cxcr2-CKO mice had worse (P < 0.05) clinical disease at 12 days postinfection (p.i.) than control mice, but otherwise, no overt differences in morbidity or mortality were observed in the experimental mice out to day 28 p.i. (Fig. 4A). Examination of the viral titers in the brains at days 5, 7, and 10 p.i. showed similar levels of virus present between tamoxifen- and vehicle-treated mice (Fig. 4B). Although the titers were higher (P < 0.05) in tamoxifen-treated animals at day 7 p.i. than in the controls, we do not believe that this reflects an overall difference in the control of viral replication, as there were no differences in viral titers between experimental groups at the other time points examined. By day 21 p.i., virus was not detected (ND) in the brains of experimental mice (Fig. 4B). There were no differences in viral titers in the spinal cords of experimental mice at days 5, 7, and 10 p.i. (Fig. 4B). Collectively, these findings indicate that Cxcr2 targeting did not compromise the control of viral replication within the CNS. Moreover, similar frequencies of CD4+ and CD8+ T cells specific for the immunodominant viral epitopes M133-144 and S510-518, respectively, as determined by tetramer staining, were detected within the brains of the experimental mice at day 7, further supporting that targeting of Cxcr2 within oligodendroglia did not impact the generation of a protective antiviral immune response (Fig. 4C). There were no differences in either total T cell subsets, neutrophils (CD11b+ Ly6G+), or macrophages (CD45hi F4/80hi) at day 7 p.i. between vehicle- and tamoxifen-treated mice (Fig. 4D). The comparable acute inflammatory response to JHMV between vehicle- and tamoxifen-treated Cxcr2-CKO mice was further supported by the observation of no significant differences in expression of proinflammatory cytokine and chemokine genes (a detailed inventory of the probes is provided in Materials and Methods) within the brain at day 7 p.i. (Fig. 4E). Collectively, these findings indicate that selectively targeting Cxcr2 within oligodendroglia does not mute neuroinflammation or impair control of JHMV replication within the CNS.
FIG 4.

Tamoxifen-treated Cxcr2-CKO mice are susceptible to JHMV-induced neuroinflammation. Cxcr2-CKO mice were treated with either tamoxifen (n = 22) or vehicle (n = 10), rested for 2 weeks, and infected i.c. with 250 PFU of JHMV. The data are representative of those from 3 independent experiments. (A) Clinical disease developed in both groups, and vehicle-treated Cxcr2-CKO mice had worse (P < 0.05) disease at 12 days p.i. than control mice, but otherwise, there were no statistically significant differences in disease severity or mortality out to day 28 p.i. between the experimental groups. (B) Viral titers in the brains and spinal cords of tamoxifen- and vehicle-treated mice at days 5, 7, and 10 p.i. *, P < 0.05. By day 21 p.i., virus was not detected (ND) in the brains of experimental mice. dpi, day postinfection. (C) No differences in the frequencies of virus-specific CD4+ and CD8+ T cells were detected, as determined by tetramer staining at day 7 p.i. (D) At day 7 p.i., similar numbers of neutrophils (Neut.), macrophages (Mac.), and T cells were detected in the brains of JHMV-infected Cxcr2-CKO mice treated with either vehicle or tamoxifen. Day 7 p.i. flow cytometric data were derived from 2 independent experiments with a minimum of 3 mice per group for each experiment. (E) RNA analysis revealed no changes in proinflammatory gene expression within the brains of the experimental groups at day 7 p.i. The data represent the average for 2 mice per group.
Cxcr2 ablation does not affect the severity of demyelination.
JHMV infection of mice results in persistent CNS infection and a chronic immune-mediated demyelinating disease. Evaluation of demyelination in JHMV-infected Cxcr2-CKO mice indicated that there were no significant differences in the severity of demyelination between vehicle- and tamoxifen-treated Cxcr2-CKO mice at days 21 (Fig. 5A to C) and 28 (Fig. 5E to G) p.i. Evaluation of immune cell infiltration into the spinal cords of tamoxifen- and vehicle-treated mice by flow cytometry at either day 21 p.i. (Fig. 5D) or day 28 p.i. (Fig. 5H) revealed no difference in CD4+ or CD8+ T cell as well as macrophage (CD45lo F4/80+) infiltration. Furthermore, examination of demyelination within the brains of JHMV-infected Cxcr2-CKO mice treated with either vehicle or tamoxifen revealed no differences in the severity of demyelination (data not shown). These findings argue that tamoxifen-mediated targeting of Cxcr2 within oligodendroglia does not alter immune cell infiltration into the spinal cords of JHMV-infected mice or influence the severity of white matter damage.
FIG 5.
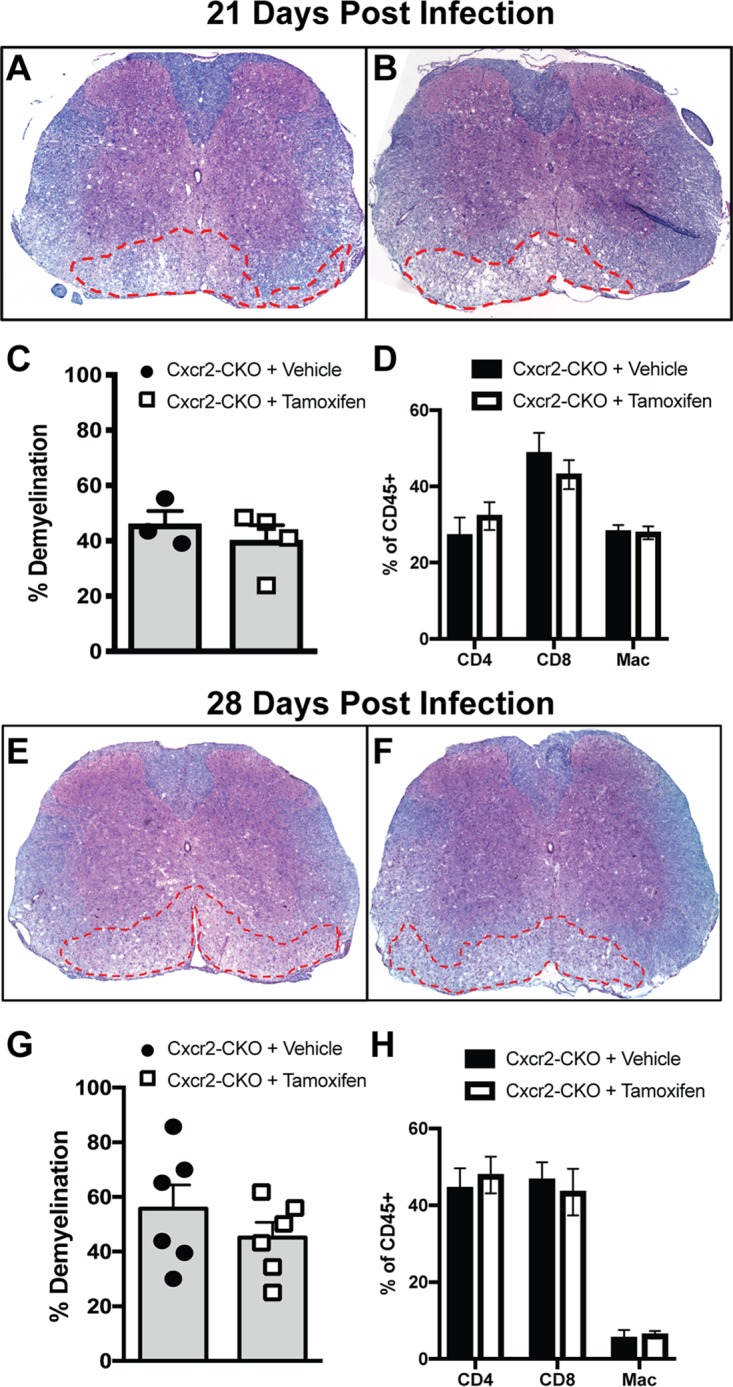
Demyelination is not affected following Cxcr2 ablation. JHMV-infected Cxcr2-CKO mice were treated with vehicle or tamoxifen and sacrificed at days 21 and 28 p.i., and spinal cords were removed to assess the severity of demyelination. Representative spinal cords from either vehicle-treated (A, E) or tamoxifen-treated (B, F) mice were collected at day 21 p.i. (A and B) or day 28 p.i. (E and F) and stained with Luxol fast blue (LFB). Quantification of the severity of demyelination revealed no differences at either day 21 p.i. (C) or day 28 p.i. (G). Flow cytometric data revealed no differences in spinal cord infiltration of T cells (CD4+ and CD8+ subsets) or macrophages (CD45hi F4/80+) at either day 21 p.i. (D) or day 28 p.i. (H). The data were derived from two independent experiments with 2 to 3 mice per group and are presented as the mean ± SEM.
Remyelination is enhanced upon silencing of CXCR2 in oligodendroglia.
Previous work has indicated that targeting of CXCR2 signaling through either administration of receptor-specific small molecules (58) or genetic targeting (25) results in increased remyelination. To determine whether remyelination was occurring following tamoxifen-mediated silencing of Cxcr2 signaling, JHMV-infected Cxcr2-CKO mice were sacrificed at day 28 p.i., and EM analysis of spinal cord sections was performed to calculate g ratios. High-magnification (×1,200) images of the spinal cord lateral white matter columns concentrated within thoracic vertebrae 6 to 10 of tamoxifen-treated and vehicle-treated control Cxcr2-CKO mice were used to evaluate axons. Representative EM images from JHMV-infected mice treated with vehicle depict axons with very little to no myelin sheath (Fig. 6A). Remyelinated axons in tamoxifen-treated JHMV-infected Cxcr2-CKO mice were identified by thin myelin sheaths, in contrast to the thicker myelin sheath detected on a normal myelinated axon (Fig. 6B). In vehicle-treated Cxcr2-CKO mice, there was an overall increase of demyelinated axons and fewer remyelinated axons compared to tamoxifen-treated Cxcr2-CKO mice. To more accurately quantitate these differences, we quantified both the g ratio and the myelin thickness in infected mice. Tamoxifen-induced ablation of Cxcr2 on oligodendroglia resulted in a g ratio (0.78 ± 0.006, n = 5 mice) significantly (P < 0.05) lower than that in control mice (0.87 ± 0.006, n = 4 mice) (Fig. 6C and D). In addition, there was a corresponding significant (P < 0.01) increase in myelin thickness in tamoxifen-treated Cxcr2-CKO mice compared to vehicle-treated mice (Fig. 6E and F). Collectively, these findings indicate that selective ablation of Cxcr2 in oligodendroglia enhances remyelination following JHMV-induced demyelination.
FIG 6.

Remyelination is increased following Cxcr2 ablation in oligodendroglia. Spinal cords were removed at day 28 p.i. from JHMV-infected mice treated with either vehicle control or tamoxifen. (A and B) Representative EM images from vehicle-treated mice depict demyelinated axons (black arrows) (A), whereas in tamoxifen-treated mice, remyelinated axons (blue arrows) were present (B). Normal myelinated axons (yellow arrows) are also shown. Magnifications, ×1,200. Analysis was performed in ventral and lateral white matter columns of spinal cords isolated from experimental mice. (C) Scatter plot depicting the g ratios of individual axons from vehicle- and tamoxifen-treated mice as a function of axonal diameter. (D) Calculation of the g ratio of vehicle-treated Cxcr2-CKO mice (0.87 ± 0.006, n = 4 mice; 297 axons randomly selected from a total of 25 fields) and tamoxifen-treated Cxcr2-CKO mice (0.78 ± 0.006, n = 5 mice; 493 axons randomly selected from a total of 34 fields) indicates a significant (P < 0.05) reduction in the g ratios in tamoxifen-treated mice compared to the vehicle-treated controls. (E) Scatter plot depicting the myelin thickness of individual axons from vehicle- and tamoxifen-treated mice as a function of axon diameter. (F) Quantification of myelin thickness in vehicle-treated mice (0.23 μm ± 0.008 μm) and tamoxifen-treated mice (0.35 μm ± 0.007 μm) indicated a significant (P < 0.01) increase in myelin thickness in tamoxifen-treated mice compared to the vehicle-treated controls. The results are derived from two independent experiments, and the data in panels D and F are presented as the mean ± SEM.
Small-molecule targeting of CXCR2 has been shown to enhance the maturation of cultured oligodendrocyte progenitor cells (OPCs) to mature myelin-producing oligodendroglia (58). To better understand how tamoxifen-mediated targeting of Cxcr2 within oligodendroglia led to an increase in remyelination, we stained for NG2, a marker associated with OPCs, and GST-π, which is normally associated with mature myelin-producing oligodendrocytes in the spinal cords of experimental mice. There was a significant (P < 0.01) decrease in NG2-positive cells in the spinal cords of tamoxifen-treated mice compared to the vehicle controls (Fig. 7A to C). Conversely, we detected an increase in the numbers of GST-π-positive cells in tamoxifen-treated mice compared to the controls (Fig. 7D to F). These findings argue that targeted ablation of Cxcr2 within oligodendrocytes increases the numbers of mature oligodendrocytes and that this correlates with a diminished pool of OPCs and, correspondingly, an increase in remyelination.
FIG 7.

Cxcr2 ablation increases the numbers of mature oligodendrocytes in JHMV-infected mice. JHMV-infected Cxcr2-CKO mice were treated with vehicle or tamoxifen, and at day 28 p.i. the animals were sacrificed and the spinal cords were removed. Representative NG2 (A, B) and GST-π (D, E) staining in control (n = 5) and tamoxifen-treated (n = 4) mice is shown. Quantification reveals reduced numbers of NG2-positive cells in tamoxifen-treated mice (C) and increased numbers of dual-positive GST-π-positive cells in tamoxifen-treated mice (F) compared to vehicle-treated mice. Data represent those from 2 independent experiments, and significance was measured using an unpaired 2-tailed Student's t test. **, P < 0.01; ***, P < 0.001. DAPI, 4′,6-diamidino-2-phenylindole.
DISCUSSION
Chemokine signaling networks that are associated with chronic CNS diseases, such as MS, or persistent viral infections are thought to amplify disease severity by attracting targeted populations of leukocytes into the CNS (59). One chemokine pathway that is emerging as important in chronic CNS disease involves the CXCR2 receptor and its cognate ELR-positive chemokine ligands CXCL1, -2, and -3 and CXCL5, -6, -7, and -8. CXCR2 is highly expressed on circulating neutrophils, enabling these cells to rapidly migrate to the blood-brain barrier (BBB) in response to CNS-derived ELR-ligand expression, whereby these cells participate in degrading components of the BBB. Evidence for a proinflammatory role of neutrophils in chronic neurologic disease is supported by the work of Segal and colleagues (8), who have shown that genetic silencing or antibody-mediated blockade of CXCR2 in mice following PLP-induced EAE results in reduced clinical disease and relapses as a result of limiting BBB permeability. Within the context of acute viral infection of the CNS, we have previously reported that antibody-mediated neutralization of CXCR2 during JHMV infection enhances disease (54). This outcome was a result of reduced neutrophil and monocyte trafficking, as these cells are critical in permeabilizing the BBB, which subsequently allows the penetration of virus-specific T cells in the parenchyma to combat viral replication.
In addition to being expressed on circulating myeloid cells, CXCR2 is also expressed on neurons (45, 46), cerebral endothelial cells (60, 61), as well as resident glia, including microglia (45, 47) and oligodendrocytes (13, 14, 22, 25, 62, 63). Further, addition of IFN-γ to cultured OPCs derived from mouse neural progenitor cells results in apoptosis, while inclusion of the CXCL1 protein blocks OPC apoptotic death (14). CXCR2 also influences OPC proliferation and differentiation (21) as well as controls the migration of spinal cord OPCs during development (22). Ablation of Cxcr2 results in a paucity of OPC numbers and structural misalignments that persist into adulthood in the mouse and manifest as reduced numbers of mature oligodendrocytes and total myelin within the white matter (23).
The functional role of CXCR2 signaling in mouse models of demyelination within the CNS is enigmatic. Some findings suggest that the CXCR2 signaling axis downregulates myelin production by oligodendrocytes (64), while we have reported that CXCR2 signaling is a survival mechanism for OPCs needed to halt the apoptosis induced by cytotoxic factors secreted during an inflammatory response (14, 37, 63, 65). With regards to CXCR2 signaling and survival, these studies were performed using either antibody targeting of CXCR2 or germ line Cxcr2−/− mice, and this opens the possibility of CXCR2 signaling on other cell types, either resident or immune cells, in augmenting experimental outcomes. Previously, members of our group (24) used bone marrow chimeric mice to partition the contribution of CXCR2 expression on either hematopoietic or CNS-derived cells. Adoptive transfer of hematopoietic cells derived from the bone marrow of Cxcr2+/+ mice into Cxcr2−/− mice enabled the study of both EAE- and cuprizone-mediated demyelination and demonstrated increased oligodendrocyte differentiation in both models of demyelination, suggesting that CXCR2 may be an inhibitory signaling cue for myelin repair (24). CXCR2 antagonism via neutralizing antibodies enhanced oligodendrocyte differentiation and clinical recovery during EAE, further supporting a detrimental role for CXCR2 signaling within the CNS in mouse models of chronic inflammatory demyelinating disease (58). Conversely, inducible overproduction of CXCL1 by astrocytes reduced EAE clinical disease, although it was unclear whether increased levels of CXCL1 directly affected oligodendrocyte biology or modulated immune cell recruitment into the CNS (65). We have recently shown that induced expression of CXCL1 resulted in increased demyelination mediated by neutrophils in both JHMV and EAE models of neurologic disease (18, 19). In addition, previous work from our laboratories employing antibody-mediated targeting of CXCR2 shows that blocking signaling in JHMV-infected mice increases the severity of demyelination, arguing for a protective role for CXCR2 in a model of virus-induced demyelination, as previously discussed (37). However, whether this is a direct effect of blocking CXCR2 signaling on oligodendroglia and/or other CNS resident cells is unknown. Given the various resident cell types within the CNS that express CXCR2 as well as inflammatory myeloid cells expressing CXCR2, it is difficult to accurately assign a specific role for CXCR2 in protecting spinal cord oligodendroglia from apoptosis using an antibody-mediated targeting approach. It may be possible that anti-CXCR2 treatment of JHMV-infected mice altered the signaling responses on other CXCR2-positive resident cells and/or infiltrating immune cells and that this contributed to the increased clinical disease severity associated with more severe demyelination and oligodendrocyte death.
Our present study used a more incisive genetic model to further our understanding of how CXCR2 signaling on oligodendrocytes affects neurologic disease in a preclinical animal model of MS. Using this genetic approach, our findings indicate that targeted deletion of Cxcr2 occurs in oligodendrocytes both in vitro and in vivo. In addition, our previous work indicates that CXCR2 expression on neutrophils is important in host defense by allowing these cells to participate in increasing the permeability of the blood-brain barrier, allowing access of virus-specific T cells into the CNS to control virus replication. However, tamoxifen treatment did not diminish CXCR2 expression on neutrophils, as determined by flow cytometry. In addition, we have previously reported that antibody targeting of CXCR2 during acute JHMV-induced disease limits neutrophil migration to the CNS, resulting in increased mortality correlating with impaired T cell access to CNS and increased viral titers (54).
Targeting of CXCR2 in Cxcr2-CKO mice was selective to oligodendroglia and did not affect myelin thickness in adult naive animals. These findings allowed us to move forward and assess how silencing of Cxcr2 on oligodendrocytes influences neuroinflammation, demyelination, and remyelination in response to intracerebral inoculation with the neurotropic virus JHMV. Targeting of CXCR2 signaling on oligodendroglia did not dampen the accumulation of either myeloid cells or lymphocytes; the infiltration of virus-specific T cells was not affected, and this correlated with the ability to efficiently control viral replication within the CNS. We also determined that expression of proinflammatory genes within the CNS was not impacted in response to tamoxifen-mediated silencing of Cxcr2 on oligodendrocytes, further supporting the notion that chemokine signaling through CXCR2 oligodendrocytes does not influence neuroinflammation. Sterile immunity is not acquired in JHMV-infected mice, and persistent virus is enriched within glial cells present within white matter tracts, resulting in an immune-mediated demyelinating disease mediated by activated T lymphocytes as well as microglia and macrophages (28). We did not detect differences in the severity of demyelination in tamoxifen-treated JHMV-infected Cxcr2-CKO mice from that in the controls, and these results are consistent with our findings that selective ablation of Cxcr2 in oligodendrocytes does not impact neuroinflammation.
We observed increased numbers of mature GST-π oligodendrocytes along with reduced numbers of NG2-positive cells representing OPCs in the spinal cords of Cxcr2-CKO mice treated with tamoxifen compared to the vehicle-treated controls. Through EM analysis of the spinal cords of the experimental mice, we determined that an increase in the remyelination of axons occurred following targeting of CXCR2 and that this correlated with an increased myelin thickness compared to that in control animals. Collectively, these findings support the concept that targeted ablation of CXCR2 on oligodendroglia in adult mice may enhance the maturation of OPCs into myelin-producing oligodendrocytes (25, 58).
Previously, members of our group (25) generated mice in which Cxcr2 was selectively ablated in oligodendrocytes. Similar to the findings for the animals that we report here, Cxcr2 was silenced within oligodendroglia in adult mice upon tamoxifen treatment (25). Employing toxin models of demyelination, the authors clearly showed enhanced remyelination in animals in which CXCR2 signaling in oligodendrocytes was inhibited, arguing for an important role for this chemokine receptor in influencing oligodendrocyte biology. Our findings would suggest that the influence of CXCR2 signaling on oligodendroglia is not model dependent, as remyelination is observed in both toxin and viral models of demyelination. Our findings are consistent with those of Kerstetter et al. (58), who showed that treatment of mice with MOG35–55-induced EAE with small-molecule antagonists specific for CXCR2 resulted in improved motor skills that correlated with diminished white matter damage associated with enhanced remyelination, arguing that CXCR2 may be a relevant therapeutic target for the treatment of demyelinating diseases. We would not argue with this conclusion but suggest that the small-molecule antagonists employed in this study may be targeting other cell types, e.g., resident glia and/or inflammatory cells, including neutrophils, resulting in improved clinical and histologic outcomes.
MATERIALS AND METHODS
Mice and tamoxifen treatment.
Plp-Cre-ER(T)::Cxcr2floc/floc mice were crossed to mice of the reporter strain B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (stock number 007909; The Jackson Laboratory) to generate Plp-Cre-ER(T)::Cxcr2floc/floc::R26-stop-Td+/− (Cxcr2-CKO) mice. Tamoxifen was prepared by resuspending it at 10 mg/ml in prewarmed sesame seed oil. Four-week-old Cxcr2-CKO mice received either 1 mg/ml tamoxifen or vehicle (control) twice daily for 5 days via intraperitoneal (i.p.) injection, rested for 2 weeks, and subsequently infected intracranially with JHMV (43, 44).
Viral infection.
Age-matched 5- to 6-week-old Cxcr2-CKO mice were infected i.c. with 250 PFU of JHMV in 30 μl of sterile Hanks balanced salt solution (HBSS); sham-infected animals received 30 μl HBSS via i.c. injection (56, 66). For viral titer analysis, one half of each brain or whole spinal cord was homogenized and used in a plaque assay as previously described (19, 67). Clinical disease severity was assessed using a previously described scoring system (19, 67). All animal experiments were approved by the University of Utah Institutional Animal Care and Use Committee per protocol no.16-09008.
Primary oligodendrocyte cultures.
Cortices from postnatal day 1 Cxcr2-CKO mice were dissected and processed according to previously published protocols (68). In brief, following removal of the meninges, cortical tissue was minced with a razor and placed in prewarmed Dulbecco modified Eagle medium (DMEM) containing papain in order to completely dissociate the tissue. Following further aspiration through a Pasteur pipette, single-cell suspensions were added to poly-d-lysine-coated culture flasks and grown for 9 days in DMEM supplemented with 10% fetal bovine serum. Th flasks were then transferred to an orbital shaker in a 5% CO2 tissue culture incubator and shaken for approximately 16 h at 220 rpm in order to remove loosely adherent oligodendroglia. Medium containing OPCs was transferred to 10-cm dishes for 30 min to remove strongly adherent astroglial contaminants. Oligodendroglia were transferred to a 1- ml conical tube and centrifuged at 300 × g for 5 min. The cells were counted and plated onto Matrigel-coated Nunc Lab-Tek II chamber slides (Thermo Fisher Scientific, Waltham, MA) at 50,000 OPCs per chamber in N2 medium supplemented with 3,3′,5-triiodo-l-thyronine sodium salt hydrate (T3; Sigma, St. Louis, MO). After 2 days, fresh medium supplemented with (Z)-4-hydroxytamoxifen (4-OHT; Sigma, St. Louis, MO) at 100 nM was used to induce Cre-mediated recombination. Cells were cultured for an additional 6 days.
Cell isolation and flow cytometry.
Flow cytometry was performed to identify inflammatory cells entering the CNS using established protocols (67, 69). In brief, single-cell suspensions were generated from tissue samples by grinding with frosted microscope slides. Immune cells were enriched via a 2-step Percoll cushion (90% and 63%), and the cells at the interface of the two Percoll layers were collected. Before staining with fluorescent antibodies, isolated cells were incubated with anti-CD16/32 Fc block (BD Biosciences, San Jose, CA) at a 1:200 dilution. Immunophenotyping was performed using commercially available antibodies specific for the following cell surface markers: F4/80 (Serotec, Raleigh, NC), CD4, CD8, Ly6G, and CD11b (BD Biosciences, San Jose, CA), and Ly6C (eBioscience, San Diego, CA). The indicated flow cytometric gating strategies were employed for following inflammatory cells isolated from the CNS: neutrophils (CD45hi CD11b+ Ly6G+), monocytes (CD45hi CD11b+ Ly6C+ Ly6G−), macrophages (CD45hi CD11b+ F4/80+), and microglia (CD45lo CD11b+ F4/80lo). Allophycocyanin (APC)-conjugated rat anti-mouse CD4 and a phycoerythrin (PE)-conjugated tetramer specific for the CD4 immunodominant epitope present within the JHMV matrix (M) glycoprotein spanning amino acids 133 to 147 (the M133–147 tetramer) were used to determine total and virus-specific CD4+ cells, respectively (19, 56); APC-conjugated rat anti-mouse CD8a and a PE-conjugated tetramer specific for the CD8 immunodominant epitope present in the spike (S) glycoprotein spanning amino acids 510 to 518 (S510–518) were used to identify total and virus-specific CD8+ cells, respectively (19, 56). Samples were analyzed using a BD LSR Fortessa X-20 flow cytometer and analyzed with FlowJo software (Tree Star Inc.).
PCR array and semiquantitative RT-qPCR.
Proinflammatory gene expression was determined using a mouse cytokine and chemokine RT2 Profiler PCR array (Qiagen Inc., Valencia, CA), which included the following genes: chemokine genes Ccl1, Ccl11, Ccl12, Ccl17, Ccl19, Ccl2, Ccl20, Ccl22, Ccl24, Ccl3, Ccl4, Ccl5, Ccl7, Cx3cl1, Cxcl1, Cxcl10, Cxcl11, Cxcl12, Cxcl13, Cxcl16, Cxcl3, Cxcl5, Cxcl9, Pf4, Ppbp, and Xcl1; interleukin/cytokine genes Il10, Il11, Il12a, Il12b, Il13, Il15, Il16, Il17a, Il17f, Il18, Il1a, Il1b, Il1rn, Il2, Il21, Il22, Il23a, Il24, Il27, Il3, Il4, Il5, Il6, Il7, Il9, Adipoq (Acrp30), Ctf1, Hc, Mif, Spp1, Tgfb2, Ccl19, Il10, Il11, Il12a, Il12b, Il13, Il18, Il2, Il22, Il23a, Il24, Il4, Il6, and Tgfb2; interferon genes Ifna2 and Ifng; growth factor genes Bmp2, Bmp4, Bmp6, Bmp7, Cntf, Csf1, Csf2, Csf3, Gpi1, Lif, Mstn, Nodal, Osm, Thpo, and Vegfa; and TNF receptor superfamily member genes Cd40lg, Cd70, Fasl, Lta, Ltb, Tnf, Tnfrsf11b, Tnfsf10, Tnfsf11, and Tnfsf13b. For reverse transcription-quantitative PCR (RTqPCR) analysis, total cDNA from the brains of JHMV-infected mice at day 7 p.i. was generated via SuperScript III reverse transcriptase (Life Technologies, Carlsbad, CA) after homogenization in the TRIzol reagent (Life Technologies, Carlsbad, CA).
Histology.
Mice were euthanized at defined times points according to approved IACUC protocol no.16-09008, and the length of the spinal cord extending from thoracic vertebrae 6 to 10 was cryoprotected in 30% sucrose, cut into 1-mm transverse blocks, processed so as to preserve the craniocaudal orientation, and subsequently embedded in OCT (VWR, Radnor, PA, USA). Eight-micrometer-thick coronal sections were cut, sections were stained with hematoxylin/eosin (H&E) in combination with Luxol fast blue (LFB), and between 4 and 8 sections per mouse were analyzed. Areas of total white matter and demyelinated white matter were determined with ImageJ software, and demyelination was scored as the percentage of total demyelination from the spinal cord sections analyzed (55, 67, 70).
Quantitative immunocytochemistry.
For cultured oligodendroglia, cells were fixed for 20 min in 4% paraformaldehyde before being blocked with species-appropriate serum. Cells were then stained with mouse anti-O1 (1:50 dilution; eBioscience, San Diego, CA) overnight at 4°C, followed by a secondary stain with fluorescently labeled secondary antibodies. Control sections were incubated with an appropriate secondary antibody in the absence of primary antibody. Cell morphology and the presence of nuclei were criteria for determining cells dual positive for tdTomato and O1. TdTomato red expression in O1-positive oligodendrocyte cultures was quantified by random selection of 10 imaging sections (×20 magnification) from three independently isolated oligodendrocyte-enriched cultures. The slides were deidentified and read independently by two investigators.
Quantitative immunohistochemistry.
Spinal cords (ranging from thoracic vertebrae 6 to 10) were removed from experimental mice at defined times p.i. and processed as described above. For immunohistochemical analysis, 8-μm sections were desiccated at room temperature for 2 h before beginning the staining process. The slides were then washed in phosphate-buffered saline (PBS) and blocked with species-appropriate serum for 1 h at room temperature. Rabbit anti-GST-π (1:1,000 dilution; MBL Life Science, Woburn, MA), rabbit anti-Olig2 (1:500 dilution; Millipore Corp., Temecula, CA), rabbit anti-NG2 (1:200 dilution; Millipore Corp., Temecula, CA), mouse anti-MAP2 (1:200 dilution; Sigma-Aldrich, St. Louis, MO), and rabbit anti-IBA1 (1:500 dilution; Wako Chemicals, Inc., Richmond, VA) were used to stain the slides overnight at 4°C, the slides were subsequently washed in PBS, and appropriate secondary fluorescently conjugated antibodies were used for detection of targeted antigens. Control sections were incubated with appropriate secondary antibody in the absence of primary antibody.
Quantitative analysis of NG2- and GST-π-positive cells was performed by analyzing spinal cords from Cxcr2-CKO mice treated with tamoxifen (n = 4) from two independent experiments and performed in a blind fashion, and the results were read by two investigators. Coronal spinal cord sections (ranging from 4 to 10 sections per mouse) were used to count cells positive for either NG2 or GST-π in each experimental group. Microscopy was performed on a Nikon A1 confocal laser microscope (University of Utah Cell Imaging Core Facility). To phenotype tdTomato-positive cells, spinal cords from tamoxifen-treated mice (n = 5) were used to detect cells dual positive for tdTomato and cellular markers for neurons (MAP2), macrophage/microglia (IBA1), or oligodendroglia (Olig2 and GST-π). Coronal spinal cord sections (ranging from 2 to 5 sections per mouse) were used to count dual-positive cells.
EM.
For electron microscopy (EM) analysis of spinal cords (ranging from thoracic vertebrae 6 to 10), experimental mice were sacrificed and underwent cardiac perfusion with 0.1 M cacodylate buffer containing 2% paraformaldehyde–2.5% glutaraldehyde. Serial ultrathin sections of spinal cords embedded in Epon epoxy resin were stained with uranyl acetate-lead citrate and analyzed as previously described (36, 55, 56, 71). Images at ×1,200 magnification were analyzed for the g ratio and myelin sheath thickness. In adult animals, there is a relationship between axon circumference and total myelin sheath thickness (number of lamellae), expressed by the g ratio (axon diameter/total fiber diameter); in remyelination, this relationship changes such that myelin sheaths are abnormally thin for the axons that they surround (72). An abnormally thin myelin sheath relative to the axonal diameter was used as the criterion for oligodendrocyte remyelination. The absence of a myelin sheath was used as the criterion for demyelination. Analysis was performed in ventral and lateral white matter columns of spinal cords isolated from experimental mice. For vehicle-treated mice, a total of 297 axons were counted (n = 4 mice) from a total of 25 randomly selected fields, and for tamoxifen-treated mice, a total of 493 axons were counted (n = 5 mice) from a total of 34 randomly selected fields. EM images were analyzed using ImageJ software.
Statistics.
For RT-qPCR quantification, the fold change in expression was determined by normalizing the expression of each sample to that of β-actin and then quantifying the fold change in expression relative to that in naive mice. A log-rank (Mantel-Cox) test was used for survival curve analysis. Comparisons of two groups were analyzed by two-tailed Student’s t tests unless otherwise indicated. Data are reported as the mean ± standard error of the mean (SEM), with a P value of <0.05 being considered significant.
ACKNOWLEDGMENTS
T.E.L. was supported by funding from the National Institutes of Health (NIH; grant R01 NS041249), the National Multiple Sclerosis Society (NMSS) Collaborative Research Center (grant CA-1607-25040), The Ray and Tye Noorda Foundation, and The McCarthey Family Foundation. B.S.M. was supported by NIH training grant 5T3232A1007319. L.L.D. was supported by postdoctoral fellowship FG20105A1 from the NMSS. R.M.R. and L.L. were supported by NIHR grant R01-NS32151.
We gratefully acknowledge Thomas Fielder and the UC Irvine transgenic core facility for assistance in generating the transgenic animals used for these studies. We also thank Sarah Winn for excellent technical assistance.
We declare no competing financial interests.
REFERENCES
- 1.Steinman L. 2014. Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol 32:257–281. doi: 10.1146/annurev-immunol-032713-120227. [DOI] [PubMed] [Google Scholar]
- 2.Holman DW, Klein RS, Ransohoff RM. 2011. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta 1812:220–230. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelhardt B, Ransohoff RM. 2012. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol 33:579–589. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. 2000. Resistance to experimental autoimmune encephalomyelitis in mice lacking the Cc chemokine receptor (Ccr2). J Exp Med 192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. 1993. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J 7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 6.Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. 1995. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol 155:5003–5010. [PubMed] [Google Scholar]
- 7.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. 2000. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson T, Kroenke M, Rao P, Lane TE, Segal B. 2008. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 205:811–823. doi: 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rotzschke O, Falk K. 2005. CCR6 expression defines regulatory effector/memory-like cells within the CD25+CD4+ T-cell subset. Blood 105:2877–2886. doi: 10.1182/blood-2004-07-2505. [DOI] [PubMed] [Google Scholar]
- 10.Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. 2009. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 11.Horuk R. 2009. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 12.Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. 2000. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest 80:617–653. doi: 10.1038/labinvest.3780067. [DOI] [PubMed] [Google Scholar]
- 13.Omari KM, John G, Lango R, Raine CS. 2006. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia 53:24–31. doi: 10.1002/glia.20246. [DOI] [PubMed] [Google Scholar]
- 14.Tirotta E, Ransohoff RM, Lane TE. 2011. CXCR2 signaling protects oligodendrocyte progenitor cells from IFN-gamma/CXCL10-mediated apoptosis. Glia 59:1518–1528. doi: 10.1002/glia.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horuk R, Martin AW, Wang Z, Schweitzer L, Gerassimides A, Guo H, Lu Z, Hesselgesser J, Perez HD, Kim J, Parker J, Hadley TJ, Peiper SC. 1997. Expression of chemokine receptors by subsets of neurons in the central nervous system. J Immunol 158:2882–2890. [PubMed] [Google Scholar]
- 16.Flynn G, Maru S, Loughlin J, Romero IA, Male D. 2003. Regulation of chemokine receptor expression in human microglia and astrocytes. J Neuroimmunol 136:84–93. doi: 10.1016/S0165-5728(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Li M, Spangler LC, Spear C, Veenstra M, Darnall L, Chang C, Cotleur AC, Ransohoff RM. 2013. Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis 51:587–595. doi: 10.1002/dvg.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grist JJ, Marro BS, Skinner DD, Syage AR, Worne C, Doty DJ, Fujinami RS, Lane TE. 2018. Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment. Eur J Immunol 48:1199–1210. doi: 10.1002/eji.201747442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marro BS, Grist JJ, Lane TE. 2016. Inducible expression of CXCL1 within the central nervous system amplifies viral-induced demyelination. J Immunol 196:1855–1864. doi: 10.4049/jimmunol.1501802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson S, Tani M, Strieter RM, Ransohoff RN, Miller RH. 1998. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J Neurosci 18:10457–10463. doi: 10.1523/JNEUROSCI.18-24-10457.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filipovic R, Zecevic N. 2008. The effect of CXCL1 on human fetal oligodendrocyte progenitor cells. Glia 56:1–15. doi: 10.1002/glia.20582. [DOI] [PubMed] [Google Scholar]
- 22.Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH. 2002. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 110:373–383. doi: 10.1016/S0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- 23.Padovani-Claudio DA, Liu LP, Ransohoff RM, Miller RH. 2006. Alterations in the oligodendrocyte lineage, myelin, and white matter in adult mice lacking the chemokine receptor CXCR2. Glia 54:471–483. doi: 10.1002/glia.20383. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Darnall L, Hu T, Choi K, Lane TE, Ransohoff RM. 2010. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J Neurosci 30:9074–9083. doi: 10.1523/JNEUROSCI.1238-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Spangler LC, Prager B, Benson B, Hu B, Shi S, Love A, Zhang C, Yu M, Cotleur AC, Ransohoff RM. 2015. Spatiotemporal ablation of CXCR2 on oligodendrocyte lineage cells: role in myelin repair. Neurol Neuroimmunol Neuroinflamm 2:e174. doi: 10.1212/NXI.0000000000000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergmann CC, Lane TE, Stohlman SA. 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol 4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skinner D, Marro BS, Lane TE. 2018. Chemokine CXCL10 and coronavirus-induced neurologic disease. Viral Immunol 32:25–37. doi: 10.1089/vim.2018.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hosking MP, Lane TE. 2009. The biology of persistent infection: inflammation and demyelination following murine coronavirus infection of the central nervous system. Curr Immunol Rev 5:267–276. doi: 10.2174/157339509789504005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lane TE, Buchmeier MJ. 1997. Murine coronavirus infection: a paradigm for virus-induced demyelinating disease. Trends Microbiol 5:9–14. doi: 10.1016/S0966-842X(97)81768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glass WG, Chen BP, Liu MT, Lane TE. 2002. Mouse hepatitis virus infection of the central nervous system: chemokine-mediated regulation of host defense and disease. Viral Immunol 15:261–272. doi: 10.1089/08828240260066215. [DOI] [PubMed] [Google Scholar]
- 31.Glass WG, Lane TE. 2003. Functional analysis of the CC chemokine receptor 5 (CCR5) on virus-specific CD8+ T cells following coronavirus infection of the central nervous system. Virology 312:407–414. doi: 10.1016/S0042-6822(03)00237-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glass WG, Liu MT, Kuziel WA, Lane TE. 2001. Reduced macrophage infiltration and demyelination in mice lacking the chemokine receptor CCR5 following infection with a neurotropic coronavirus. Virology 288:8–17. doi: 10.1006/viro.2001.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Held KS, Chen BP, Kuziel WA, Rollins BJ, Lane TE. 2004. Differential roles of CCL2 and CCR2 in host defense to coronavirus infection. Virology 329:251–260. doi: 10.1016/j.virol.2004.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosking MP, Lane TE. 2010. The role of chemokines during viral infection of the CNS. PLoS Pathog 6:e1000937. doi: 10.1371/journal.ppat.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE. 2000. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol 165:2327–2330. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- 36.Liu MT, Keirstead HS, Lane TE. 2001. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol 167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- 37.Hosking MP, Tirotta E, Ransohoff RM, Lane TE. 2010. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS One 5:e11340. doi: 10.1371/journal.pone.0011340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. 1996. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A 93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feil R, Wagner J, Metzger D, Chambon P. 1997. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 40.Metzger D, Clifford J, Chiba H, Chambon P. 1995. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci U S A 92:6991–6995. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Riesterer C, Ayrall AM, Sablitzky F, Littlewood TD, Reth M. 1996. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res 24:543–548. doi: 10.1093/nar/24.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mallon BS, Shick HE, Kidd GJ, Macklin WB. 2002. Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci 22:876–885. doi: 10.1523/JNEUROSCI.22-03-00876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feil S, Valtcheva N, Feil R. 2009. Inducible Cre mice. Methods Mol Biol 530:343–363. doi: 10.1007/978-1-59745-471-1_18. [DOI] [PubMed] [Google Scholar]
- 44.Leone DP, Genoud S, Atanasoski S, Grausenburger R, Berger P, Metzger D, Macklin WB, Chambon P, Suter U. 2003. Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells. Mol Cell Neurosci 22:430–440. doi: 10.1016/S1044-7431(03)00029-0. [DOI] [PubMed] [Google Scholar]
- 45.Lin YF, Liu TT, Hu CH, Chen CC, Wang JY. 2018. Expressions of chemokines and their receptors in the brain after heat stroke-induced cortical damage. J Neuroimmunol 318:15–20. doi: 10.1016/j.jneuroim.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 46.Xu J, Zhu MD, Zhang X, Tian H, Zhang JH, Wu XB, Gao YJ. 2014. NFkappaB-mediated CXCL1 production in spinal cord astrocytes contributes to the maintenance of bone cancer pain in mice. J Neuroinflammation 11:38. doi: 10.1186/1742-2094-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryu JK, Cho T, Choi HB, Jantaratnotai N, McLarnon JG. 2015. Pharmacological antagonism of interleukin-8 receptor CXCR2 inhibits inflammatory reactivity and is neuroprotective in an animal model of Alzheimer's disease. J Neuroinflammation 12:144. doi: 10.1186/s12974-015-0339-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, Duncan JA III, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MGH, Barres BS. 2016. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehrlich M, Mozafari S, Glatza M, Starost L, Velychko S, Hallmann AL, Cui QL, Schambach A, Kim KP, Bachelin C, Marteyn A, Hargus G, Johnson RM, Antel J, Sterneckert J, Zaehres H, Scholer HR, Baron-Van Evercooren A, Kuhlmann T. 2017. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc Natl Acad Sci U S A 114:E2243–E2252. doi: 10.1073/pnas.1614412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitew S, Hay CM, Peckham H, Xiao J, Koenning M, Emery B. 2014. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 276:29–47. doi: 10.1016/j.neuroscience.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 52.Doerflinger NH, Macklin WB, Popko B. 2003. Inducible site-specific recombination in myelinating cells. Genesis 35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- 53.Tansey FA, Cammer W. 1991. A pi form of glutathione-S-transferase is a myelin- and oligodendrocyte-associated enzyme in mouse brain. J Neurochem 57:95–102. doi: 10.1111/j.1471-4159.1991.tb02104.x. [DOI] [PubMed] [Google Scholar]
- 54.Hosking MP, Liu L, Ransohoff RM, Lane TE. 2009. A protective role for ELR+ chemokines during acute viral encephalomyelitis. PLoS Pathog 5:e1000648. doi: 10.1371/journal.ppat.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanc CA, Grist JJ, Rosen H, Sears-Kraxberger I, Steward O, Lane TE. 2015. Sphingosine-1-phosphate receptor antagonism enhances proliferation and migration of engrafted neural progenitor cells in a model of viral-induced demyelination. Am J Pathol 185:2819–2832. doi: 10.1016/j.ajpath.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L, Coleman R, Leang R, Tran H, Kopf A, Walsh CM, Sears-Kraxberger I, Steward O, Macklin WB, Loring JF, Lane TE. 2014. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Rep 2:825–837. doi: 10.1016/j.stemcr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mei F, Lehmann-Horn K, Shen Y-A, Rankin KA, Stebbins KJ, Lorrain DS, Pekarek K, Sagan SA, Xiao L, Teuscher C, von Budingen HC, Wess J, Lawrence JJ, Green AJ, Fancy SP, Zamvil SS, Chan JR. 2016. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5:e18246. doi: 10.7554/eLife.18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH. 2009. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol 220:44–56. doi: 10.1016/j.expneurol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Charo IF, Ransohoff RM. 2006. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 60.Wu F, Zhao Y, Jiao T, Shi D, Zhu X, Zhang M, Shi M, Zhou H. 2015. CXCR2 is essential for cerebral endothelial activation and leukocyte recruitment during neuroinflammation. J Neuroinflammation 12:98. doi: 10.1186/s12974-015-0316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang R, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho KL, Morshead C, Chopp M. 2004. Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J Cereb Blood Flow Metab 24:441–448. doi: 10.1097/00004647-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 62.Omari KM, John GR, Sealfon SC, Raine CS. 2005. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain 128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- 63.Tirotta E, Kirby LA, Hatch MN, Lane TE. 2012. IFN-gamma-induced apoptosis of human embryonic stem cell derived oligodendrocyte progenitor cells is restricted by CXCR2 signaling. Stem Cell Res 9:208–217. doi: 10.1016/j.scr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 64.Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM. 2010. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Omari KM, Lutz SE, Santambrogio L, Lira SA, Raine CS. 2009. Neuroprotection and remyelination after autoimmune demyelination in mice that inducibly overexpress CXCL1. Am J Pathol 174:164–176. doi: 10.2353/ajpath.2009.080350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE. 2010. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A 107:11068–11073. doi: 10.1073/pnas.1006375107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blanc CA, Rosen H, Lane TE. 2014. FTY720 (fingolimod) modulates the severity of viral-induced encephalomyelitis and demyelination. J Neuroinflammation 11:138. doi: 10.1186/s12974-014-0138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Meara RW, Ryan SD, Colognato H, Kothary R. 2011. Derivation of enriched oligodendrocyte cultures and oligodendrocyte/neuron myelinating co-cultures from post-natal murine tissues. J Vis Exp 2011:3324. doi: 10.3791/3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lane TE, Liu MT, Chen BP, Asensio VC, Samawi RM, Paoletti AD, Campbell IL, Kunkel SL, Fox HS, Buchmeier MJ. 2000. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol 74:1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dickey LL, Worne CL, Glover JL, Lane TE, O’Connell RM. 2016. MicroRNA-155 enhances T cell trafficking and antiviral effector function in a model of coronavirus-induced neurologic disease. J Neuroinflammation 13:240. doi: 10.1186/s12974-016-0699-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Totoiu MO, Nistor GI, Lane TE, Keirstead HS. 2004. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp Neurol 187:254–265. doi: 10.1016/j.expneurol.2004.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith KJ, Bostock H, Hall SM. 1982. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci 54:13–31. doi: 10.1016/0022-510X(82)90215-5. [DOI] [PubMed] [Google Scholar]