

The shock-and-kill approach consists of the reactivation of HIV-1 replication from latency using latency-reversing agents (LRAs), followed by the elimination of reactivated virus-producing cells. The cellular transcription factor NF-κB is considered a master mediator of HIV-1 escape from latency induced by LRAs. Nevertheless, a systemic activation of NF-κB in HIV-1-infected patients resulting from the combined administration of different LRAs could represent a potential risk, especially in the case of a prolonged treatment. We demonstrate here that conventional treatments with bryostatin-1 and hexamethylenebisacetamide (HMBA) or ionomycin synergistically reactivate HIV-1 from latency, even under conditions where NF-κB activation is repressed. Our study provides a molecular proof of concept for the use of anti-inflammatory drugs, like aspirin, capable of inhibiting NF-κB in patients under combination antiretroviral therapy during the shock-and-kill approach, to avoid potential autoimmune and inflammatory disorders that can be elicited by combinations of LRAs.
KEYWORDS: aspirin, HIV-1, LRA, NF-κB, c-Jun, shock and kill
ABSTRACT
Current combination antiretroviral therapies (cART) are unable to eradicate HIV-1 from infected individuals because of the establishment of proviral latency in long-lived cellular reservoirs. The shock-and-kill approach aims to reactivate viral replication from the latent state (shock) using latency-reversing agents (LRAs), followed by the elimination of reactivated virus-producing cells (kill) by specific therapeutics. The NF-κB RelA/p50 heterodimer has been characterized as an essential component of reactivation of the latent HIV-1 long terminal repeat (LTR). Nevertheless, prolonged NF-κB activation contributes to the development of various autoimmune, inflammatory, and malignant disorders. In the present study, we established a cellular model of HIV-1 latency in J-Lat CD4+ T cells that stably expressed the NF-κB superrepressor IκB-α 2NΔ4 and demonstrate that conventional treatments with bryostatin-1 and hexamethylenebisacetamide (HMBA) or ionomycin synergistically reactivated HIV-1 from latency, even under conditions where NF-κB activation was repressed. Using specific calcineurin phosphatase, p38, and MEK1/MEK2 kinase inhibitors or specific short hairpin RNAs, c-Jun was identified to be an essential factor binding to the LTR enhancer κB sites and mediating the combined synergistic reactivation effect. Furthermore, acetylsalicylic acid (ASA), a potent inhibitor of the NF-κB activator kinase IκB kinase β (IKK-β), did not significantly diminish reactivation in a primary CD4+ T central memory (TCM) cell latency model. The present work demonstrates that the shock phase of the shock-and-kill approach to reverse HIV-1 latency may be achieved in the absence of NF-κB, with the potential to avoid unwanted autoimmune- and or inflammation-related side effects associated with latency-reversing strategies.
IMPORTANCE The shock-and-kill approach consists of the reactivation of HIV-1 replication from latency using latency-reversing agents (LRAs), followed by the elimination of reactivated virus-producing cells. The cellular transcription factor NF-κB is considered a master mediator of HIV-1 escape from latency induced by LRAs. Nevertheless, a systemic activation of NF-κB in HIV-1-infected patients resulting from the combined administration of different LRAs could represent a potential risk, especially in the case of a prolonged treatment. We demonstrate here that conventional treatments with bryostatin-1 and hexamethylenebisacetamide (HMBA) or ionomycin synergistically reactivate HIV-1 from latency, even under conditions where NF-κB activation is repressed. Our study provides a molecular proof of concept for the use of anti-inflammatory drugs, like aspirin, capable of inhibiting NF-κB in patients under combination antiretroviral therapy during the shock-and-kill approach, to avoid potential autoimmune and inflammatory disorders that can be elicited by combinations of LRAs.
INTRODUCTION
Combination antiretroviral therapy (cART) has shown a high efficacy in reducing the HIV-1 viral load to an undetectable level (1, 2) but is unable to eradicate an established infection (3) because of maintenance of latent HIV-1 provirus in long-lived, resting memory CD4+ T cell reservoirs (4–11). The shock-and-kill approach to HIV cure proposes the reactivation of proviral transcription from its dormant state (shock) using potent drug combinations, defined as latency-reversing agents (LRAs). This induction must take place in the presence of cART in order to inhibit the newly produced virus from further infecting cells, followed by the killing of cells in which reactivation has taken place (12).
During HIV-1 replication or escape from latency, initial HIV-1 transcription exclusively depends upon cellular transcription factors, before Tat viral protein accumulates (13). Subsequently, the newly synthesized Tat binds the CycT1 components of the P-TEFb elongation complex, allowing viral transcription to proceed (14). Despite the fact that binding sites for cellular transcription factors are scattered throughout the HIV-1 long terminal repeat (LTR) promoter region and downstream, the cis-acting nuclear factor κB (NF-κB) enhancer appears to be essential for the inducible reactivation from latency in a primary CD4+ T central memory (TCM) cell latency model (15).
NF-κB is one of the main cellular transcription factors involved in HIV-1 production and likewise plays a central role in innate and adaptive immune responses (16, 17) by binding consensus sequences in the enhancer region (18, 19). Five members of the family, C-Rel, RelA (p65), RelB, NF-κB1 (p50 and its precursor, p105), and NF-κB2 (p52 and its precursor, p100), exist as homodimers or heterodimers in quiescent or unstimulated cells, in association with the inhibitory inhibitor of nuclear factor κB (IκB), in particular, IκB-α, which maintains the cytoplasmic localization of NF-κB (20). Moreover, the transcriptionally inactive p50/p50 NF-κB homodimer, which possesses a lower affinity to IκB-α (21), is present in the nucleus of latently infected cells, where it binds the LTR enhancer, further contributing to the repression of viral transcription (22). During T cell receptor (TCR) stimulation, protein kinase C (PKC) θ is activated, and subsequently, IκB-α is phosphorylated at serine 32 and 36 by the IκB kinase α/β/γ (IKK-α/β/γ) complex (23, 24). IκB-α is then ubiquitinated and degraded by the proteasome, releasing RelA/p50, which accumulates in the nucleus, binds DNA, and activates cellular target genes and HIV-1 transcription (18, 20, 25).
It is known that the combination of P-TEFb-releasing agents, such as hexamethylenebisacetamide (HMBA), and PKC agonists, such as bryostatin-1, provide a synergistic latency-reversing activity, resulting in NF-κB and P-TEFb activation more potent than that achieved with the corresponding individual drug treatments (26). Similarly, the triggering of the Ca2+ calcineurin pathway with the Ca2+ ionophore ionomycin results in the activation of the CDK9 kinase of P-TEFb in T cells (27). Moreover, the concomitant PKC stimulation results in a cooperative effect allowing a strong nuclear translocation of RelA and a full transactivation on HIV-1 (28).
Bryostatin-1 is a natural macrolide lactone that acts as a potent activator of PKC and mitogen-activated protein (MAP) kinases (MAPK), particularly p38 (29, 30). Bryostatin-1 stimulates the nuclear translocation of NF-κB (31) and also induces AP1, resulting in the cooperative transcriptional initiation on the LTR enhancer together with NF-κB (32). Bryostatin-1 also has an indirect role in the stimulation of elongation in resting CD4+ T cells by increasing the levels of the P-TEFb elongation factor (31).
HMBA, although originally studied as an antineoplastic agent, is a recognized activator of P-TEFb through the stimulation of the phosphoinositide 3 kinase/AKT (PI3K/AKT) transduction pathway that allows the release of P-TEFb from 7SK small nuclear ribonucleoproteins (snRNPs) (33). HMBA also controls P-TEFb release by activating calcineurin and PP1α phosphatases (34), resulting in HIV-1 transcriptional elongation after 48 to 72 h of treatment in HIV-1 T cell latency models (35). A synergistic effect on HIV-1 reactivation from latency is obtained by the combined use of HMBA and bryostatin-1 both in T cell models of latency and in HIV-1 patient-derived cells (26), making them potential candidates for employment in antilatency therapeutic approaches.
Because aberrant NF-κB activation contributes to the development of various autoimmune, inflammatory, and malignant disorders (36), systemic activation of NF-κB in HIV-1-infected patients resulting from the combined administration of different LRAs could represent a potential risk, especially in the case of prolonged treatment. To explore potential alternate NF-κB-independent reactivation strategies, we engineered the T cell J-Lat 10.6 latency model (37) to express the NF-κB superrepressor IκB-α 2NΔ4 (17, 38) and tested different combinations of latency reversal treatments based on simultaneous Ca2+/calcineurin pathway activation, P-TEFb release, and PKC stimulation. The rationale for the use of these combinations is based on the possibility to determine a synergistic reactivation of HIV-1 replication characterized by both the initiation and elongation of transcription, as already demonstrated in cell lines, primary CD4+ T cell latency models, and in HIV-1-infected patient-derived cells (26, 34, 39, 40).
Apart from NF-κB, different transcription factors, like the nuclear factor of activated T cells (NFAT), interferon regulatory factor 1 (IRF-1), and the activator of protein 1 (AP1), were shown to be involved in HIV-1 LTR transcription and replication (40). Therefore, in order to identify the transcription factor potentially responsible for HIV-1 escape from latency in the engineered cells, where NF-κB is inhibited, we focused on AP1, considering its ability to act independently from NF-κB in stimulating HIV-1 transcription (41). Moreover, in the HIV-1 latency model resembling that in TCM cells, the inhibition of p38, one of the AP1-inducing kinases (42), resulted in a significant inhibition of viral reactivation (15). Differently from AP1, IRF-1 mainly potentiates NF-κB transcriptional activity, rather than acting on its own (43), whereas previous studies appear to rule out the involvement of NFAT in CD4+ T cell models with the TCM phenotype upon combined treatment with activators of the Ca2+ pathway and PKC activators (28).
Due to the fact that AP1 determines activation of HIV-1 LTR transcription from the κB sites in the enhancer region but without direct binding to DNA (41), we also explored the possibility that NF-κB p50 may contribute to AP1 binding, since inhibitory p50/p50 dimers were shown to bind the enhancer κB sites (22) and a p50 physical interaction with c-Jun was also reported (44). The present study demonstrates that NF-κB is dispensable for the synergistic reactivation of HIV-1 from latency with the combination of Ca2+/calcineurin and PKC activators. c-Jun was identified here to be a crucial factor required for the reactivation of HIV that was able to physically interact with p50 and to bind to the κB sites in the HIV-1 LTR enhancer together with p50 in the presence of NF-κB inhibition. Furthermore, acetylsalicylic acid (ASA), a potent inhibitor of NF-κB activator kinase IKK-β and of NF-κB (45, 46), was used to inhibit NF-κB activation in a primary CD4+ T central memory (TCM) cell latency model without affecting reactivation by the combination of bryostatin-1 and HMBA.
RESULTS
Ca2+/calcineurin stimulators and the PKC activator bryostatin-1 synergistically reactivate HIV-1 transcription independently of NF-κB.
To evaluate HIV-1 reactivation in the absence of NF-κB activation, J-Lat cells (37) were engineered to express the NF-κB superrepressor IκB-α 2NΔ4 (17, 38), an expression construct that converts the activating phosphorylation sites at Ser32 and Ser36 to the inert alanine residues (Ala32 and Ala36). As a consequence, IκB-α cannot be phosphorylated or degraded in response to NF-κB-activating stimuli. In addition, a 22-amino-acid deletion (Δ4) in the PEST domain stabilizes the protein by reducing its physiological turnover; because of its shorter size, IκB-α 2NΔ4 expression can be easily distinguished from the expression of the wild-type (wt) IκB-α (17, 38).
Initially, to identify the most highly inducible subclones of J-Lat cells, different subclones of J-Lat, the 6.3, 8.4, 9.2, 10.6, and 15.4 subclones, were subjected to phytohemagglutinin L (PHA-L) treatment for 72 h (Fig. 1A). The 10.6 clone resulted in the highest percentage of reactivating cells, similar to the levels obtained by Bosque and Planelles in a primary TCM model (15), and therefore was chosen to select for superrepressor IκB-α 2NΔ4 expression (Fig. 1B). After PHA-L stimulation, a significant decrease in the percentage of reactivating cells was observed in IκB-α 2NΔ4 cells compared to control (neomycin [Neo]) cells after treatment (Fig. 1C), and as shown in Fig. 1D, wild-type IκB-α degradation was obtained in control cells but not in IκB-α 2NΔ4-expressing cells after PHA-L treatment (2 h).
FIG 1.

Generation and characterization of the J-Lat 10.6 cell line stably expressing the NF-κB superrepressor IκB-α 2NΔ4. (A) Reactivation analysis of different J-Lat cell clones. For each experimental point, a total of 0.2 × 106 cells of 6.3, 8.4, 9.2, 10.6, and 15.4 J-Lat clones were treated for 72 h with 2 μg/ml PHA-L and subjected to fluorescence-activated cell sorter (FACS) analysis. (B) Expression of the NF-κB superrepressor in J-Lat 10.6 cells. For each experimental point, 50 μg of WCEs from a total of 1 × 106 cells was subjected to SDS-PAGE on a 10% polyacrylamide gel and to Western blotting analysis using a specific anti-IκB-α Ab. (C) Expression of the NF-κB superrepressor results in a decrease in the level of reactivation from latency in a significant percentage of PHA-L-treated 2NΔ4 versus Neo cells. For each experimental point, a total of 0.2 × 106 J-Lat 10.6 Neo and 2NΔ4 cells treated as described in the legend to panel A were subjected to FACS analysis. (D) PHA-L treatment results in the degradation of IκB-α in control Neo cells but not in cells expressing IκB-α 2NΔ4. For each experimental point, a total of 1 × 106 cells were treated for 2 h with 2 μg/ml PHA-L, and 50 μg of WCEs was subjected to SDS-PAGE and to Western blotting analysis as described in the legend to B. (A and C) The graphs show the level of reactivation of virus transcription, measured by the percentage of cells expressing the green fluorescent protein (GFP). Data from 4 separate experiments are presented as box plots indicating the median and the interquartile range. Statistical analysis was made using one-way ANOVA (posttest, the Newman-Keuls test) upon transformation of percentage values with the logit function (***, P < 0.001). (B and D) Actin expression represents a control of equal protein loading.
Two combinations of compounds were tested on J-Lat 10.6 Neo and IκB-α 2NΔ4 cells; stimulation of the Ca2+/calcineurin pathway with either ionomycin or HMBA and activation of PKC with bryostatin-1 were achieved (26, 34, 39, 40). The combinations of bryostatin-1 with either ionomycin (Fig. 2A) or HMBA (Fig. 2B) synergistically increased the percentage of cells reactivating HIV-1 provirus transcription in both Neo and 2NΔ4 cells, despite an overall decrease in reactivation in the presence of the NF-κB superrepressor. To rule out the possibility that the synergistic reactivation obtained in the 2NΔ4 cell population was due to a residual activation of NF-κB in cells not expressing the superrepressor, the C11 2NΔ4 cell clone was isolated after limiting dilution. As shown in Fig. 2C, comparable percentages of cells reactivating HIV-1 transcription were obtained for both the 2NΔ4 cell population and the C11 cell clone expressing 2NΔ4 (Fig. 2D).
FIG 2.

Synergistic reactivation of HIV-1 transcription in both J-Lat 10.6 Neo and 2NΔ4 cells by different LRA combinations. (A) Treatment of J-Lat 10.6 Neo and 2NΔ4 cells with the combination of ionomycin plus bryostatin-1 results in a synergistic HIV-1 reactivation from latency. For each experimental point, 0.2 × 106 cells were treated with ionomycin at a concentration of 0.5 μg/ml 72 h before FACS analysis and with bryostatin-1 at a concentration of 20 nM 24 h before FACS analysis. (B) Treatment of J-Lat 10.6 Neo and 2NΔ4 cells with the combination of HMBA plus bryostatin-1 results in a synergistic reactivation from latency. For each experimental point, 0.2 × 106 cells were treated with HMBA at a concentration of 1 mM 72 h before FACS analysis and with bryostatin-1 at a concentration of 20 nM 24 h before FACS analysis. (C) Treatment of a single cell clone with ionomycin plus bryostatin-1 results in a synergistic HIV-1 reactivation from latency comparable to that obtained with the parental 2NΔ4 cell population. For each experimental point, 0.2 × 106 cells were treated with ionomycin at a concentration of 0.5 μg/ml 72 h before FACS analysis and with bryostatin-1 at a concentration of 20 nM 24 h before FACS analysis. (D) Expression of the NF-κB superrepressor in the C11 2NΔ4 cell clone. From a total of 1 × 106 cells for each group (Neo, 2NΔ4, and C11 2NΔ4 cells), 50 μg of WCEs was subjected to SDS-PAGE on a 10% polyacrylamide gel and to Western blotting analysis using a specific anti-IκB-α Ab. Actin expression represents a control of equal protein loading. (A to C) CTR, control; I, ionomycin; H, HMBA; B, bryostatin-1. An equal amount of dimethyl sulfoxide (DMSO) was present at each experimental point in each panel at any time. The graphs show the level of reactivation of virus replication, measured by the percentage of cells expressing GFP. Data from 4 separate experiments are presented as box plots indicating the median and the interquartile range. Statistical analysis was made using the two-tailed paired t test (*, P < 0.05; **, P < 0.01).
c-Jun expression is essential for viral reactivation from latency induced by the simultaneous triggering of the PKC and Ca2+/calcineurin pathways.
Next, the potential involvement of the c-Jun transcription factor in the synergistic reactivation of HIV-1 was determined. As shown in Fig. 3A and C, bryostatin-1 and ionomycin or HMBA led to a strong comparable early increase in c-Jun protein expression in both control and 2NΔ4 cells, whereas c-Jun expression was only mildly detected after treatment with bryostatin-1 alone. The active form of c-Jun, phosphorylated at Ser63, was also preferentially detected with the combined treatment but with a strong decrease in 2NΔ4 cells compared to control cells (Fig. 3A and C, top). Accordingly, bryostatin-1 plus ionomycin or HMBA highly increased late green fluorescent protein (GFP) expression, which was used as a measure of HIV-1 reactivation, while a lower increase was observed with bryostatin-1 alone (Fig. 3B and D). In support of the role of c-Jun in HIV-1 reactivation, increased early c-Jun nuclear accumulation was observed only after the combination treatment (Fig. 3E and F). To confirm the involvement of the PKC and Ca2+/calcineurin pathways in mediating the reactivating effect of LRA combinations, J-Lat 10.6 IκB-α 2ΝΔ4 cells were treated with the p38 inhibitor SB202190, which reduced the percentage of reactivating cells induced by bryostatin-1 plus either ionomycin or HMBA (Fig. 4A and B, respectively), and cyclosporine (CsA), which also inhibited HIV-1 reactivation following the combined LRA treatment (Fig. 4C and D). Each inhibitor treatment resulted in an incomplete inhibition of reactivation mediated by bryostatin-1 alone or by the combined treatments, suggesting that kinases other than p38 induced by PKC activation could have a role. Therefore, to verify this hypothesis, treatment with PD98059, a selective and potent inhibitor of MAP kinase kinases (MAPKK), MEK1, and MEK2, was carried out and a lower, but still significant, inhibition of HIV-1 reactivation was determined following treatment with the combination of bryostatin-1 plus ionomycin (Fig. 4E) or bryostatin-1 plus HMBA (Fig. 4F).
FIG 3.

HIV-1 reactivation from latency induced by bryostatin-1 coupled with either ionomycin or HMBA determines a synergistic increase of active c-Jun transcription factor and its nuclear accumulation even in the absence of NF-κB. (A) Combined treatment of J-Lat 10.6 Neo and 2NΔ4 cells with bryostatin-1 and ionomycin leads to an early synergistic accumulation of active c-Jun transcription factor. For each experimental point, a total of 1 × 106 cells were treated with ionomycin at a concentration of 0.5 μg/ml for 52 h and 48 h before 20 nM bryostatin-1 treatment, which was carried out for the subsequent 4 h. (B) The synergistic activation of c-Jun upon combined treatment of J-Lat 10.6 Neo and 2NΔ4 cells with bryostatin-1 plus ionomycin is lost at a late time point, but synergistic reactivation from latency still occurs. For each experimental point, a total of 1 × 106 cells were treated with ionomycin at a concentration of 0.5 μg/ml for 72 h and 48 h before 20 nM bryostatin-1 treatment, which was carried out for the subsequent 24 h. (C) Combined treatment of J-Lat 10.6 Neo and 2NΔ4 cells with bryostatin-1 plus HMBA leads to an early synergistic accumulation of active c-Jun transcription factor. For each experimental point, a total of 1 × 106 cells were treated with HMBA at a 1 mM concentration for 52 h and 48 h before 20 nM bryostatin-1 treatment, which was carried out for the subsequent 4 h. (D) Upon bryostatin-1 plus HMBA combined treatment of J-Lat 10.6 Neo and 2NΔ4 cells, the synergistic activation of c-Jun is lost at a late time point, but synergistic reactivation from latency still occurs. For each experimental point, a total of 1 × 106 cells were treated with HMBA at a 1 mM concentration for 72 h and 48 h before 20 nM bryostatin-1 treatment, which was carried out for the subsequent 24 h. (E) Synergistic nuclear accumulation of c-Jun is obtained upon combined bryostatin-1 and ionomycin treatment of both Neo and 2NΔ4 cells. For each experimental point, a total of 10 × 106 cells were treated as described in the legend to panel A. (F) Synergistic nuclear accumulation of c-Jun is obtained upon combined bryostatin-1 and HMBA treatment of both Neo and 2NΔ4 cells. For each experimental point, a total of 10 × 106 cells were treated as described in the legend to panel C. (A to D) For each experimental point, 50 μg of WCEs was subjected to SDS-PAGE on a 10% polyacrylamide gel and to Western blotting analysis using specific anti-c-Jun Ser63, c-Jun, GFP, and actin Abs. (E and F) For each experimental point, 25 μg of NEs was subjected to SDS-PAGE and Western blotting analysis as described in the legend to panels A to D. (A to F) GFP expression represents the readout for HIV-1 reactivation in the J-Lat system; actin and USF2 protein expressions represent controls of equal WCE and NE loadings, respectively. C, control; I, ionomycin; H, HMBA; B, bryostatin-1.
FIG 4.

MAPK and Ca2+ pathway-specific inhibitors reduce reactivation of viral transcription, correlating with the reduction in c-Jun accumulation in J-Lat 10.6 2NΔ4 cells. (A) Inhibition of p38 MAP kinase determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and ionomycin. For each experimental point, 0.2 × 106 cells were treated with ionomycin (0.5 μg/ml) and with bryostatin-1 (20 nM) for 72 h and 24 h before FACS analysis, respectively, and with SB202190 (50 μM) for 1 h before bryostatin-1 treatment. (B) Inhibition of p38 MAP kinase determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and HMBA. For each experimental point, 0.2 × 106 cells were treated with HMBA (1 mM) and with bryostatin-1 (20 nM) for 72 h and 24 h before FACS analysis, respectively, and with SB202190 (50 μM) for 1 h before bryostatin-1 treatment. (C) Calcineurin inhibition determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and ionomycin. A total of 0.2 × 106 cells were treated with ionomycin and bryostatin-1 as described in the legend to panel A before FACS analysis and with cyclosporine (CsA) at 0.5 μg/ml, performed 1 h before ionomycin treatment and repeated 1 h before bryostatin-1 treatment. (D) Calcineurin inhibition determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and HMBA. A total of 0.2 × 106 cells were treated with HMBA and bryostatin-1 as described in the legend to panel B before FACS analysis and with cyclosporine at 0.5 μg/ml, performed 1 h before HMBA treatment and repeated 1 h before bryostatin-1 treatment. (E) Inhibition of MEK1/2 MAP kinases determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and ionomycin. For each experimental point, 0.2 × 106 cells were treated with ionomycin (0.5 μg/ml) and with bryostatin-1 (20 nM) for 72 h and 24 h before FACS analysis, respectively, and with PD98059 (25 μM) for 1 h before bryostatin-1 treatment. (F) Inhibition of MEK1/2 MAP kinases determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and HMBA. For each experimental point, 0.2 × 106 cells were treated with HMBA (1 mM) and with bryostatin-1 (20 nM) for 72 h and 24 h before FACS analysis, respectively, and with PD98059 (25 μM) for 1 h before bryostatin-1 treatment. (G) SB202190, CsA, and PD98059 are able to reduce c-Jun protein levels in J-Lat 10.6 2NΔ4 cells stimulated with ionomycin or HMBA plus bryostatin-1. For each experimental point, a total of 1 × 106 cells were treated with a combination of ionomycin (0.5 μg/ml) or HMBA (1 mM) plus bryostatin-1 (20 nM) with HMBA or ionomycin, both of which were administered 48 h before bryostatin-1; CsA (0.5 μg/ml) was given 1 h before ionomycin or HMBA treatment and again 1 h before bryostatin-1 treatment; SB202190 (50 μM) was given 1 h before bryostatin-1 treatment; PD98059 (25 μM) was given 1 h before bryostatin-1 treatment. (A to E) CTR, control; I, ionomycin; H, HMBA; B, bryostatin-1. An equal amount of dimethyl sulfoxide (DMSO) was present at each experimental point at any time (A to F), and different DMSO controls were used to normalize different volume dilutions of each inhibitor compound (G). The graphs show the level of reactivation of virus transcription, measured by the percentage of cells expressing GFP (A to F). Data from 4 separate experiments are presented as box plots indicating the median and the interquartile range. Statistical analysis was made using the two-tailed paired t test (*, P < 0.05; **, P < 0.01). (E) WCEs (50 μg) were collected 4 h after bryostatin-1 treatment, separated by SDS-PAGE on a 10% polyacrylamide gel, and subjected to Western blotting analysis using specific Abs against c-Jun and actin Abs.
The possibility that CsA, SB202190, or PD98059 could have an effect on c-Jun expression induced by LRA combinations in IκB-α 2NΔ4 cells was also verified. As shown in Fig. 4G, a dramatic decrease in c-Jun expression was observed in the cells following treatment with SB202190 compared to that following dimethyl sulfoxide (DMSO) treatment. A less dramatic, but still evident, decrease in c-Jun expression was obtained upon CsA or PD98059 treatment under the same experimental conditions, demonstrating a correlation between the early inhibition of c-Jun and the inhibition of viral reactivation from latency.
To confirm the importance of c-Jun in reactivation, J-Lat 10.6 2NΔ4 cells were engineered to stably express control and c-Jun short hairpin RNAs (shRNAs). The combinations of bryostatin-1 with either ionomycin (Fig. 5A) or HMBA (Fig. 5B) resulted in a significantly lower percentage of 2NΔ4 cells expressing c-Jun shRNA than the percentage of control cells expressing scrambled shRNA that reactivated HIV-1 provirus transcription.
FIG 5.
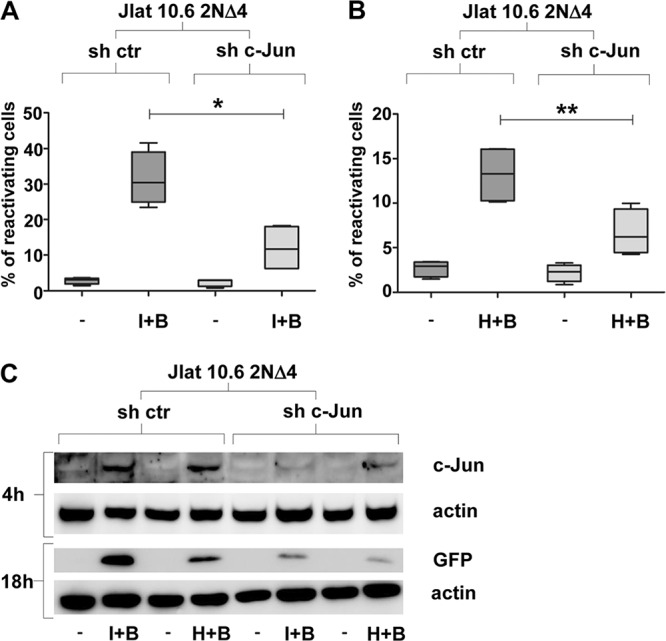
c-Jun is a crucial factor mediating HIV-1 reactivation from latency in J-Lat 10.6 2NΔ4 cells. (A and B) Inhibition of c-Jun expression by stable genetic knockdown determines a significant reduction of the percentage of J-Lat 10.6 2NΔ4 cells reactivating from latency upon combined stimulation with bryostatin-1 and ionomycin. For each experimental point, 0.2 × 106 2NΔ4 cells expressing control shRNA (sh ctr) and c-Jun-specific shRNA (sh c-Jun) were treated with ionomycin (A) at a concentration of 0.5 μg/ml or with HMBA (B) at a concentration of 1 mM for 72 h before FACS analysis and with bryostatin-1 (A and B) at a concentration of 20 nM for 24 h before FACS analysis. The graphs show the level of reactivation of virus transcription, measured by the percentage of cells expressing GFP. Data from 4 separate experiments are presented as box plots indicating the median and the interquartile range. Statistical analysis was made using the two-tailed paired t test (*, P < 0.05; **, P < 0.01). (C) Stable genetic knockdown of c-Jun in J-Lat 10.6 2NΔ4 cells results in the inhibition of HIV-1 reactivation from latency. A total of 1 × 106 cells stably expressing control or c-Jun-specific shRNA were treated with a combination of ionomycin (0.5 μg/ml) or HMBA (1 mM) plus bryostatin-1 (20 nM) with HMBA or ionomycin, both of which were administered 48 h before bryostatin-1 treatment. WCEs (50 μg) were collected 4 h and 18 h after bryostatin-1 treatment, separated by SDS-PAGE on a 10% polyacrylamide gel, and subjected to Western blotting analysis using specific Abs against c-Jun, GFP, and actin. Different DMSO controls were used to normalize different volume dilutions of each inhibitor compounds. (A to C) ctr, control; I, ionomycin; H, HMBA; B, bryostatin-1.
As shown in Fig. 5C (top), combined treatment induced early c-Jun accumulation in control shRNA-expressing cells, while cells expressing c-Jun shRNAs showed a very low level of c-Jun protein, corresponding to the late reduction in GFP expression, which was used as a measurement of viral reactivation (Fig. 5C, bottom).
c-Jun cooperates with NF-κB p50 in HIV-1 LTR activation.
In the absence of an active NF-κB dimer (i.e., RelA/p50) in J-Lat 10.6 2NΔ4 cells, we next examined the hypothesis that c-Jun interaction with the transcriptionally inactive NF-κB p50 subunit contributed to latency reactivation. Both c-Jun and p50 accumulated in the nucleus and specifically bound to the HIV-1 enhancer element following the combined treatments (Fig. 6A and B). As a control, RelA binding was also monitored by use of an oligonucleotide DNA pulldown assay; as expected, the combined treatments did not result in the nuclear accumulation of RelA or binding to the LTR enhancer (Fig. 6A and B).
FIG 6.

HIV-1 enhancer mediates c-Jun-p50 LTR transcriptional activation by combined ionomycin or HMBA and bryostatin-1 treatments in J-Lat 10.6 2NΔ4 cells. (A and B) C-Jun and p50 bind the LTR enhancer DNA sequences upon combined ionomycin (A) or HMBA (B) and bryostatin-1 treatment of J-Lat 10.6 2NΔ4 cells. A total of 25 × 106 J-Lat 10.6 2NΔ4 cells were treated with ionomycin (0.5 μg/ml) or HMBA (1 mM) for 48 h before bryostatin-1 treatment (20 nM). Nuclear extracts (NEs) were obtained 4 h after bryostatin-1 treatment and subjected (400 μg) to a DNA affinity binding assay using a double-stranded DNA sequence corresponding to wild-type (top) or mutant (bottom) κB HIV-1 LTR enhancer. (Right) Proteins capable of binding the enhancer sequence were separated by SDS-PAGE on a 10% polyacrylamide gel and subjected to Western blotting using specific Abs for c-Jun, c-Fos, and RelA. (Middle) Twenty micrograms of NE from the same experimental points was used as an input control in the Western blotting assay with the same Abs used in the DNA affinity binding assay plus anti-USF2. (Left) WCEs from the same experimental points (30 μg) were also used as controls in the Western blotting assay with the same Abs used in the DNA affinity binding assay plus anti-actin. I, ionomycin; H, HMBA; B, bryostatin-1; PD, pulldown. (C) wt IκB-α is not degraded in J-Lat 10.6 2NΔ4 cells treated with the combination of ionomycin or HMBA plus bryostatin-1. For each experimental point, a total of 1 × 106 cells were treated for 48 h with ionomycin at a concentration of 0.5 μg/ml or with HMBA at a concentration of 1 mM and then treated for 4 h (left) or 18 h (right) with bryostatin-1 (20 nM), and 50 μg of WCEs was subjected to SDS-PAGE on a 10% polyacrylamide gel and to Western blotting analysis using anti-IκB-α Abs. (D) C-Jun and p50 cooperate in activating transcription at the HIV-1 LTR κB binding sites in T cells. Jurkat IκB-α 2NΔ4 cells were transiently transfected with c-Jun and p50 expression vectors (400 ng each), luciferase reporter constructs containing wt and mutant HIV-1 LTR κB enhancer sequences (100 ng each), the pNull renilla reporter vector (100 ng), and the empty vector to normalize the total amount of transfected DNA. Data are presented as the fold luciferase induction (FOI), with the histogram plot indicating the average and standard deviation for each experimental condition. Statistical analysis was made using one-way ANOVA (posttest, the Newman-Keuls test) (***, P < 0.001; n.s., not significant). Data represent those from 3 separate experiments. (E) A physical interaction between c-Jun and p50 occurs in J-Lat 10.6 2NΔ4 cells upon combined treatments with HMBA (top) or ionomycin (bottom) plus bryostatin-1. J-Lat 10.6 2NΔ4 cells (10 × 106) were treated with ionomycin (0.5 μg/ml) or HMBA (1 mM) for 48 h before bryostatin-1 treatment (20 nM). WCEs were obtained 4 h after bryostatin-1 treatment and subjected (1 mg) to immunoprecipitation using agarose-conjugated anti-p50 Abs. Immunoprecipitated proteins and 50 μg of WCEs from the same experimental points, used as input controls, were subjected to SDS-PAGE on a 10% polyacrylamide gel and to Western blotting analysis using specific anti c-Jun and p50 Abs.
The inhibition of NF-κB activation was further confirmed in J-Lat 10.6 2NΔ4 cells at early (4 h) and later (18 h) times following bryostatin-1 stimulation in the combined treatments by showing no wt IκB-α degradation (Fig. 6C).
To confirm the ability of both c-Jun and p50 to cooperate in stimulating LTR enhancer transcription in the absence of active NF-κB, a luciferase assay was performed with wt and mutant LTR κB reporters; transient co-overexpression of p50 and c-Jun in Jurkat T cells stably expressing IκB-α 2NΔ4 (47) synergistically activated luciferase from the wild-type but not the mutated LTR enhancer (Fig. 6D).
Moreover, the ability of c-Jun to directly interact with p50 was demonstrated by c-Jun coimmunoprecipitation using anti-p50 antibodies (Abs) upon treatment of 2NΔ4 cells with either HMBA or ionomycin plus bryostatin-1 (Fig. 6E).
Chemical inhibition of NF-κB activation by ASA does not affect reactivation of latent HIV-1 in J-Lat 10.6 cells and primary central memory CD4+ T cells upon bryostatin-1 and HMBA combined treatment.
In order to determine if chemical inhibition of the NF-κB pathway could reproduce the genetic repression of NF-κB obtained with the IκB-α 2NΔ4 construct, we employed acetylsalicylic acid (ASA), known to inhibit tumor necrosis factor alpha (TNF-α)-induced IKK-β and NF-κB activation (45). The level of inhibition of the TNF-α-mediated NF-κB activation obtained with the expression of the NF-κB superrepressor in 2NΔ4 versus Neo control cells (Fig. 7, lanes 1 to 4) was reproduced in 10.6 parental cells upon ASA treatment (Fig. 7, lanes 5 to 7). In contrast, the reactivation effect of bryostatin-1 with HMBA was not significantly affected by ASA (Fig. 7, lanes 8 to 10).
FIG 7.
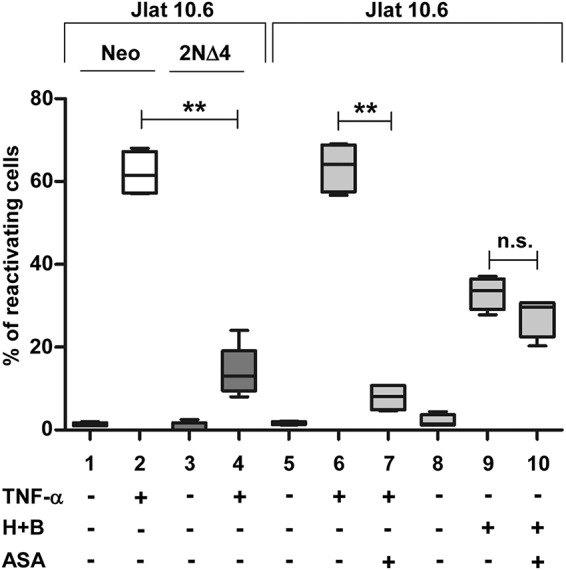
Acetylsalicylic acid (ASA) is unable to inhibit reactivation from latency by HMBA plus bryostatin-1 in J-Lat 10.6 cells, despite its ability to inhibit TNF-α-mediated NF-κB activation. ASA inhibits reactivation from latency in J-Lat 10.6 cells induced by NF-κB-inducing cytokine TNF-α but not HMBA plus bryostatin-1 at a level comparable to the expression of the NF-κB superrepressor IκB-α 2NΔ4. For each experimental point, 0.2 × 106 J-Lat 10.6 parental cells or J-Lat 10.6 Neo and 2NΔ4 cells were treated with TNF-α (10 ng/ml) or with HMBA (1 mM), given 72 h before FACS analysis, plus bryostatin-1 (20 nM), given 24 h before FACS analysis. ASA (2.5 mM) was given 1 h before bryostatin-1. Statistical analysis was made using one-way ANOVA (posttest, the Newman-Keuls test) upon transformation of the percentage values with the logit function (**, P < 0.01; n.s., not significant).
Given that treatment with the combination of bryostatin-1 and HMBA was effective for latency reversal in in vitro and in ex vivo CD4+ T cell models of HIV-1 reactivation (26), we tested the same drug combination in a primary CD4+ TCM HIV-1 latency model (28, 48–50). As shown in Fig. 8B, stimulation of CD4+ TCM cells with bryostatin-1 and HMBA resulted in a 5-fold induction of luciferase activity, comparable to the reactivation obtained with anti-CD3/CD28, whereas treatment with the p38 kinase inhibitor SB202190 resulted in significant inhibition of activity. In contrast, induction of luciferase activity was still obtained with bryostatin-1 and HMBA in the presence of ASA, with only minimal inhibition being seen. The levels of HIV-1 reactivation in the CD4+ TCM model were mirrored by the levels of c-Jun and HIV-1 Gag accumulation (Fig. 8C).
FIG 8.

Acetylsalicylic acid (ASA) is unable to inhibit reactivation from latency by HMBA plus bryostatin-1 in a CD4+ TCM cell HIV-1 latency model. (A) Schematic of HIV-1 reactivation from the CD4+ TCM cell latency model experiment (O/N, overnight). (B) ASA only mildly interferes with HIV-1 reactivation from latency obtained with HMBA plus bryostatin-1 treatment of the CD4+ TCM cell latency model. For each experimental point, 5 × 105 CD4+ TCM cells latently infected with luciferase HIV-1 were treated with HMBA (1 mM) plus bryostatin-1 (20 nM) with or without ASA (2.5 mM), as described in the legend to panel A. ASA and SB202190 (50 μM) were used 2 h before bryostatin-1 treatment, where indicated. As a positive control for reactivation from latency, anti-CD3/CD28 Ab-conjugated Dynabeads were used (25 μl/1 × 106 cells) to stimulate the cells and were given at the same time as bryostatin-1. The luciferase assay was performed upon lysis of cells using 100 μl of Bright-Glo substrate. Statistical analysis was made using one-way ANOVA (posttest, Newman-Keuls test) (*, P < 0.05; **, P < 0.01; n.s., not significant). (C) c-Jun expression correlates with the expression of HIV-1 Gag, used as a measurement of HIV-1 escape from latency obtained with HMBA plus bryostatin-1 treatment of the CD4+ TCM cell latency model. WCEs (20 μg) derived from the same experimental points described in the legend to panel C were collected 4 h after bryostatin-1 treatment, separated by SDS-PAGE on a 10% polyacrylamide gel, and subjected to Western blotting analysis using specific Abs against c-Jun, HIV-1 Gag, and actin.
DISCUSSION
The major obstacle to HIV-1 eradication from infected patients resides in the capacity of the virus to establish latency in T cell reservoirs. Among several innovative approaches proposed to counteract HIV-1 persistence, the shock-and-kill strategy consists of stimulating latently infected cells to reactivate viral replication in the presence of cART, followed by killing via virus-mediated cytotoxicity or ad hoc therapeutics. Among the different CD4+ T cell subsets that can behave as an HIV-1 cellular reservoir (51), there is the long-lived central memory CD4+ T cell (TCM) compartment (4). Latent proviruses present in cells resembling the quiescent CD4+ TCM phenotype reactivate less efficiently when these cells are stimulated with single LRAs used at concentrations achievable in patients than when they are stimulated by T cell receptor engagement (15, 39, 52).
NF-κB activation has been shown to mediate, in part, reactivation from latency following stimulation of a CD4+ T cell HIV-1 latency model, as well as HIV-1-infected patient-derived cells, with different LRA combinations made of PKC activators plus Ca2+ pathway stimulators, P-TEFb activators, or BRD4 inhibitors (26, 28). Nevertheless, the use of specific inhibitors of the NF-κB pathway is unable to block HIV-1 reactivation from quiescence in an HIV-1 latency model resembling TCM cells stimulated by TCR engagement or PHA (15).
The use of LRA combinations that activate NF-κB for a prolonged period in the host may result in generalized immune activation and inflammation. Therefore, we evaluated HIV-1 reactivation from latency in the absence of NF-κB activation, initially using the J-Lat cell clone 10.6, which displayed a highly inducible level of GFP expression after reactivation (Fig. 1A) to a level comparable to that of the primary TCM model described by Bosque and Planelles (15). Two cell lines were generated by retroviral transduction of 10.6 cells, with one stably expressing the NF-κB superrepressor IκB-α 2NΔ4 (17, 38) and the other representing control cells with the vector alone.
Treatment with HMBA and bryostatin-1, combining an activator of the elongation factor P-TEFb and an activator of NF-κB through PKC, respectively, resulted in the synergistic activation of HIV-1, particularly in cells repressed for NF-κB activation. In cells expressing IκB-α 2NΔ4, a stringent maintenance of latency was observed, as shown by the absence of GFP expression in unstimulated cells and an overall decrease in GFP accumulation in stimulated cells (Fig. 3), as well as by the percentage of cells reactivating from latency (Fig. 2). The involvement of calcineurin in the action of the bryostatin-1 plus HMBA combination but not on bryostatin-1 treatment alone was demonstrated in IκB-α 2NΔ4 cells with the specific chemical inhibitor CsA (Fig. 4D). These results demonstrate that HMBA activates the Ca2+/calcineurin pathway, as previously suggested (34). As a positive control for activation of the same pathway, experiments were performed in parallel using the ionophore ionomycin in combination with bryostatin-1, resulting in a similar outcome (Fig. 4C). In order to maximize the induction of the Ca2+ pathway and the synergism, cells were treated with ionomycin or HMBA 48 h before bryostatin-1 treatment for an overall treatment time of 72 h, given that, especially for HMBA, a treatment of at least 48 h is required for the optimal activation of calcineurin and LTR transcription in CD4+ T cells (35). Regarding bryostatin-1, a treatment time of 18 to 24 h was sufficient to observe GFP expression in stimulated cells, given the ability of this compound to rapidly and potently activate PKC (Fig. 2 and 3).
Speculating on which transcription factors were responsible for HIV-1 escape from latency in the engineered cells, nuclear factor of activated T cells (NFAT), interferon regulatory factor 1 (IRF-1), and activator of protein 1 (AP1) have all been described to be capable of cooperating with NF-κB to promote HIV-1 transcription and replication (40). Nevertheless, we did not observe the binding of NFATC1 and NFATC2 to the HIV-1 LTR enhancer by an oligonucleotide DNA pulldown assay, despite their nuclear localization (data not shown); these results are in agreement with those of previous studies that ruled out a role for NFAT in HIV-1 reactivation (28).
On the other hand, AP1 appeared to be particularly relevant, since both NF-κB and AP1 have been shown to act independently and cooperatively in stimulating HIV-1 transcription from the LTR enhancer (41). Moreover, in TCM cells, the inhibition of p38, one of the AP1-inducing kinases (42), resulted in a significant inhibition of viral reactivation (15). Synergistic accumulation of c-Jun at 4 h but not at 18 h (Fig. 3A and C), coupled with the matching 18-h accumulation of GFP, suggests that c-Jun is involved in early events in transcriptional reactivation. Moreover, the levels of the activated form of c-Jun (Fig. 3A and C), phosphorylated at Ser63 (53), correlated with the percentage of cells reactivating from latency shown in Fig. 2, except for the combined treatments, where the increased nuclear accumulation of c-Jun (Fig. 3E and F) accounted more robustly for the higher reactivation observed. A stronger decrease in c-Jun Ser63 phosphorylation is also observed in 2NΔ4 cells than in control cells, linking NF-κB with c-Jun activation signal transduction pathways.
A correlation between inhibition of latent proviral reactivation obtained with the SB202190, CsA, and PD98059 inhibitors and inhibition of c-Jun expression was also observed (compare Fig. 4A to F with Fig. 4G). The crucial role of c-Jun was further demonstrated by knockdown experiments using J-Lat 10.6 IκB-α 2NΔ4 cells expressing either a specific shRNA targeting c-Jun or a scrambled shRNA control; in c-Jun shRNA-expressing cells, a lack of early c-Jun accumulation compared to that in control cells was observed following the combined treatments, correlating with an inhibition of viral reactivation, detected as a strong decrease in late GFP expression (Fig. 5C), and with a significant decrease in the percentage of cells reactivating viral transcription from latency (Fig. 5A and B).
Despite the fact that binding sites for cellular transcription factors are scattered throughout the HIV-1 LTR region and downstream (13), the HIV-1 enhancer was shown to be the only essential LTR region capable of mediating viral reactivation (15). Moreover, data from the literature (54) revealed that AP1 binding sites close to the enhancer able to significantly affect HIV-1 transcription were not present in the subtype B proviruses that were used in this work.
At this point, the LTR enhancer sequence itself was analyzed for the TGACTCA AP1 consensus sequence (55). Since no such sequence was detected, the hypothesis that the binding of c-Jun to the enhancer could be mediated by another protein was tested. The NF-κB family member p50, already known to bind the enhancer as a homodimer in quiescent T cells (22) and also to interact with c-Jun (44), was considered a potential mediator of the c-Jun transcriptional effect. Indeed, following stimulation with ionomycin or HMBA plus bryostatin-1, c-Jun and p50 simultaneously bound the LTR enhancer, but no binding of these factors was detected when κB sites were mutated (Fig. 6A and B). Moreover, in Jurkat parental T cells expressing IκB-α 2NΔ4, the cooperative stimulation of subtype B LTR-mediated luciferase reporter gene transcription was obtained with the transient co-overexpression of c-Jun and p50 but not with the expression of c-Jun alone, in accordance with literature data (54), while the synergism was totally abolished using a κB mutant reporter (Fig. 6D). Coimmunoprecipitation experiments further strengthen the conclusion that, in our system, c-Jun physically interacts with p50 upon combined treatments (Fig. 6E).
In agreement with this hypothesis, it has been demonstrated that IκB-α binds to p50/p50 homodimers with a lower affinity than to p50/RelA heterodimers or RelA/RelA homodimers (21), and therefore, it is feasible that in IκB-α 2NΔ4 cells, p50/p50 binds to the LTR even when NF-κB activation is inhibited.
In order to mimic the inhibition of NF-κB obtained with the IκB-α 2NΔ4 construct compared to control Neo cells, treatment with an average ASA concentration of 2.5 mM, measured in the serum of patients treated with aspirin for chronic inflammatory diseases (56), was performed in J-Lat 10.6 parental cells. In fact, ASA, among its many function, also behaves as a potent inhibitor of TNF-α-induced IKK-β and, therefore, of IκB-α phosphorylation and NF-κB activation when given to cells at a concentration as low as 1 mM and for a time as short as 2 h (45). As shown in Fig. 7, TNF-α-mediated reactivation from latency was inhibited in IκB-α 2NΔ4 cells as well as in 10.6 cells treated with ASA. In contrast, only a mild and not significant inhibition was obtained by ASA treatment of 10.6 cells stimulated with bryostatin-1 plus HMBA.
The ability of the combined bryostatin-1 plus HMBA treatment to determine HIV-1 reactivation from latency without the requirement of NF-κB activation was confirmed in the primary CD4+ TCM cell HIV-1 latency model that we set up here (Fig. 8A). In accordance with the data shown in Fig. 7, ASA treatment of latently infected CD4+ TCM cells determined only a mild reduction of HIV-1 reactivation from latency (Fig. 8B), paralleled by the corresponding levels of the c-Jun and HIV-1 Gag proteins (Fig. 8C). On the other hand, treatment with the p38 kinase inhibitor SB202190 determined an almost complete inhibition of virus reactivation and Gag accumulation and the return of c-Jun protein levels back to control levels (Fig. 8B and C).
Data derived from studies in which LRA activity was assessed in primary model systems were not always reproduced in ex vivo experiments using cells from patients (39, 57). Therefore, our data, showing the ability of the combined treatment with a PKC activator like bryostatin-1 and a calcineurin/P-TEFb inducer like HMBA to determine reactivation of HIV-1 transcription from latency in CD4+ TCM cells in an NF-κB-independent manner, need to be confirmed in an ex vivo model that isolates the same type of cells utilized in our primary in vitro model. The setup of such a model could be challenging, though, due to limitations derived from the small amount of CD4+ TCM cells obtainable from aviremic patients and the small percentage of latently infected cells within this cell population.
In conclusion, this work led to modification of the well-characterized J-Lat cell model (37) to generate a tool that was used to produce a proof of concept for the capacity of PKC activators, together with calcineurin/P-TEFb inducers, to reactivate HIV-1 from its dormant state without the requirement for active NF-κB. Similar systems may permit the identification of active compounds with lower NF-κB-stimulating properties, being less risky for patients. One of the goals of the shock-and-kill strategy is to uncouple HIV-1 latency reversal from subsequent adverse reactions usually elicited by T cell activation by using immunosuppressive agents (58). In this respect, aspirin is also able to attenuate immune activation in HIV-1-infected patients on antiretroviral therapy (59). Since bryostatin-1 plus HMBA activated both NF-κB (26) and c-Jun in CD4+ T cells and c-Jun activation could also be associated with detrimental effects, we propose the use of this drug combination in the shock-and-kill approach together with a drug like ASA that could at least minimize the risk of a generalized immune activation and inflammation without affecting reactivation of HIV-1 replication from latency.
MATERIALS AND METHODS
Cell cultures and treatments.
J-Lat full-length cell clone 10.6, originally developed by Jordan and colleagues (37), was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. Jurkat IκB-α 2NΔ4 cells were described previously (47). Cells were grown in RPMI 1640 (Bio-Whittaker, Cambrex Bio Science, Verviers, Belgium) complemented with 10% inactivated fetal bovine serum (FBS), l-glutamine (1 mg/ml), and antibiotics (100 IU/ml penicillin and 100 μg/ml streptomycin). Phoenix-Ampho packaging cells (ATCC) were grown in Dulbecco modified Eagle medium (Bio-Whittaker, Cambrex Bio Science) supplemented with 10% FBS, l-glutamine (1 mg/ml), and antibiotics as described above.
Ionomycin (Sigma-Aldrich, Merck KGaA, Darmstadt, German) was used at 0.5 μg/ml, bryostatin-1 (Sigma-Aldrich) was used at 20 nM, hexamethylenebisacetamide (HMBA; Sigma-Aldrich) was used at 1 mM, tumor necrosis factor alpha (TNF-α; Pepro Tech EC Ltd., London, United Kingdom) was used at 10 ng/ml, phytohemagglutinin L (PHA-L; Sigma-Aldrich Merck KGaA) was used at 2 μg/ml, anti-CD3/CD28 Ab-conjugated T cell expander Dynabeads (Invitrogen Dynal AS, Oslo, Norway) were used at a 1:1 ratio with CD4+ TCM cells, SB202190 (Sigma-Aldrich, Merck KGaA) was used at 50 μM, cyclosporine (CsA; LKT Laboratories Inc., St. Paul, MN, USA) was used at 0.5 μg/ml, PD98059 (Cayman Chemical, Ann Arbor, MI, USA) was used at 25 μM, and acetylsalicylic acid (ASA; Carlo Erba Reagents srl, Milan, Italy) was used at 2.5 mM.
Plasmids.
The pMSCVneo and pMSCVneo IκB-α 2NΔ4 retroviral vectors were previously generated (38); pSIREN RetroQ sh c-Jun and pSIREN RetroQ sh control were generated by subcloning a synthetic double-stranded DNA containing one c-Jun and the corresponding scrambled shRNA sequences, respectively (60), followed by a polymerase III (Pol III) terminator, the 7SK promoter, a second set of shRNAs targeting c-Jun and scrambled corresponding sequences (61), and another Pol III terminator into the pSIREN RetroQ retroviral vector using the BamHI and EcoRI restriction enzymes; all RNA Pol III promoter and shRNA sequences were obtained by de novo gene synthesis (GenScript USA Inc., Piscataway, NJ). Control CMVBL empty vector and CMVBL p50 were previously described (43), while CMVBL c-Jun was obtained by subcloning into the CMVBL vector the cDNA of human c-Jun obtained by de novo gene synthesis (GenScript USA Inc.), using HindIII and EcoRI.
Luciferase reporter constructs containing wt and mutant HIV-1 LTR κB enhancer sequences were previously described (43); a pNull renilla reporter vector was a gift of Keiko Ozato (NIH, Bethesda, MD, USA).
Retrovirus production.
Packaging cells were transiently transfected at 80% confluence by adding chloroquine (Sigma-Aldrich Merck KGaA) to a final concentration of 25 μM. Plasmids pMSCVneo and pMSCVneo IκB-α 2NΔ4 (for transduction of J-Lat 10.6 cells) or pSIREN-RetroQ puro sh ctr (control) and pSIREN-RetroQ puro sh c-Jun (for transduction of J-Lat 10.6 2NΔ4 cells) were transfected by the calcium phosphate method. Medium was replaced 24 h after transfection and again 8 h later. After an additional 24 h, medium containing retroviruses was collected (1 ml), centrifuged at 500 × g for 10 min, and used to transduce J-Lat 10.6 cells (with PMSCVneo and pMSCVneo IκB-α 2NΔ4) and J-Lat 10.6 IκB-α 2NΔ4 cells (pSIREN-RetroQ puro sh ctr and pSIREN-RetroQ puro sh c-Jun).
Infection of target cells.
J-Lat 10.6 or J-Lat 10.6 IκB-α 2NΔ4 cells were infected twice at 24-h intervals in the presence of 8 mg/ml Polybrene (Sigma-Aldrich Merck KGaA); at 24 h after the last transduction, the cells were seeded in RPMI 1640 supplemented with 10% FBS, glutamine, and antibiotics upon complete removal of the transduction medium following cell centrifugation at 400 × g for 10 min. Selection with 2,000 μg/ml of the antibiotic G418 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) or 2 μg/ml of puromycin (Sigma-Aldrich Merck KGaA) was started 5 days later. After 14 days, a population of resistant J-Lat 10.6 cells was obtained.
Isolation of C11 2NΔ4 cell clone by limiting dilutions.
When growing cells of the 10.6 2NΔ4 cell population reached a density of 0.8 × 106/ml, they were initially diluted 1:20 (final density, 4 × 104 cells) and subsequently diluted 1:10 (final density, 4 × 103 cells). One hundred microliters of the last dilution (400 cells) was seeded in one well of a 96-well plate and further diluted 1:2 12 times in the 96-well plate, reaching a final cell density of 0.2 cell/well. The C11 clone was selected as a single cell growing at the 0.4-cell/well dilution.
FACS analysis.
J-Lat cells were seeded at a density of 0.5 × 106 cells/ml in 48-well plates, the medium was completely changed, and the cells were treated with different latency-reversing agents (LRAs) and specific, inhibitors as described in each figure legend.
The percentage of GFP-positive cells within the viable portion of analyzed cells was calculated by fluorescence-activated cell sorter (FACS) analysis using a BD FACSCalibur flow cytometer. GFP-positive cells were identified as those expressing a fluorescence level over a threshold value determined using parental Jurkat cells not expressing GFP and not stimulated. The software CellQuestPro was used for data analysis.
Transient-transfection and reporter gene assays.
Transfection experiments were performed by using the JetPEI reagent (Polyplus Transfection SA, Illkirch, France) according to the manufacturer's instructions. Briefly, Jurkat IκB-α2NΔ4 T cells were grown to reach a density of about 1 × 106 cells/ml and then seeded at a density of 0.5 × 106 cells/ml in 24-well plates (1.5 ml for each experimental condition). For each well, 1 μg of DNA was diluted into 50 μl of 150 mM NaCl and 4 μl of jetPEI was diluted into 50 μl of 150 mM NaCl. Next, 50 μl of this jetPEI solution were added to the 50-μl DNA solution. The 100-μl jetPEI-DNA mixture was then incubated for 15 min at room temperature and finally added dropwise to each well. Transfected cells were grown in 1.5 ml for 40 to 44 h, lysed, and assayed for luciferase activity (Promega, Madison, WI, USA). All DNA solutions also contained 50 ng of the pNull renilla reporter vector (used as an internal control for transfection efficiency). The amounts of transfected DNA were normalized by using empty vectors. Dual-luciferase activity was evaluated using a Lumat LB9501 luminometer (E&G Berthold, Bad Wildbad, Germany).
Western blotting and coimmunoprecipitation analysis.
J-Lat cells were seeded at a density of 0.5 × 106 cells/ml in 6-well plates, the medium was completely changed, and the cells were treated with different LRAs and specific inhibitors as described in each figure legend. Whole-cell extracts (WCEs) and/or nuclear extracts (NEs) were obtained as previously described (62). The expression of specific proteins in equal amounts of WCEs and/or NEs was analyzed by denaturing SDS-PAGE. After the transfer of separated proteins onto nitrocellulose membranes, blocking of nonspecific binding sites was obtained after incubation for 1 h at room temperature with a solution of 5% no-fat dry milk in 1× phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST). The membranes were then incubated for 18 h at 4°C with the primary antibody of interest diluted in 5% no-fat dry milk in 1× PBST, washed 6 times for 5 min each time in 1× PBST, incubated for 1 h at room temperature with the secondary antibody conjugated with peroxidase (horseradish peroxidase [HRP]; Calbiochem), washed again 6 times for 5 min each time in 1× PBST, and then submitted to enhanced chemiluminescence analysis (Immobilon Western Chemiluminescent HRP substrate; Millipore, Merck KGaA).
Coimmunoprecipitation experiments were carried out using 60 μl of anti-p50 NF-κB agarose-conjugated Ab (catalog number sc-1190; Santa Cruz Biotechnology, Dallas, TX, USA) for each experimental point (30 μl of packed beads). The beads were washed twice with 1× PBS without Ca2+ and Mg2+ upon centrifugation for 30 s at 1,000 × g at 4°C and then incubated with 1 mg of WCE in 500 ml of NP-40 lysis buffer complemented with protease and phosphatase inhibitors (62) for 18 h at 4°C. Subsequently, the beads were washed 4 times with complemented NP-40 lysis buffer, as described above. After a final wash, the supernatant was aspirated and discarded and the beads were resuspended in 20 μl of 4× electrophoresis sample buffer and subjected to Western blotting analysis as described above using anti-c-Jun (H-79; catalog number sc-1694; Santa Cruz Biotechnology) and anti-p50 NF-κB (NLS; catalog number sc-114; Santa Cruz Biotechnology) Abs. The resulting images from the chemiluminescence reactions were obtained by use of a BioSpectrum imaging system (UVP; Ultra-Violet Products Ltd., Upland, CA, USA). The following primary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) were used in the experiments: anti-GFP (B-2; catalog number sc-9996), anti-c-Jun (H-79; catalog number sc-1694), anti-RelA NF-κB (A; catalog number sc-109), anti-p50 NF-κB (NLS; catalog number sc-114), anti-IκB-α (H4; catalog number sc-1643), antiactin (I-19; catalog number sc-1616), and anti-USF2 (N-18; catalog number sc-861). Other primary Abs used were anti-HIV-p24 core protein AB006 (Polymun Scientific, Klosterneuburg, Austria) and anti-phospho-c-Jun Ser63 II (Cell Signaling Technology, Leiden, The Netherlands).
DNA affinity binding assay.
J-Lat cells were seeded at a density of 0.5 × 106 cells/ml in a 175-cm2 flask, the medium was completely changed, and the cells were treated with different LRAs as described in each figure legend. Whole-cell extracts (WCEs) and/or nuclear extracts (NEs) were obtained as previously described (62). Biotinylated oligonucleotides corresponding to the wild-type κB LTR enhancer region (sequence, 5′-ACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAG-3′) and to a version of it mutated in the two κB sites (indicated in boldface in the sequence 5′-ACAAGGGACTATCCGCTGGGGACTATCCAGGGAG-3′) were chemically synthesized (Eurofins MWG/Operon, Ebersberg, Germany) and annealed in 1× STE buffer (sodium chloride-Tris-EDTA) containing 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, and 2 mM EDTA. Thirty picomoles of double-stranded biotinylated DNA was mixed with 400 μg of nuclear protein extracts in 200 μl of binding buffer containing 10 mM Tris-HCl (pH 7.5), 0.5 mM dithiothreitol, 0.5 mM EDTA, 1 mM MgCl2, and 50 mM NaCl in the presence of 4% glycerol and 6 μg of poly(dI · dC). Subsequently, a 25-min incubation was performed at room temperature. The oligonucleotide-protein complex was bound to magnetic beads (Streptavidin MagneSphere; Promega) and mixed by rotatory movement for 30 min at 4°C and subsequently at room temperature for 10 min. The collected beads were washed, and the bound proteins were eluted by boiling in the presence of a loading buffer. The eluted proteins were separated by 10% SDS-PAGE and subjected to Western blotting. The DNA affinity binding assay nitrocellulose filters were incubated with anti-c-Jun (catalog number sc-1694), anti-p50 (catalog number sc-114), and anti-RelA (catalog number sc-109) antibodies from Santa Cruz Biotechnology. The input nitrocellulose filters were also incubated with anti-actin (I-19; catalog number sc-1616) and anti-USF2 (N-18; catalog number sc-861) antibodies (Santa Cruz Biotechnology).
CD4+ TCM cell isolation.
Human peripheral blood mononuclear cells (PBMCs) from freshly collected buffy coats obtained from healthy voluntary blood donors (Blood Bank of University “La Sapienza,” Rome, Italy) were isolated by density gradient centrifugation with the Lympholyte-H reagent (Cedarlane Laboratories, Burlington, Canada). Informed consent was given for the anonymous leftovers of the collected buffy coats for research purposes. Central memory CD4+ T cells were accordingly obtained in two steps with a CD4+ central memory T cell isolation kit (Miltenyi Biotech GmbH, Bergisch Gladbach, Germany). Briefly, in the first step, human memory CD4+ T cells were isolated by depletion of non-CD4+ T cells and naive CD4+ T cells (negative selection); in the second step, CD4+ central memory T cells were isolated by positive selection with CD197 (CCR7)-phycoerythrin (PE) antibody. After isolation, the cells were maintained at 4°C for 18 h in RPMI 1640 medium (Bio-Whittaker, Cambrex Bio Science) containing 10% FBS and antibiotics, to minimize potential T cell activation induced by CCR7-mediated positive selection. Then, the cells were seeded at 2 × 106 cells/ml and cultured in RPMI 1640 medium containing 20% FBS, antibiotics, and recombinant interleukin-7 (IL-7; Pepro Tech EC Ltd., London, United Kingdom), added at 5 ng/ml, for 72 h before infection. The signature of the TCM cells after isolation and, subsequently, after IL-7 stimulation prior to HIV-1 infection was assessed by flow cytometry (FACSCanto II flow cytometer; BD Biosciences, Franklin Lakes, NJ, USA) using the following antibodies: CD4-V450, CD27-allophycocyanin (APC)-H7 (BD Biosciences), CD197 (CCR7)-PE-cyanine 7 (PE-Cy7), CD45RA Brilliant Violet 510 (V500; BioLegend, San Diego, CA, USA), and CD62L-allophycocyanin (APC) (ImmunoTools GmbH, Friesoythe, Germany). Data were analyzed using FlowJo, version X.0.7, software (TreeStar, Inc., Ashland, OR, USA). TCM cells reached the following purity after isolation and after IL-7 stimulation prior to HIV-1 infection: >97% and 99%, respectively, for CD4 cells; >92% and 95%, respectively, for CD27 cells; >88% and >90%, respectively, for CCR7 cells; and <2% and <6%, respectively, for CD45RA cells.
Plasmids, virus production, infection of CD4+ TCM cells, and reactivation experiments.
Our model is based on CD4+ TCM cells, obtained by initial negative selection of CD4+ T memory cells from PBMCs, followed by a subsequent positive selection for CCR7-expressing cells; the resulting CD4+ TCM cells were then subjected to homeostatic proliferation by IL-7, a cytokine known to favor virus integration in vitro and persistent infection in vivo. Latent infection is obtained by spinoculation with a pseudotyped HIV-1 luciferase virus able to complete a single round of infection only (Fig. 8A). Env-defective HIV-1 genome pSVC2.1 vpr+ vpu+ nef+ ΔBglII rev− Luc plasmid (kindly provided by C. Parolin, Padua University, Padua, Italy) was generated from the pSVCBc2.1 plasmid containing the complete proviral genome of HIV-1 HXBc2 (vif positive and vpu, vpr, and nef negative) (J. Sodroski, Harvard University, Boston, MA) by inserting the vpu, vpr, and nef sequences and deleting the BglII-BglII env region. Then, a luciferase reporter gene (Luc), obtained from the pGL3-control vector, was cloned into the pSP73 plasmid using HindIII-XbaI digestion and subsequently digested with PvuII, blunted, and ligated to BamHI linkers. The fragment bearing the luciferase gene generated by BamHI digestion of the pSP73-Luc+ plasmid was inserted in the pSVC2.1 vpr+ vpu+ nef+ ΔBglII vector, with the resulting inactivation of the rev gene. The pREV and pSVIII expression vectors were kindly provided by C. Parolin (University of Padua, Padua, Italy) and J. Sodroski (Harvard University, Boston, MA), respectively.
To produce recombinant HIV-1 virions, human embryonal kidney (HEK) 293 cells were cotransfected with 12 μg of pSVC 2.1 vpr+ vpu+ nef+ ΔBglII rev− Luc, 5 μg of pREV, and 6 μg of pSVIII envelope glycoprotein with a ProFection mammalian transfection system-calcium phosphate (Promega). Cell supernatants carrying progeny pseudotyped virions were harvested at 48 h after transfection, clarified, filtrated, aliquoted, and stored at −80°C. Virus production in the cell-free supernatants was normalized for reverse transcriptase (RT) activity as previously described (63) and used to infect TZM-bl cells for titration. The 50% tissue culture infectious dose (TCID50), calculated by the Reed-Muench method, of each virus batch was determined as follows: quadruplicate serial 5-fold dilutions of pseudovirus were made in 96-well black culture plates in a total volume of 100 μl of growth medium for a total of 11 dilution steps. A total of 1 × 104 cells in 100 μl of growth medium containing 40 μg/ml DEAE-dextran were added to each well, and the plates were incubated at 37°C. After 48 h of incubation, 100 μl of culture medium was removed from each well, 100 μl of Bright-Glo reagent (Promega) was added to the cells, and then luminescence was measured by considering relative luminescence units (RLU) of >2.5 times the background to be a positive result.
For infection of CD4+ TCM cells, 1 × 104 TCID50 units of virus were used to infect 5 × 105 cells. Spinoculations were performed in 15-ml Falcon conical tubes in volumes of 200 μl or less. Cells and virus were centrifuged at 1,200 × g for 2 h at 20°C, washed twice to remove the unbound virus, and resuspended in fresh RPMI 1640 medium supplemented with 20% FBS, antibiotics, and IL-7 for 72 h before the reactivation experiments. Cells were plated in 48-well plates at concentrations of 5 × 105 cells/ml in the presence of raltegravir (Asta Tek Inc. Bristol, PA, USA) at 30 μM for 1 h. Then, the cells were treated with HMBA and bryostatin-1 and with inhibitors (ASA or SB202190). The cells were harvested, washed one time with PBS, and lysed in 100 μl of Bright-Glo reagent. Luminescence was measured using a Victor luminometer (PerkinElmer, Waltham, MA, USA). Wells producing relative luminescence units (RLU) of >2.5 times the background were scored as positive.
Statistical analysis.
Statistical analysis of the experiments conducted by FACS was made in part through the two-tailed paired t test and in part by one-way analysis of variance (ANOVA), followed by appropriate post hoc tests. An appropriate transformation of the percentage values through a logit function was performed where necessary. The data were graphically represented by the use of box plots indicating the median and the interquartile range. Statistical analysis of the results of the luciferase experiments was performed using ANOVA, followed by an appropriate post hoc test. A P value of <0.05 was considered statistically significant. The software used was GraphPad Prism for Windows (version 5.0).
ACKNOWLEDGMENTS
This work was supported in part by the AIDS program 2009-2012 of the Italian Ministry of Health to M. Sgarbanti. A special thanks goes to Angela Battistini for partially financing this work by ISS intramural research grant 524/2013-2015.
Many thanks go to Andrea Savarino for scientific support and personal encouragement throughout many years. Thanks go to Flavia Mancini for technical support.
C. Acchioni performed the majority of the experiments, participated in the experimental design, analyzed the data, and contributed to writing the manuscript; A. L. Remoli, G. Marsili, M. Acchioni, and I. Nardolillo contributed to performing the oligonucleotide pulldown assays, Western blotting, and luciferase assays and analyzing the data; G. Marsili also generated Jurkat cells expressing the IκB-α 2NΔ4 NF-κB superrepressor; A. L. Remoli contributed to writing the manuscript; R. Orsatti prepared the plasmids used throughout the work; S. Farcomeni and E. Palermo performed FACS analysis to provide purity data about the isolated CD4+ TCM cells; E. Perrotti, M. L. Barreca, S. Sabatini, and S. Sandini contributed with reagents and critical readings of the manuscript and provided substantial analysis and interpretation of the experiments; C. Parolin provided the HIV-1 pseudotyped luciferase virus system used in the CD4+ TCM cellular model. R. Lin originally designed the IκB-α 2NΔ4 NF-κB superrepressor and critically read the manuscript; A. Borsetti produced the HIV-1 pseudotyped luciferase virus, performed the infection of CD4+ TCM cells by spinoculation, and provided a substantial analysis and interpretation of the experiments; J. Hiscott originally provided the IκB-α 2NΔ4 NF-κB construct and performed a major critical reading and revision of the manuscript; M. Sgarbanti designed and supervised the work, engineered J-Lat cells to express the IκB-α 2NΔ4 NF-κB superrepressor and c-Jun shRNAs, designed the CD4+ TCM cellular model, contributed to the experimental design, analyzed the data, and wrote the manuscript.
We declare no conflict of interest.
REFERENCES
- 1.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med 337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 2.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ Jr, Feinberg JE, Balfour HH Jr, Deyton LR, Chodakewitz JA, Fischl MA. 1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med 337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 4.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel M-R, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy J-P, Haddad EK, Sékaly R-P. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 6.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 8.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 9.Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, Kuruppu J, Yazdani J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA. 2004. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J Virol 78:1160–1168. doi: 10.1128/jvi.78.3.1160-1168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, Rabi SA, Laird GM, Kim M, Hosmane NN, Yang HC, Zhang H, Margolick JB, Li L, Cai W, Ke R, Flavell RA, Siliciano JD, Siliciano RF. 2017. Transcriptional reprogramming during effector-to-memory transition renders CD4(+) T cells permissive for latent HIV-1 infection. Immunity 47:766–775.e3. doi: 10.1016/j.immuni.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 12.Hamer DH. 2004. Can HIV be cured? Mechanisms of HIV persistence and strategies to combat it. Curr HIV Res 2:99–111. doi: 10.2174/1570162043484915. [DOI] [PubMed] [Google Scholar]
- 13.Colin L, Van Lint C. 2009. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA. 1990. HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J 9:4145–4153. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosque A, Planelles V. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. 1993. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J 12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwon H, Pelletier N, DeLuca C, Genin P, Cisternas S, Lin R, Wainberg MA, Hiscott J. 1998. Inducible expression of IkappaBalpha repressor mutants interferes with NF-kappaB activity and HIV-1 replication in Jurkat T cells. J Biol Chem 273:7431–7440. doi: 10.1074/jbc.273.13.7431. [DOI] [PubMed] [Google Scholar]
- 18.Alcami J, Lain de Lera T, Folgueira L, Pedraza MA, Jacque JM, Bachelerie F, Noriega AR, Hay RT, Harrich D, Gaynor RB. 1995. Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J 14:1552–1560. doi: 10.1002/j.1460-2075.1995.tb07141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen BK, Feinberg MB, Baltimore D. 1997. The kappaB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J Virol 71:5495–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 21.Phelps CB, Sengchanthalangsy LL, Huxford T, Ghosh G. 2000. Mechanism of I kappa B alpha binding to NF-kappa B dimers. J Biol Chem 275:29840–29846. doi: 10.1074/jbc.M004899200. [DOI] [PubMed] [Google Scholar]
- 22.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isakov N, Altman A. 2012. PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front Immunol 3:273. doi: 10.3389/fimmu.2012.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng J, Montecalvo A, Kane LP. 2011. Regulation of NF-kappaB induction by TCR/CD28. Immunol Res 50:113–117. doi: 10.1007/s12026-011-8216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabel G, Baltimore D. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 26.Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, Van Driessche B, Gatot JS, Cherrier T, Pianowski LF, Gama L, Schwartz C, Vila J, Burny A, Clumeck N, Moutschen M, De Wit S, Peterlin BM, Rouzioux C, Rohr O, Van Lint C. 2015. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog 11:e1005063. doi: 10.1371/journal.ppat.1005063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramakrishnan R, Dow EC, Rice AP. 2009. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J Leukoc Biol 86:1345–1350. doi: 10.1189/jlb.0509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan JK, Bhattacharyya D, Lassen KG, Ruelas D, Greene WC. 2013. Calcium/calcineurin synergizes with prostratin to promote NF-kappaB dependent activation of latent HIV. PLoS One 8:e77749. doi: 10.1371/journal.pone.0077749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson TJ, Alkon DL. 2009. Neuroprotective versus tumorigenic protein kinase C activators. Trends Biochem Sci 34:136–145. doi: 10.1016/j.tibs.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Curiel RE, Garcia CS, Farooq L, Aguero MF, Espinoza-Delgado I. 2001. Bryostatin-1 and IL-2 synergize to induce IFN-gamma expression in human peripheral blood T cells: implications for cancer immunotherapy. J Immunol 167:4828–4837. doi: 10.4049/jimmunol.167.9.4828. [DOI] [PubMed] [Google Scholar]
- 31.Pérez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Muñoz-Fernandez MA, Moreno S, Castor TP, Calzado MA, Muñoz E. 2010. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr HIV Res 8:418–429. doi: 10.2174/157016210793499312. [DOI] [PubMed] [Google Scholar]
- 32.De Lorenzo MS, Yamaguchi K, Subbaramaiah K, Dannenberg AJ. 2003. Bryostatin-1 stimulates the transcription of cyclooxygenase-2: evidence for an activator protein-1-dependent mechanism. Clin Cancer Res 9:5036–5043. [PubMed] [Google Scholar]
- 33.Contreras X, Barboric M, Lenasi T, Peterlin BM. 2007. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 3:1459–1469. doi: 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, Zhou Q. 2008. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev 22:1356–1368. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klichko V, Archin N, Kaur R, Lehrman G, Margolis D. 2006. Hexamethylbisacetamide remodels the human immunodeficiency virus type 1 (HIV-1) promoter and induces Tat-independent HIV-1 expression but blunts cell activation. J Virol 80:4570–4579. doi: 10.1128/JVI.80.9.4570-4579.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park MH, Hong JT. 2016. Roles of NF-kappaB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5:E15. doi: 10.3390/cells5020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sgarbanti M, Arguello M, tenOever BR, Battistini A, Lin R, Hiscott J. 2004. A requirement for NF-kappaB induction in the production of replication-competent HHV-8 virions. Oncogene 23:5770–5780. doi: 10.1038/sj.onc.1207707. [DOI] [PubMed] [Google Scholar]
- 39.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. 2013. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog 9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Remoli AL, Marsili G, Battistini A, Sgarbanti M. 2012. The development of immune-modulating compounds to disrupt HIV latency. Cytokine Growth Factor Rev 23:159–172. doi: 10.1016/j.cytogfr.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Yang X, Chen Y, Gabuzda D. 1999. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-kappaB. J Biol Chem 274:27981–27988. doi: 10.1074/jbc.274.39.27981. [DOI] [PubMed] [Google Scholar]
- 42.Muthumani K, Choo AY, Hwang DS, Premkumar A, Dayes NS, Harris C, Green DR, Wadsworth SA, Siekierka JJ, Weiner DB. 2005. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood 106:2059–2068. doi: 10.1182/blood-2005-03-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sgarbanti M, Remoli AL, Marsili G, Ridolfi B, Borsetti A, Perrotti E, Orsatti R, Ilari R, Sernicola L, Stellacci E, Ensoli B, Battistini A. 2008. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J Virol 82:3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Qin J, Lan L, Li N, Wang C, He P, Liu F, Ni H, Wang Y. 2014. M-CSF cooperating with NFkappaB induces macrophage transformation from M1 to M2 by upregulating c-Jun. Cancer Biol Ther 15:99–107. doi: 10.4161/cbt.26718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin MJ, Yamamoto Y, Gaynor RB. 1998. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 46.Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O, Ludwig S. 2007. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol 9:1683–1694. doi: 10.1111/j.1462-5822.2007.00902.x. [DOI] [PubMed] [Google Scholar]
- 47.Sgarbanti M, Marsili G, Remoli AL, Ridolfi B, Stellacci E, Borsetti A, Ensoli B, Battistini A. 2004. Analysis of the signal transduction pathway leading to human immunodeficiency virus-1-induced interferon regulatory factor-1 upregulation. Ann N Y Acad Sci 1030:187–195. doi: 10.1196/annals.1329.024. [DOI] [PubMed] [Google Scholar]
- 48.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. 2012. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 7:e30176. doi: 10.1371/journal.pone.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coiras M, Bermejo M, Descours B, Mateos E, García-Pérez J, López-Huertas M-R, Lederman MM, Benkirane M, Alcamí J. 2016. IL-7 induces SAMHD1 phosphorylation in CD4+ T lymphocytes, improving early steps of HIV-1 life cycle. Cell Rep 14:2100–2107. doi: 10.1016/j.celrep.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vandergeeten C, Fromentin R, DaFonseca S, Lawani MB, Sereti I, Lederman MM, Ramgopal M, Routy JP, Sekaly RP, Chomont N. 2013. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 121:4321–4329. doi: 10.1182/blood-2012-11-465625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sengupta S, Siliciano RF. 2018. Targeting the latent reservoir for HIV-1. Immunity 48:872–895. doi: 10.1016/j.immuni.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novis CL, Archin NM, Buzon MJ, Verdin E, Round JL, Lichterfeld M, Margolis DM, Planelles V, Bosque A. 2013. Reactivation of latent HIV-1 in central memory CD4(+) T cells through TLR-1/2 stimulation. Retrovirology 10:119. doi: 10.1186/1742-4690-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, Henderson MA, Luen SJ, Mansour M, Kershaw MH, Trapani JA, Neeson PJ, Salgado R, McArthur GA, Balko JM, Beavis PA, Darcy PK, Loi S. 2017. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat Commun 8:606. doi: 10.1038/s41467-017-00728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Sluis RM, Derking R, Breidel S, Speijer D, Berkhout B, Jeeninga RE. 2014. Interplay between viral Tat protein and c-Jun transcription factor in controlling LTR promoter activity in different human immunodeficiency virus type I subtypes. J Gen Virol 95:968–979. doi: 10.1099/vir.0.059642-0. [DOI] [PubMed] [Google Scholar]
- 55.Vogt PK. 2001. Jun, the oncoprotein. Oncogene 20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- 56.Hardman JG, Limbird LE, Molinoff PB, Ruddon RW (ed). 1996. Goodman and Gilman’s the pharmacological basis of therapeutics, 9th ed McGraw-Hill, New York, NY. [Google Scholar]
- 57.Knights H. 2017. A critical review of the evidence concerning the HIV latency reversing effect of disulfiram, the possible explanations for its inability to reduce the size of the latent reservoir in vivo, and the caveats associated with its use in practice. AIDS Res Treat 2017:8239428. doi: 10.1155/2017/8239428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin AR, Pollack RA, Capoferri A, Ambinder RF, Durand CM, Siliciano RF. 2017. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J Clin Invest 127:651–656. doi: 10.1172/JCI89552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Brien M, Montenont E, Hu L, Nardi MA, Valdes V, Merolla M, Gettenberg G, Cavanagh K, Aberg JA, Bhardwaj N, Berger JS. 2013. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: a pilot study. J Acquir Immune Defic Syndr 63:280–288. doi: 10.1097/QAI.0b013e31828a292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT, Reddy SA, Sabapathy K. 2007. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ 14:218–229. doi: 10.1038/sj.cdd.4401946. [DOI] [PubMed] [Google Scholar]
- 61.Peng TL, Chen J, Mao W, Song X, Chen MH. 2009. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol 10:27. doi: 10.1186/1471-2121-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sgarbanti M, Marsili G, Remoli AL, Stellacci E, Mai A, Rotili D, Perrotti E, Acchioni C, Orsatti R, Iraci N, Ferrari M, Borsetti A, Hiscott J, Battistini A. 2014. IkappaB kinase epsilon targets interferon regulatory factor 1 in activated T lymphocytes. Mol Cell Biol 34:1054–1065. doi: 10.1128/MCB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ridolfi B, Titti F, Fulgenzi D, Maggiorella MT, Tinari A, Superti F, Parolin C, Ensoli B, Borsetti A. 2004. Infection of a simian B cell line by human and simian immunodeficiency viruses. AIDS Res Hum Retroviruses 20:723–732. doi: 10.1089/0889222041524652. [DOI] [PubMed] [Google Scholar]