

Abstract
Monoterpenes represent a class of hydrocarbons consisting of two isoprene units. Like many other terpenes, monoterpenes emerge mainly from vegetation, indicating their significance in both atmospheric chemistry and pharmaceutical and food industries. The atmospheric recycling of monoterpenes constitutes a major source of secondary organic aerosols. Therefore, this contribution focuses on the mechanism and kinetics of atmospheric oxidation of five dominant monoterpenes (i.e., limonene, α-pinene, β-pinene, sabinene, and camphene) by singlet oxygen. The reactions are initiated via the ene-type addition of singlet oxygen (O21Δg) to the electron-rich double bond, progressing favorably through the concerted reaction mechanisms. The physical analyses of the frontier molecular orbitals agree well with the thermodynamic properties of the selected reagents, and the computed reaction rate parameters. The reactivity of monoterpenes with O21Δg follows the order of α-pinene > sabinene > limonene > β-pinene > camphene, i.e., α-pinene and camphene retain the highest and lowest reactivity toward singlet oxygen, with rate expressions of k(T) (M–1 s–1) = 1.13 × 108 exp(−48(kJ)/RT(K)) and 6.93 × 108 exp(−139(kJ)/RT(K)), respectively. The effect of solvent on the primary reaction pathways triggers a slight reduction in energy, ranging between 12 and 34 kJ/mol.
1. Introduction
Plants emit nearly 98% of the total non-methane volatile organic compounds into the atmosphere, 20% of which comprises monoterpenes.1 Monoterpenes represent a class of C10 members of terpene hydrocarbons, the biogenic (naturally occurring) volatile organic compounds (BVOC) with high chemical reactivity and annual global emission rate of between 128 and 450 Tg per year.2Figure 1 summarizes the photosensitised oxidation products of the selected monoterpenes detected and characterized in literature.3−7 The chemical structure of monoterpenes features two isoprene units, constituting the major component of essential oils in various plant matters and can be isolated from trees for anti-inflammatory and antimicrobial drug synthesis.8 An alternative source of monoterpene emission includes woody household products.9
Figure 1.

Oxidation products of limonene, α-pinene, and β-pinene by O21Δg.3−7
Limonene and pinene are among the most abundant monoterpenes operating in the global tropospheric chemistry,10,11 being produced in relatively noticeable quantities by vegetation such as aromatic plants, flowers, and leaves. Limonene arises mainly from young plant leaves, and its formation rate reduces rapidly based on the age and extent of oxidation of the leaves.12 Moreover, this monocyclic monoterpene is significantly utilized in medicinal chemistry and disease treatment, due to its antitumor and antibacterial activity, and dietary formulations.13,14 Over 80% of the total monoterpene emission from a Monterey pine (Pinus radiata) comprises α- and β-pinene,15 and it has also been evidenced that about 50% of the emitted monoterpene from forests and tree species in the United States consists of α-pinene.16 Other examples of bicyclic monoterpenes are camphene and sabinene, which are minor constituents of many essential oils from plants such as turpentine, rosemary, ginger, and valerian.
Tropospheric hydrocarbons such as monoterpenes have relatively high molecular weights, thus their atmospheric oxidation gives rise to semivolatile organic compounds and secondary organic aerosol (SOA). SOAs originate primarily from the oxidation of BVOCs including monoterpenes. Subsequently, SOA plays a significant part in climate change and global radiation imbalance due to their involvement in the absorption and scattering of solar radiation. For instance, SOA are significant constituents of atmospheric fine particulate matters (PM2.5) as well as various haze pollution episodes.17 Monoterpenes find further applications in aromatization of cleaning products, paintings, air fresheners, and flavoring agents due to their pleasant fragrance. Therefore, the risk of accumulation of atmospheric oxidation products (i.e., SOA) could be substantial in enclosed, poorly ventilated spaces.
The maximum rate of singlet O2 formation by energy-transfer mechanism in a polluted atmosphere is approximated to be 4 × 10–12 mol L–1 s–1, which corroborates the significant role of such reactive oxygen species as atmospheric oxidants.18 Reactions of singlet oxygen with electron-rich acceptors such as olefins, dienes, and aromatic compounds are grouped into [4 + 2]-cycloadditions, [2 + 2]-cycloadditions, and the so-called ene reactions. Ene reaction is based on the interaction of 1O2 with an unsaturated compound containing an allylic hydrogen, during which the allylic hydrogen is abstracted in association with a reorganization of the bonding to give allylhydroperoxides.19 Oxidation of substrates with conjugated double bonds by singlet oxygen is feasible through [4 + 2]-cycloaddition, resulting in the synthesis of endoperoxides. Furthermore, the [2 + 2]-cycloaddition of the singlet oxygen to one double bond results in 1,2-dioxetane, and apparently olefins with unreachable allylic hydrogen atoms tend to give [2 + 2]-adducts.20 However, it should be noted that all of the reactions can compete for the same substance if the molecular structure allows it.21
Previous research efforts had investigated the oxidation of monoterpenes to SOA by OH radical,22,23 ozone,24−26 hydrogen peroxide,27−29 and nitrogen oxides,30−32 reporting the yield of the corresponding carbonyl compounds as the predominant products as a result of the oxidative cleavage of the C=C bonds.33 However, the role of a highly reactive singlet molecular oxygen in the photo-oxidation of monoterpenes has not been properly addressed in the literature. Some experimental studies elucidating the product distribution of dye-sensitized oxidation of terpenoid biogenic hydrocarbons (i.e., limonene, α-, and β-pinene) in different media demonstrated the formation of organic aerosols.3,4,34,35 The aim of this contribution is to report modes of reactions between singlet oxygen and monoterpenes with a prime focus on deriving kinetic parameters.
2. Results and Discussion
2.1. Mechanism and Kinetics of Singlet Oxidation of Monoterpenes
Due to the extreme electrophilic nature of singlet oxygen, the introduction of oxygen atoms into monoterpenes during photo-oxidation will occur at the molecular site where the Fukui function for an electrophilic attack (f–1) displays its maximum value. This parameter indicates the most reactive site of chemical systems for electrophilic substitution reactions. According to Figure 2, the f–1 indices are the largest at >C=C< sites associated with the higher photo-oxidation reactivity of nucleophile substrates therein.
Figure 2.

Local electrophilic Fukui indices (f–1) of reactive monoterpenes.
The interaction between the nucleophile’s HOMO and the electrophile’s LUMO plays a vital role in elucidating the reaction dynamics, as though the larger LUMO–HOMO energy gaps lead to the decelerated chemical reactivity. EHOMO for selected monoterpenes and ELUMO for singlet oxygen are shown in Figure 3. Apparently, α-pinene appears to be most reactive toward singlet oxygen, whereas camphene exhibits the least reactivity among all. Based on the electron cloud distribution in Figure 3, the valence orbital electrons highly engulf the π-bond, justifying the Fukui indices results (Figure 2) to designate >C=C< as the most favorable molecular site for electrophilic attacks.
Figure 3.
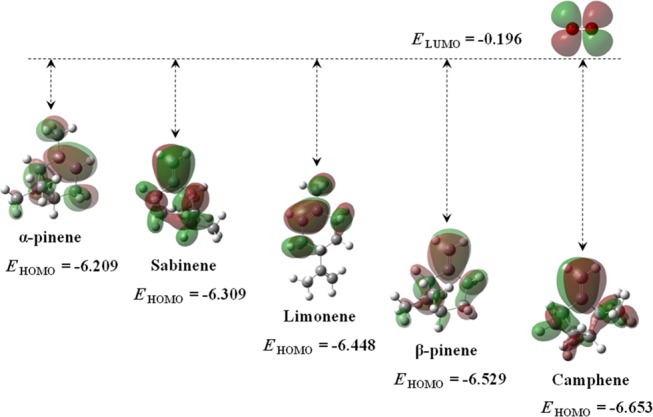
Electron cloud distribution and energy levels of HOMO of monoterpenes and LUMO of singlet oxygen. Energy values are in eV unit.
Table 1 lists the global hardness (η), softness (S), electronegativity (χ), chemical potential (μ), and electrophilicity index (ω) of the species derived from the computed HOMO and LUMO energy levels, enabling the predictions of the respective chemical characteristics of the species. Camphene remains the hardest reagent with the highest electronegativity and the lowest chemical potential. Apparently, the higher electronegativity signifies the lower chemical activity, all of which connote a higher oxidation resistance.36 Thus, the following trend of chemical reactivity of the species is predicted herein: α-pinene > sabinene > limonene > β-pinene > camphene. The stated reactivity sequence accords well with the arrangement of the energy gap between LUMO of O21Δg and HOMO of monoterpene species in Figure 3, i.e., the smaller the energy gap, the higher the reactivity.
Table 1. HOMO/LUMO Energies, Electronegativities (χ), Hardnesses (η), Softness (S), and Electrophilicity Index (ω) of the Monoterpene Species Obtained at the B3LYP 6-311+g(d,p) Level of Calculationsa.
| EHOMO | ELUMO | ΔELUMO–HOMO | χ | μ | η | S | ω | |
|---|---|---|---|---|---|---|---|---|
| limonene | –6.448 | –0.055 | 6.393 | 3.252 | –3.252 | 3.196 | 0.313 | 1.654 |
| α-pinene | –6.209 | 0.017 | 6.226 | 3.096 | –3.096 | 3.113 | 0.321 | 1.540 |
| β-pinene | –6.529 | 0.005 | 6.534 | 3.262 | –3.262 | 3.267 | 0.306 | 1.628 |
| camphene | –6.653 | –0.007 | 6.646 | 3.330 | –3.330 | 3.323 | 0.301 | 1.669 |
| sabinene | –6.309 | –0.063 | 6.246 | 3.186 | –3.186 | 3.123 | 0.320 | 1.625 |
Values are in eV.
The following sections present the mechanisms of reaction of each of the aforementioned monoterpenes with singlet oxygen, discussing in detail the energy potentials and kinetics features of the reactions.
2.1.1. Limonene
Figure 4 displays the optimized structure of limonene. According to the acquired reactivity indices in Figure 2, the most vulnerable site for electrophilic attack rests on the trisubstituted double bond (C1=C2). Therefore, the plausible reaction mechanisms for singlet oxidation of limonene should involve the ene reaction and [2 + 2]-cycloaddition. The latter failed to locate a genuine transition state despite our best efforts. Thus, the overall reaction merely involves ene-type reaction of O21Δg with limonene’s unconjugated double bond to form allylic hydroperoxides. Yet, our computational model failed to optimize related diradicals as potential intermediates in the ene reactions of limonene photo-oxidation, prompting to suggest a concerted mechanism for the ene-type addition of singlet oxygen to the limonene structure.
Figure 4.
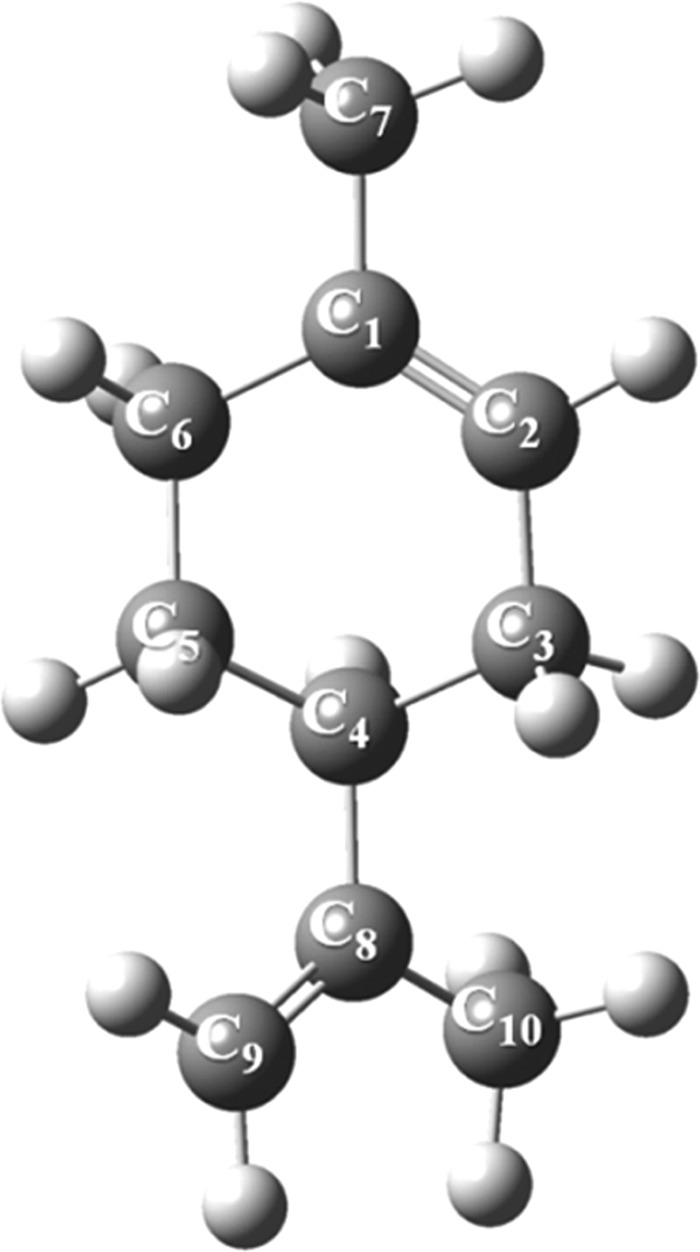
Optimized structure of limonene computed at the B3LYP/6-311+g(d,p) level of theory.
As illustrated in the proposed reaction mechanism in Figure 5, singlet oxygen clings to the limonene’s cyclohexene ring at the C2 atom, bearing analogous barrier enthalpies of TS2 and TS3 around 40 kJ/mol. Hydrogen abstraction from the methylene group (in P3) appears relatively energetically more favorable as compared to that from the methyl group (in P2). The C1 atom represents an alternative reactive spot for the addition of singlet oxygen enduring a relatively higher enthalpy barrier of TS1 (50 kJ/mol). In accordance to the computed Fukui indices in Figure 2, this enthalpic behavior is justifiable as the C2 position is the most favorable site for an electrophilic attack.
Figure 5.

Reaction mechanism of the singlet oxidation of limonene. The enthalpies are obtained at the 6-311+g(d,p) level of theory and are reported in kJ/mol.
Although the electrophilic Fukui functions demonstrate that the likelihood of the C8=C9 site to be attacked by electrophiles is negligible, the corresponding transition state values (TS5 and TS6) compare well with the transition states for the attack on the C1=C2 in-ring double bond. As previously illustrated in Figure 1, the literature suggests that the product distribution from the photo-oxidation of limonene consists of terpene alcohols that are being reduced from P1, P2, and P3 hydroperoxides, whose RO–OH bond efficiently undergoes a cleavage. Table 2 lists the reaction rate coefficient for the bimolecular reactions involved in the interaction of limonene with the singlet oxygen, fitted to the Arrhenius equation of k(T) = Ae–Ea/RT at a high pressure limit for the temperature range of 300 and 600 K.
Table 2. Kinetic Parameters of Limonene Interaction with O21Δg.
| reaction | A (s–1 or cm3 molecule–1 s–1) | Ea (kJ/mol) |
|---|---|---|
| limonene + O2 1Δg → P1 | 1.53 × 10–13 | 57 |
| limonene + O2 1Δg → P2 | 1.87 × 10–13 | 48 |
| limonene + O2 1Δg → P3 | 9.71 × 10–13 | 49 |
| limonene + O2 1Δg → P5 | 3.43 × 10–13 | 54 |
| limonene + O2 1Δg → P6 | 2.25 × 10–13 | 49 |
The branching ratios for the bimolecular reaction channels are evaluated based on the Arrhenius parameters for ki(T) given in Table 2. Figure 6 plots the ki/∑ ki, verifying channel TS3 as the dominant reaction pathway and P3 as the major primary product according to the reaction network. In a qualitative agreement with the experimental results by Chalchat et al.,37 P3 constituted ∼40% of the initial product yield from the photochemical hydroperoxidation of limonene in the presence of oxygen. In general, photo-oxidation of the products of limonene features −OOH/–OH/–O substitution at para position.7,37 This is consistent with our prediction of P3 as the likely dominant initial intermediate.
Figure 6.

Plot of branching ratios of different reaction pathways as a function of temperature (K).
A subsequent H-transfer step into the outer OH group in the P3 intermediate liberates a water molecule and forms the experimentally detected product of carvone (P4) via an accessible energy barrier of only 30 kJ/mol.
In addition to carvone, terpene alcohols constitute the major experimentally detected products from the photo-oxidation of limonene. As illustrated above, these molecules most likely arise from a radical-induced mechanism that begins with the fission of the O–OH bonds in the P1–P6 intermediates. For instance, the four para-substituted terpene alcohols account for 60% of the total product yields from the photo-oxidation of limonene.37 Clearly, these compounds may originate from the scission of the O–OH bond in the predicted dominant P3 intermediate followed by H abstraction by the phenoxy-type O atom and structural arrangements. Products feature ipso substitution, most likely directly stemming from the P1 moiety or via an intramolecular transfer of the OOH group along the reaction P3 → P1. Such a step proceeds without encountering a reaction as an intrinsic barrier in a thermodynamically neutral reaction.
2.1.2. Pinene
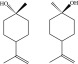
Figures 7,8, and 9 depict the optimized structures of α- and β-pinene. The f–1 indices (Figure 2) reveal C2 in α-pinene and C1 in β-pinene structures as the most preferred sites of interaction with electrophilic species. We were unable to locate the transition structures for the [2 + 2]-cycloaddition of singlet oxygen to pinene, certifying that the formation of hydroperoxides as the sole light-induced oxidation product of α- and β-pinene stems from ene-type reaction.38,39
Figure 7.

Structures of para-substituted terpene alcohols.
Figure 8.
Structures of ipso-substituted terpene alcohols.
Figure 9.

Optimized structures of α-pinene (left) and β-pinene (right) computed at B3LYP/6-311+g(d,p) level of theory.
The reaction mechanisms of the singlet oxidation of α- and β-pinene are displayed in Figures 10 and 11, respectively. The figures reveal that the ene reaction proceeds with the addition of singlet oxygen to the in-ring C2 atom of α-pinene and the abstraction of allylic hydrogen from the methyl group via concerted or stepwise mechanisms. The stepwise TS2 barrier overshoots the concerted TS1 by 20 kJ/mol, leaving the concerted mechanism as the dominating reaction pathway for the formation of P1 hydroperoxide. The product P1 is also attainable through a stepwise channel via a facile transformation of the diradical (P2) to hydroperoxide (P1).40 Jefford et al.4 recorded similar observation, identifying P1 as the utmost product from the 1O2-initiated atmospheric oxidation of α-pinene.
Figure 10.

Reaction mechanism of the singlet oxidation of α-pinene. The enthalpies are obtained at the 6-311+g(d,p) level of theory and are in kJ/mol.
Figure 11.

Reaction mechanism of the singlet oxidation of β-pinene. The enthalpies are obtained at the 6-311+g(d,p) level of theory and are in kJ/mol.
Moreover, as shown in Figure 11, the ene-type addition of the singlet oxygen to C1 atom within a β-pinene structure (the one with the highest Fukui function value) occurs similarly in both stepwise and concerted fashions through TS1 (57 kJ/mol) and TS4 (49 kJ/mol) steps. Apparently, the concerted mechanism (TS4) is the governing reaction channel triggering the formation of P2 hydroperoxide as the major product of the singlet oxidation of β-pinene. The stepwise TS1 channel, on the other hand, gives rise to the production of a diradical P1, which further branches into two exit routes, resulting in two types of hydroperoxide adducts, P2 and P3. The product P2 resides in a significant well-depth in reference to the entrance channel, affirming its greater stability as compared to P3 hydroperoxide. Besides, TS2 signifies a trivial transition barrier leading to the formation of the main product P2.
Table 3 presents the fitted Arrhenius parameters. According to the computed reaction rate coefficients, the α-pinene + O21Δg reaction proceeds predominantly by the ene addition of the singlet delta oxygen to C2 atom to form P1 hydroperoxide in a concerted mechanism. Oxidation of β-pinene by 1O2 similarly proceeds through a concerted channel, giving rise to the formation of P2 hydroperoxide via an enthalpy barrier of 49 kJ/mol.
Table 3. Kinetic Parameters of α- and β-Pinene Interaction with O21Δg.
| reaction | A (s–1 or cm3 molecule–1 s–1) | Ea (kJ/mol) |
|---|---|---|
| α-pinene + O2 1Δg → P1 | 2.51 × 10–13 | 57 |
| α-pinene + O2 1Δg → P2 | 3.50 × 10–13 | 63 |
| P2 → P1 | 3.19 × 10+12 | 12 |
| β-pinene + O2 1Δg → P1 | 1.18 × 10–12 | 64 |
| P1 → P2 | 2.93 × 10+12 | 25 |
| P1 → P3 | 1.16 × 10+12 | 45 |
| β-pinene + O2 1Δg → P3 | 2.99 × 10–13 | 56 |
Data in Table 3 afforded the estimation of the branching ratios of the bimolecular reaction channels for α- and β-pinene as shown in Figure 12. In view of this finding, the concerted TS1 channel remains the dominant reaction pathway for the singlet oxidation of α-pinene, and there exists a moderate decline in the branching ratio value as the temperature increases, reflecting the reverse effect of temperature on the reaction rate constant of the concerted mechanism of the singlet oxygen addition to α-pinene.
Figure 12.

Plots of branching ratios of α-pinene (top) and β-pinene (bottom) at different temperatures (K).
In a similar fashion, the concerted TS4 step leads to the reaction pathways for the singlet oxidation of β-pinene and the calculated branching ratios show that the contribution of the concerted mechanism (k4) noticeably diminishes as the temperature increases. Nonetheless, at atmospheric relevant temperatures (i.e., 300 K), reaction of β-pinene with singlet oxygen largely ensues via the concerted mechanism.
2.1.3. Camphene and Sabinene
There exists a resemblance between the structure of camphene (Figure 13) and β-pinene owing to the presence of an exocyclic double bond. Therefore, the addition of a singlet oxygen to camphene in an ene reaction mode occurs at the C1 position via a stepwise mechanism.
Figure 13.
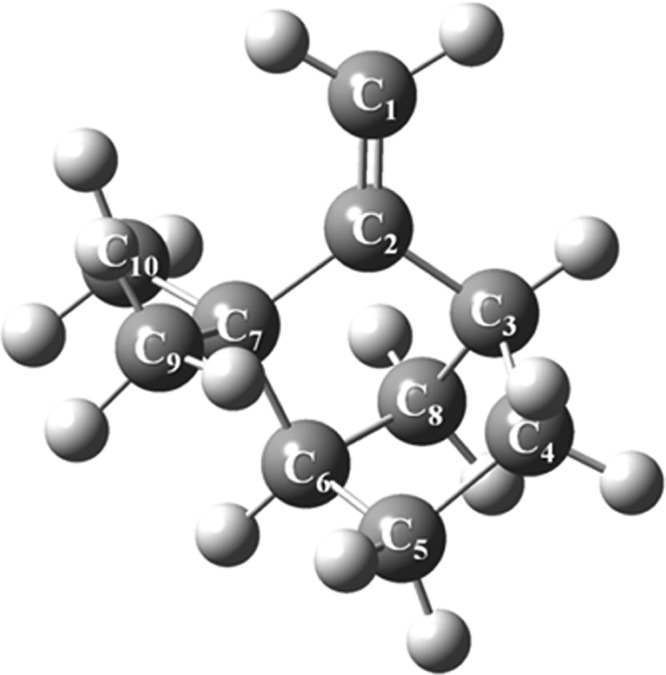
Optimized structure of camphene computed at the B3LYP/6-311+g(d,p) level of theory.
According to the mechanism of the singlet-oxygen-initiated atmospheric oxidation of camphene shown in Figure 14, the initial reaction between camphene and singlet oxygen comprises O21Δg addition to the terminal carbon atom of C1 via sizable thermal enthalpy (TS1) amounting to 132 kJ/mol. The resulting highly unstable diradical (P2) is then transformed to the P2 hydroperoxide adduct through a readily accessible transition state TS2 through an enthalpic barrier of 30 kJ/mol.
Figure 14.

Reaction mechanism of the singlet oxidation of camphene. The enthalpies are obtained at the 6-311+g(d,p) level of theory and are in kJ/mol.
Table 4 assembles the fitted high-pressure limiting Arrhenius parameters. The relatively high activation barrier of camphene photo-oxidation (Ea = 139 kJ/mol) correlates well with its high LUMO–HOMO energy gap (Table 1) and proves its slight tendency to undergo 1O2-induced oxidation process.
Table 4. Arrhenius Parameters for Camphene Oxidation by Singlet Oxygen.
| reaction | A (s–1 or cm3 molecule–1 s–1) | Ea (kJ/mol) |
|---|---|---|
| camphene + O2 1Δg → P1 | 1.15 × 10–12 | 139 |
| P1 → P2 | 3.58 × 10+12 | 33 |
Furthermore, sabinene, as illustrated in Figure 15, represents a bicyclic monoterpene and encompasses an exocyclic double bond. The sabinene’s terminal double bond is characterized as an exclusive electron-rich molecular site and could readily be attacked by reactive oxygen species such as O21Δg.
Figure 15.
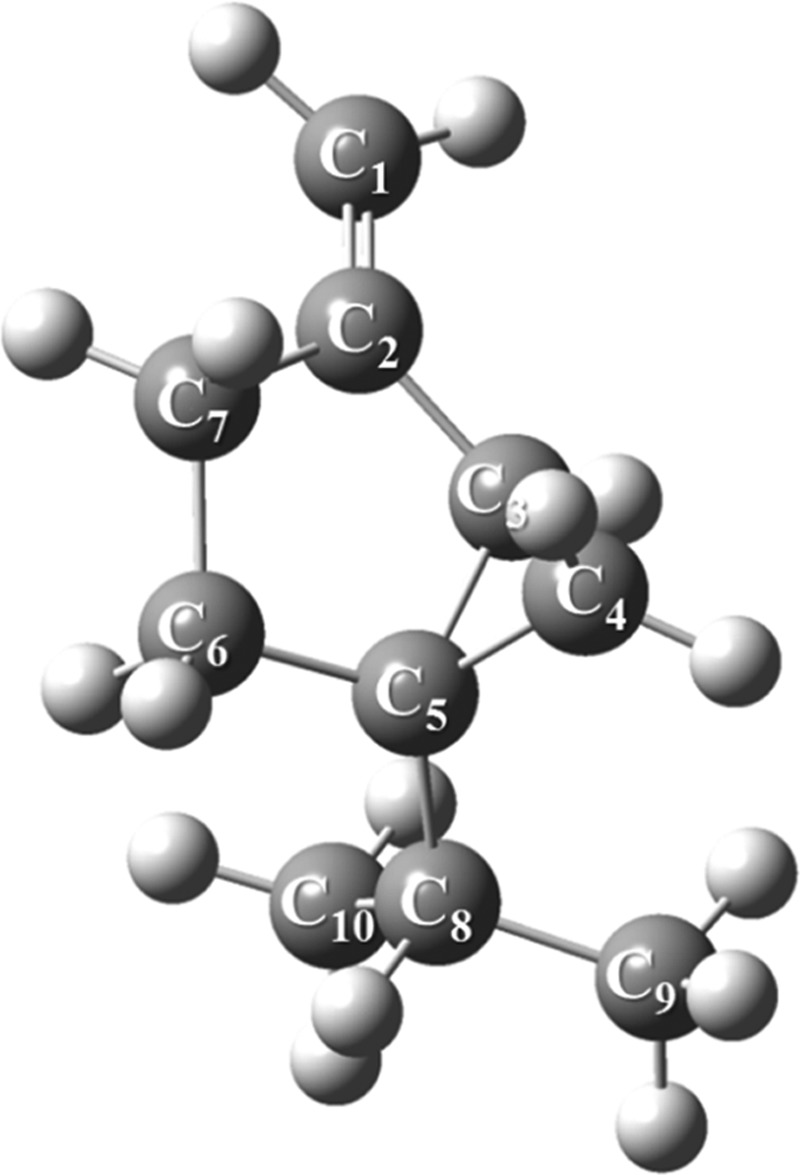
Optimized structure of sabinene computed at the B3LYP/6-311+g(d,p) level of theory.
Introduction of singlet oxygen to the sabinene structure via ene reaction takes place at the C1 site through a concerted bimolecular reaction bearing a slight thermal enthalpy barrier of 55 kJ/mol (Figure 16). The oxidation process proceeds with an activation energy of 62 kJ/mol as obtained from the first-order Arrhenius equation fitted for the temperature range of 300–600 K (Table 5).
Figure 16.
Reaction mechanism of the singlet oxidation of sabinene. The enthalpies are obtained at the 6-311+g(d,p) level of theory and are in kJ/mol.
Table 5. Arrhenius Parameters for Sabinene Oxidation by Singlet Oxygen.
| reaction | A (s–1 or cm3 molecule–1 s–1) | Ea (kJ/mol) |
|---|---|---|
| sabinene + O2 1Δg → P1 | 5.12 × 10–13 | 62 |
The resulting hydroperoxide (P1) would presumably be reduced to the corresponding alcohol (I) and aldehyde (II) via the O–O bond fission stimulated by thermal or catalytic effects.41
2.2. Effect of Solvent on Singlet Oxidation of Cyclic Monoterpenes
This section considers the influence of solvent in the relevant process involving the interaction of singlet oxygen with monoterpenes rather than atmospheric oxidation. The selected monoterpenes are nearly insoluble in water42 and thus methanol served as the solvent herein. The polarizable continuum model43 simulates the aqueous medium. In essence, this method relies on the representation of a solute molecule as a charge distributor located inside a cavity surrounded by a continuous dielectric medium, which is polarized as a result of the point charge distribution on the cavity surface.
According to Figures 17 and 18, the effect of solvent is revealed through a systematic energy reduction in the barrier enthalpies of the transition states, intermediates, and final products along the major channels of the singlet oxygen interaction with each monoterpene substrate. The reduced enthalpies (involving solvent effect) with reference to the gas-phase system range from 12 kJ/mol (P1 value in α-pinene) to 34 kJ/mol (P7 value in limonene). Although the reaction barrier enthalpies are influenced by solvent effects, the product distribution and reaction channels remain intact.
Figure 17.

Reduction of hydroperoxide to terpene alcohol and aldehyde.
Figure 18.

Potential energy diagrams of major monoterpenes + O21Δg reaction with (red) and without (black) solvent (methanol) effects.
3. Conclusions
Introduction of singlet oxygen to the cyclic monoterpenes exclusively follows the ene reaction pathway to yield the corresponding allylic hydroperoxides. Limonene and sabinene photo-oxidation transpire through concerted mechanisms, whereas singlet oxidation of β-pinene and camphene proceed via stepwise mechanism, resulting in the formation of diradical intermediates. In the case of α-pinene, the concerted channel ensues the most energetically favorable pathway. In terms of reactivity, α-pinene and sabinene exhibit the highest reactivity, while camphene is kinetically evidenced to be the lowest reactive species incurring relatively high activation energy of 139 kJ/mol.
4. Methodology
Gaussian 09 program44 deployed the unrestricted density functional theory in acquiring optimized energies and geometries of the reacting species due to its accuracy in computing singlet biradical properties.45,46 For that reason, we utilize the B3LYP functional with the extended 6-311+g(d,p)47 basis set. A simple approximate spin-projection (AP) scheme48,49 served to correct the final energies of species displaying biradical characters. For this, the approximate spin-projected energy (EAP) has been derived from the energies of the broken-symmetry (EBS) and pure high-spin (EHS) states according to eq 1.
| 1 |
where fAP denotes the spin-projection factor
| 2 |
and ⟨S2⟩HS and ⟨S2⟩BS signify the expectation values of the spin contamination pertinent to the pure high-spin and broken-symmetry states, respectively. We verified the transition structures via intrinsic reaction coordinate calculations. ChemRate software50 facilitated the calculation of the reaction rate constants, within the temperature range of 300–600 K based on the Arrhenius equation. The electrophilic Fukui indices, serving as indicators for molecular site’s reactivity toward electrophilic addition reactions, are quantified by Dmol3 code51 in Material Studio package at the B3LYP functional while applying a double numerical plus d-functions atomic basis set.
Acknowledgments
This study has been supported by grants of computing time from the National Computational Infrastructure (NCI) in Canberra and the Pawsey Supercomputing Centre (iVEC) in Perth. N.Z. acknowledges the Iraqi government for awarding PhD scholarships.
The authors declare no competing financial interest.
References
- Guenther A.; Geron C.; Pierce T.; Lamb B.; Harley P.; Fall R. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ. 2000, 34, 2205–2230. 10.1016/S1352-2310(99)00465-3. [DOI] [Google Scholar]
- Steinbrecher R. In Emission of VOCs from Selected European Ecosystems: The State of the Art, The Proceedings of EUROTRAC Symposium, 1994; pp 448–454.
- Ohloff G.Singlet Oxygen: A Reagent in Organic Synthesis. In Organic Synthesis; Elsevier, 1975; pp 481–502. [Google Scholar]
- Jefford C. W.; Boschung A. F.; Moriarty R. M.; Rimbault C. G.; Laffer M. H. The Reaction of Singlet Oxygen with α-and β-Pinenes. Helv. Chim. Acta 1973, 56, 2649–2659. 10.1002/hlca.19730560748. [DOI] [Google Scholar]
- Goldberg M. C.; Cunningham K. M.; Aiken G. R.; Weiner E. R. The aqueous photolysis of α-pinene in solution with humic acid. J. Contam. Hydrol. 1992, 9, 79–89. 10.1016/0169-7722(92)90051-F. [DOI] [Google Scholar]
- Clark B. Jr.; Jones B.; Iacobucci G. Characterization of the hydroperoxides derived from singlet oxygen oxidation of (+)-limonene. Tetrahedron 1981, 37, 405–409. 10.1016/0040-4020(81)85078-8. [DOI] [Google Scholar]
- Foote C. S.; Wexler S.; Ando W. Chemistry of singlet oxygen III. Product selectivity. Tetrahedron Lett. 1965, 6, 4111–4118. 10.1016/S0040-4039(01)99574-7. [DOI] [Google Scholar]
- Rufino A. T.; Ribeiro M.; Judas F.; Salgueiro L. G.; Lopes M. C.; Cavaleiro C.; Mendes A. F. Anti-inflammatory and chondroprotective activity of (+)-α-pinene: structural and enantiomeric selectivity. J. Nat. Prod. 2014, 77, 264–269. 10.1021/np400828x. [DOI] [PubMed] [Google Scholar]
- Wolkoff P.; Clausen P.; Wilkins C.; Nielsen G. Formation of strong airway irritants in terpene/ozone mixtures. Indoor Air 2000, 10, 82–91. 10.1034/j.1600-0668.2000.010002082.x. [DOI] [PubMed] [Google Scholar]
- Almatarneh M. H.; Elayan I. A.; Poirier R. A.; Altarawneh M. The ozonolysis of cyclic monoterpenes: a computational review. Can. J. Chem. 2018, 96, 281–292. 10.1139/cjc-2017-0587. [DOI] [Google Scholar]
- Atkinson R. Gas-phase tropospheric chemistry of organic compounds: a review. Atmos. Environ., Part A 1990, 24, 1–41. 10.1016/0960-1686(90)90438-S. [DOI] [Google Scholar]
- Gershenzon J.; McConkey M. E.; Croteau R. B. Regulation of monoterpene accumulation in leaves of peppermint. Plant Physiol. 2000, 122, 205–214. 10.1104/pp.122.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim I. A.; Harris R. B.; Ritenbaugh C. Citrus peel use is associated with reduced risk of squamous cell carcinoma of the skin. Nutr. Cancer 2000, 37, 161–168. 10.1207/S15327914NC372_7. [DOI] [PubMed] [Google Scholar]
- Mukhtar Y. M.; Adu-Frimpong M.; Xu X.; Yu J. Biochemical significance of limonene and its metabolites: future prospects for designing and developing highly potent anticancer drugs. Biosci. Rep. 2018, 38, BSR20181253 10.1042/BSR20181253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juuti S.; Arey J.; Atkinson R. Monoterpene emission rate measurements from a Monterey pine. J. Geophys. Res.: Atmos. 1990, 95, 7515–7519. 10.1029/JD095iD06p07515. [DOI] [Google Scholar]
- Geron C.; Rasmussen R.; Arnts R. R.; Guenther A. A review and synthesis of monoterpene speciation from forests in the United States. Atmos. Environ. 2000, 34, 1761–1781. 10.1016/S1352-2310(99)00364-7. [DOI] [Google Scholar]
- Liu J.; Chu B.; Chen T.; Liu C.; Wang L.; Bao X.; He H. Secondary organic aerosol formation from ambient air at an urban site in Beijing: effects of OH exposure and precursor concentrations. Environ. Sci. Technol. 2018, 52, 6834–6841. 10.1021/acs.est.7b05701. [DOI] [PubMed] [Google Scholar]
- Pitts J. N. Jr.; Khan A. U.; Smith E. B.; Wayne R. P. Singlet oxygen in the environmental sciences. Singlet molecular oxygen and photochemical air pollution. Environ. Sci. Technol. 1969, 3, 241–247. 10.1021/es60026a004. [DOI] [PubMed] [Google Scholar]
- Bayer P.; Pérez-Ruiz R.; Jacobi von Wangelin A. Stereoselective Photooxidations by the Schenck Ene Reaction. ChemPhotoChem 2018, 2, 559–570. 10.1002/cptc.201800058. [DOI] [Google Scholar]
- Clennan E. L. Synthetic and mechanistic aspects of 1, 3-diene photooxidation. Tetrahedron 1991, 47, 1343–1382. 10.1016/S0040-4020(01)86413-9. [DOI] [Google Scholar]
- Maranzana A.; Ghigo G.; Tonachini G. Diradical and peroxirane pathways in the [π2+ π2] cycloaddition reactions of 1Δg dioxygen with ethene, methyl vinyl ether, and butadiene: a density functional and multireference perturbation theory study. J. Am. Chem. Soc. 2000, 122, 1414–1423. 10.1021/ja990805a. [DOI] [Google Scholar]
- Hakola H.; Arey J.; Aschmann S. M.; Atkinson R. Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes. J. Atmos. Chem. 1994, 18, 75–102. 10.1007/BF00694375. [DOI] [Google Scholar]
- Atkinson R. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 1986, 86, 69–201. 10.1021/cr00071a004. [DOI] [Google Scholar]
- Pinto D. M.; Tiiva P.; Miettinen P.; Joutsensaari J.; Kokkola H.; Nerg A.-M.; Laaksonen A.; Holopainen J. K. The effects of increasing atmospheric ozone on biogenic monoterpene profiles and the formation of secondary aerosols. Atmos. Environ. 2007, 41, 4877–4887. 10.1016/j.atmosenv.2007.02.006. [DOI] [Google Scholar]
- Johnson D.; Rickard A. R.; McGill C. D.; Marston G. The influence of orbital asymmetry on the kinetics of the gas-phase reactions of ozone with unsaturated compounds. Phys. Chem. Chem. Phys. 2000, 2, 323–328. 10.1039/a908701j. [DOI] [Google Scholar]
- Oliveira R. d. M.; Bauerfeldt G. Ozonolysis reactions of monoterpenes: a variational transition state investigation. J. Phys. Chem. A 2015, 119, 2802–2812. 10.1021/jp5129222. [DOI] [PubMed] [Google Scholar]
- Carari D. M.; da Silva M. J. Fe (NO 3) 3-Catalyzed Monoterpene Oxidation by Hydrogen Peroxide: An Inexpensive and Environmentally Benign Oxidative Process. Catal. Lett. 2014, 144, 615–622. 10.1007/s10562-013-1189-x. [DOI] [Google Scholar]
- de Correa C. M. Kinetics of limonene epoxidation by hydrogen peroxide on PW-Amberlite. J. Mol. Catal. A: Chem. 2002, 185, 269–277. 10.1016/S1381-1169(02)00077-8. [DOI] [Google Scholar]
- da Silva M. J.; Vieira L. M.; Oliveira A. A.; Ribeiro M. C. Novel effect of palladium catalysts on chemoselective oxidation of β-pinene by hydrogen peroxide. Monatsh. Chem. 2013, 144, 321–326. 10.1007/s00706-012-0875-5. [DOI] [Google Scholar]
- Pandis S. N.; Paulson S. E.; Seinfeld J. H.; Flagan R. C. Aerosol formation in the photooxidation of isoprene and β-pinene. Atmos. Environ., Part A 1991, 25, 997–1008. 10.1016/0960-1686(91)90141-S. [DOI] [Google Scholar]
- Wildt J.; Mentel T.; Kiendler-Scharr A.; Hoffmann T.; Andres S.; Ehn M.; Kleist E.; Müsgen P.; Rohrer F.; Rudich Y.; et al. Suppression of new particle formation from monoterpene oxidation by NOx. Atmos. Chem. Phys. 2014, 14, 2789–2804. 10.5194/acp-14-2789-2014. [DOI] [Google Scholar]
- Atkinson R. Kinetics and mechanisms of the gas-phase reactions of the NO3 radical with organic compounds. J. Phys. Chem. Ref. Data 1991, 20, 459–507. 10.1063/1.555887. [DOI] [Google Scholar]
- Hakola H.; Arey J.; Aschmann S. M.; Atkinson R. Product formation from the gas-phase reactions of OH radicals and O 3 with a series of monoterpenes. J. Atmos. Chem. 1994, 18, 75–102. 10.1007/BF00694375. [DOI] [Google Scholar]
- Foote C. S. Photosensitized oxygenations and the role of singlet oxygen. Acc. Chem. Res. 1968, 1, 104–110. 10.1021/ar50004a002. [DOI] [Google Scholar]
- You K.; Yin D.; Mao L.; Liu P.; Luo H. A. Selective photosensitized oxidation and its catalytic regulation of monoterpene with molecular oxygen in different reaction media. J. Photochem. Photobiol., A 2011, 217, 321–325. 10.1016/j.jphotochem.2010.10.026. [DOI] [Google Scholar]
- Gao Z.; Yang Z.; Li Y.; Deng A.; Luo Y.; Li H.-W. Improving the phase stability and cycling performance of Ce 2 Ni 7-type RE–Mg–Ni alloy electrodes by high electronegativity element substitution. Dalton Trans. 2018, 47, 16453–16460. 10.1039/C8DT03644F. [DOI] [PubMed] [Google Scholar]
- Chalchat J.; Chiron F.; Garry R. P.; Lacoste J.; Sautou V. Photochemical hydroperoxidation of terpenes. Antimicrobial activity of α-pinene, β-pinene and limonene hydroperoxides. J. Essent. Oil Res. 2000, 12, 125–134. 10.1080/10412905.2000.9712059. [DOI] [Google Scholar]
- Kenney R.; Fisher G. Preparation of trans-Pinocarveol and Myrtenol. Ind. Eng. Chem. Prod. Res. Dev. 1973, 12, 317–319. 10.1021/i360048a011. [DOI] [Google Scholar]
- Hennig H.; Rehorek D.; Stich R.; Weber L. Photocatalysis induced by light-sensitive coordination compounds. Pure Appl. Chem. 1990, 62, 1489–1494. 10.1351/pac199062081489. [DOI] [Google Scholar]
- Maranzana A.; Ghigo G.; Tonachini G. The 1Δg Dioxygen Ene Reaction with Propene: A Density Functional and Multireference Perturbation Theory Mechanistic Study. Chem. - Eur. J. 2003, 9, 2616–2626. 10.1002/chem.200204522. [DOI] [PubMed] [Google Scholar]
- Davies A. G.; Feld R. 133. Organic peroxides. Part VI. A stereochemical investigation of the preparation and reactions of 1-phenylethyl hydroperoxide. J. Chem. Soc. 1956, 665–670. 10.1039/jr9560000665. [DOI] [Google Scholar]
- Lide D. R.CRC Handbook of Chemistry and Physics; CRC Press, 2004; Vol. 85. [Google Scholar]
- Barone V.; Cossi M.; Tomasi J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. 10.1063/1.474671. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, A.1; Gaussian, Inc: Wallingford, CT, 2009. [Google Scholar]
- Gräfenstein J.; Cremer D. The combination of density functional theory with multi-configuration methods–CAS-DFT. Chem. Phys. Lett. 2000, 316, 569–577. 10.1016/S0009-2614(99)01326-3. [DOI] [Google Scholar]
- Cramer C. J.; Thompson J. Quantum chemical characterization of singlet and triplet didehydroindenes. J. Phys. Chem. A 2001, 105, 2091–2098. 10.1021/jp004379n. [DOI] [Google Scholar]
- Montgomery J. A. Jr.; Ochterski J. W.; Petersson G. A. A complete basis set model chemistry. IV. An improved atomic pair natural orbital method. J. Chem. Phys. 1994, 101, 5900–5909. 10.1063/1.467306. [DOI] [Google Scholar]
- Yamaguchi K.; Takahara Y.; Fueno T.; Houk K. Extended Hartree-Fock (EHF) theory of chemical reactions. Theor. Chim. Acta 1988, 73, 337–364. 10.1007/BF00527740. [DOI] [Google Scholar]
- Yamaguchi K.; Okumura M.; Mori W.; Maki J.; Takada K.; Noro T.; Tanaka K. Comparison between spin restricted and unrestricted post-Hartree—Fock calculations of effective exchange integrals in Ising and Heisenberg models. Chem. Phys. Lett. 1993, 210, 201–210. 10.1016/0009-2614(93)89124-Z. [DOI] [Google Scholar]
- Mokrushin V.; Bedanov V.; Tsang W.; Zachariah M.; Knyazev V.. ChemRate, version 1.19; NIST: Gaithersburg, MD, 2002.
- Delley B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. 10.1063/1.1316015. [DOI] [Google Scholar]