

Abstract
Enzymes in the prolyl oligopeptidase family possess unique structures and substrate specificities that are important for their biological activity and for potential biocatalytic applications. The crystal structures of Pyrococcus furiosus (Pfu) prolyl oligopeptidases (POP) and the corresponding S477C mutant were solved to 1.9 and 2.2 Å resolution, respectively. The wild type enzyme crystallized in an open conformation, indicating that this state is readily accessible, and it contained bound chloride ions and a prolylproline ligand. These structures were used as starting points for molecular dynamics simulations of Pfu POP conformational dynamics. The simulations showed that large-scale domain opening and closing occurred spontaneously, providing facile substrate access to the active site. Movement of the loop containing the catalytically essential histidine into a conformation similar to those found in structures with fully-formed catalytic triads also occurred. This movement was modulated by chloride binding, providing a rationale for experimentally observed activation of POP peptidase catalysis by chloride. Thus, the structures and simulations reported in this study, combined with existing biochemical data, provide a number of insights into POP catalysis.
Keywords: prolyl oligopeptidase, protease, serine hydrolase, x-ray crystallography, molecular dynamics simulation
INTRODUCTION
Prolyl oligopeptidases (POPs) hydrolyze small peptide substrates following proline residues.1–3 This specificity leads to the involvement of POP family enzymes in a range of important biological processes2 and biocatalytic applications4. With molecular weights of ~70–80 kDa, POPs are significantly larger than many prototypical proteases like trypsin or subtilisin (25–30 kDa), suggesting that the additional bulk of the former may contribute to their unique substrate specificity.3,5 Indeed, structural studies have established that POPs are comprised of a ~30 kDa peptidase domain containing a Ser-His-Asp catalytic triad topped by a ~40 kDa 7-bladed β-propeller domain that restricts substrate access to a large active site cavity.3,6–10 Experimental and computational studies have suggested that this domain undergoes large scale conformational changes that regulate substrate access to its active site.
While extensive structural characterization of bacterial and mammalian POPs exists, no structures for archaeal POPs have been reported. Particularly notable in this regard, given the extensive biophysical characterization dedicated to it,11–15 is the POP from the hyperthermophilic archaeon, Pyrococcus furiosus (Pfu). Pfu POP is at most 28% identical to previously crystallized POPs, which are up to 44% identical, suggesting that Pfu POP could possess properties not present in these enzymes. Indeed, functional differences have been noted and most extensively discussed relative to porcine (Sus scrofa) POP. While porcine POP has melting points of 44.6 °C and 52.8 °C (pH 8), for example, Pfu POP melts at 91.7 °C and 109.5 °C (pH 8.4)15 and exhibits maximum activity at 85 °C11. Both enzymes are activated by NaX (X = Cl, Br, etc.), but the nature of their response to [NaX] differs. Finally, while peptidase catalysis by both porcine and Pfu POP proceeds via the general mechanism shown in Fig. 1A, substrate entry into the active sites of these enzymes and inter-domain conformational changes associated with this process appear to involve important differences (Fig. 1B).16 These issues and our interest in biocatalytic applications of Pfu POP17,18 led us to solve the structures of this enzyme and its S477C mutant. The structures obtained, along with molecular dynamics simulations based on them, comparative analysis of previously reported POP structures, and existing biochemical data, have resolved several previously debated aspects of Pfu POP structure and function3,13,15.
Figure 1.

A) General scheme for peptidase catalysis involving an enzyme (E) and a peptide substrate (S) to generate two product peptides (P1 and P2) via enzyme-substrate (ES) and enzyme-acyl (EA) intermediates. B) Potential domain opening and closing during Pfu POP peptidase catalysis.
MATERIALS AND METHODS
Standard cloning procedures and site directed mutagenesis.
The Pfu POP gene, cloned into a pET 11c vector, was obtained from Prof. Harold Schreier (UMBC). Cysteine and alanine mutations were introduced into the gene at position S477 by site directed overlap extension PCR.19 Two separate PCRs were performed as outlined in the supporting information, each using a perfectly complementary flanking primer (Table S1) at the 5’ and 3’ end of the sequence and a mutagenic primer. The resulting two overlapping fragments that contained the base pair substitution were then assembled in a second PCR to give the full-length gene.
PCR amplified fragments and pET11C were digested with NdeI and BamHI enzymes in recommended buffers at 37 °C for 2 hours. Digested DNA was purified by agarose gel extraction before ligation. Ligation reactions were conducted using a molar ratio of 1:3 (plasmid: insert) in 10 μL reaction mix. The reactions were incubated at 16 °C overnight, desalted, and transformed into E. coli DH5 cells. Cells were recovered in SOC media for 1 hour at 37 °C and spread onto LB Ampicillin plates (6.25 g LB powder mix, 4 g agar, 250 mL DDI water, 0.1 mg/mL Ampicillin). Plates were incubated at 37 °C overnight, and single colonies that appeared overnight were tested for the desired POP gene by colony PCR. Clones containing the desired insert were used to inoculate LB broth containing 0.10 mg/mL Ampicillin and grown overnight at 37 °C, 250 rpm. Recombinant plasmid DNA from these overnight grown cultures was isolated using miniprep kit from Qiagen (Valencia, CA) and verified via sequencing at the U Chicago sequencing facility using T7 forward and T7 reverse primers (Table S1).
POP expression and purification.
Single colonies of E. coli BL21 (DE3) cells harboring either pET11C-POPS477A or pET11C-POPS477C were used to inoculate 5 mL of 2YT/ampicillin. The culture was incubated overnight at 37 °C with constant shaking at 250 rpm. On the following day, 5 mL of the overnight culture was used to inoculate 500 mL of fresh 2YT/ampicillin in a 5 L Erlenmeyer flask. The culture was incubated at 37 °C, 250 rpm, and protein expression was induced by adding 1 mM IPTG when OD600 reached 1. The induced culture was incubated for 12 hours, and then the cells were harvested by centrifugation at 4 °C, 3000g, for 20 minutes. Cell pellets were re-suspended in 20 mM phosphate buffer (pH 6.5) and lysed via sonication (40 amplitude, 30 second burst, 20-minute total process). Cell lysate was clarified at 16000g, 4 °C for 30 minutes, and the resulting supernatant was heated at 80 °C for 15 minutes. After cooling, the precipitated proteins were removed by centrifugation (16000g, 4 °C for 30 minutes). The supernatant was further purified by ion-exchange chromatography on Mono Q columns in 20 mM phosphate buffer, pH 6.5, using a linear gradient of 10–30% buffer containing 0.5 M NaCl. Fractions of the purified proteins were collected and the homogeneity of the protein was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). Purified protein was buffer exchanged to 10 mM Tris (pH 7.5) and measured by Pierce® BCA Protein Assay Kit as recommended.
POP crystallization.
Crystals of native wild-type and S477C Pyrococcus furiosus prolyl oligopeptidase were grown at 20°C using the hanging-drop vapor-diffusion technique. A highly purified (>95%, determined by SDS-PAGE) protein solution of 8 mg/mL (determined by A280) in H2O was used. Crystallization drops contained 1 μL protein solution with 1 μL of well solution containing 30% PEG 8K and 100 mM Tris•HCl pH 9.0. Diffraction quality crystals were obtained after 4 months of incubation. Crystals were harvested and cryoprotected in a solution of 15% glycerol in well solution. These were then flash frozen in liquid nitrogen and mounted for data collection. Details regarding data collection and structure refinement are provided in the supporting information.
Molecular dynamics simulations.
Initial configurations for the WT and S477C simulations were taken from the corresponding crystal structures, while the IHBT system was constructed by fusing the covalent inhibitor from a crystal structure of the porcine POP-inhibitor complex (1QFS) to the WT structure. MD simulations were performed for the wildtype (WT), inhibitor ZPR bound (IHBT), and the mutant (S477C) Pfu POP enzymes. The chains with a lower B-factor in the crystal structures of the wildtype and the mutant in this report were used to generate the simulation systems (chain B for WT and chain A for S477C). Residual pKa calculations were performed using PROPKA3.120 and the protonation states of the protonatable residues were assigned based on the calculated pKa values. The wildtype chain B was aligned to other POP crystal structures that are closed with inhibitor bound to model the coordinates of the covalently linked inhibitor ZPR. All the three structures (i.e., WT, IHBT, and S477C) were bathed in 0.15M KCl solution using the Solvator Module in CHARMM-GUI21. Three replicas for each system were generated by using different random seeds to distribute the ions during the solvation process.
Once the simulation systems were generated, they were subjected to equilibrating simulations at 300 K and 358.15 K. The equilibration took two stages. In the first stage, harmonic positional restraints were imposed on the non-hydrogen atoms in the protein and were gradually released during 675 ps of the simulation. The system was then equilibrated without restraints for an additional 10 ns. The equilibration simulations were performed using OpenMM6.2 simulation package21,22 on GeForce GTX 780 Ti GPU processors. After equilibration, the systems were simulated for 1.44 μs each using the special-purpose supercomputer Anton designed for long time scale simulations23 with the exception for systems WT_2, WT_3, IHBT_2, and IHBT_3. These systems were simulated for 2.88 μs each.
The input files for the OpenMM equilibration simulations are taken from CHARMM-GUI Quick MD Simulator module21. These simulations are performed in an NPT ensemble with the temperature set to either 300 K or 358.15 K and the isotropic pressure set to 1 atm. The Langevin thermostats with a damping coefficient of 1 ps−1 are used to keep the temperature constant. The OpenMM package uses a Monte Carlo algorithm to adjust the size of the periodic box, therefore simulating the effect of constant pressure. The van der Waals interactions were smoothly switched off at 10–12 Å by a force switching function24 and the electrostatic interactions were calculated using the particle-mesh Ewald method with a mesh size of ~1 Å25.
The production simulations on Anton are performed with the NPT ensemble. The Nosé-Hoover thermostat26 was used to maintain the system temperature. The lengths of all bonds involving hydrogen atoms were constrained using M-SHAKE27. The cutoff of the van der Waals and short-range electrostatic interactions was set to ~12 Å as suggested by the guesser script. Long-range electrostatic interactions were evaluated using the k-space Gaussian split Ewald method28 with a 64 × 64 × 64 mesh. The r-RESPA integration method29 was employed and long-range electrostatics were evaluated every 6 fs. The additive C36 force field was used in all the simulations performed here30–33. The force field parameters for the covalently linked ZPR inhibitor were generated using the GAAMP server34. Additional details regarding simulations of the loop containing H592 and of chloride binding are provided in the supporting information.
RESULTS
Pfu POP Structural Overview
The structures of wild type Pfu POP and the corresponding S477C mutant were solved at 1.9 Å and 2.2 Å resolution (Table 1). As observed in previously reported POP structures,2 both Pfu POP and the S447C mutant possess a two-domain architecture involving a peptidase domain with an α/β-hydrolase fold capped by a 7-bladed β-propeller domain (Fig. 2A–C). The N-terminus of the enzyme (residues 1–47) consists of an α-helical segment that wraps around the C-terminal peptidase domain (residues 367–616). The intermediate segment (residues 48–366) comprises the β-barrel domain. The first and seventh blade of the propeller domain (residues 48–83 and 321–366, respectively) are joined by a number of hydrogen bonding and ion-pairing interactions in addition to hydrophobic interactions between the blade surfaces. The peptidase and β-barrel domains are joined covalently via a hinge region comprised of residues 47–50 and residues 361–367. An inter-domain angle of ~30° allows access to a large host cavity at the domain interface. Residues comprising the catalytic triad reside at the peptidase/barrel interface, with S/C477 located centrally within the peptidase domain and D560 and H592 located in flexible loops at the peptidase periphery (Fig. 2D).
Table 1.
X-ray Diffraction Data Collection and Refinement Statisticsa
| Pfu POP (5T88) | S477C (6CAN) | |
|---|---|---|
| Data collection | ||
| wavelength (Å) | 0.98 | 0.98 |
| space group | P21 | P21 |
| unit cell dimensions | ||
| a, b, c (Å) | 55.5, 176.8, 57.9 | 56.3, 178.8, 59.3 |
| α, β, γ (°) | 90, 106.0, 90 | 90, 104.4, 90 |
| res. limit (Å) | 50–1.9 (1.97–1.9) | 100–2.2 (2.28–2.2) |
| I/Sigma(I) | 12.3 (1.5) | 9.5(1.2) |
| Rpim (%) | 5.9(48.3) | 7.1(44.0) |
| highest shell resol. CC1/2(%) | 57.5 | 53.0 |
| completeness (%) | 99.9(100.0) | 93.7(74.6) |
| redundancy | 9.7 (9.3) | 4.5(3.5) |
| Refinement | ||
| resolution range (Å) | 47.1 – 1.9 | 19.5 – 2.2 |
| Rwork (%) | 19.3 | 19.7 |
| Rfree (%) | 24.7 | 25.1 |
| R (working + test) (%) | 19.4 | 19.7 |
| no. of reflections | 83441 | 53366 |
| Model | ||
| no. of amino acids | 1232 | 1232 |
| no. of H2O mol. | 946 | 842 |
| no. Cl− ions | 4 | 6 |
| no. residues: | ||
| in generously allowed region | 12 | 13 |
| in disallowed regions | 0 | 0 |
| Stereochemical ideality | ||
| bonds (Å) | 0.01 | 0.01 |
| angles (°) | 1.44 | 1.24 |
| dihedral angles (°) | 11.39 | 16.92 |
| planarity (Å) | 0.01 | 0.00 |
Value in parentheses are for the outer shell.
Figure 2.

A-C) Annotated ribbon diagram of Pfu POP showing inter-domain angle (θ), blade (b1–7) and hinge locations, and key active site residues and Cl− ions. D) expanded view of POP active site showing residues in the catalytic triad, bound chloride ions, and the Pro-Pro ligand (omitted in A-C for clarity).
Inter-domain Loop Composition and Structure
The composition and conformation of loops at the domain interface of different POPs possess many unique features,35 and notable differences between the loops in these enzymes and Pfu POP are like-wise apparent (Fig. S1). Extensive ion pairing is observed between residues located in these loop regions in closed POP structures,7–10,13 but only a single inter-domain ion pair is present in the Pfu POP crystal structures. The loop containing the catalytic histidine (H592, residues 588–597) possesses a relatively high average B-factor in both structures (46.8/64.0 Å2 versus 32.4/45.9 Å2 for the full chains of Pfu and S477C, respectively, Table S1), the density of the main chain and the residues of this loop were apparent in the observed electron density (Fig. 3A, S11). This finding is notable because the histidine loop is disordered in all previously reported structures of POPs that crystallized in their open form9,10. Finally, the loop connecting β-strands 2 and 3 of blade 3 of the POP β-propeller domain (residues 158–169) is significantly shorter than in previously characterized homologues, and it is folded back onto the surface of the β-propeller domain (Fig. 3B). This conformation is enforced by two sequential proline residues (P168 and P169) and a salt bridge between the end of the loop (D164) and the β-propeller domain (R172). This orientation contrasts with other POP structures in which residues near the end of this loop often exhibit hydrogen-bonding and/or salt bridging interactions with the peptidase domain that “latch” the enzyme shut.9
Figure 3.

Electron density (contoured at 1.0 σ) for key features in the PfU POP crystal structure, including: A) the loop containing His592, B) the “latch loop”, C) a bound prolylproline ligand and bound chloride ions.
Prolylproline and Chloride Binding within Pfu POP.
The active site of Pfu POP exhibits many features common to other POPs crystallized to date.3 The hydrophobic proline-binding site (S1), comprised of F404, W518, and Y522, is conserved among all previously crystalized POPs,3 and the active-site serine (S477) is predictably positioned within a GXSXGG motif (Fig. 3C)36. Notably, however, both chains of the Pfu POP structure contain electron density consistent with a bound prolylproline ligand, albeit with a significantly higher B-factor (58.0 Å2) than the main chains (32.4 Å2). The source of this ligand could not be determined, but we speculate that it could result from POP-catalyzed cleavage of other proteins or itself during the purification process. The orientation of this ligand is similar to proline-based inhibitors in previously reported crystal structures of porcine POP in a closed conformation (Fig. S10).37 In these structures, the proline residue binds in a hydrophobic S1 pocket while the carbonyl of the P1-P2 amide bond forms a hydrogen bond with R643. Similarly, in the Pfu POP structure, the C-terminal proline carboxylate of the prolylproline ligand forms an ion pair with the homologous R562 residue.
The Pfu POP crystal structures also possess multiple Cl− ions bound within the active site (Fig. 3C, Tris•HCl was present in the crystallization solutions). WT Pfu POP contains two Cl− ions bound via opposing arginine residues (R476 and R600) proximal to the active site serine. Each Cl− is coordinated in a bidentate, end-on fashion to one arginine and in a monodentate, side-on fashion to the second with B-factors of 45 and 20 Å2, respectively. In the S477C mutant, both of these sites are occupied by Cl− ions with B-factors of 58 and 39 Å2, and an additional chloride is also bound in the P1 pocket with a B-factor of 53 Å2.
MD Simulations of Pfu POP Domain Dynamics
Microsecond MD simulations were carried out to investigate the domain dynamics of Pfu POP at 300 K and its optimal temperature for peptidase catalysis (358.15 K). Simulations were conducted in triplicate on wildtype Pfu POP (WT), WT containing a covalently linked CbzProProlinal inhibitor (IHBT, Fig. S1), and the inactivated S477C mutant of WT (S477C), all hereafter referred to as “apo” states because they lack a covalently attached cofactor. The simulations show that the apo systems open and close spontaneously as the peptidase and the propeller domains pivot about a hinge region (Fig. 1B). An inter-domain angle (θ) was defined (Fig. 1A, S2) to quantify domain opening, and the time evolution of 6 was evaluated at both 300K and 358.15K (Figs. 4 and S3). The open/closed transition occurs at a 6 of ~17–23°, and θ ranges from 8–55°. At 358.15 K, the average θ is 20.0° ± 1.8°, 24.4° ± 4.2°, and 23.3° ± 0.9° in the WT, IHBT, and S477C simulations, respectively (Table S2). In general, the high temperature simulations showed slightly increased fluctuation of θ and more open/close transitions.
Figure 4.
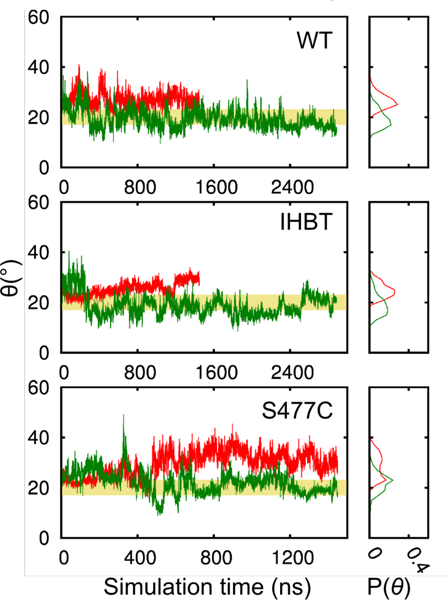
The open angle and its distribution in the MD simulations at 300 K (red) and 358.15 K (green) in replica 2 of the WT, IHBT, and S477C systems. The yellow banner indicates the intermediate open angle values between the open and the closed states.
Inter-domain opening renders the active site solvent-accessible, which is readily visualized in trajectories from the IHBT simulations (Fig. 5A and B). To quantitate this opening, the solvent-accessible area between each propeller blade and the peptidase domain was calculated for the apo systems (Fig. S4). This analysis shows that the largest opening occurs between blade 3 of the propeller domain and the peptidase domain (Tables S3 and S4). In the WT simulations, the average solvent accessible area is 219 ± 74 Å2, even at the most constricted region along the blade 3 opening axis. Large fluctuations in the loop spanning D505-Y522 in the peptidase domain, opposite to blade 3, were also observed (Fig. S5). In contrast, the narrowest opening in the central pore of the propeller domain, which has been proposed as an alternate substrate entry site10, has a solvent-accessible area of only 93 ± 24 Å2 in the WT simulations. No significant opening between blades 1 and 7 of the propeller domain was observed in any of the simulations.
Figure 5.

The cartoon and surface representation showing the largest opening between propeller (light grey) blade b3 and the peptidase domain (dark grey). The snapshots are taken from the covalently-linked inhibitor simulation IHBT_3 to illustrate (A) the closed state and (B) the open, substrate-accessible state enzyme. The bottom panel shows a surface presentation of the same snapshot. The covalently bound inhibitor is colored green.
Histidine Loop Conformational Dynamics
Large-scale domain movements are accompanied by smaller-scale conformational changes throughout the structures examined, but particularly notable are the dynamics of the histidine (H592) loop. In two simulations, the histidine loop achieves a conformation that places H592 within 4 Å from S477, its approximate orientation in crystal structures of POP enzymes in their closed forms (Fig. 6). The process of loop closing primarily involves changes in the φ - ψ back-bone dihedral angles (Fig. S6) and sequential interaction of H592 with hydrogen bond acceptors on D94, G117, A118, D119, D561, and R476 (Fig. 7). Histidine loop dynamics were also evaluated starting from a model of Pfu POP in the closed form (see supporting information). The similar φ/ψ dihedral angles distribution from the trajectories of the engineered His loop and WT systems suggest that similar dynamics are observed from either the open or closed state of Pfu POP (Fig. S6).
Figure 6.

The time evolution of the S477 and H592 distance, DS477-H592. The hydroxyl oxygen (OG) in S477 and the imidazole amide hydrogen (HE2) are used to define the distance.
Figure 7.

Sequential steps in the H-bond network H592 uses to get into the catalytic site (A-F). The contributing residues are shown in cyan sticks. The H592 look is in green and the residues H592 and S477 are colored in yellow.
Effects of Halide Binding on Histidine Loop Conformational Dynamics
As noted above, there are two Cl− binding sites in the WT Pfu POP crystal structure and three in that of the S477C mutant. The sites involving bidentate Cl− coordination by the guanidinium groups of R476 and R600, respectively, are common to both structures. The bidentate-R600 site has the lowest B-factors in the crystal structures, an intermittent binding to this site is observed in MD simulations. Starting from crystal structures with this site occupied by a Cl−, both dissociation and association of Cl− can be observed (Table S6); Cl− ions bound at other sites dissociate early in the simulations and do not again associate. Interestingly, Cl− binding to the R476/R600 site has a minor effect on the S477-H592 distance distribution in the WT and the inhibitor bound systems, but not for S477C (Fig. 8). The effect manifests itself as a shift in the distance distribution towards the lower end of the ranges observed (a shorter S477-H592 distance). The Cl− binding also affects the inter-domain angle but to a lesser extent (Fig. S7).
Figure 8.

The impact on S477-H592 distance from the binding of Cl− to the R600 site. The site is occupied when the Cl− is within 1.5 A to the crystal binding site of this Cl−. The transparent, filled curves show the normalized population (Pnorm) distributions of DS477-H592 computed from the entire trajectory at 358.15 K. The thick, colored lines are the Pnorm distributions of DS477-H592 drawn from when Cl− is bound to the R600 site. The three replicas are shown in red, green, and blue.
DISCUSSION
Structural Origins of Pfu POP Stability
The stability of Pfu POP makes this enzyme a promising candidate for biocatalytic applications, and the crystal structures reported herein provide a number of insights into the origins of its stability. Comparing the molecular weights of Pfu POP (70.8 kDa) to those of the other POPs crystallized to date (76–83 kDa) highlights the relatively small size of the former. Aligning the Pfu POP sequence to those of other POPs crystalized to date shows that this smaller size results primarily from shortened loop regions between structurally conserved β-sheets in the propeller domain (Fig. S8). The structures of Pfu POP and the corresponding S477C mutant also have an average of 0.10–0.11 ion pairs/residue versus 0.061–0.090 for the other (mesophilic) POP enzymes crystalized to date (Table 2). Shortened loop regions and increased ion pair content have both been proposed to contribute to the stability of hyperthermophilic enzymes.38,39
Table 2.
Structural data for previously crystalized POPs.
| POPa | MWb | Additional Residuesc |
Ion Pairsd | Inter domain Angle (°)d |
||
|---|---|---|---|---|---|---|
| Peptidase domain |
Propeller domain |
Per residue |
Inter- domain |
|||
| Pfu | 70824 | - | - | 0.11 | 1 | 29 |
| S477C | 70840 | - | - | 0.11 | 0 | 32 |
| Pfu_mod | 70824 | - | - | 0.084 | 5 | 19 |
| M. xanthus | 76848 | 14 | 28 | 0.086 | 5 | 21 |
| R. typhi | 83074 | 27 | 54 | 0.061 | 3 | 23 |
| S. scrofa | 80770 | 27 | 40 | 0.076 | 4 | 22 |
| H. sapiens | 80700 | 27 | 40 | 0.061 | 3 | 22 |
| A. caviae_c | 76467 | 19 | 24 | 0.078 | 2 | 25 |
| A. caviae_o | 0.070 | 0 | 47 | |||
| S. capsulata | 78435 | 11 | 34 | 0.064 | 0 | 58 |
Pfu chain B from 5T88; Pfu S477C chain B from 6CAN; Pfu_mod from a previously reported homology model; M. xanthus chain A from 2BKL; R. typhi from 4HVT; S. Scrofa from 1QFS; H. sapiens from 3DDU; A. caviae_c from 3IVM; A. caviae_o from 3IUJ; S. capsulata from 1YR2.
Determined using the ExPASy server.
relative to Pfu.
Determined using VMD or UCSF Chimera as described in the SI.
Conformational Dynamics of Pfu POP
Despite the stability of Pfu POP, earlier kinetic analyses of this enzyme suggested that it might undergo significant conformational changes during catalysis.13–15 Both open9,10 and closed7–10,13 conformations of previous POP crystal structures or homology models can be distinguished by aligning their peptidase domains to that of the Pfu POP crystal structures (RMSD = 0.18–1.06 Å2, Fig. S8). Using the inter-domain angle (θ) definition described above, closed conformations for previously studied structures have θ < 25°, while θ values of 47° and 58° were reported for open structures. At 29 and 32°, θ values for Pfu POP fall between these extremes, suggesting that it crystalized in an intermediate conformation. The single inter-domain ion pair in Pfu POP is also intermediate between these enzymes (Table 2), suggesting that ions pairs that could contribute to the stability of the closed form of the enzyme have only partially formed.
While crystal structures show that different domain angles can be achieved in different structures (Fig. S1), and both NMR spectros-copy40,41 and MD simulations have been used to study conformational dynamics in individual POPs, none of the previous MD studies reported spontaneous domain opening or closing during simulations16,35,42–45. The microsecond MD simulations conducted herein show, for the first time, spontaneous conversion between the open and closed conformations of POP molecules (Fig. 3 and S3). The peptidase and propeller domains move largely as rigid bodies with important exceptions outlined below. Average θ values are similar to those observed in the crystal structures (Tables 2 and S2), but θ values ranging from 8–55° are sampled, consistent with the notion that the 29 or 32° angles observed in the crystal structures are intermediate in nature. The inter-domain ion pairs observed in the crystal structures are observed in all MD simulations, and a negative correlation exists between the number of ion pairs and the inter-domain angle (Fig. S9). The covalently bound ZPR ligand in the IHBT system resulted in slightly larger θ values at 358.15 K, with an average 6 of 24.4° ± 4.2° compared to the 20.0° ± 1.8° in the apo structure. These values fall near the upper end of the ~17–23° θ range that defines the open/closed boundary for Pfu POP, which contrasts some-what with experimental findings for porcine POP that showed inhibitor binding favors the closed form of the enzyme.46 This difference could reflect either fundamental differences in how the dynamics of Pfu and porcine POP respond to ZPR binding or insufficient simulation time to sample the lowest energy substrate-bound closed state of Pfu POP.
Large-scale domain dynamics of POPs have been proposed to be modulated by a so-called “latching loop” (i.e., R158-P169 in Pfu POP). Previously reported POP structures show this loop bound to the peptidase domain in closed structures or disordered in open structures.7,10 The “latching” action of the loop observed in these structures is primarily mediated through polar and ionic interactions. A homology model of Pfu POP built from the porcine structure predicted that this loop would extend to the peptidase domain,13 contrary to the conformation observed in the Pfu POP crystal structures. The conformation of the “latching loop” in Pfu POP remains relatively constant throughout all MD simulations and makes minimal contact with the hydrolase domain in the closed structures from the simulations. There is no persisting pattern of interactions between the “latching loop” and the hydrolase domain, which could contribute to the apparent favorability of the open form of Pfu POP.
The conformation of the histidine (H592) loop also plays a pivotal role in POP peptidase catalysis. In the other POP crystal structures reported to date, the histidine loop was modeled only in closed structures7,9 and omitted from open structures due to poor electron density.9 This trend holds for the structures of most other POP family enzymes, although the histidine loop of oligopeptidase B from T. Brucei47 was resolved in the structures of both the open and closed conformations. While the closed form of Pfu POP was not observed in any of the crystal structures, overlaying the structures obtained with those for other closed POPs shows that H592 would have to travel 7–12 Å to achieve an orientation similar to the closed structures (Fig. S1). The structure of Pfu POP thus offers a rare view47 of the position of the catalytic histidine in a POP prior to formation of a competent catalytic triad.
Two approaches were used to examine the conformational dynamics involved in forming active site configurations analogous to those observed in closed POP structures. In the first, triplicate MD simulations were performed on the WT, IHBT, and S477C systems as discussed in the His Loop Conformational Dynamics section above. During these simulations, a number of open-to-closed transitions were observed (Figs. 3 and S3), but only two of these events led to the formation of active site configurations analogous to those observed in the structures of closed, inhibitor-bound POPs (Figs. 9 and S1). The slight structural deviation from the closed structures of these covalently inhibited structures is in part due to the fact that their formation requires bond breakage and formation, including changes in the protonation state of H592. This residue was deprotonated in the beginning of the simulation based on its calculated pKa value of 6.1. The starting structure does not, however, capture the electrostatic environment near H592 when catalytic triad formation is near complete. Indeed, the calculated pKa of the corresponding histidine is raised to around 7 in crystal structures with a fully formed catalytic triad (Fig 9). Nonetheless, competent-like active site configurations were only seen in snapshots of closed POP structures with small open angles, suggesting that domain closing shifts the conformational distribution of the H592 loop to an orientation suitable for catalysis. In the second approach, model structures of Pfu POP in its closed form were generated using the H592 loop ϕ/ψ angle information from the crystal structures of POP in closed state. The MD simulations of these model structures without any inhibitors bound show that the H592 loop rapidly moves away from the closed conformation to adopt an orientation similar to that seen in the open conformation. The observed dynamics of these model systems after the H592 loop moves away from the active site are similar to those observed in the WT simulations according to the ϕ/ψ angle distributions for both systems (Fig. S6), providing a clear picture of the large-scale inter-domain opening/closing that occurs in Pfu POP.
Figure 9.

Overlay of closed crystal structures with a simulation snapshot with a closed H592 loop. Important residues are shown with sticks. The pKa values of the residues that correspond to H592 are 6.6, 6.6/6.9, 7.2, and 6.9 for 1QFS, 2BKL, 3IVM, and 4HVT. The pKa values are calculated using the program PROPKA3120.
Effects of Pfu POP Dynamics on Peptidase Catalysis
Crystal structures and MD simulations for different POPs suggest that these enzymes undergo large conformational changes, but the extent to which these changes influence POP catalysis (e.g. substrate binding, formation of a competent active site, etc.) remains an open question. Several findings from previous studies provide key insights into different steps of Pfu POP peptidase catalysis (Fig. 1A): 1) The rate-limiting step in peptide hydrolysis differs at lower temperatures (25–40 °C) versus higher temperatures (40–95 °C), according to observed non-linearity in a plot of ln(kcat/T) vs (1/T).15 2) A 70–80-fold increase in k1 was observed from 25–55 °C, suggesting a large kinetic barrier for formation of an enzyme-substrate complex at lower temperatures.15 3) A plot of kcat versus temperature for hydrolysis of Z-Gly-Pro-pNA was linear between the ranges reported (60–90 °C), indicating that a single elementary step was rate limiting over this range.15 4) Peptide hydrolysis at high temperatures (56 and 85 °C) exhibited a solvent isotope effect, implicating a rate-limiting chemical step in this range.13,15 5) Lack of a leaving group effect at high temperatures (55–75 °C) was observed, which, in conjunction with points 3 and 4, suggests that k3 (chemical hydrolysis of the acyl intermediate) is rate-limiting at high temperatures.15 6) A sigmoidal increase in kcat/KM as a function of [NaX] (X = Cl, Br, F) and no change in kcat was observed for Pfu POP up to ~2M NaX, suggesting that halide binding activates Pfu POP hydrolase activity (~2–3 fold) via a decrease in Km.13 Thus, while a chemical step is rate limiting at high temperatures, substrate binding, and any conformational changes associated with it, are rate limiting below 40 °C.
Based on these findings, it was previously hypothesized that Pfu POP has a stable, closed structure at low temperature, and that the high barrier to substrate binding arises from a high-barrier conformational change between open- and closed-states (Fig. 10A).15 The crystal structure of Pfu POP shows that an open form of the enzyme is in fact readily accessible, that the enzyme possesses several Cl− binding sites, and that a dipeptide can bind within the open active site (Fig. 3C). MD simulations allowed for observation of the open/closed transition, and the same dynamics are observed in simulations of WT, a model of closed WT, and IHBT (Figs. 4 and S3). These findings are consistent with low barrier binding equilibria involving substrate, Cl−, and the open and closed forms of Pfu POP, in which formation of substrate/Cl−-bound open (observed by X-ray crystallography) and closed (observed in MD simulations) forms of the enzyme can be generated. The previously noted rate limiting formation of the ES complex below 40 °C could result not from domain opening but from conversion of a closed, substrate-bound form of the enzyme to one in which the catalytic triad is fully formed and poised for attack of substrate (Fig. 10B)48. While such a state is beyond the ability of classical MD simulations to model, conformations resembling those of closed structures with covalent inhibitors bound were observed in simulations of dynamics at 358.15 K but not at 300 K, consistent with the idea that accessing this state is rate limiting at low temperatures.
Figure 10.

Conceptual summary of updated models for A) the inter-domain angle of the Pfu POP, B) the rate limiting step of Pfu POP peptidase catalysis, C) substrate entry into Pfu POP, and D) effects of chloride binding within Pfu POP (S = substrate).
The Pfu POP crystal structures and MD simulations also provide insight into substrate entry into the enzyme (Fig. 10C). Specifically, the 219 ± 74 Å2 opening at the narrowest region between blade 3 and the peptidase domain can accommodate a peptide chain (WT Replica 2 in Table S4). The loop lining the edge of this opening within the peptidase domain (D505-Y522) exhibits large fluctuations in the MD simulations (Fig. S5). Fluctuation of this loop could act to facilitate substrate entry into the POP enzyme interior. While a small opening (71–112 Å2 at 358.15K, Table S5) is observed at the pore lined by the seven p-blades in the propeller domain, this would only be sufficient for entry of peptides containing small residues, contrary to the reported substrate scope. No opening is observed between the propeller blades 1 and 7 (b1/b7), so substrate entry from this region is also unlikely. Entry through the much wider opening between the propeller and peptidase domains therefore seems most reasonable for Pfu POP.
Finally, the observation of three distinct halide binding sites between WT and S477C Pfu POP structures (Fig. 2) is qualitatively consistent with the aforementioned activation by halide.13 Halide binding to R476 and R600 in particular provides a structural rationale for the impact of halide concentration on POP activity (Fig. 10D). Forming the catalytic triad in Pfu POP requires a large conformational change (Fig. 6) to position H592 between D560 and S477, which move independent of one another. The presence of R476 immediately before to S477 and of R600 in the helix adjacent to the loop containing H592 provides a means to orient the secondary structures containing these residues upon halide binding. By securing the orientation of these secondary structures, the salt bridge helps position S477 and H592 to form the catalytic triad. This hypothesis is consistent with the results of MD simulations showing shorter H592-S477 distances and smaller inter-domain angles (Figs. 3, 6, and S3) when chloride is bound versus when it is not. This mechanistic proposal, which involves active site pre-organization to facilitate substrate binding, would be reflected in Km, consistent with the kinetic analyses noted above and reminiscent of previous work on angiotensin converting enzyme49.
CONCLUSION
The crystal structures of WT Pfu POP and its S477C mutant reveal that both enzymes adopt an open structure with an intermediate inter-domain angle relative to other POP enzymes reported to date. MD simulations show that the conformation observed in the crystal structures is indeed intermediate between much larger extremes that can be sampled by the enzyme. The observed dynamics show that substrates can access the Pfu POP active site via the inter-domain opening, which lacks a “latch” present in other structures, while other previously proposed openings are much smaller and show no significant opening during MD simulations.
The Pfu POP crystal structures provide rare glimpses of the loop containing the catalytic histidine (H592) in an open POP structure. MD simulations show that H592 can access conformations analogous to those observed in previously reported structures of POP-inhibitor complexes. The structure of WT Pfu POP also contains a bound prolylproline ligand, showing that substrate-like compounds remain bound in the open form of the enzyme. Up to three bound chloride ions were observed in the different Pfu POP structures, and those bound to R476 and R600 provide a rationale for previously observed halide activation involving active site preorganization. MD simulations show that chloride binding at these sites alters both histidine loop conformation and the inter-domain angle to favor a more closed form of the enzyme.
Together, these results resolve a number of questions in the literature regarding native Pfu POP structure, inter-domain dynamics, substrate entry, and activation by halide ions. Specifically, our analysis reveals the importance of facile inter-domain opening, rate-limiting EA formation or hydrolysis, and halide-induced active site preorganization during Pfu POP catalysis. We anticipate that these findings will facilitate further development of biocatalytic applications involving Pfu POP.17,18
Supplementary Material
ACKNOWLEDGMENT
This work was supported by, or in part by, the U.S. Army Research Laboratory and the U. S. Army Research Office under contract/grant numbers W911NF-14–1-0334 and 66796-LS-RIP (to J.C.L.) and W911NF-18–1-0200 (to J.C.L. and B.R.), and the NSF under CAREER Award CHE-1351991 (to J.C.L.), and The David and Lucile Packard Foundation (to J.C.L.). This work is based on research conducted at the APS on the NE-CAT beamlines, which are supported by a grant from the NIGMS (P41 GM103403) from the NIH. The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). Use of the APS, an Office of Science User Facility operated for the U.S. DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02–06CH11357. This research was supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative. The Indiana METACyt Initiative at IU was also supported in part by Lilly Endowment, Inc.
ABBREVIATIONS
- Pfu
pyrococcus furiousus
- POP
prolyl oligopeptidase
Footnotes
Supporting Information
Complete experimental and computational procedures, supplemental tables and figures, and crystallographic data are available. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Polgár L The Prolyl Oligopeptidase Family. Cell Mol Life Sci 2002, 59 (2), 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Szeltner Z; Polgar L Structure, Function and Biological Relevance of Prolyl Oligopeptidase. Curr. Protein Pept. Sci. 2008, 9 (1), 96–107. [DOI] [PubMed] [Google Scholar]
- (3).Rea D; Fülöp V Structure-Function Properties of Prolyl Oligopeptidase Family Enzymes. Cell Biochem. Biophys. 2006, 44 (3), 349–365. [DOI] [PubMed] [Google Scholar]
- (4).Ehren J; Govindarajan S; Moron B; Minshull J; Khosla C Protein Engineering of Improved Prolyl Endopeptidases for Celiac Sprue Therapy. Prot. Eng. Des. Sel. 2008, 21 (12), 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Walter R; Shlank H; Glass JD; Schwartz IL; Kerenyi TD Leucylglycinamide Released From Oxytocin by Human Uterine Enzyme. Science 1971, 173 (3999), 827–829. [DOI] [PubMed] [Google Scholar]
- (6).Kaushik S; Sowdhamini R Distribution, Classification, Domain Architectures and Evolution of Prolyl Oligopeptidases in Prokaryotic Lineages. BMC Genomics 2014, 15 (1), 985–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fülöp V; Böcskei Z; Polgar L Prolyl Oligopeptidase: an Unusual B-Propeller Domain Regulates Proteolysis. Cell 1998, 94 (2), 161–170. [DOI] [PubMed] [Google Scholar]
- (8).Haffner CD; Diaz CJ; Miller AB; Reid RA; Madauss KP; Hassell A; Hanlon MH; Porter DJT; Becherer JD; Carter LH Pyrrolidinyl Pyridone and Pyrazinone Analogues as Potent Inhibitors of Prolyl Oligopeptidase (POP). Bioorg. Med. Chem. Lett. 2008, 18 (15), 4360–4363. [DOI] [PubMed] [Google Scholar]
- (9).Shan L; Mathews II; Khosla C Structural and Mechanistic Analysis of Two Prolyl Endopeptidases: Role of Interdomain Dynamics in Catalysis and Specificity. Proc. Natl. Acad. Sci. U.S.A. 2005, 102 (10), 3599–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li M; Chen C; Davies DR; Chiu TK Induced-Fit Mechanism for Prolyl Endopeptidase. J. Biol. Chem. 2010, 285 (28), 21487–21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Harwood VJ; Denson JD; Robinson-Bidle KA; Schreier HJ Overexpression and Characterization of a Prolyl Endo-peptidase From the Hyperthermophilic Archaeon Pyrococcus Furiosus. J. Bacteriol. 1997, 179 (11), 3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Harwood VJ; Schreier HJ Prolyl Oligopeptidase From Pyrococcus Furiosus. Meth. Enzymol. 2001, 330, 445–454. [DOI] [PubMed] [Google Scholar]
- (13).Harris MN; Madura JD; Ming L-J; Harwood VJ Kinetic and Mechanistic Studies of Prolyl Oligopeptidase From the Hyperthermophile Pyrococcus Furiosus. J. Biol. Chem. 2001, 276 (22), 19310–19317. [DOI] [PubMed] [Google Scholar]
- (14).Juhasz T; Szeltner Z; Polgar L Properties of the Prolyl Oligopeptidase Homologue From Pyrococcus Furiosus. FEBS Letters 2006, 580 (14), 3493–3497. [DOI] [PubMed] [Google Scholar]
- (15).Juhasz T; Szeltner Z; Polgar L Truncated Prolyl Oligopeptidase From Pyrococcus Furiosus. Proteins 2007, 69 (3), 633–643. [DOI] [PubMed] [Google Scholar]
- (16).Kaushik S; Etchebest C; Sowdhamini R Decoding the Structural Events in Substrate-Gating Mechanism of Eukaryotic Prolyl Oligopeptidase Using Normal Mode Analysis and Molecular Dynamics Simulations. Proteins 2014, 82 (7), 1428–1443. [DOI] [PubMed] [Google Scholar]
- (17).Yang H; Swartz AM; Park H-J; Srivastava P; Ellis-Guardiola K; Upp DM; Lee G; Belsare K; Gu Y; Zhang C; Moellering RE; Lewis JC Evolving Artificial Metalloenzymes via Random Mutagenesis. Nat. Chem. 2018, 10 (3), 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gu Y; Guardiola KE; Srivastava P; Lewis JC Preparation, Characterization, and Oxygenase Activity of a Photocatalytic Artificial Enzyme. Chem Bio Chem 2015, 16 (13), 1880–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Heckman KL; Pease LR Gene Splicing and Mutagenesis by PCR-Driven Overlap Extension. Nat Protoc 2007, 2 (4), 924–932. [DOI] [PubMed] [Google Scholar]
- (20).Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2010, 7 (2), 525–537. [DOI] [PubMed] [Google Scholar]
- (21).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: a Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
- (22).Eastman P; Friedrichs MS; Chodera JD; Radmer RJ; Bruns CM; Ku JP; Beauchamp KA; Lane TJ; Wang L-P; Shukla D; Tye T; Houston M; Stich T; Klein C; Shirts MR; Pande VS OpenMM 4: a Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2012, 9 (1), 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shaw DE; Dror RO; Salmon JK.; Grossman J; Mackenzie KM; Bank JA; Young C; Deneroff MM; Batson B; Bowers KJ. Proceedings of the Conference on High Performance Computing Networking Storage and Analysis; Acm: 2009. [Google Scholar]
- (24).Steinbach PJ; Brooks BR New Spherical-Cutoff Methods for Long-Range Forces in Macromolecular Simulation. J. Comput. Chem. 1994, 15 (7), 667–683. [Google Scholar]
- (25).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. The Journal of Chemical Physics 1995, 103 (19), 8577–8593. [Google Scholar]
- (26).Martyna GJ; Klein ML; Tuckerman M Nose-Hoover Chains: the Canonical Ensemble via Continuous Dynamics. The Journal of Chemical Physics 1992, 97 (4), 2635–2643. [Google Scholar]
- (27).Kräutler V; van Gunsteren WF; Hünenberger PH A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22 (5), 501–508. [Google Scholar]
- (28).Shan Y; Klepeis JL; Eastwood MP; Dror R O.; Shaw, D. E. Gaussian Split Ewald: a Fast Ewald Mesh Method for Molecular Simulation. The Journal of Chemical Physics 2005, 122 (5), 054101–054114. [DOI] [PubMed] [Google Scholar]
- (29).Tuckerman M; Berne BJ; Martyna GJ Reversible Multiple Time Scale Molecular Dynamics. The Journal of Chemical Physics 1992, 97 (3), 1990–2001. [Google Scholar]
- (30).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone Φ, Ψ and Side-Chain X 1and X 2Di-hedral Angles. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. The Journal of Chemical Physics 1983, 79 (2), 926–935. [Google Scholar]
- (32).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKe-rell AD Jr; Pastor RW . Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B 2010, 114 (23), 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL Jr; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiorkiewicz-Kuczera J; Yin D; Karplus M All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins f. J Phys Chem B 1998, 102 (18), 3586–3616. [DOI] [PubMed] [Google Scholar]
- (34).Huang L; Roux B Automated Force Field Parameterization for Nonpolarizable and Polarizable Atomic Models Based on Ab Initio Target Data. J. Chem. Theory Comput. 2013, 9 (8), 3543–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Szeltner Z; Juhasz T; Szamosi I; Rea D; Fülöp V; Modos K; Juliano L; Polgar L Biochimica Et Biophysica Acta. BBA-Proteins and Proteomics 2013, 1834 (1), 98–111. [DOI] [PubMed] [Google Scholar]
- (36).Ollis DL; Cheah E; Cygler M; Dijkstra B; Frolow F; Franken SM; Harel M; Remington SJ; Silman I; Schrag J; Sussman JL; Verschueren KHG; Goldman A The A/B Hydrolase Fold. Prot. Eng. Des. Sel. 1992, 5 (3), 197–211. [DOI] [PubMed] [Google Scholar]
- (37).Fülöp V; Szeltner Z; Renner V; Polgar L Structures of Prolyl Oligopeptidase Substrate/Inhibitor Complexes. Use of Inhibitor Binding for Titration of the Catalytic Histidine Residue. J. Biol. Chem. 2001, 276 (2), 1262–1266. [DOI] [PubMed] [Google Scholar]
- (38).Vieille C; Zeikus GJ Hyperthermophilic Enzymes: Sources, Uses, and Molecular Mechanisms for Thermostability. Microbiol Mol Biol R 2001, 65 (1), 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sterpone F; Melchionna S Thermophilic Proteins: Insight and Perspective From in Silico Experiments. Chem. Soc. Rev. 2012, 41 (5), 1665–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kichik N; Tarrago T; Claasen B; Gairi M; Millet O; Giralt E 15N Relaxation NMR Studies of Prolyl Oligopeptidase, an 80 kDa Enzyme, Reveal a Pre-Existing Equilibrium Between Different Conformational States. 2011, 12 (18), 2737–2739. [DOI] [PubMed] [Google Scholar]
- (41).Tarrago T; Claasen B; Kichik N; Rodriguez-Mias RA; Gairi M; Giralt E A Cost-Effective Labeling Strategy for the NMR Study of Large Proteins: Selective 15N-Labeling of the Tryptophan Side Chains of Prolyl Oligopeptidase. Chem Bio-Chem 2009, 10 (17), 2736–2739. [DOI] [PubMed] [Google Scholar]
- (42).Kaszuba K; Rog T; Danne R; Canning P; Fülöp V; Juhasz T; Szeltner Z; Pierre JFS; Garcia-Horsman A; Männistö PT; Karttunen M; Hokkanen J; Bunker A Molecular Dynamics, Crystallography and Mutagenesis Studies on the Substrate Gating Mechanism of Prolyl Oligopeptidase. Biochimie 2012, 94 (6), 1398–1411. [DOI] [PubMed] [Google Scholar]
- (43).Kaushik S; Sowdhamini R Structural Analysis of Prolyl Oligopeptidases Using Molecular Docking and Dynamics: Insights Into Conformational Changes and Ligand Binding. PLoS ONE 2011, 6 (11), e26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).St-Pierre J-F; Karttunen M; Mousseau N; Róg T; Bunker A Use of Umbrella Sampling to Calculate the Entrance/Exit Pathway for Z-Pro-Prolinal Inhibitor in Prolyl Oligopeptidase. J. Chem. Theory Comput. 2011, 7 (6), 1583–1594. [DOI] [PubMed] [Google Scholar]
- (45).Kotev M; Lecina D; Tarragó T; Giralt E; Guallar V Unveiling Prolyl Oligopeptidase Ligand Migration by Comprehensive Computational Techniques. Biophysj 2015, 108 (1), 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).López A; Herranz-Trillo F; Kotev M; Gain M; Guallar V; Bernadó P; Millet O; Tarragó T; Giralt E Active-Site-Directed Inhibitors of Prolyl Oligopeptidase Abolish Its Conformational Dynamics. ChemBioChem 2016, 17 (10), 913–917. [DOI] [PubMed] [Google Scholar]
- (47).Canning P; Rea D; Morty RE; Fülöp V. Crystal Structures of Trypanosoma Brucei Oligopeptidase B Broaden the Paradigm of Catalytic Regulation in Prolyl Oligopeptidase Family Enzymes. PLoS ONE 2013, 8 (11), e79349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Smith AJT; Mueller R; Toscano MD; Kast P; Hellinga HW; Hilvert D; Houk KN Structural Reorganization and Preorganization in Enzyme Active Sites: Comparisons of Experimental and Theoretically Ideal Active Site Geometries in the Multistep Serine Esterase Reaction Cycle. J. Am. Chem.Soc. 2008, 130 (46), 15361–15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu X; Fernandez M; Wouters MA; Heyberger S; Husain A Arg1098 Is Critical for the Chloride Dependence of Human Angiotensin I-Converting Enzyme C-Domain Catalytic Activity. J. Biol. Chem. 2001, 276 (36), 33518–33525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.