

Abstract
Bengal tigers (Panthera tigris tigris) serve a pivotal role as an apex predator in forest ecosystems. To increase our knowledge on factors impacting the viability and health of this endangered species, we studied the gut microbiota in 32 individual Bengal tigers from three geographically separated areas (Chitwan National Park (CNP), Bardia National Park (BNP) and Suklaphanta Wildlife Reserve (SWR)) in Nepal, using noninvasive genetic sampling methods. Gut microbiota influence the immune system, impact various physiological functions, and modulates metabolic reactions, that ultimately impact the host health, behavior and development. Across the tiger populations in Nepal, we found significant differences in the composition of microbial communities based on their geographic locations. Specifically, we detected significant differences between CNP and the other two protected areas (CNP vs BNP: pseudo t = 1.944, P = 0.006; CNP vs SWR: pseudo t = 1.9942, P = 0.0071), but no differences between BNP and SWR. This mirrors what has been found for tiger gene flow in the same populations, suggesting gut microbiota composition and host gene flow may be linked. Furthermore, predictive metagenome functional content analysis (PICRUSt) revealed a higher functional enrichment and diversity for significant gut microbiota in the Chitwan tiger population and the lowest enrichment and diversity in Suklaphanta. The CNP tiger population contained higher proportions of microbiota that are associated with predicted functions relevant for metabolism of amino acid, lipid, xenobiotics biodegradation, terpenoides and polyketides than the SWR population. We conclude the tiger population structure, gut microbiota profile and associated functional metabolic categories are correlated, with geographically most separated CNP and SWR tiger population having the most distinct and different host genotype and microbiota profiles. Our work dramatically expands the understanding of tiger microbiota in wild populations and provides a valuable case study on how to investigate genetic diversity at different hierarchical levels, including hosts as well as their microbial communities.
Introduction
Gut microbiota are a complex community of microorganisms in the intestinal tract that has co-evolved with the host [1, 2] playing an important role in maintaining the host’s health. Gut microbial communities shape the immune system, impact various physiological functions, and modulate metabolic reactions that ultimately impact the host health, fitness, behavior, digestion and development [3–5]. The composition of gut microbiota are largely determined by several intrinsic and extrinsic factors such as the host’s environment, health status, genotype, dietary habits, age, sex, social relationships and disease prevalence [6–10]. For example, the composition of gut microbiota in wildlife may change in response to anthropogenic stresses such as the loss and fragmentation of host habitat [11–14]. Habitat fragmentation could alter the microbiota directly via changes in diet and/or exposure to human associated microbes [15] or indirectly via changes in host genetic structure [16, 17]. The indirect effects of host genetics on gut microbial community structure or ‘phylosymbiosis’ are poorly understood in wild animal populations and are likely interact with other factors such as shifts in diet [17]. Untangling the relative importance of these direct or indirect effects is difficult in wild animal populations (i.e, different populations have different diets) but crucial given the importance of the gut microbiota to the health of individuals and populations.
In the last decade, spurred by technological advances in DNA sequencing, multiple studies have described gut microbiota of various terrestrial and aquatic species, including humans, primates, whales and other mammals [18–20]. Gut microbiome composition is critical for the host’s health and disturbances in the bacterial microbiota might, for example, result in immunological dysregulation that may underlie disorders such as inflammatory bowel disease, Crohn's disease, and ulcerative colitis [21, 22]. The mammalian immune system which appears to control microbes, in fact, might be controlled by the microbes themselves [23]. For example, by stimulating the immune system and the development of gut structure, gut microbes play a crucial role in the regulation of host health by aiding in the defense against invading pathogens and providing nutritional benefit to the host such as the production of short chain fatty acids and vitamin B12 [24]. As microbial communities inhabiting wildlife species greatly affect host health, nutrition, physiology and immune systems, understanding gut microbial community dynamics is increasingly considered crucial for successful wildlife conservation and management programs [7, 25, 26].
Globally, the population of wild tigers is declining dramatically due to widespread habitat loss and fragmentation, prey depletion, illegal hunting and various infectious diseases [27–30]. Within Nepal, habitat loss and fragmentation have forced extant tigers to divide into distinct geographically separated populations in Nepal, which has been extensively studied using long-term field data [31] and noninvasive genetic sampling [32]. The Bengal tiger’s main habitat in Nepal is restricted to five protected areas along the Terai Arc Landscape (TAL), including Chitwan National Park (CNP), Parsa Wildlife Reserve (PWR), Bardia National Park (BNP), Banke National Park (BaNP), and Suklaphanta Wildlife Reserve (SWR) [33, 34]. The TAL has experienced significant land use changes in the recent past [28, 34]. Human settlements surround and encroach into tiger habitat degrading natural areas and potentially increasing levels of environmental stress for tigers. This region has also experienced severe socio-political unrest, which included 10 year civil war during the Maoist insurgency, that has negatively impacted fragile ecosystems with weakened wildlife conservation programs [35, 36]. Considering the degree of environmental degradation in conjunction with habitat loss and fragmentation over the last century [32], it is vital to take a multidimensional and interdisciplinary approach to monitoring and managing the health of wild tiger subpopulations.
The extent to which habitat loss and fragmentation alter the gut microbiota and in turn impact the health of endangered wildlife is largely unknown. Small isolated wildlife populations may not only have low genetic diversity but also have a low gut microbial diversity with an altered functionality that could adversely impact the health of these animals and potentially increase the risk of local extinction [17]. For example, the Red colobus monkey (Procolobus gordonorum), an endangered species, residing around human settlements seemed to have reduced gut microbial diversity compared to a population found in a wild habitat [12]. In our previous work, we identified 120 individual tigers based on field-collected fecal samples using eight microsatellite markers and found that tigers in SWR had the lowest genetic diversity and were the most isolated in terms of gene flow compared to the other parks. Tigers from CNP and BNP had similar levels of genetic diversity even though CNP is geographically distant from BNP and SWR [32]. Based on this, we hypothesized that SWR might have the least diverse microbiota compared to the other tiger populations. However, as anthropogenic effects can substantially perturb the microbiota of wildlife [37], we expect that tigers in the CNP may have a unique gut microbiome composition as this park receives much higher human visitation compared to the other parks. The aim of our study is to examine structural and functional diversity of gut microbial communities in tiger populations of TAL (Fig 1).
Fig 1. Scat sample collection sites for tiger baseline genetic database under Nepal Tiger Genome Project (NTGP, 2011–2013).

We identified 120 individual tigers using 8 microsatellite markers from TAL (SWR = 19, BNP = 32, CNP = 69). A total of 70 tiger scat samples from 32 identified individual tigers (CNP = 12; BNP = 12; SWR = 8) were randomly selected for gut microbiota analysis.
As part of the Nepal Tiger Genome Project (NTGP) [38], we conducted one of the largest microbiota surveys of a wild carnivore spanning three populations with different degrees of connectivity and human visitation. We take advantage of data of likely prey species to help untangle the drivers of microbial community structure and assess what role phylosymbiosis plays in structuring the tiger microbiota. This study increased our knowledge of tiger gut microbiota and the information could contribute towards the development of a more comprehensive strategy to conserve and manage wild tiger populations occurring across fragmented landscapes.
Results
Composition of tiger gut microbiota across different protected sites of Nepal
16S rRNA amplicon sequencing of tiger scat and soil samples (tiger, n = 70; soil, n = 8) targeting the hypervariable V4 region of 16S rRNA gene generated a total of 4,385,688 sequences, among which tiger samples consisted of 2,985,814 sequences and soil samples consisted of 1,399,874 sequences. For 70 tiger samples from 32 individual tigers, the mean number of sequences per sample was 42,654 (range: 1,614–95,553). Similarly, for 8 soil samples, the mean number of sequences per sample was 174,984 (range: 134,283–235,580). After rarefaction, four samples having less than 10,000 sequences were filtered out and excluded from further analyses (S1 Table and S1 Fig). The gut microbiota communities characterized via Operational Taxonomic Unit (OTU), with ≥ 97% nucleotide sequence identity, differed significantly between soil and tiger samples (PERMANOVA; Unweighted Unifrac: pseudo F = 12.69, P = 0.001; Weighted Unifrac: pseudo F = 15.52, P = 0.001) (Fig 2). Overall composition of highly abundant microbiota of tiger samples are very similar across the three regions (Fig 3). Overall, the most dominant phyla detected in the gut microbiota of tigers were Proteobacteria (37.1% +/- 8.49E-02), Firmicutes (30.1% +/- 8.54E-02), Bacteroidetes (16.1% +/- 5.48E-02), Fusobacteria (12.3% +/- 6.47E-02), and Actinobacteria (2.8% +/- 1.40E-02) (Table 1 and Fig 3). The major microbial phyla present in soil were Proteobacteria (33% +/- 6.9E-02), Acidobacteria (19% +/- 3.2E-02), Actinobacteria (9% +/- 2.3E-02) and Bacteroidetes (9% +/- 7.8E-03) (Table 2 and Fig 4). Acidobacteria, Verrucomicrobia, Chloroflexi, Planctomycetes, Cyanobacteria and Gemmatimonadetes are only observed in soil samples (Table 2). Fusobacteria were only found in tiger samples. Bacteroidetes, Firmicutes, Proteobacteria and Actinobacteria are common in both soil and tiger samples (Tables 1 and 2). We also compared gut microbiota profile between samples of same individuals (n = 6) collected at various times and observed slightly different microbiota profiles of identified phyla (S2–S7 Figs).
Fig 2. Principal Coordinate Analysis (PCoA) of soil (all sites, n = 8; CNP, n = 3; BNP, n = 5) and tiger fecal samples (all sites, n = 32; CNP, n = 12; BNP, n = 12; SWR, n = 8).

Gut microbiota profiles for soil samples are distinct from fecal samples indicating that cross-contamination between these two sample sources is unlikely.
Fig 3. Gut microbiota diversity in tiger populations of Nepal.

Relative abundance of top five microbial phyla and their subsequent genera identified in tiger fecal samples collected across three protected areas (CNP, BNP, SWR) within TAL.
Table 1. Relative abundance of gut microbiota composition in Bengal tigers across three protected areas within TAL.
The 10 most abundant bacterial phyla detected in tiger fecal samples collected across three major protected sites (CNP, BNP, SWR) within the Terai Arc Landscape of Nepal.
| Phylum | CNP (Stdev) | BNP (Stdev) | SWR (Stdev) | Total (Mean +/- error) |
|---|---|---|---|---|
| Proteobacteria | 40.5% (2.97E-01) | 43.3% (3.55E-01) | 27.4% (3.01E-01) | 37.1% (8.49E-02) |
| Firmicutes | 27.8% (2.85E-01) | 22.9% (2.11E-01) | 39.5% (2.38E-01) | 30.1% (8.54E-02) |
| Bacteroidetes | 22.4% (2.19E-01) | 12.8% (1.46E-01) | 13.1% (1.30E-01) | 16.1% (5.48E-02) |
| Fusobacteria | 4.9% (6.86E-02) | 16.8% (1.75E-01) | 15.2% (1.53E-01) | 12.3% (6.47E-02) |
| Actinobacteria | 2.8% (4.16E-02) | 1.4% (2.50E-02) | 4.2% (5.61E-02) | 2.8% (1.40E-02) |
| Tenericutes | 1.3% (7.03E-02) | 2.2% (8.96E-02) | 0.1% (2.07E-03) | 1.2% (1.03E-02) |
| TM7 | 0.03% (6.05E-04) | 0.5% (1.87E-02) | 0.2% (4.01E-03) | 0.2% (2.09E-03) |
| Unassigned;Other | 0.1% (1.20E-03) | 0.1% (2.23E-03) | 0.1% (5.53E-04) | 0.1% (2.48E-04) |
| Verrucomicrobia | 0.1% (4.05E-03) | 0.004% (9.05E-05) | 0.01% (2.94E-04) | 0.04% (5.51E-04) |
| All Other Categories (Remaining Phyla) | 0.001% (3.86E-05) | 0.002% (5.20E-05) | 0.01% (1.43E-04) | 0.003% (2.15E-05) |
Table 2. Relative abundance of microbiota composition of soil samples collected at two protected areas (all sites n = 8; CNP, n = 3; BNP, n = 5) within TAL.
| Phylum | CNP (Stdev) | BNP (Stdev) | Total (Mean +/- error) |
|---|---|---|---|
| Proteobacteria | 28.1% (1.11E-01) | 37.8% (4.34E-02) | 32.9% (6.86E-02) |
| Acidobacteria | 21.3% (9.64E-02) | 16.8% (2.47E-02) | 19.1% (3.21E-02) |
| Actinobacteria | 7.5% (1.78E-02) | 10.8% (5.03E-02) | 9.1% (2.30E-02) |
| Bacteroidetes | 9.4% (3.27E-02) | 8.3% (2.29E-02) | 8.8% (7.80E-03) |
| Verrucomicrobia | 5.9% (8.12E-03) | 5.4% (1.15E-02) | 5.6% (3.48E-03) |
| Chloroflexi | 5.4% (7.45E-03) | 4.5% (3.68E-03) | 4.9% (6.35E-03) |
| Planctomycetes | 4.8% (8.67E-03) | 4.5% (7.45E-03) | 4.6% (2.60E-03) |
| Cyanobacteria | 3.9% (5.10E-02) | 2.8% (1.81E-02) | 3.4% (8.04E-03) |
| Gemmatimonadetes | 2.7% (8.81E-04) | 2.8% (4.57E-03) | 2.8% (3.36E-04) |
| Unassigned;Other | 2.2% (3.95E-03) | 0.8% (6.49E-03) | 1.5% (9.89E-03) |
| Nitrospirae | 1.4% (5.58E-03) | 1.2% (5.51E-03) | 1.3% (1.78E-03) |
| Firmicutes | 0.7% (5.45E-03) | 1.2% (7.00E-03) | 1.0% (3.33E-03) |
| TM7 | 0.8% (7.47E-03) | 0.3% (4.83E-04) | 0.6% (3.49E-03) |
| WS3 | 0.3% (2.40E-03) | 0.7% (3.05E-03) | 0.5% (2.47E-03) |
| OD1 | 0.7% (4.75E-03) | 0.2% (3.40E-03) | 0.5% (3.60E-03) |
| Armatimonadetes | 0.5% (4.12E-03) | 0.3% (1.11E-03) | 0.4% (1.41E-03) |
| Elusimicrobia | 0.6% (4.49E-03) | 0.3% (1.27E-03) | 0.4% (1.83E-03) |
| OP3 | 0.5% (3.46E-03) | 0.3% (3.15E-03) | 0.4% (2.05E-03) |
| Chlorobi | 0.4% (1.61E-03) | 0.2% (1.29E-03) | 0.3% (1.32E-03) |
| All Other Categories (Remaining Phyla) | 2.7% (1.61E-02) | 1.0% (9.05E-03) | 1.8% (1.23E-02) |
Fig 4. Soil microbiota biodiversity examined at two TAL sites.
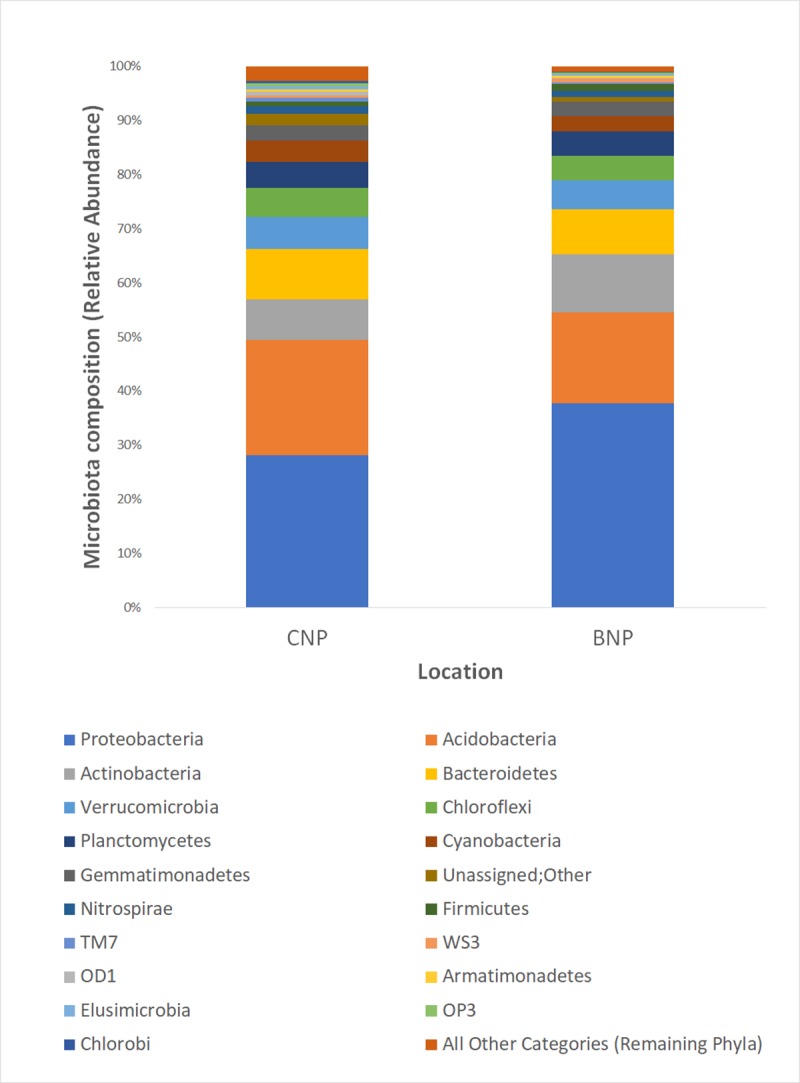
Relative abundance of microbial phyla detected in soil samples (n = 8) collected at CNP (n = 3) and BNP (n = 5).
All the raw sequences associated with this study have been deposited at figshare repository and can be publicly assessed using the link: https://doi.org/10.6084/m9.figshare.8010389.v1
Alpha-diversity (within population) measures of tiger gut microbiota
Overall, there was no significant difference in alpha diversity (within population) assessed in fecal microbiota samples collected from three tiger populations independent of the index used (Chao1 and ACE metrics Shannon’s index, Simpson’s index [39], Inverted Simpson and Fisher’s indexes [40]) (Fig 5).
Fig 5. Alpha-diversity in tiger gut microbial communities is similar across different regions studied.

Beta-diversity (between population) of the tiger gut microbiota
Our beta diversity analysis (between population) revealed significant differences in the phylogenetic and taxonomic composition of microbial communities across the tiger populations. We compared both phylogenetic and taxonomic diversity to test if differences between microbial beta-diversity across these tiger populations were driven by evolutionary history (i.e., different lineages present) or taxonomic membership (i.e., similar lineages present in each community, but different species detected, as described in [41] for the utility of comparing both diversity measures). Canonical analysis of principal coordinates (CAP) of weighted UniFrac distances indicated that microbial communities clustered based on their geographic locations with moderate levels of overlap (Fig 6). One-way PERMANOVA on weighted UniFrac distances found that the clustering across all areas was significant (pseudo F = 3.086, P = 0.006) with pairwise tests showing significant differences between CNP and the other two protected areas (CNP vs BNP: pseudo t = 1.944, P = 0.006; CNP vs SWR: pseudo t = 1.994, P = 0.007), but no significant differences between SWR and BNP (t = 0.782, P = 0.606) (Fig 6A). Furthermore, there was a greater beta phylogenetic diversity between CNP compared to SWR (PERMDISP, pseudo t = 2.72, P = 0.015), but not between CNP and BNP (PERMDISP, pseudo t = 1.92, P = 0.083) or BNP and SWR (PERMDISP, pseudo t = 0.79, P = 0.797). CAP confirmed these results by correctly assigning samples to each respective group most of the time (57%; null allocation success = 0.33) (Fig 6A).
Fig 6. Canonical analysis of principal (CAP) coordinates showing differences in microbiota composition across three protected sites within TAL.

PERMANOVA of both phylogenetic composition (weighted UniFrac distance) (a) and taxonomic composition (Bray-Curtis similarity) (b) similarity measures illustrated that CNP had differing microbial composition and overall greater beta diversity.
We found similar patterns when we analyzed taxonomic beta-diversity based on Bray-Curtis similarity with significant clustering of tiger populations (PERMANOVA, pseudo F = 2.712, P = 0.0003) and similar pair-wise contrasts (CNP vs SWR: pseudo t = 2.056, P = 0.003; CNP vs BNP: t = 1.512, P = 0.005; BNP vs SWR: pseudo t = 1.114, P = 0.216). CNP had higher taxonomic beta-diversity than other protected sites studied here (PERMDISP, CNP vs BNP, pseudo t = 2.36, P = 0.031, CNP vs SWR: pseudo t = 3.511, P = 0.001), however, there was no difference in beta-diversity between BNP or SWR (PERMDISP, pseudo t = 1.21, P = 0.27). CAP correctly assigned samples to each population in most cases (63%, null allocation success = 0.33) (Fig 6B).
Statistical analysis for bacterial abundance in tiger fecal microbiota
Analysis of differences in abundance, based on the F test (with Benjamini and Hochberg control for false discovery rate [42]) identified significant differences in three bacterial phyla (Fusobacteria, TM7 and Thermi) across the three protected sites. Phyla Fusobacteria and TM7 separated samples collected in CNP from a combined group of samples from BNP and SWR (adjusted P = 0.01, fdr = 0.028). The abundance of the phylum Thermi, also referred to as Deinococcus-Thermus, was significantly different in SWR samples from samples collected in BNP and CNP (adjusted P = 0.072, fdr = 0.168).
This inference motivated a more in-depth analysis with representative sequences obtained for twenty of the most prevalent genera (based on presence/absence). A statistical analysis with F-test and a Benjamini and Hochberg correction for false discovery identified significant differences in Comamonas, Collinsella, and Fusobacterium across CNP, BNP, and SWR with significant adjusted P values and acceptable rates of false discovery (fdr) (Table 3).
Table 3. Statistical analysis for most prevalent microbial phyla and genera in gut microbiota found in three sub-populations of tigers in Nepal.
| Phylum | Genus | Relative Abundance (StDev) | Adj P | False discovery rate (fdr) | ||
|---|---|---|---|---|---|---|
| CNP | BNP | SWR | ||||
| Proteobacteria | Comamonas | 2.47% (2.68E-02) | 0.36% (1.99E-02) | 0.26% (7.89E-03) | 0.03 | 0.024 |
| Actinobacteria | Collinsella | 0.61% (2.32E-02) | 0.61% (9.05E-03) | 2.93% (5.22E-02) | 0.03 | 0.024 |
| Fusobacteria | Fusobacterium | 3.49% (6.37E-02) | 16.73% (1.66E-01) | 15.79% (1.51E-01) | 0.109 | 0.064 |
Predictive metabolic functions associated with tiger gut microbiota
The PICRUSt analysis of all obtained OTUs using multiple group statistical ANOVA test suggested that there are notable divergences in predicted functional categories among the gut microbiota across tiger populations of CNP, BNP and SWR (P value < 0.05) based on eight functional categories as listed in the Table 4. Likewise, pairwise Welch’s t-test performed on the mean proportion of all the functional categories showed 13 functional pathway differences between CNP vs. SWR (P value < 0.05) (Fig 7A), and two in CNP vs. BNP (P value < 0.05) (Fig 7B). There was no significant functional pathways difference between BNP and SWR.
Table 4. Predictive functional profiling of microbial communities using multiple-group comparative PICRUSt analysis (P-value corrected < 0.05).
| Functional categories | P-values | P-values (corrected) | CNP | BNP | SWR | |||
|---|---|---|---|---|---|---|---|---|
| mean rel. freq. (%) | std. dev. (%) | mean rel. freq. (%) | std. dev. (%) | mean rel. freq. (%) | std. dev. (%) | |||
| Metabolism of Other Amino Acids | 2.46E-05 | 1.01E-03 | 1.7586 | 0.1840 | 1.6343 | 0.1300 | 1.5277 | 0.1463 |
| Lipid Metabolism | 5.11E-05 | 1.05E-03 | 3.3852 | 0.4784 | 3.0074 | 0.2283 | 2.9102 | 0.2805 |
| Xenobiotics Biodegradation and Metabolism | 2.37E-04 | 3.23E-03 | 2.6593 | 0.7133 | 2.1849 | 0.4225 | 1.9963 | 0.3467 |
| Metabolism of Terpenoids and Polyketides | 4.61E-04 | 4.72E-03 | 1.8282 | 0.2626 | 1.6218 | 0.1454 | 1.6080 | 0.1672 |
| Transport and Catabolism | 4.38E-03 | 2.24E-02 | 0.3019 | 0.0999 | 0.2527 | 0.0633 | 0.2222 | 0.0656 |
| Amino Acid Metabolism | 6.04E-03 | 2.75E-02 | 9.8148 | 0.6008 | 9.2886 | 0.6104 | 9.4261 | 0.4714 |
| Replication and Repair | 1.13E-02 | 4.23E-02 | 7.5835 | 0.7039 | 7.6596 | 0.7293 | 8.1723 | 0.5950 |
| Nucleotide Metabolism | 1.19E-02 | 4.05E-02 | 3.5190 | 0.3695 | 3.6291 | 0.3409 | 3.8212 | 0.2764 |
Fig 7. Mean proportions of predictive metabolic functional categories between tiger populations based on pair-wise comparison- functional categories are more diverse between CNP and SWR than CNP and BNP.

(a) Significant functional categories identified between CNP and SWR. (b) Significant functional categories identified between CNP and BNP.
Overall significant differences were observed in predictive metabolic functions for the tiger populations studied across three protected sites within TAL. The pair-wise relationship between samples based on functional analysis corresponded to the pair-wise relation between samples based on fecal microbiota structure (PROCRUSTES, m2 = 0.72, P = 0.0009). This analysis further underscores the geo-location specificity with BNP samples overlapping with well separated CNP and SWR samples.
The Welch’s t-test between CNP and SWR identified higher proportions of predicted functional categories related to “amino acid metabolism”, “lipid metabolism”, “xenobiotics biodegradation and metabolism”, and “metabolism of terpenoides and polyketides” in CNP samples. While SWR samples had higher proportions of “energy metabolism”, “carbohydrate metabolism”, “metabolism of cofactors and vitamins” and “nucleotide metabolism” categories in comparison with CNP (Fig 7A). Similarly, the Welch’s t-test between CNP and BNP identified lower proportions of “lipid metabolism” and “metabolism of terpenoids and polyketides” in BNP samples than the CNP samples.
Discussion
We found that whilst microbial alpha diversity did not differ significantly between tiger populations, phylosymbiosis among other factors most likely played a role in shaping tiger microbiota as their composition and beta diversity mirrored the host genetic patterns as observed in our previous study [32]. This supports the theory that host evolutionary background plays an important role in shaping the bacterial gut communities [17]. However, the unique compositional and functional signature of gut microbiota detected in tigers from CNP, which represents the protected site most heavily affected by human development and disturbance, although speculative, possibly shows that anthropogenic impact may also contribute to shaping microbial gut profiles in tigers by influencing biological and environmental pathways which enable bacteria within microbiota and the genes they carry to spread between different biomes [43]. Our findings have critical implications for overall tiger's health, highlighting the importance of microbiome studies in comprehensive species conservation and management efforts.
Tiger genetics, diet or human exposure in explaining gut microbial composition
Our study shows notable differences in microbiota diversity observed between CNP and the two other protected areas, as opposed to the differences observed between BNP and SWR, giving an indication that the microbiota diversity in CNP is unique from that of BNP and SWR. This mirrors the results from our previous study where we have observed limited gene flow between tiger population from CNP with the other two protected areas (BNP and SWR) [32]. This could be due to several different factors, including higher levels of human microbiota influence on wild tigers at the CNP site. Also, habitat fragmentation, differences in diet and limited gene flow in tigers between CNP and other sites may be contributing to CNP’s unique microbiota profiles. In contrast, genetic connectivity for tigers between BNP and SWR habitats, which had more similar microbiota profiles, is supported by the known presence of wildlife corridors. Although this study is the first of its kind in wild tigers, a study in an endangered primate, red colobus monkey, showed a direct correlation between higher habitat fragmentation and reduced gut microbiota diversity, which had some profound implications on health and long-term viability of the species [12]. Low population numbers in some tiger populations and increased levels of habitat loss and fragmentation may contribute towards lowering of genetic diversity in host species, which in turn can also adversely impact gut microbiota. In the TAL region of Nepal, there has been a 97% increase in agriculture and settlement areas in the past 200 years and forested areas decreased by 47% between the 19th and 20th centuries [32]. In our study, we found some significant differences in gut microbial composition in three geo-spatially separated tiger population. These differences were highest between CNP and SWR tiger populations which are geographically most separated (Fig 1). Differences in habitat including differences in prey composition and tiger densities, as well as interactions across the fragmented population of these protected areas, might have an additional role in shaping such microbiota composition and diversity.
Although numerous studies have shown the effect of dietary habits on the composition of the gut microbiota [44, 45], most of them focus on structural dietary differences such as a protein-rich versus a polysaccharide-rich diet, which seems to be not relevant for a strict carnivore such as tiger. The overall gut microbiota profile in tigers that we observed in our study were similar in composition with findings reported in various other microbiome research done on mammals [19, 46–48], including carnivore species for which we have prepared a graph (S8 Fig) demonstrating their microbiota composition profiles derived from the respective research [47, 49–56] (S8 Fig). Relatively few studies on the link between gut microbiota and diet have been conducted in wild animals [17]. However, one study showed that in black howler monkeys habitat fragmentation was correlated with a less diverse diet and correspondingly less diverse gut microbiota [13].
Tiger diet composition in Nepal has only been sparsely studied, but the main prey species are chital (Axis axis), sambar deer (Cervus unicolor), hog deer (A. porcinus), barking deer (Muntiacus muntjak) and wild boar (Sus scrofa) [57]. Other species, such as swamp deer (C. duvauceli), gaur (Bos gaurus) and langur (Semnopithecus entellus), may represent a smaller part of tiger diet and also livestock may play a role in tiger diet, notably on the edges of protected areas [58, 59]. Given the habitat characteristics of the three protected areas included in this study, available diet seems to be similar across the sites. This is corroborated by a study estimating prey density as presented by Dhakal et al. [60]. BNP has an overall higher prey density, but in all cases chital makes up the vast majority of available tiger prey. In SWR sambar or barking deer were not detected, although they are relatively common at both the CNP and BNP sites. However, nilgai (Boselaphus tragocamelus) was detected relatively often at SWR, whereas this species was only seen twice during the study at Bardia and it does not occur at CNP. Although we cannot rule out that there are slight differences in dietary composition between the studied areas, we hypothesize that there are no major differences between the diets of the individual tiger populations, which would explain the observed differences in microbiota content. However, tiger diet and variation between the different tiger populations in Nepal should be subject to further investigation.
Gut microbiota and functional metabolic implications
PICRUSt based predictive metabolic functionalities in tiger population revealed higher functional enrichment in the CNP tiger population for most categories, whereas the SWR tiger population had the lowest levels of enrichment in comparison with gut microbiota from other sites. We found significant differences in two functional categories among CNP vs BNP (Fig 7B), and 13 categories among CNP vs SWR (Fig 7A), while functional categories did not differ significantly between BNP and SWR sites (Fig 7). Predictive metabolic profiles are just rough indicators of possible functional implication of microbiota present. In conclusion, we observed that the tiger population structure, gut microbiota profile and associated functional metabolic categories are correlated, with geographically most separated CNP and SWR population having the most distinct and different host genotype and microbiota profiles.
This study further highlights the necessity of a more comprehensive systems biology based approach to assess the conservation status of the species by monitoring and maintaining genetic diversity of the host and its associated microbiota. We also encourage further investigation of various extrinsic and intrinsic factors that might influence gut microbiota and its influence on tiger health.
Application of gut microbiota in conservation
Microbial analyses hold a great potential in uncovering information on host population dynamics, however studies in wild carnivores are scant. Such information can be used to preserve host biodiversity and develop effective conservation and management strategies. Microbiota is closely linked to health and hence, microbial phylogenies can be used as signatures of disease transmission and has potential for monitoring population health, density, movement, and dispersal [26].
Methods
Methods for host genetic analysis
Genetic database of wild tiger in Nepal and fecal DNA sampling
NTGP created Nepal’s first comprehensive tiger genetic reference database by collecting and analyzing fecal samples (n = 770) from the TAL (December, 2011- March, 2012) (Fig 1), which included all the known habitat of tigers in Nepal. The TAL has a sub-tropical monsoonal climate and mixed deciduous vegetation ranging from alluvial floodplain grasslands communities to Climax Sal (Shorea robusta) forests and includes five protected areas, among which SWR (28°50′25″N 80°13′44″E), BNP(28°23′N 81°30′E), and CNP (27°30'0.00" N 84°40'0.12" E) are the major tiger habitats (Fig 1).
Putative tiger fecal (scat) samples were collected from protected areas and connecting wildlife corridors across the TAL-Nepal [32]. Ninety-eight grid cells each measuring 15 X 15 km (225 km2, sampling unit) were sampled using opportunistic field surveys. A few grams from the upper surfaces of the scat were removed and stored at room temperature in sterile 2-ml vials filled with DETs buffer (dimethyl sulphoxide saline solution) [61] at 1:4 volume scat-to-solution ratio following field sampling protocols by Wultsch, Waits [62].
DNA extraction, species identification, and individual identification
We extracted DNA from scat samples using a commercially available QIAmp DNA Stool Kit (QIAGEN Inc., Germany) following the manufacturer’s instructions. Each batch of DNA extraction included a negative extraction control. Extracted DNA was stored at -20°C. Tigers were identified using PCR assay that used tiger specific mtDNA Cytochrome-b (CYT-B) primers [63]. Individual tigers were identified by microsatellite analysis using a panel of eight microsatellite markers developed from the domestic cat (Felis catus) and tiger genomes [64–66] as described in Thapa et al. [32].
Gut microbiota analysis
We randomly selected a total of 70 scat samples from 32 unique individual tigers (n = 12, CNP; male = 9, female = 2, undetermined = 1); (n = 13, BNP; male = 6, female = 6, undetermined = 1); (n = 7, SWR; male = 4, female = 3) for gut microbiota analysis. We also selected multiple samples from 8 individual tigers (n = 2, CNP; n = 3, BNP; n = 3, SWR) (S1 Table). Soil samples were also collected from two of the study sites (n = 3, CNP; n = 5, BNP) with the goal to profile soil microbiota to assess cross-contamination between soil and fecal samples occurred.
Microbial DNA was isolated from tiger fecal and soil samples using PowerSoil DNA Isolation Kit (MoBio, Qiagen, Carlsbad, CA). DNA quality was checked by gel electrophoresis (mostly >10 kbps fragments), and DNA concentration was measured using Qubit (Invitrogen, Carlsbad, CA). We completed microbial community profiling (identification and composition) by amplifying and sequencing the hyper-variable region (V4) of the 16S rRNA from both tiger scat and soil control samples using a modified version of the protocol presented in Caporaso et. al 2012 [67], adapted for the Illumina MiSeq platform. Using a two-step polymerase chain reaction (PCR), we amplified the V4 region of the 16S rRNA using the ‘universal’ bacterial primer pairs (515F and 806R) linked to the forward and reverse Illumina flow cell adapter sequences. PCR was carried out in two steps, both using the 2X KAPA HiFi HotStart ReadyMix (KAPA Biosystems/Roche, USA) and cycling at initial denaturation at 95° C for 30sec followed by 95° C for 30sec, 55° C for30sec, 72° C for 30sec. Post cycling, samples were incubated at 72° C for 5 min, followed by a hold at 4°C. The first PCR was conducted in 25 cycles, adding a 6 bp barcode sequence to enable multiplexing. The second PCR was conducted in 8 cycles to amplify the PCR products and add the remaining full-length Illumina adapters. We purified the resulting PCR products using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA), quantified with Qubit (Invitrogen, USA), normalized, and pooled all sample libraries prior to sequencing. Paired-end sequencing (2 x 300bp) was completed on Illumina MiSeq (Illumina, Inc., San Diego, CA), using a v3 600-cycle kit according to the manufacturer’s instructions.
After sequencing and de-multiplexing, we filtered all reads by quality (q>29 across 50% of the read length with no ambiguous N base calls) and length (>75 bp). A custom Perl script was written to execute several analysis modules of QIIME, version 1.9.1 [68]. First, we joined raw paired-end Illumina fastq files by fastq-join. We discarded all OTU containing less than 10 sequences. We chose the cluster centroid for each OTU as the OTU representative sequence and taxonomically assigned each sequence using homologous searches to 16S reference sequences found in the Greengenes database [69] at greater than or equal to 96% sequence identity [12, 17, 70]. To construct a phylogenetic tree of the OTU representative sequences, we aligned sequences using PyNAST, version 1.2.2 [71] against an existing alignment of the Greengenes database. Post alignment and construction of phylogenetic trees was completed using FastTree, version 2.1 [72].
Alpha-diversity, beta-diversity estimates, and relative abundance analysis of each taxonomic group were performed after rarefaction was applied with even sub-sampling of 10,000 sequences per sample. The abundances of OTUs were normalized based on proportion and OTUs with very low variability (1e-05) were filtered out. Microbial diversity within (alpha diversity) and between tiger subpopulations (beta diversity) were obtained and visualized with QIIME and the phyloseq package in R [73, 74]. We assessed alpha diversity using several metrics (Chao1, ACE, Shannon, Simpson, InvSimpson, Fisher) [39, 40, 75–77]. Statistical evaluation of differential abundance was done with F test supplemented in the mt function in phyloseq [74]. The resulting P values were adjusted for multiple comparisons using Benjamin and Hoechberg’s false discovery method (Fig 6).
To test if gut microbiota diversity were significantly differentiated across different study sites (beta diversity), we employed Permutational Multivariate Analysis of Variance (PERMANOVA) [78], canonical analysis of principal coordinates (CAP) [79], and permutational tests of homogeneity of dispersions (PERMDISP) [80]. To test for differences in both UniFrac and Bray-Curtis similarity distances, we performed a one-way PERMANOVA and used pair-wise contrasts to examine differences between sites. We analyzed compositional differences using CAP and DPCoA (detrended principal coordinate analysis) and also used the CAP discriminant analysis to validate the PERMANOVA results (i.e., how distinct was each site in multivariate space) by assessing allocation success using the ‘leave-one-out’ procedure [79]. PERMDISP was used to compare beta diversity between sites for both metrics [80] and to test if differences detected by PERMANOVA were likely due to differences in-group dispersion. All of the above analyses were conducted in PRIMER- E PERMANOVA+[81].
Predictive metabolic functions associated with tiger gut microbiota
We used PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) [82] to predict functional roles played by the tiger gut microbiota communities. PICRUSt predicts metabolic and functional profiles of a microorganism based on known functional roles of its closely related microorganism [82]. It utilizes existing information from Integrated Microbial Genomes (IMG) database [83], which contains annotation of gene content and 16S copy number data of reference bacterial and archaeal genomes. Then by implementing extended ancestral-state reconstruction algorithm, the taxonomic composition and phylogenetic information of the observed OTUs are used in estimating the comprehensive metagenome of the microbiota community classifying their metabolic and functional categories in the KEGG Orthology (KO) classification scheme [84]. The PICRUSt predictions were subjected to statistical analyses with Statistical Analysis of Metagenomic Profiles (STAMP) [85] for identifying and characterizing significant functional categories across three subpopulations CNP, BNP, and SWR. We conducted multiple group statistical tests with ANOVA and pair-wise statistical tests using Welch’s t-test to test for statistical differences in mean proportion of functional categories among subpopulations. The P-value was adjusted by applying the Benjamini-Hochberg false discovery rate (FDR) method to correct for multiple hypotheses testing. We conducted Procrustes [86] analysis using QIIME to test correlations on beta-diversity obtained for gut microbiota and predictive microbiota functionality contents using Bray-Curtis distance metrics.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Representation of microbial biodiversity found in various carnivore species, including environmental samples (soil). For the soil and Bengal tiger, we used data from our current study. The data for Dhole1 [54], Dhole2 [49], Wolf [56], Giant panda [55], Snow leopard [47], Antarctic seals [52], Domestic cat1 [53], Domestic cat2 [50] and Cheetah [51] were compiled from other published studies.
(DOCX)
(DOCX)
Acknowledgments
We would like to thank the Department of National Parks and Wildlife Conservation and the Ministry of Forest and Soil Conservation (Nepal) for giving us permission to conduct the first genetic study of tigers in Nepal. We would also like to thank our collaborators at the Los Alamos National lab, Griffith University and the University of Idaho for providing valuable technical support and resources. And finally, this work would not have been possible without dedication and hard work from our team at the Center for Molecular Dynamics Nepal.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Funded by DBK USAID FOG AID-367-G-11-00001 United States Agency for International Development.
References
- 1.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320(5883):1647–51. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quercia S, Candela M, Giuliani C, Turroni S, Luiselli D, Rampelli S, et al. From lifetime to evolution: timescales of human gut microbiota adaptation. Frontiers in microbiology. 2014;5:587 10.3389/fmicb.2014.00587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer F, Bäckhed F. The gut microbiota—masters of host development and physiology. Nature Reviews Microbiology. 2013;11(4):227–38. 10.1038/nrmicro2974 [DOI] [PubMed] [Google Scholar]
- 4.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–70. 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–7. 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- 6.Zoetendal EG, Akkermans AD, Akkermans-van Vliet WM, de Visser JAG, de Vos WM. The host genotype affects the bacterial community in the human gastronintestinal tract. Microbial ecology in health and disease. 2001;13(3):129–34. [Google Scholar]
- 7.Redford KH, Segre JA, Salafsky N, del Rio CM, McAloose D. Conservation and the microbiome. Conservation Biology. 2012;26(2):195–7. 10.1111/j.1523-1739.2012.01829.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48. 10.1016/j.cell.2006.02.017 [DOI] [PubMed] [Google Scholar]
- 9.Degnan PH, Pusey AE, Lonsdorf EV, Goodall J, Wroblewski EE, Wilson ML, et al. Factors associated with the diversification of the gut microbial communities within chimpanzees from Gombe National Park. Proceedings of the National Academy of Sciences. 2012;109(32):13034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tung J, Barreiro LB, Burns MB, Grenier J-C, Lynch J, Grieneisen LE, et al. Social networks predict gut microbiome composition in wild baboons. Elife. 2015;4:e05224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez A, Petrzelkova KJ, Burns MB, Yeoman CJ, Amato KR, Vlckova K, et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell reports. 2016;14(9):2142–53. 10.1016/j.celrep.2016.02.013 . [DOI] [PubMed] [Google Scholar]
- 12.Barelli C, Albanese D, Donati C, Pindo M, Dallago C, Rovero F, et al. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: implications for conservation. Scientific reports. 2015;5:14862 10.1038/srep14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. The ISME journal. 2013;7(7):1344–53. 10.1038/ismej.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwab C, Cristescu B, Northrup J, Stenhouse G, Gänzle M. Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS One. 2011;6(12):e27905 10.1371/journal.pone.0027905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez A, Petrzelkova K, Yeoman CJ, Vlckova K, Mrázek J, Koppova I, et al. Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Molecular ecology. 2015;24(10):2551–65. 10.1111/mec.13181 [DOI] [PubMed] [Google Scholar]
- 16.Brucker RM, Bordenstein SR. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution: International Journal of Organic Evolution. 2012;66(2):349–62. [DOI] [PubMed] [Google Scholar]
- 17.Kohl KD, Varner J, Wilkening JL, Dearing MD. Gut microbial communities of American pikas (Ochotona princeps): Evidence for phylosymbiosis and adaptations to novel diets. Journal of Animal Ecology. 2018;87(2):323–30. 10.1111/1365-2656.12692 [DOI] [PubMed] [Google Scholar]
- 18.Phillips CD, Phelan G, Dowd SE, McDONOUGH MM, Ferguson AW, Delton Hanson J, et al. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Molecular ecology. 2012;21(11):2617–27. 10.1111/j.1365-294X.2012.05568.x [DOI] [PubMed] [Google Scholar]
- 19.Menke S, Wasimuddin Meier M, Melzheimer J, Mfune JK, Heinrich S, et al. Oligotyping reveals differences between gut microbiomes of free-ranging sympatric Namibian carnivores (Acinonyx jubatus, Canis mesomelas) on a bacterial species-like level. Frontiers in microbiology. 2014;5:526 10.3389/fmicb.2014.00526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amato K. Co-evolution in context: the importance of studying gut microbiomes in wild animals. Microbiome Science and Medicine. 2013;1(1). [Google Scholar]
- 21.Podolsky DK. The current future understanding of inflammatory bowel disease. Best practice & research Clinical gastroenterology. 2002;16(6):933–43. [DOI] [PubMed] [Google Scholar]
- 22.Targan SR, Karp LC. Defects in mucosal immunity leading to ulcerative colitis. Immunological reviews. 2005;206(1):296–305. [DOI] [PubMed] [Google Scholar]
- 23.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nature Reviews Immunology. 2009;9(5):313–23. 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suchodolski JS. Intestinal microbiota of dogs and cats: a bigger world than we thought. Veterinary Clinics of North America: Small Animal Practice. 2011;41(2):261–72. 10.1016/j.cvsm.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahrndorff S, Alemu T, Alemneh T, Lund Nielsen J. The microbiome of animals: implications for conservation biology. International journal of genomics. 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stumpf R, Gomez A, Amato K, Yeoman CJ, Polk J, Wilson B, et al. Microbiomes, metagenomics, and primate conservation: New strategies, tools, and applications. Biological Conservation. 2016;199:56–66. [Google Scholar]
- 27.Seidensticker J. Saving wild tigers: a case study in biodiversity loss and challenges to be met for recovery beyond 2010. Integrative zoology. 2010;5(4):285–99. 10.1111/j.1749-4877.2010.00214.x . [DOI] [PubMed] [Google Scholar]
- 28.Dinerstein E, Loucks C, Wikramanayake E, Ginsberg J, Sanderson E, Seidensticker J, et al. The fate of wild tigers. BioScience. 2007;57(6):508–14. [Google Scholar]
- 29.Barber‐Meyer SM, Jnawali SR, Karki JB, Khanal P, Lohani S, Long B, et al. Influence of prey depletion and human disturbance on tiger occupancy in Nepal. Journal of Zoology. 2013;289(1):10–8. [Google Scholar]
- 30.Kenney JS, Smith JL, Starfield AM, McDougal CW. The long‐term effects of tiger poaching on population viability. Conservation Biology 1995. October 1;9(5):1127–33. 1995;9(5):1127–33. [DOI] [PubMed] [Google Scholar]
- 31.Smith JL, Ahern SC, McDougal C. Landscape analysis of tiger distribution and habitat quality in Nepal. Conservation Biology 1998. December 1;12(6):1338–46. 1998;12(6):1338–46. [Google Scholar]
- 32.Thapa K, Manandhar S, Bista M, Shakya J, Sah G, Dhakal M, et al. Assessment of genetic diversity, population structure, and gene flow of tigers (Panthera tigris tigris) across Nepal's Terai Arc Landscape. PLOS ONE. 2018;13(3):e0193495 10.1371/journal.pone.0193495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wikramanayake E, McKnight M, Dinerstein E, Joshi A, Gurung B, Smith D. Designing a conservation landscape for tigers in human‐dominated environments. Conservation Biology. 2004;18(3):839–44. [Google Scholar]
- 34.Kanagaraj R, Wiegand T, Kramer‐Schadt S, Anwar M, Goyal SP. Assessing habitat suitability for tiger in the fragmented Terai Arc Landscape of India and Nepal. Ecography 2011. December 1;34(6):970–81. 2011;34(6):970–81. [Google Scholar]
- 35.Treves AaKUK. Human-carnivore conflict and perspectives on carnivore management worldwide. Conservation Biology. 2003;17:1491–9. [Google Scholar]
- 36.Zimmermann A, Baker N, Inskip C, Linnell JC, Marchini S, Odden J, et al. Contemporary views of human–carnivore conflicts on wild rangelands. Wiley-Blackwell, Oxford: 2010. [Google Scholar]
- 37.Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, Al-Ghalith GA, et al. Captivity humanizes the primate microbiome. Proceedings of the National Academy of Sciences. 2016:201521835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.NTGP/CMDN. Nepal Tiger Genome Project—Final Report 2014, Center for Molecular Dynamics-Nepal. Center for Molecular Dynamics-Nepal, 2014.
- 39.Simpson E. Measurement of diversity. Nature. 1949. [Google Scholar]
- 40.Fisher RA, Corbet AS, Williams CB. The relation between the number of species and the number of individuals in a random sample of an animal population. The Journal of Animal Ecology. 1943:42–58. [Google Scholar]
- 41.Fountain-Jones NM, Jordan GJ, Burridge CP, Wardlaw TJ, Baker TP, Forster L, et al. Trophic position determines functional and phylogenetic recovery after disturbance within a community. Functional Ecology. 2017;31(7):1441–51. 10.1111/1365-2435.12845 [DOI] [Google Scholar]
- 42.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological). 1995:289–300. [Google Scholar]
- 43.da Costa PM, Loureiro L, Matos AJ. Transfer of multidrug-resistant bacteria between intermingled ecological niches: the interface between humans, animals and the environment. International journal of environmental research and public health. 2013;10(1):278–94. 10.3390/ijerph10010278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. nature. 2012;486(7402):222 10.1038/nature11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker AA, Janssens GP, Snauwaert C, Hesta M, Huys G. Integrated community profiling indicates long-term temporal stability of the predominant faecal microbiota in captive cheetahs. PLoS One. 2015;10(4):e0123933 10.1371/journal.pone.0123933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Liu G, Chen L, Sha W. Composition and diversity of the bacterial community in snow leopard (Uncia uncia) distal gut. Annals of microbiology. 2015;65(2):703–11. [Google Scholar]
- 48.Handl S, Dowd S, Garcia-Mazcorro J, Steiner J, Suchodolski J. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS microbiology ecology. 2011;76(2):301–10. 10.1111/j.1574-6941.2011.01058.x [DOI] [PubMed] [Google Scholar]
- 49.Chen L, Zhang H, Liu G, Sha W. First report on the bacterial diversity in the distal gut of dholes (Cuon alpinus) by using 16S rRNA gene sequences analysis. Journal of applied genetics. 2016;57(2):275–83. 10.1007/s13353-015-0319-0 [DOI] [PubMed] [Google Scholar]
- 50.Duarte AM, Jenkins TP, Latrofa MS, Giannelli A, Papadopoulos E, de Carvalho LM, et al. Helminth infections and gut microbiota–a feline perspective. Parasites & vectors. 2016;9(1):625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Menke S, Melzheimer J, Thalwitzer S, Heinrich S, Wachter B, Sommer S. Gut microbiomes of free‐ranging and captive Namibian cheetahs: Diversity, putative functions and occurrence of potential pathogens. Molecular ecology. 2017;26(20):5515–27. 10.1111/mec.14278 [DOI] [PubMed] [Google Scholar]
- 52.Nelson TM, Rogers TL, Carlini AR, Brown MV. Diet and phylogeny shape the gut microbiota of Antarctic seals: a comparison of wild and captive animals. Environmental microbiology. 2013;15(4):1132–45. 10.1111/1462-2920.12022 [DOI] [PubMed] [Google Scholar]
- 53.Tun HM, Brar MS, Khin N, Jun L, Hui RK-H, Dowd SE, et al. Gene-centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. Journal of microbiological methods. 2012;88(3):369–76. 10.1016/j.mimet.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu X, Zhang H, Chen J, Shang S, Wei Q, Yan J, et al. Comparison of the fecal microbiota of dholes high-throughput Illumina sequencing of the V3–V4 region of the 16S rRNA gene. Applied microbiology and biotechnology. 2016;100(8):3577–86. 10.1007/s00253-015-7257-y [DOI] [PubMed] [Google Scholar]
- 55.Xue Z, Zhang W, Wang L, Hou R, Zhang M, Fei L, et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. MBio. 2015;6(3):e00022–15. 10.1128/mBio.00022-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H, Chen L. Phylogenetic analysis of 16S rRNA gene sequences reveals distal gut bacterial diversity in wild wolves (Canis lupus). Molecular biology reports. 2010;37(8):4013–22. 10.1007/s11033-010-0060-z [DOI] [PubMed] [Google Scholar]
- 57.Khadka BB, Lamichhane BR, Aryal N. High prey density observed in village evacuated area: A case study from Padampur of Chitwan National Park. 2016. [Google Scholar]
- 58.Baniya RK, Baniya CB, Mou P, Ge J. Prey Selection By Tiger (Panthera Tigris Tigris) In Shuklaphanta Wildlife Reserve Nepal. 2017. [Google Scholar]
- 59.Shrestha MK. Relative ungulate abundance in a fragmented landscape: implications for tiger conservation: university of Minnesota St. Paul; 2004. [Google Scholar]
- 60.Dhakal M, Karki M, Jnawali S, Subedi N, Pradhan N, Malla S, et al. Status of tigers and prey in Nepal. Department of National Park and Wildlife Conservation, Kathmandu, Nepal: 2014. [Google Scholar]
- 61.Seutin G, White BN, Boag PT. Preservation of avian blood and tissue samples for DNA analyses. Canadian Journal of Zoology. 1991;69(1):82–90. 10.1139/z91-013 [DOI] [Google Scholar]
- 62.Wultsch C, Waits L, Hallerman E, Kelly M. Optimizing Collection Methods for Noninvasive Genetic Sampling of Neotropical Felids. Wildlife Society Bulletin. 2015;39(2):403–12. [Google Scholar]
- 63.Bhagavatula J, Singh L. Genotyping faecal samples of Bengal tiger Panthera tigris tigris for population estimation: a pilot study. BMC genetics. 2006;7:48 10.1186/1471-2156-7-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menotti-Raymond M, David VA, Lyons LA, Schäffer AA, Tomlin JF, Hutton MK, et al. A genetic linkage map of microsatellites in the domestic cat (Felis catus). Genomics. 1999;57(1):9–23. 10.1006/geno.1999.5743 [DOI] [PubMed] [Google Scholar]
- 65.Sharma R, Stuckas H, Moll K, Khan I, Bhaskar R, Goyal S, et al. Fourteen new di‐and tetranucleotide microsatellite loci for the critically endangered Indian tiger (Panthera tigris tigris). Molecular ecology resources. 2008;8(6):1480–2. 10.1111/j.1755-0998.2008.02292.x [DOI] [PubMed] [Google Scholar]
- 66.Menotti-Raymond MA, David VA, Wachter LL, Butler JM, O’Brien SJ. An STR forensic typing system for genetic individualization of domestic cat (Felis catus) samples. J Forensic Sci. 2005;50(5):1061–70. [PubMed] [Google Scholar]
- 67.Caporaso J, Lauber C, Walters W, Berg-Lyons D, Lozupone C, Turnbaugh P, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences. 2011;108(Supplement 1):4516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso J, Kuczynski J, Stombaugh J, Bittinger K, Bushman F, Costello E, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7(5):335–6. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeSantis T, Hugenholtz P, Larsen N, Rojas M, Brodie E, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72(7):5069–72. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He Y, Caporaso JG, Jiang X-T, Sheng H-F, Huse SM, Rideout JR, et al. Stability of operational taxonomic units: an important but neglected property for analyzing microbial diversity. Microbiome. 2015;3(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caporaso J, Bittinger K, Bushman F, DeSantis T, Andersen G, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–7. 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price M, Dehal P, Arkin A. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one. 2010;5(3):e9490 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.RDCT. R Development Core Team (2014). R: a language and environment for statistical computing (R Foundation for Statistical Computing; ). 2014. [Google Scholar]
- 74.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8(4):e61217 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shannon C, Weaver W. The mathematical theory of communication (Urbana, IL: University of Illinois Press IL; 1949. [Google Scholar]
- 76.Chao A. Nonparametric estimation of the number of classes in a population. Scandinavian Journal of statistics. 1984:265–70. [Google Scholar]
- 77.Chao A. Species estimation and applications. Encyclopedia of statistical sciences. 2005. [Google Scholar]
- 78.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26(1):32–46. 10.1111/j.1442-9993.2001.01070.pp.x [DOI] [Google Scholar]
- 79.Anderson MJ, Willis TJ. Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology. 2003;84(2):511–25. [Google Scholar]
- 80.Anderson MJ, Ellingsen KE, McArdle BH. Multivariate dispersion as a measure of beta diversity. Ecology letters. 2006;9(6):683–93. 10.1111/j.1461-0248.2006.00926.x [DOI] [PubMed] [Google Scholar]
- 81.Anderson M, Gorley RN, Clarke RK. Permanova+ for Primer: Guide to Software and Statisticl Methods: Primer-E Limited; 2008. [Google Scholar]
- 82.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31(9):814–21. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic acids research. 2011;40(D1):D115–D22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 2011;40(D1):D109–D14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–4. 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gower JC. Generalized procrustes analysis. Psychometrika. 1975;40(1):33–51. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Representation of microbial biodiversity found in various carnivore species, including environmental samples (soil). For the soil and Bengal tiger, we used data from our current study. The data for Dhole1 [54], Dhole2 [49], Wolf [56], Giant panda [55], Snow leopard [47], Antarctic seals [52], Domestic cat1 [53], Domestic cat2 [50] and Cheetah [51] were compiled from other published studies.
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.