

Abstract
Drug resistance is a rapidly emerging concern, thus prompting the development of novel therapeutics or combinatorial therapy. Currently, combinatorial therapy targets are based on knowledge of drug mode of action and/or resistance mechanisms, constraining the number of target proteins. Unbiased genome-wide screens could reveal novel genetic components within interaction networks as potential targets in combination therapies. Testing this, in the context of antimicrobial resistance, we implemented an unbiased genome-wide screen, performed in Saccharomyces cerevisiae expressing a Candida glabrata PDR1+ gain-of-function allele. Gain-of-function mutations in this gene are the principal mediators of fluconazole resistance in this human fungal pathogen. Eighteen synthetically lethal S. cerevisiae genetic mutants were identified in cells expressing C. glabrata PDR1+. One mutant, lacking the histone acetyltransferase Gcn5, was investigated further. Deletion or drug-mediated inhibition of Gcn5 caused a lethal phenotype in C. glabrata cells expressing PDR1+ alleles. Moreover, deletion or drug-mediated inactivation of Gcn5, inhibited the emergence of fluconazole-resistant C. glabrata isolates in evolution experiments. Thus, taken together, the data generated in this study provides proof of concept that synthetically lethal genetic screens can identify novel candidate proteins that when therapeutically targeted could allow effective treatment of drug-resistant infections.
Author summary
Life threatening infections are an increasing reality. Multi-drug resistant bacteria e.g. MRSA are present in nearly all hospitals, and community acquired TB is often recalcitrant to treatment. Less well known, but causing four to five times more deaths in the UK than MRSA, are fungi in the genus Candida. These are commonly associated with mucosal infections such as “thrush”, but are responsible for > 400,000 life-threatening infections worldwide every year, especially in the immunosuppressed patient population. One of the principle pathogens is Candida glabrata. This fungus grows as a single celled yeast, and alarmingly is highly resistant to both host innate defences and many clinically used antifungal drugs. Even with the best possible medical care, infections with this yeast cause ~50% mortality and alarmingly the incidence of C. glabrata infections is steadily climbing. This demands that novel antifungal therapies are developed that can target drug resistant fungal infections such as C. glabrata. We have identified genes that prevent the growth of fungal cells that express gain-of-function mutations in Pdr1, a key mediator of antifungal resistance in C. glabrata. We also provide proof of concept that targeting a protein essential for Pdr1 function significantly inhibits the emergence of antifungal drug resistance. Thus such an approach could provide a powerful tool in developing treatments for drug resistant infections.
Introduction
Drug resistance has emerged as a huge problem in many areas of medicine from cancer to infectious diseases [1, 2, 3, 4]. This is leading to the development of novel therapeutic strategies. Multi-target therapies are gaining ground, where combinations of drugs targeting different components of disease networks are deployed with the expectation of reduced toxicity, emergence of resistance, and off-target effects [5, 6, 7]. Combinatorial therapies involving an antibiotic and a second drug either targeting the same pathway, another cellular function, and/or specific mechanisms of antimicrobial drug resistance have shown promise as therapeutic regimens to treat antimicrobial drug resistant infections [8]. A major impediment to this approach is the characterization of the adjunctive targets. To date most adjunctive therapy targets have been selected based on previous biological knowledge of drug mode of action and/or mechanisms of resistance. This severely constrains the number of proteins that can be targeted for adjunctive therapy. In this study, we hypothesized that unbiased genome-wide screens can reveal previously unknown proteins that could be targeted for adjunctive therapy. This has recently been demonstrated in the context of cancer, where Cas9 mediated genome editing was used to target chromatin regulatory domains in a murine acute myeloma cell line, identifying six known drug targets, and a further 19 genes that are essential in this cancer cell line [9]. In relation to antimicrobial resistant infection, we rationalised that the characterization of mutations that genetically interact with alleles conferring drug resistance could reveal novel proteins that could be therapeutically targeted to allow effective treatment of antimicrobial drug resistant infections.
Fungi are important agents of infectious disease, causing more deaths annually than either malaria or TB [10]. In this context, Candida species are the fourth most commonly isolated species from nosocomial blood stream infections, causing life threatening disease in individuals with AIDs, patients recovering from surgical procedures or major burns, and those undergoing chemotherapy and organ transplant. Systemic fungal infections are currently very difficult to diagnose, and even with best practice management mortality rates are generally higher than for bacterial disease [11]. Furthermore, the effectiveness of the drugs used to treat fungal infections is decreasing, as antifungal drug resistance is rapidly emerging. Antifungal resistance has been reported in environmental fungal isolates suggesting a reservoir of resistant strains [12,13]. There is an urgent clinical and economic need for new cost effective treatment options, including novel therapeutics.
Candida glabrata ranks second after Candida albicans as the most common yeast pathogen of humans. It is responsible for many opportunistic infections in immunocompromised individuals, which are associated with a high mortality rate. The incidence of C. glabrata infections has grown rapidly over the last 20 years, and is responsible for ~25% of systemic candidiasis cases [14,15]. The reason for this increasing incidence of C. glabrata infection is not fully understood, but it is well established that this species has a higher innate tolerance to commonly administered azole antifungals, in particular fluconazole (FLZ), the principle therapeutic option for Candida infections. For instance, C. glabrata populations have an average MIC to fluconazole (FLZ) of 4 μg/ml compared to 0.125 μg/ml for C. albicans populations [16–18]. Alarmingly, C. glabrata is also adept at rapidly acquiring drug resistance [19]. MIC values of 64 μg/ml are found in up to 30% of C. glabrata isolates, and thus are often resistant to FLZ therapy [20,21].
One of the principle mediators of FLZ resistance and acquired resistance are gain of function mutations in the PDR1 (CAGL0A00451g) gene (PDR1+), which encodes a transcriptional activator of genes encoding drug efflux pumps [22]. To date, many PDR1+ mutations have been described that mediate azole resistance (Table 1). These PDR1+ mutations cause amino acid changes across all four functional domains of the transcription factor: the transcriptional activation domain, the regulatory domain, the middle homology region, and the activation domain (Fig 1). PDR1 is up-regulated during systemic infections [23,24], and is induced in response to combinatorial stresses encountered in vivo. C. glabrata strains harbouring PDR1+ mutations exhibit increased virulence [18] implying adaptation within the host to antifungals may itself enhance the ability of C. glabrata to cause disease. In this study, we have performed an unbiased genetic screen to identify mutants that are synthetically lethal with PDR1+ fluconazole resistant C. glabrata cells, having adopted the approach of a combination of genome-wide screens [25] and mutant construction to identify C. glabrata loss of function mutations that interact to impact negatively on the growth in combination of with specific FLZ resistant alleles. We then used prior knowledge to identify which of the proteins encoded by these genetic interactors can be targeted therapeutically, either using known or newly discovered small molecule inhibitors, to treat FLZ resistant C. glabrata.
Table 1. PDR1 clinical isolates and location of gain-of-function mutation used in this study.
| Clinical Isolate name | PDR1 allele |
|---|---|
| DSY486 | WT* |
| DSY489 | L328F |
| DSY562 | WT* |
| DSY565 | L280F |
| DSY2253 | WT* |
| DSY2254 | D1082G |
| DSY529 | WT* |
| DSY530 | E1083Q |
| DSY738 | WT* |
| DSY739 | R376W |
| DSY726 | WT* |
| DSY727 | D876Y |
| DSY753 | WT* |
| DSY754 | Y584C |
| DSY2234 | WT* |
| DSY2235 | T607S |
| DSY3629 | P822L |
| DSY2726 | F853Q |
| DSY2746 | I373V |
| DSY2725 | Y285C |
| DSY1169 | V785D |
| DSY1180 | D1089Y |
| DSY2257 | N691D |
| DSY2268 | S316I |
| DSY1185 | R592G |
| DSY2279 | G583S |
| DSY2271 | D261G |
| DSY2273 | R293I |
| DSY756 | S343F |
| DSY2315 | R376G |
| DSY2277 | R592S |
* refers to the parental strain of gain-of-function mutations that arose after FLZ exposure.
Fig 1. Schematic of PDR1 (CAGL0A00451g).
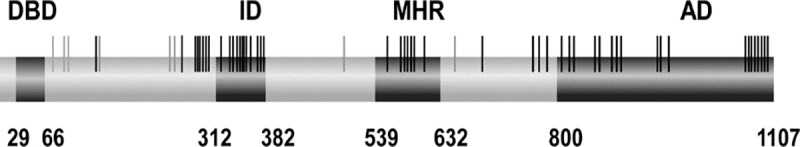
The 4 different domains are indicated by the dark grey boxes – DBD – DNA binding domain; ID – inhibitory domain; MHR- middle homology region and AD – activation domain. Each of the individual light grey and black vertical lines represents a PDR1 gain of function mutation that has been identified in a clinical isolates resulting in resistance and/or tolerance to azoles.
Mutations are termed ‘synthetically lethal’ if either mutation alone has no impact on cellular viability, but in combination result in cellular death. Our hypothesis, was that by inhibiting the product of a gene whose deletion is synthetically lethal with PDR1+ alleles will allow targeting of FLZ resistant C. glabrata. To test this prediction we set to address two questions; what are the synthetic lethal/synthetic sick interaction partners of PDR1+ alleles, and, as proof of concept, can any of these be targeted to prevent the emergence of azole resistant C. glabrata? Therefore the primary aim of this work is to identify pathways that could be targeted to prevent the emergence of antifungal drug resistance. In this study we have taken this methodology to identify conserved synthetic genetics interactions across a PDR1+ allele, then used this data to identify small molecule inhibitors of these synthetic interactors (namely GCN5 inhibitor ɣ-butyrolactone) and determined if they can be used to treat FLZ resistant C. glabrata.
Results & discussion
SDL-SGA screen of C. glabrata PDR1+
Synthetic Genetic Array (SGA) screening is not currently possible in C. glabrata, as the technique relies on high throughput mating. Hence to identify PDR1+ synthetic genetic interactions, we used the model yeast S. cerevisiae as a surrogate with a view that key synthetic lethal interactions would subsequently be confirmed in C.glabrata. To initiate the characterization of C. glabrata PDR1+ synthetic genetic interaction network, we performed a synthetic dosage lethal (SDL)-SGA experiment [26], to identify synthetic interactions with a PDR1+L280F allele described in a clinical C. glabrata isolate DYS565 (Fig 2) [19,21]. This particular allele was chosen due to its poor clinical outcomes and high FLZ MIC [27]. The DYS565 strain has a FLZ MIC of 128 μg/ml and a G840C (L280F) mutation (PDR1+L280F), whereas the parental strain, DYS562, obtained from the same patient is relatively FLZ sensitive (MIC 8 μg/ml) and contains a wild-type PDR1 allele. The PDR1+L280F was amplified by PCR, sequence verified, cloned and transformed into an S. cerevisiae MATa SGA starter strain, with the endogenous copy of PDR1 deleted and then mated to the entire MATα knock-out collection. This genome-wide SGA screen was performed in triplicate and all double mutants were visually scored for growth. A total of 144 negative genetic interactions were identified with the C. glabrata PDR1+L280F allele (Fig 2, S1 Table), of which 22 were synthetically lethal (S2 Table) and the remainder caused significant reductions in growth. Of the 22 synthetic lethal interactions four were also lethal with wild-type PDR1 i.e. elp2, elp4, elp6 and pdr5. Elp2, Elp4 and Elp6 are all components of the elongator complex, while Pdr5 is a multidrug transporter involved in pleiotropic drug responses. Thus 18 strains had specific lethal interactions with C. glabrata PDR1+L280Fand included genes with functions related to drug transport (ERG5, EAF1), and others transcription factors (e.g. PDR3, PDR8, STE12 and UME6). In the case of the synthetic sick interactions identified in both screens, CgPDR1 (104 interactions) and PDR1+L280F (105 interactions), 90 were common to both screens with 14 unique to CgPDR1 and 15 unique to PDR1+L280F (S1 Table and Fig 2).
Fig 2. Genetic interaction network map of C. glabrata PDR1WT and PDR1L280F.

Genome-wide synthetic interaction SGA screens were performed using query strains expressing either the wild type or PDR1+L280F C. glarbata ORF. Genes are represented by nodes that are colour coded corresponding to their cellular roles (www.yeastgenome.org and www.candidagenome.org) and/or assigned through review of the literature. Interactions are represented by edges. A comprehensive list of all interactions can be found in the supplementary information.
To determine if these genetic interactions were maintained with different PDR1+ gain of function alleles, we performed tailored SGA screens. Specifically, the previous interactions identified from the PDR1+L280F screen were sub-arrayed to determine if the synthetic lethal interactions were common to other gain of function alleles. Four other gain-of-function alleles were selected; S316I, L1391I, E555K and F817S, covering the four main functional domains of Pdr1 (S3 Table, S1 Fig and Fig 1). From this refined screen, we were confident in following GCN5 as our proof of principle gene of interest for chemical inhibition, due to its synthetically lethal interaction with the additional gain of function mutants screens and when chemically inhibited in a series of gain of function mutants we were able to induce lethality in the strains (Fig 3).From these additional screens, we identified 9 SL interactions that were common to all gain of function genes tested; DUG1, EA1, ELP4, GLC5, HEK2, PDR5, PDR8, STE12 and STE2. These genes offer potential further targets for chemical inhibition in future studies, across a variety of molecular functions. In S. cerevisiae GCN5 encodes a component of the ADA and SAGA histone acetyltransferase complexes.
Fig 3. FLZ resistant clinical isolates of C. glabrata containing PDR1+ alleles are sensitive to ɣ-butyrolacetone.

C. glabrata FLZ resistant clinical isolates containing PDR1+ mutations were grown on synthetic complete (top panel) or synthetic complete +2mM ɣ-butylroacetone (bottom panel). The ɣ-butylroacetone was synthetic lethal to 20/31 FLZ resistant C. glabrata clinical isolates containing PDR1+ alleles. Plate layout showing which gain-of-function mutants screened shown below the gel images.
Deletion of GCN5 is synthetically lethal in C. glabrata PDR1+ cells
To test our hypothesis that drug targeting of lethal interactors, identified above, would abolish survival of drug resistant PDR1+ cells, we focussed on GCN5 for two reasons. Firstly, deletion of GCN5 was synthetically lethal with the five gain-of-function PDR1+ alleles tested and, secondly, there is a well-characterized specific inhibitor of the Gcn5 HAT, γ-butyrolactone. Thus, if our hypothesis is correct, prevention of Gcn5 function through γ-butyrolactone treatment, should kill C. glabrata cells harbouring the drug resistant PDR1+L280Fallele.
As a first step, we confirmed that expression of PDR1+L280F in a C. glabrata gcn5 null mutant background was lethal (Fig 4). To achieve this PDR1+L280F was placed under the control of the methionine repressible promoter in pCU-MET3 [28] and transformed into a C. glabrata gcn5 pdr1 double null mutant. The induction of PDR1+L280F in this strain resulted in a loss of viability, thus confirming that the synthetic lethal interaction identified in S. cerevisiae is conserved in C. glabrata.
Fig 4. Confirmation that expression of PDR1+L280F in C. glabrata gcn5Δ cells is synthetically lethal.
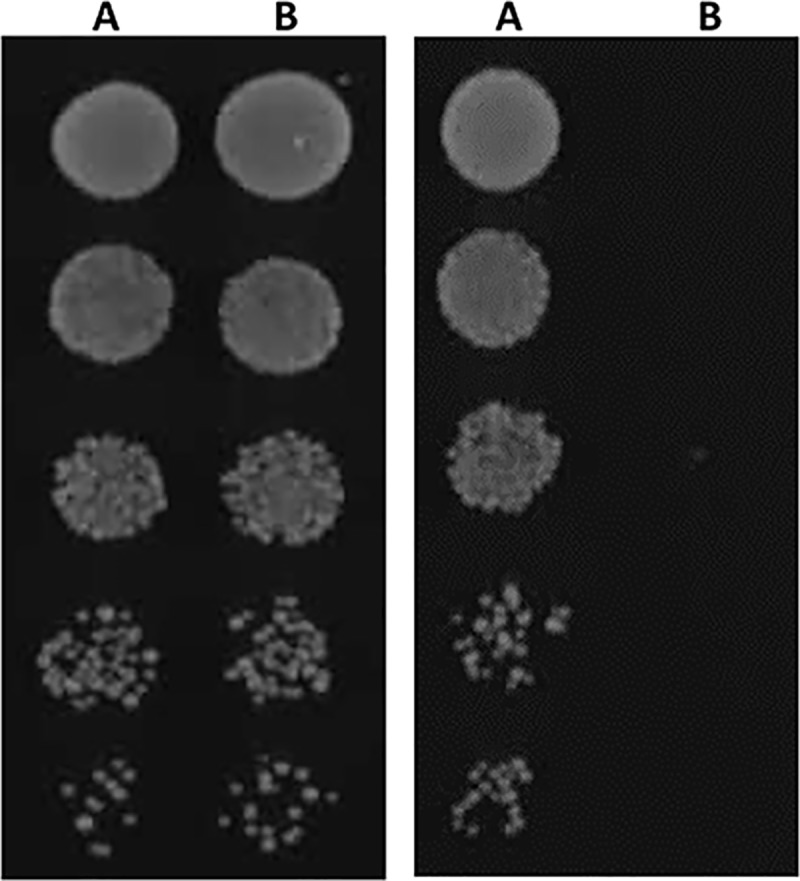
A C. glabrata gcn5 pdr1 strain was transformed with pCU-MET3 (A) or pCU-MET3 containing PDR1L280F (B) and cultured on synthetic complete media (left panel) or synthetic complete media lacking methionine (right panel). Induction of PDR1L280F is lethal in C. glabrata gcn5 cells thus confirming the synthetic lethal interaction identified in S. cerevisiae.
Once we had confirmed that loss of Gcn5 function is lethal in C. glabrata cells expressing the PDR1+L280F fluconazole resistant allele, we then determined the impact of chemically inhibiting Gcn5. Notably, the addition of 2mM γ-butyrolactone, the chemical inhibitor of Gcn5, prevented the growth of S. cerevisiae pdr1Δ strains expressing C. glabrata PDR1L280F, the clinical FLZ resistant C. glabrata strain DYS565 expressing PDR1L280F, and an engineered C. glabrata lab strain (BG2 derivative) in which the wild-type PDR1 allele was replaced with PDR1L280F (Fig 4). Collectively, these data demonstrate that targeting Gcn5, a synthetically lethal interacting partner of PDR1L280F identified in S. cerevisiae, renders both this species and the orthologous C. glabrata mutant inviable, strongly supporting the proposition that targeting synthetic lethal interactions offers a new paradigm for the treatment of drug resistant infection.
To further explore this concept, we investigated whether the addition of ɣ-butyrolactone would prevent and/or inhibit the growth of addition FLZ-resistant clinical isolates with different gain of function mutations in PDR1 (Table 1). Notably, ɣ-butyrolactone prevented the growth of 20/31 clinical isolates screened (Fig 4). This demonstrates that the chemical inactivation of the Gcn5 protein is synthetically lethal in approximately 65% of the PDR1+ FLZ resistant alleles tested.
Does the deletion of synthetic lethal genes, or chemical inactivation of their encoded proteins reduce the emergence of FLZ resistance in C. glabrata?
Finally, we examined whether targeting synthetic lethal interactions could minimise the emergence of FLZ resistance utilising an experimental evolution approach [29]. Using such an approach allowed for the observation of the impact of their deletion on the emergence of FLZ resistance. C. glabrata wild-type and gcn5 null strains, together with wild-type cells in which the function of Gcn5 was chemically inhibited, were exposed to doubling dilutions of FLZ and the emergence of resistance monitored. Our working hypothesis was that FLZ resistance would emerge at a much-reduced rate and to a lower level in strains that had synthetic lethal genes deleted or chemically inactivated (in this case GCN5), compared to wild-type C. glabrata.
As each propagation was made to the next round of selection, the PDR1 gene was sequenced to determine in each condition, and at which cycle, gain of function mutations started to arise in the populations and to what region of the gene they mapped to. For the wild type strain BG2, after going through three rounds of exposure to FLZ and up to a concentration of 8μg/ml, we identified the appearance of the first gain of function mutation in the PDR1 allele (Fig 5A). This mutation was located in the activation domain of the gene. As the exposure to FLZ continued for a further 7 cycles, there was a noted increase in the number of gain of function mutations arising in the wild type strain. This was also linked to the increase in FLZ concentration. Following the 10 cycles of propagation in FLZ, we had identified 50 previously described gain of function mutations in PDR1 (Fig 5B).
Fig 5. Evolution of wildtype, gcn5 null and gcn5 chemically inhibited C. glabrata cells in the presence of fluconazole.

C. glabrata cells grown in increasing concentrations of FLZ, with the PDR1 gene sequenced after each round to determine when and in which domain gain of function mutations are identified in. (A) Schematic of experiment. 10 individual flasks of C. glabrata cells – wildtype, Δgcn5 and chemically inhibited Gcn5 were exposed to increased concentrations of FLZ. The flask where inhibition of growth was first observed was used as the started culture for the subsequence round of drug exposure until 10 rounds of drug exposure was completed. At each pitching of cells, PDR1 was sequenced to identified when a gain of function mutation first emerged. (B) In wildtype C. glarbata cells, after 3 rounds of exposure to increasing FLZ concentrations, PDR1 mutations were identified and mapped to the activation domain. (C) C. glabrata Δgcn5 cells, after 6 rounds of exposure to increasing FLZ concentrations, PDR1 gain of function mutations were isolated and mapped to the activation domain and the middle homology domain. (D) C. glabrata cells that had Gcn5p chemically inhibited through with the addition of ɣ-, after 7 rounds of exposure to increasing FLZ concentrations, the emergence of PDR1 gain of function mutations was observed in the activation domain and DNA binding domain. The number of gain of function mutations observed is dramatically reduced in both the Δgcn5 and chemically inhibited Gcn5 FLZ exposures.
In the case of the gcn5 null strain (Fig 5C), and the chemically inhibited gcn5 strain (Fig 5D), a gain of function mutation was not observed in PDR1 until 7 rounds of propagation in FLZ. In the gcn5 null mutant, the T2450C (F817S) mutation in the activation domain (Table 2) was the first observed mutation, whereas T2575 (F859L) was the first mutation identified in the chemically inhibited strain. From this data, it is possible to determine that the evolution of C. glabrata Δgcn5 mutants in the presence of FLZ inhibits the emergence of gain of function mutations in PDR1.
Table 2. Gain-of-function mutations in the activation domain of CgPDR1 observed during evolution in presence of FLZ.
| T2450C (F817S) |
| C2465T (P822L) |
| T2558C (F853S) |
| T2575C (F859L) |
| G2626T (D876Y) |
| G2827A (G943S) |
| T2837C (L946S) |
| T2842A (F948I) |
| A3229G (N1077D) |
| G3235A (G1079R) |
| G3236T (G1079V) |
| C3239T (T1080I) |
| A3245G (D1082G) |
| G3265T (D1089Y) |
| T3278C (L1093P) |
| G3296C (G1099A) |
Concluding remarks
In this proof of concept study we have demonstrated that the identification of synthetic lethal genetic interactions with alleles that confer antifungal drug resistance is a valuable approach to identify pathways that could be targeted to prevent the emergence of drug resistance. By employing SGA analysis we identified a number of genetic mutations that were synthetically lethal with PDR1+ gain-of-function alleles. Focussing on one specific mutation, that in the histone acetyltransferase Gcn5, we could show that deletion or chemical inactivation of Gcn5 significantly inhibited the emergence of PDR1+ gain-of-function alleles in evolution experiments. Histone modifications modulate the packing of chromatin, this level of packing is critical for gene transcription, as the cellular machinery must have access to promoters to allow for transcription. As previously stated GCN5 in S. cerevisiae is known to be a component of the ADA and SGA complexes, therefore we propose that in C. glabrata clinical isolate with gain of function mutations in PDR1, it is acting as a gene silencer thus resulting in the synthetic lethal phenotype. The combination of the interaction between GCN5 and PDR1gain of function may be resulting in the inhibition of histone acetyltransferases and DNA damage events resulting from drug exposure leading to cell death. This control of chromatin remodelling processes may provide a target for novel drug therapies. Future work employing similar genetic approaches could be powerful in identifying additional targets that could halt the emergence of drug resistant strains.
Methods
Strain generation
PDR1 genes were PCR amplified from their relevant strains, wildtype from BG2 (i.e. no point mutations) and DSY565, the clinical isolate containing the PDR1+ gain of function mutation L280F. PCR products were sequence verified prior to cloning into the Gateway system. Final destination plasmids were transformed into S. cerevisiae strain Y7092[30], using standard LiAc transformation protocols[31].
Candida glabrata PDR1 synthetic genetic interaction screens
The deletion mutant array was manipulated using a Singer RoTor HAD (Singer Instruments). For the genome wide PDR1 and PDR1+L280F synthetic genetic screens, the MATα query strain Y7092 [30] was transformed with either pDEST426-ccdB-GPD-PDR1 or pDEST426-ccdB-GPD- PDR1+L280F. The resulting query strains were mated to the entire MATa deletion mutant array and the SGA methodology was used as previously described to maintain the plasmid [26]. All genome-wide screens were performed in triplicate at 30°C with growth visually scored for lethality (SL), slow growth (synthetic sick SS) or suppression (SUP). Putative genetic interactions were identified in a minimum of two out of three replicates. These putative interactions were then confirmed in S. cerevisiae and C. glabrata. (Confirmed genetic interactions are listed in Supporting information, Fig 2).
Confirmation in Candida glabrata
To confirm the SGA screens performed in S. cerevisiae, we recapitulated the SL phenotypes in C. glabrata. The gcn5 pdr1 null mutant was generated using standard deletion protocol [29], followed by transformation of the plasmid containing the PDR1 or PDR1+L280F under the control of the MET3 promoter [31].
Dot assays with FLZ and γ-butyrolactone
Dot assays were performed by spotting 5 μl of 10-fold serial dilutions (OD600 = 0.1, 0.01, 0.001, 0.0001) onto specified media, and sealed plates were incubated at 37°C. All dot assay experiments were repeated using three different isolates of each strain. FLZ at concentrations 16–64μg/ml and ɣ-butyrolactone at 2mM were used in screening plates.
Evolution of C. glabrata strains in the presence of fluconazole
To examine the effect of FLZ treatment on the genome of C. glabrata strains, we performed a series of control evolution experiments. We took C. glabrata strains; BG2, Δgcn5, and chemically inhibited BG2, and inoculated into doubling dilution of FLZ from 0-256 μg/ml, in synthetic complete medium at 37°C for 24 hours. The culture at the highest FLZ dose with obvious growth was used to propagate the next FLZ gradient. This was performed for ten cycles with each strain being tested in ten technical replicates. The same workflow was followed for the chemically inhibited gcn5 strain to determine the impact on the emergence of resistance via the addition of γ-butyrolactone to the media. The chemical inhibitor was combined with FLZ. For both experimental regimens the level of FLZ was inferred, for each strain, as the concentration from which propagation was made to the next round of selection. The driving hypothesis for this section of work was that FLZ resistance would emerge at a reduced rate and to a lower level in the strains that have had gcn5 deleted or chemically inhibited compared to wild-type C. glarbata.
Supporting information
Genome-wide synthetic interaction SGA screens were performed using query strains expressing PDR1+L139I, PDR1+E555K, PDR1+F817S or PDR1+S316I C. glarbata ORFs. Genes are represented by nodes that are colour coded corresponding to their cellular roles (www.yeastgenome.org and www.candidagenome.org) and/or assigned through review of the literature. Interactions are represented by edges. A comprehensive list of all interactions can be found in the supplementary S3 Table.
(TIFF)
A comprehensive list of interactions from SGA screens for PDR1 wildtype and PDR1 gain-of-function mutation L280F.
(XLSX)
A functional analysis of the genetic interactions for PDR1+L280F. The interactions have been broken down into the separate phenotypic subsets of synthetic lethal and synthetic sick interactions followed by a further breakdown of those unique to the gain-of-function mutation L280F.
(XLSX)
Breakdown of the genetic interactins for 4 different gain-of-function mutations in C. glabrata, the interactions are characterised as either SL – synthicaly lethal or SS – synthetically sick phenotype.
(XLSX)
Acknowledgments
The authors wish to thank Dominique Sanglard for providing the clinical isolates used in this study and his support, and to Janet Quinn for proof reading the manuscript and her support. This work is dedicated to our late friend, supervisor and mentor (DOD 19 March 2018), Prof Ken Haynes. We would like to thank the Candida research community for their support in continuing with this work.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Klein EY. Antimalarial drug resistance: A review of the biology and strategies to delay emergence and spread. Int J Antimicrob Agents [Internet]. Elsevier B.V.; 2013;41(4):311–7. Available from: 10.1016/j.ijantimicag.2012.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler G, Rasmussen MD, Lin MF, Santos M a S, Sakthikumar S, Munro C a, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature [Internet]. Nature Publishing Group; 2009. June 4;459(7247):657–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19465905 10.1038/nature08064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gow N a R, Brown AJP, Odds FC. Fungal morphogenesis and host invasion. Curr Opin Microbiol [Internet]. 2002. August;5(4):366–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12160854 [DOI] [PubMed] [Google Scholar]
- 4.Kaloriti D, Tillmann A, Cook E, Jacobsen M, You T, Lenardon M, et al. Combinatorial stresses kill pathogenic Candida species. Med Mycol [Internet]. 2012. October [cited 2013 Apr 12];50(7):699–709. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3483063&tool=pmcentrez&rendertype=abstract 10.3109/13693786.2012.672770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. Nature Publishing Group; 2012. July;30(7):679–92. 10.1038/nbt.2284 [DOI] [PubMed] [Google Scholar]
- 6.Nosten F, White NJ. Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg. 2007;77(SUPPL. 6):181–92. [PubMed] [Google Scholar]
- 7.Richman DD. HIV chemotherapy. Nature. 2001;410(6831):995–1001. 10.1038/35073673 [DOI] [PubMed] [Google Scholar]
- 8.Worthington RJ, Melander C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol [Internet]. Elsevier Ltd; 2013;31(3):177–84. Available from: 10.1016/j.tibtech.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–7. 10.1038/nbt.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. Hidden Killers: Human Fungal Infections. 2012;4(165):1–10. [DOI] [PubMed] [Google Scholar]
- 11.Falagas ME, Apostolou KE, Pappas VD. Attributable mortality of candidemia: A systematic review of matched cohort and case-control studies. Eur J Clin Microbiol Infect Dis. 2006;25(7):419–25. 10.1007/s10096-006-0159-2 [DOI] [PubMed] [Google Scholar]
- 12.Pfaller MA. Antifungal drug resistance: Mechanisms, epidemiology, and consequences for treatment. Am J Med [Internet]. Elsevier Inc.; 2012;125(1 SUPPL.):S3–13. Available from: 10.1016/j.amjmed.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 13.Alexander BD, Johnson MD, Pfeiffer CD, Jiménez-Ortigosa C, Catania J, Booker R, et al. Increasing echinocandin resistance in candida glabrata: Clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis. 2013;56(12):1724–32. 10.1093/cid/cit136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. Candida bloodstream infections: Comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008-2009. Antimicrob Agents Chemother. 2011;55(2):561–6. 10.1128/AAC.01079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hajjeh RA, Sofair AN, Harrison LH, Lyon GM, Arthington-Skaggs BA, Mirza SA, et al. Incidence of Bloodstream Infections Due to Candida Species and In Vitro Susceptibilities of Isolates Collected from 1998 to 2000 in a Population-Based Active Surveillance Program. J Clin Microbiol. 2004;42(4):1519–27. 10.1128/JCM.42.4.1519-1527.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vale-Silva L, Ischer F, Leibundgut-Landmann S, Sanglard D. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in candida glabrata, control adherence to host cells. Infect Immun. 2013;81(5):1709–20. 10.1128/IAI.00074-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One. 2011;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009. January;5(1):e1000268 10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tumbarello M, Sanguinetti M, Trecarichi EM, La Sorda M, Rossi M, de Carolis E, et al. Fungaemia caused by Candida glabrata with reduced susceptibility to fluconazole due to altered gene expression: Risk factors, antifungal treatment and outcome. J Antimicrob Chemother. 2008;62(6):1379–85. 10.1093/jac/dkn381 [DOI] [PubMed] [Google Scholar]
- 20.Borst A, Raimer MT, Warnock DW, Morrison CJ, Arthington-Skaggs BA. Rapid acquisition of stable azole resistance by Candida glabrata isolates obtained before the clinical introduction of fluconazole. Antimicrob Agents Chemother. 2005;49(2):783–7. 10.1128/AAC.49.2.783-787.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanglard D, Ischer F, Bille J. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob Agents Chemother. 2001;45(4):1174–83. 10.1128/AAC.45.4.1174-1183.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vermitsky J-P, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol [Internet]. 2006;61(3):704–22. Available from: 10.1111/j.1365-2958.2006.05235.x [DOI] [PubMed] [Google Scholar]
- 23.Fukuda Y, Tsai HF, Myers TG, Bennett JE. Transcriptional profiling of Candida glabrata during phagocytosis by neutrophils and in the infected mouse spleen. Infect Immun. 2013;81(4):1325–33. 10.1128/IAI.00851-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaur R, Ma B, Cormack BP. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci [Internet]. 2007;104(18):7628–33. Available from: 10.1073/pnas.0611195104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006. February;21(3):319–30. 10.1016/j.molcel.2005.12.011 [DOI] [PubMed] [Google Scholar]
- 26.Usher J, Thomas G, Haynes K. Utilising established SDL-screening methods as a tool for the functional genomic characterisation of model and non-model organisms. FEMS Yeast Res. 2015;15(8):1–8. [DOI] [PubMed] [Google Scholar]
- 27.Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglarnd D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One. 2011; 9;6(3):e17589 10.1371/journal.pone.0017589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zordan RE, Ren Y, Pan S-J, Rotondo G, Peñas AD Las, Iluore J, et al. Expression Plasmids for Use in Candida glabrata. G3; Genes|Genomes|Genetics [Internet]. 2013;3(10):1675–86. Available from: 10.1534/g3.113.006908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shou C, Bhardwaj N, Lam HYK, Yan K-K, Kim PM, Snyder M, et al. Measuring the evolutionary rewiring of biological networks. PLoS Comput Biol [Internet]. 2011. January [cited 2011 Jun 11];7(1):e1001050 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3017101&tool=pmcentrez&rendertype=abstract 10.1371/journal.pcbi.1001050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hin A, Tong Y, Evangelista M, Parsons AB, Xu H, Bader GD, et al. Systematic Genetic Analysis with Ordered Arrays of Yeast Deletion Mutants. 2001;294(December):2364–9. [DOI] [PubMed] [Google Scholar]
- 31.Gietz RD, Schiestl RH. Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2(1):35–7. 10.1038/nprot.2007.14 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome-wide synthetic interaction SGA screens were performed using query strains expressing PDR1+L139I, PDR1+E555K, PDR1+F817S or PDR1+S316I C. glarbata ORFs. Genes are represented by nodes that are colour coded corresponding to their cellular roles (www.yeastgenome.org and www.candidagenome.org) and/or assigned through review of the literature. Interactions are represented by edges. A comprehensive list of all interactions can be found in the supplementary S3 Table.
(TIFF)
A comprehensive list of interactions from SGA screens for PDR1 wildtype and PDR1 gain-of-function mutation L280F.
(XLSX)
A functional analysis of the genetic interactions for PDR1+L280F. The interactions have been broken down into the separate phenotypic subsets of synthetic lethal and synthetic sick interactions followed by a further breakdown of those unique to the gain-of-function mutation L280F.
(XLSX)
Breakdown of the genetic interactins for 4 different gain-of-function mutations in C. glabrata, the interactions are characterised as either SL – synthicaly lethal or SS – synthetically sick phenotype.
(XLSX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.