

Abstract
Purpose of Review
We reviewed the current literature on the roles of the Wnt antagonists sclerostin (Sost) and sclerostin-containing domain protein 1 (Sostdc1) on bone homeostasis, the relationship of the hypoxia-inducible factor (Hif) and von Hippel-Lindau (Vhl) pathways on Sost expression, and how changes in bone induced by depletion of Sost, Sostdc1, and Vhl affect hematopoietic cells.
Recent Findings
B cell development is adversely affected in Sost-knockout mice and is more severely affected in Vhl-knockout mice. Inflammation in the Sost−/− bone microenvironment could alter hematopoietic stem cell behavior. Sostdc1−/− mice display defects in natural killer cell development and cytotoxicity.
Summary
Depletion of Sost and Sostdc1 have effects on immune cell function that warrant investigation in patients receiving Wnt antagonist-depleting therapies for treatment of bone diseases. Additional clinical applications for manipulation of Wnt antagonists include cancer immunotherapies, stem cell transplantation, and directed differentiation to immune lineages.
Keywords: Wnt, Wnt antagonists, Hematopoiesis, Immunology, Cell differentiation, Osteoimmunology
Introduction
Osteoblast, osteocyte, and osteoclast cells maintain bone homeostasis by building, supporting, and breaking down bone tissue, respectively. The Wnt signaling pathway plays a major role in the regulation of bone homeostasis and is a strategic target for the treatment of bone diseases such as osteoporosis [1–3]. Wnt signaling also plays a role in hematopoiesis and immune cell development [4, 5], but the mechanisms of crosstalk between the skeletal and hematopoietic systems is incompletely understood. Here, we briefly review the Wnt signaling pathway and discuss how deficiencies or depletion of the Wnt antagonists sclerostin (Sost) and sclerostin domain-containing protein-1 (Sostdc1) affect bone homeostasis. In turn, we review current findings that suggest changes in bone homeostasis induced by altering expression of these Wnt antagonists (directly by targeted knockout strategies, or indirectly via manipulation of hypoxia pathway genes) influence hematopoietic stem cell fate and immune cell development and function (Table 1).
Table 1.
Summary of effects of selected Wnt signaling antagonists on the skeletal and hematopoietic systems
| Antagonist | Binding partners | Regulator of bone formation | Loss or gain of function model | Effects on bone | Immune or osteolineage cells expressing antagonist in mice | Effects on immune cells | References |
|---|---|---|---|---|---|---|---|
| Sostdc1 | Lrp4 Lrp5/6 BMPs | Negative | Knockout mouse | Increased cortical bone mineral density, increased marrow area, enhanced bone fracture healing | Bulk CD4+ and CD8+ T cells, MSCs and periosteal cells, CD4+PD-1+ T cells in spleen, TFH and TFR cells in Peyer’s Patch and peripheral lymph nodes | NK cells: maturation block and reduced cytotoxicity B cells: Increased memory B cell frequencies |
[6, 7, 8••, 9••, 10•, 11] |
| Sost | Lrp5/6 | Negative | Knockout mouse | Increased bone mass and total cortical area, decreased BM cavity. | Osteoblasts, osteocytes, chondrocytes, MSCs | B cells: differentiation and functional defects. Thl7 cells: Promotes differentiation in vitro Tregs: decreased in vitro. |
[12–15] |
Canonical and Non-canonical Wnt Pathways
The canonical Wnt/β-catenin pathways and non-canonical Wnt signaling pathways play important roles in bone tissue development, hematopoiesis, and immune cell development [4, 5, 16–18]. β-catenin is a key mediator of canonical Wnt signaling, where it is first sequestered in the cytoplasm by its interactions with Axin, GSK3, and APC proteins, and β-catenin activity is regulated by ubiquitin-initiated degradation [19]. Upon activation of Wnt signaling, β-catenin is translocated into the nucleus, and it cooperates with TCF1 and LEF1 co-activator proteins to control the transcription of Wnt target genes (Fig. 1a, left). Although binding of canonical Wnt ligands (such as Wnt3a, Wnt1, Wnt8) to Wnt receptors initiates the signal transduction pathways, the extent and intensity of this activation is influenced by multiple factors, including Wnt receptors, co-receptors, Wnt chaperones [20], and soluble Wnt antagonists [5, 21, 22]. Non-canonical Wnt signaling pathway is activated through ligands such as Wnt5a and Wnt11, and results in the phosphorylation of JNK, which subsequently translocates to the nucleus for gene transcription (Fig. 1a, right). Wnt signaling in bone may also be regulated by microenvironmental conditions, such as hypoxia, via Hif1α [23–26] (Fig. 1b and c).
Fig. 1.
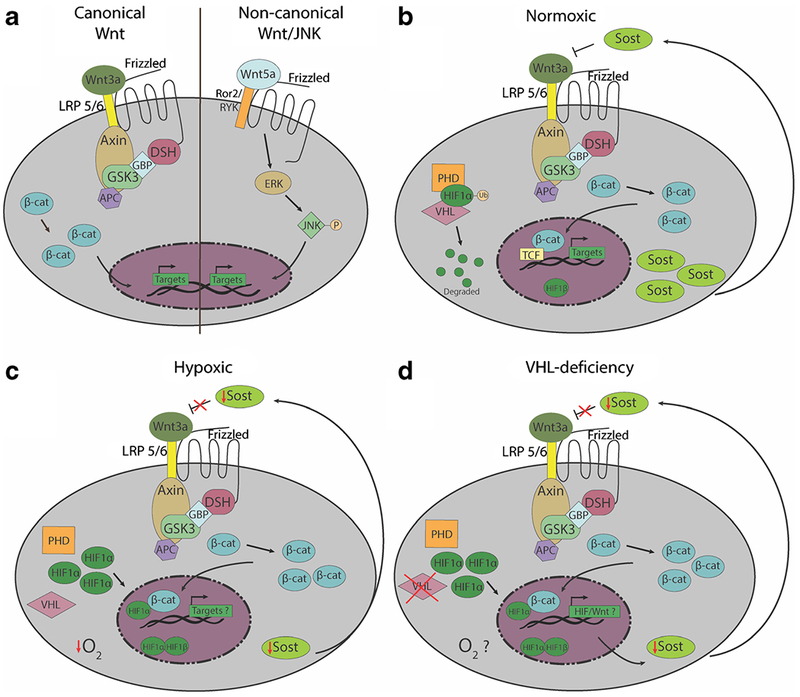
Overview of Wnt signaling pathways and the regulation by Wnt antagonists and the bone microenvironment. a Schematic of canonical (left) and non-canonical (right) Wnt pathways. In canonical Wnt signaling, binding of ligands like Wnt3a to Frizzled family receptors results in the release of β-catenin from the Axin/GSK3/APC/DSH/GBP complex, allowing β-catenin to translocate from the cytoplasm into the nucleus for Wnt target gene transcription. In the non-canonical Wnt/JNK pathway, β-catenin is not utilized. Non-canonical Wnt ligands such as Wnt5a also bind to Frizzled receptors, but utilize the Ror2/Ryk coreceptors to activate downstream phosphorylation of JNK by ERK. Phosphorylated JNK then translocates into the nucleus to activate Wnt target gene transcription. b Under normoxic conditions, PHDs hydroxylate HIF1α, allowing von Hippel-Lindau (Vhl) protein to ubiquitinate Hif1a, which is subsequently degraded. In normal conditions SOST binds to LRP 5/6 and antagonizes Wnt3a c Under hypoxic conditions, PHDs do not hydroxylate HIF1α, and VHL protein is inactivated. Therefore, HIF1α is not ubiquitinated or degraded, and is translocated into the nucleus to bind HIF1β to activate transcription of hypoxia-induced genes. Genetos et al. demonstrated that under hypoxic conditions, SOST is decreased in osteoblasts, which could lead to an increase in canonical Wnt signaling. There is evidence that Hif1α and β-catenin can act as co-activators, but how hypoxia affects Wnt signaling is not well understood. d In Dmp-Cre driven Vhl-knockout mice, which have been used as a model to study the effects of Hif1α stabilization in osteocytes in vivo, Sost expression is diminished. In turn, this decrease may may result in increased canonical Wnt signaling and promote the building of bone mass. However, whether the osteocytes or the other cells within the bone and marrow microenvironments of Vhl-conditional knockout are truly hypoxic awaits further investigation.
The Role of Wnt in Hematopoiesis and Immune Function
The regulation of adult hematopoiesis is a complex process, with contributions from many signaling pathways, including Wnt, Notch, FGF, and Hedgehog [27]. These pathways are involved in the differentiation, self-renewal, and quiescence of hematopoietic stem cells (HSCs). Early studies demonstrated that overexpression of a constitutively active form of β-catenin increased the proliferation and repopulation capacity of transplanted HSCs in mice [28, 29]. Subsequent work reported that overexpression of stable β-catenin led to exhaustion of long-term HSCs and a disruption in multilineage differentiation [30]. Mechanisms by which Wnt signaling regulates hematopoiesis in a dose-dependent fashion has more recently been revealed [31]. Low levels of Wnt signaling cause HSCs to display increased reconstitution ability, but higher levels of Wnt signaling cause failure of donor HSCs to reconstitute irradiated recipients. This lack ofreconstitution potential in HSCs is due to increased differentiation and loss of sternness [31]. The distinct role of the non-canonical Wnt pathway in hematopoiesis and lymphopoiesis has also been described [32]. However, how Wnt antagonists differentially regulate canonical and non-canonical Wnt pathways, and if Wnt antagonists secreted by cells of the skeletal lineage can influence Wnt signaling activation in adjacent hematopoietic and immune cells, have not been fully addressed.
The Role of Wnt Antagonists in Skeletal and Immune Systems
Sclerostin
The neutralization of sclerostin results in increased bone mass in mice and humans through facilitation of Wnt signaling [33]. Sclerostin (Sost) is a protein secreted primarily by osteocytes that negatively regulates bone formation [34] through inhibition of canonical Wnt signaling in osteoblasts [35–37]. Sclerostin binds to the Wnt co-receptors Lrp4, Lrp5, and Lrp6 and inhibits downstream Wnt signaling [38, 39]. A Wnt target gene of interest is osteoprotegerin (OPG), an inhibitor of bone resorption. In Sost−/− mice, where Wnt signaling is enhanced in bone, OPG is increased, and bone resorption decreases [40]. Sost also plays a role in the inhibition of the non-canonical Wnt/JNK pathway [41]. Sost is expressed in hypertrophic chondrocytes and is highly expressed in damaged cartilage, suggesting its role in bone and cartilage remodeling. Through Sost expression, restoration of anabolic genes normally inhibited by Wnt5a is achieved, allowing full metabolic activity of chondrocytes in cartilage lesion mending after mechanical stress.
Cain et al. were the first to demonstrate a role of Sost in the regulation of immune cell development, using global Sost−/− mice (MGI:3797839). The number of B lymphocytes in the Sost−/− bone marrow was reduced, due to impaired B lymphocyte survival [12]. Importantly, this effect was mediated in an indirect, cell-extrinsic fashion, as B lymphocytes do not express Sost, and wild-type bone marrow cells transplanted into Sost−/− mice displayed impaired B cell development. Further analysis demonstrated that the bone marrow stromal cells in Sost−/− mice expressed low levels of CXCL12, a critical B cell growth factor, which might contribute to the defect in B cell development. Sost is also expressed by osteoblasts and mesenchymal stem cells and identified the contributions of Sost in specific osteolineage cells using conditional Sost-knockout mice (SostiCOIN/iCOIN, MGI:5544793) [42••]. Surprisingly, the deletion of Sost in osteocytes (using Dmp-Cre) did not affect B lymphocyte development, but the deletion of Sost in MSCs (using Prxl-Cre) resulted in a decrease in the B cell precursors and immature B cell subsets. However, this decrease was not as pronounced as the global Sost−/− mice, indicating another cell that expresses Sost is involved in B cell development. Taken together, these findings imply that patients receiving sclerostin-depleting therapies for osteoporosis might suffer from B cell defects. Functional analysis of B cells in Sost−/− mice supports that B cell responses to antigens may be altered [13], but this has yet to be confirmed in humans.
Current studies are investigating whether exposure of hematopoietic progenitors to Sost-deficient microenvironments influences their differentiation. Transplantation of wild-type hematopoietic stem and progenitor cells (HSPC) into Sost−/− recipient mice suggest that the Sost−/− bone marrow niche tends to favor hematopoietic differentiation to the myeloid lineages. Further, extramedullary hematopoiesis in the spleen is clearly evident in aged Sost knockout mice (Manilay et al., unpublished data). These changes in hematopoiesis may be explained through Wnt5a overexpression. Staal and colleagues discovered that Wnt5a overexpression resulted in a marked increase in myeloid cells in the bone marrow and spleen, whereas Wnt3a overexpression resulted in an increase in lymphopoiesis [32]. In addition to the already known decrease in B cell lymphopoiesis, it is possible that lack of Sost in the bone influences myelopoiesis in mice. Increases in myeloid cells, and decreases in lymphoid cells, are indicative of an inflammatory aging hematopoietic development, and may lead to weakened immune responses. Although mice and patients with mutations in the Sost gene exhibit similar bone marrow cavity occlusion, as observed in Sost−/− mice, it is unclear from the literature whether hematopoiesis has been monitored in these patients [43–45]. Our findings in Sost−/− mice indicate that patients receiving anti-sclerostin antibodies may have altered developmental hematopoiesis in the bone marrow and spleen. This could have serious implications for patients with already diminished immune systems like the elderly, who are the primary recipient population for osteoporosis therapeutics.
von Hippel-Lindau and Hypoxia-Induced Factors
Hypoxia is a state in which the supply of oxygen is insufficient for normal cell function. Hypoxia interferes with oxygen transport and the synthesis of ATP, causing cell damage and cell death when ATP production fails to meet energy demands. One key cellular response to counter the hypoxic state is the triggering of hypoxia-inducible factor (Hif)-mediated gene expression. The Hif signaling pathway has a central role in oxygen homeostasis and it responds accordingly to oxygen tension. In normoxic conditions, prolyl hydroxylase-domain proteins (PHDs) hydroxylate hypoxia-inducible factor alpha (Hif1 α) allow von Hippel-Lindau (Vhl) protein to ubiquitinate Hif1α and Hif2a for degradation [46, 47] (Fig. 1b). Under hypoxic conditions, PHD function is inhibited and Vhl protein is inactivated, resulting in HIF1α accumulation and activation of hypoxia-response genes in the nucleus (Fig. 1c). There are over 100 known direct HIF target genes with roles in diverse biological pathways such as angiogenesis, cell proliferation, redox homeostasis, and apoptosis [48–50].
Hypoxia slows the processes of angiogenesis and osteogenesis during fracture healing and bone formation [51, 52], but also promotes osteoblast differentiation into osteocytes [53], and can stimulate osteoclast formation [54]. HIF stabilization may be a therapeutic option for treating bone fractures [55, 56] and osteoporosis [57–59], but the underlying molecular mechanism remains poorly understood. Vhl plays an important role regulating HIF expression, and disruption of VHL in bone cells leads to improper bone homeostasis. For example, Vhl depletion in osteochondral progenitor cells and osteocalcin-positive osteoblasts leads to an increase in bone mass through an increase in osteoblast number [60, 61] Depletion of Vhl in osteoblasts also had accelerated bone repair and had implications for skeletal regeneration [62]. Furthermore, disrupting Vhl in osteoblasts induces expression of β-catenin, revealing the mechanism by which Vhl/Hif pathway affects bone formation through the Wnt pathway [63]. Recent work in Vhl-conditional knockout (Vhlfl/fl; Dmp1-Cre) mice shows that Sost expression is decreased in the absence of Vhl [63, 64••] (Fig. 1d), but whether this change in Sost expression is due to actual reduction in oxygen tension awaits further investigation.
It is interesting that the global Sost−/− and osteocytic-specific Vhl-knockout mice share some similarities in their bone and B cell phenotypes (although the effects are more severe in the Vhl-knockout). The mechanism by which Vhl/Hif pathways and Wnt pathways interact in normoxic and hypoxic bone microenvironments is not fully elucidated, but there are several lines of evidence that support they are dependent on each other. HIF1α can directly bind to the promoter sequence of Sost [65]. Enhanced HIF1α expression through PHD2 depletion in osteocytes decreased sclerostin expression and enhanced Wnt/β-catenin signaling [66•]. In vitro, hypoxia decreases Sost expression in cultured osteoblasts and osteocytes through BMP [67], but contradictory results indicated that hypoxia can also induce Sost expression in osteoblasts [65]. These discrepancies could perhaps be explained by the different cell lines used in these in vitro studies. In vivo, deletion of Vhl in osteoblasts results in activation of HIF1α, and a decrease in Sost-positive osteocytes [63]. Vhl deletion in osteocytes in vivo also results in a decrease in Sost [64••].
Osteoblasts also serve a supportive function in the maintenance of hematopoiesis and B lymphocytes [5, 68]. The bone marrow microenvironment is hypoxic, which is crucial for normal hematopoiesis [69]. Heterogeneities of local pO2 exist within the bone marrow [70]. However, the implications of these variations in oxygen tension in hematopoietic stem cells and hematopoiesis remain uncharacterized. How hypoxia, Vhl, and Wnt signaling crosstalk regulates bone homeostasis and B cell development is an area of active research. The role of HIF and its regulation of the immune system has been extensively reviewed, but the mechanism of Vhl in specific immune cell lineages has not fully been addressed [71]. Localized hypoxia and HIF stabilization are normal features of germinal centers. Development of robust antibody responses from conventional B lymphocytes (a.k.a. B-2 cells) is influenced by the relatively low oxygen levels in the germinal centers of the spleen and lymph nodes [72]. Cell-intrinsic deletion of Vhl in B cells in mice (Vhlfl/fl; ERT2-Cre) stabilizes Hif1α levels and affects B cell function by impairing cell proliferation, antibody class-switching, generation of high affinity antibodies, and antibody responses [72]. Analyses by Loots et al. suggest that specific deletion of Vhl in osteocytic cells results in cell-extrinsic changes that do not support development and survival of B-2 cells. For example, in the bones of Vhtfl/fl; Dmp1-Cre mice, the number of hematopoietic cells is severely reduced, and B-2 B cell development is stunted. These mice also display splenomegaly, partly due to a movement of hematopoietic progenitors from the bone marrow to the spleen. Despite this increase in splenic hematopoiesis, the numbers of mature conventional B-2 cells is still reduced in the spleen [64••]. These data suggest that Vhl in osteocytic cells regulates B-2 cell development, but further studies are necessary to investigate the mechanisms underlying these observations.
Sostdc1
Sclerostin domain-containing protein-1 (Sostdc1), also known as Wise, Ectodin, Usag-1, and Sost-like, has been widely studied in the framework of tooth development, kidney disease, hair follicle formation, bone fracture, and cancers [73–76]. Sostdc1 and Sost share 55% protein sequence homology and both antagonize Wnt signaling by binding Lrp5 and Lrp6, whereas Sostdc1 additionally antagonizes BMP signaling and preferentially binds to Lrp4 [6, 7]. Sostdc1−/− (Sostdc1tm1.1(KOMP)vlcg, MGI:5695910) mice have increased cortical bone area (TA) and bone mineral density (BMD), suggesting that Sostdc 1, like Sost, are both negative regulators of bone formation [8••]. However, Sostdc1−/− mice have enlarged bone marrow areas, which is contrary to Sost-deficient mice, which have diminished bone marrow area [8••, 12]. Consistent with these findings, we observed an increase in bone marrow and spleen cellularity in Sostdc1−/− mice compared to wild-type control mice [9••]. Sostdc1−/− mice display enhanced callus bone formation and remodeling after 28-day post-fracture, with an increase of mesenchymal stem cells (MSCs) and mature osteoblasts at the site of fracture. In addition, Sostdc1 expression was highly expressed in the periosteal region of mice femora and MSCs during bone fracture, suggesting that Sostdc1 may prompt MSCs out of quiescence and promote bone healing [8••].
New work is revealing how deletion of Sostdc1 in the bone microenvironment may affect immune cell development. Contrary to observations in Sost- and VHL-knockout mice, B cell development in Sostdc1−/− mice is normal. However, Millan et al. have revealed a novel role for Sostdc1 in natural killer (NK) cell maturation and function [9••]. NK cells are lymphocytes from the innate immune system that eliminate virally infected and cancerous cells. As a function of age, Sostdc1−/− mice have progressively diminished numbers of mature NK cells in bone marrow and spleens, indicating a partial block in NK cell maturation. NK cells recognize target cells by Ly49 receptors that are involved in cellular self- and non-self-recognition [77, 78]. NK cells from Sostdc1−/− mice express an altered Ly49 repertoire and are hyporesponsive against MHC-I-deficient cell targets in vitro and in vivo. Consistent with Sostdc1’s known role in antagonizing Wnt signaling, Wnt/β-catenin signaling is increased in NK cells from Sostdc1−/− mice. These data suggest that Sostdc1 is required to modulate Wnt/β-catenin signaling in NK cells for maturation and function. Sostdc1 is not expressed in NK cells themselves, and studies by Millan et al. suggest that Sostdc1 expressed by both non-hematopoietic (MSCs) and hematopoietic cells (CD4+ and CD8+ T lymphocytes) regulate NK cell maturation, Ly49 repertoire expression, and NK cell cytotoxicity.
It is now evident that specialized T lymphocytes express Sostdc1, but the interaction between T cells and NK cells in maturation and function is understudied. For example, T follicular helper (TFH) and T follicular regulatory (TFR) cells are found in the peripheral lymph node (pLN) and Peyer’s Patches (PP). These cells are a small subset of CD4+ T cells that express high levels of Sostdc1 [10•]. Additionally, TFR cells in the PP secrete NK cell regulatory cytokines, IL-2 and IL-21, which play an important role in NK cell maturation and function. Analysis of the PP in Sostdc1−/− mice further demonstrated a decrease in memory B cell frequency and fecal IgA secretion, but no differences were observed in TFH and TFR cell frequencies. Additionally, CD4+PD-1+ T cells with memory phenotype (CD44HighCD62LLow) expresses high levels of Sostdc1 in the spleen of aged mice [11]. In vitro anti-CD3 and anti-CD28 stimulation of CD4+ PD-1+ T cells result in an increase of Sostdc1, Srr1, and osteopontin [11]. It is yet to be determined if CD4+ PD-1+ T cells, TFH and TFR cells, directly or indirectly regulate NK cell maturation and function via Sostdc1.
Future Directions
Can Wnt Antagonists Be Used for HSC Expansion and to Direct Hematopoietic Differentiation In Vitro?
An important goal in hematopoietic studies is expansion of patient HSCs ex vivo for different hematological treatments such as bone marrow transplantation. It has been proposed that exploitation of highly evolutionarily conserved pathways such as Wnt signaling could be used in this instance [79, 80]. By increasing canonical Wnt signaling through inhibition of Wnt antagonists like SOST, it might be possible to manipulate patient hematopoietic stem cells ex vivo towards expansion without differentiation. Wnt signaling in conjunction with organoid stem cell culture systems has made recent advances in maintaining and expanding other stem cell types in culture [81]. An organoid system is defined as a culture system with self-renewing cells that are self-organizing and differentiate into multiple cell types that recapitulate normal development. Organoid-initiating cells often express the Wnt target gene Lgr5 and these cells can be maintained in culture for long periods of time when provided with the favorable conditions and cytokines [81]. However, Lgr5 is expressed at very low levels in adult hematopoietic stem and progenitor cells. It is expressed, however, at slightly higher levels in HSCs derived from the embryonic aorta-gonad-mesonephros (AGM) region and fetal liver. These embryonically derived hematopoietic stem cells only are able to reconstitute myeloid and lymphoid lineages for a short period of time of 2 months [82]. Titration of soluble canonical and non-canonical Wnt ligands and Wnt antagonists might be useful to optimize the conditions for the expansion of long-lived self-renewing HSCs or directed differentiation to specific hematopoietic lineages in vitro [32, 83, 84], for subsequent in vivo transplantation.
Recent studies have also shown that secretion of Dkk1, another Wnt antagonist protein, from bone marrow-derived osteoprogenitors, promoted hematopoietic regeneration directly through inhibition of HSC quiescence as well as indirectly through EGF secretion by BM endothelial cells [85•]. Dkk1 inhibition increases Sost expression, which suggests a compensatory role for Sost in the absence of other Wnt antagonist [86]. Similarly, the activation of Wnt signaling through inhibition of Sost increases Dkk1 expression and limits bone formation in a negative feedback loop; therefore, perhaps combinations of Wnt antagonists could be used directly to promote hematopoietic regeneration in patients [87].
Could Wnt Antagonists Be Used to Improve Cancer Immunotherapies?
Natural killer cells provide an immediate response for the control of tumors and virally infected cells, which makes them an attractive source for immunotherapies. Current NK cell immunotherapies include engineering NK cells with chimeric antigen receptors (CAR), which had milder side effects in patients as compared to the CAR-T cell-based therapies [88]. However, CAR-NK immunotherapies are limited by small NK cell numbers, survival, and proliferation once administered to the patient [88]. Increasing evidence now suggests Wnt/β-catenin signaling may promote cancer progression by negatively regulating immune cell cytotoxicity [89–91]. Our studies have demonstrated that Sostdc1−/− mice have altered bone marrow and splenic microenvironments and defective NK cell cytotoxicity against MHC-I-deficient targets. In the absence of Sostdc1, Wnt/β-catenin signaling in NK cells is increased [9••]. Cytotoxic T cells (CD3+CD8+) are also negatively regulated by Wnt/β-catenin signaling [89]. Taken together, these data support that activation of Wnt/β-catenin signaling in NK and T cells reduces their potential cytotoxicity towards tumor cell targets. Thus, it is possible that enhancing the expression of Sostdc1 and decreasing Wnt/β-catenin signaling in the bone marrow and spleen of cancer patients may promote the immune microenvironment to support robust functional T and NK cell responses against cancerous cells. Furthermore, modulating the levels of Sostdc1 may be used jointly with the already accessible CAR-NK cell-based therapy to increase the success of this emerging technology.
Can Hypoxia That Is Induced in the Bone Be Used with Wnt Antagonists to Regulate Immune Cell Differentiation?
The crosstalk between hypoxia, bone, immune cells, and metabolic regulation could present a novel therapeutic approach to immune deficiencies. Hypoxia also has implications on glucose homeostasis and little is known about the metabolic pathways used by osteoblast lineage cells. Glucose metabolism and glycolysis is important during osteoblast differentiation and for HIF-driven bone formation [92, 93]. Aerobic glycolysis is important for osteoblast differentiation and bone formation regulated by Wnt and HIF pathways [92, 94]. A recent study showed that disrupting Vhl in osteoblasts leads to an increase in glucose tolerance and hypoglycemia [95••], altering the whole-body glucose homeostatic process. These findings indicate that metabolic regulation in osteoblasts can have effects beyond the bone environment, and might be controlled by hypoxia via the Vhl/Hif pathway In turn, changes in osteoblast cellular metabolism could impact immune cell fate, function, and glycolytic gene expression in hypoxic environment through the Hif pathway [96]. Vhl depletion in osteocytes has a detrimental effect on B lymphocyte development [64••], but the mechanism underlying requires additional investigation. Whether the whole-body glucose dysregulation in mice with Vhl-deficient osteoblasts has an impact on immune cells and response, and whether it can be prevented by overexpression of Wnt antagonists (like Sost), remains to be explored.
Conclusion
Basic biomedical and clinical research has demonstrated a clear role for the Wnt antagonists in the control of bone homeostasis, a therapeutic strategy that is already being applied to osteoporosis. Although these treatments are promising, there are subtle defects in the immune cell development in Sost-deficient and Sostdc1-deficient mice that may worsen over time and affect immune responses to pathogens and transformed cells. The changes in the bone microenvironment induced by depletion of Wnt antagonists and changes in oxygen tension may have effects in hematopoietic stem cells that could reduce their capacity to replenish the blood system or create new hematopoietic niches outside of the bone. Future studies to investigate mechanisms controlling the crosstalk between the skeletal and immune systems should help to identify solutions for possible side effects of Wnt antagonist-depleting therapies for osteoporosis. It also provide additional avenues to influence hematopoietic differentiation in vitro and in vivo.
Acknowledgments
The authors thank Dr. Damian Genetos and Dr. Gabriela Loots for their comments on the manuscript.
Funding Information This work was supported by University of California (UC), Merced faculty research funding, University of California Cancer Research Coordinating Committee Grant CRC-15380532, National Institutes of Health Award 1R15HL121786-01A1, Halcyon-Dixon Trust award to JOM, and UC Graduate Student Fellowships to AM, CD, and BC.
Footnotes
This article is part of the Topical Collection on Skeletal Biology and Regulation
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards and institutional approvals.
Conflict of Interest Betsabel Chicana, Cristine, Alberto Millan, and Jennifer Manilay declare no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Canalis E Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9(10):575–83. [DOI] [PubMed] [Google Scholar]
- 2.Canalis E Management of endocrine disease: novel anabolic treatments for osteoporosis. Eur J Endocrinol. 2018;178(2):R33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langdahl BL, Libanati C, Crittenden DB, Bolognese MA, Brown JP, Daizadeh NS, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet. 2017;390(10102): 1585–94. [DOI] [PubMed] [Google Scholar]
- 4.Richter J, Traver D, Willert K. The role of Wnt signaling in hematopoietic stem cell development. Crit Rev Biochem Mol Biol. 2017;52(4):414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cain CJ, Manilay JO. Hematopoietic stem cell fate decisions are regulated by Wnt antagonists: comparisons and current controversies. Exp Hematol. 2013;41(1):3–16. [DOI] [PubMed] [Google Scholar]
- 6.Laurikkala J, Kassai Y, Pakkasjarvi L, Thesleff I, Itoh N. Identification of a secreted BMP antagonist, ectodin, integrating BMP, FGF, and SHH signals from the tooth enamel knot. Dev Biol. 2003;264(1):91–105. [DOI] [PubMed] [Google Scholar]
- 7.Ahn Y, Sanderson BW, Klein OD, Krumlauf R. Inhibition of Wnt signaling by wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development. 2010;137(19): 3221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.••.Collette NM, Yee CS, Hum NR, Murugesh DK, Christiansen BA, Xie L, et al. Sostdc1 deficiency accelerates fracture healing by promoting the expansion of periosteal mesenchymal stem cells. Bone. 2016;88:20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used genetic knockout mice to outline the role of Sostdc1 as a negative regulator of bone fracture repair and identified Sostdc1 expression in periosteal cells and MSCs and their role in bone fracture repair. Furthermore, Sostdc1−/− mice were found to have enlarged cortical bones with higher bone marrow density.
- 9.••.Millan A, Elizaldi S, Lee E, Aceves J, Murugesh D, L GG, et al. Sostdc1 regulates natural killer cell maturation and cytotoxicity. J. of Immunology, 2019, in press [DOI] [PubMed] [Google Scholar]; This study demonstrates that Sostdc1 plays a crucial role in NK cell maturation and cytotoxicity, and reveals Sostdc1 from both hematopoietic and non-hematopoietic sources differentially regulate on NK cell development.
- 10.•.Georgiev H, Ravens I, Papadogianni G, Halle S, Malissen B, Loots GG, et al. Shared and unique features distinguishing follicular T helper and regulatory cells of peripheral lymph node and Peyer’s Patches. Front Immunol. 2018;9:714. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found Sostdc1 expression in CD4+ T follicular helper (TFH) and T follicular regulatory (TFR) cells in the peripheral lymph node and Peyer’s Patches, and revealed that lack of Sostdc1 in mice increases frequencies of memory B cells increases fecal IgA secretion.
- 11.Shimatani K, Nakashima Y, Hatton M, Hamazaki Y, Minato N. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc Natl Acad Sci U S A. 2009;106(37):15807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cain CJ, Rueda R, McLelland B, Collette NM, Loots GG, Manilay JO. Absence of sclerostin adversely affects B-cell survival. J Bone Miner Res. 2012;27(7):1451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chow A, Mason J, Coney L, Bajwa J, Zaslavsky A, Pellman Y, et al. Sclerostin deficiency alters peripheral B lymphocyte responses in mice. BioRxiv 2018. 10.1101/357772. [DOI] [Google Scholar]
- 14.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.You L, Chen L, Pan L, Peng Y, Chen J. SOST gene inhibits osteogenesis from adipose-derived mesenchymal stem cells by inducing Th17 cell differentiation. Cell Physiol Biochem. 2018;48(3):1030–40. [DOI] [PubMed] [Google Scholar]
- 16.Divieti Pajevic P, Krause DS. Osteocyte regulation of bone and blood. Bone. 2019;119:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi Y, Thirukonda GJ, Nakamura Y, Koide M, Yamashita T, Uehara S, et al. Wnt16 regulates osteoclast differentiation in conjunction with Wnt5a. Biochem Biophys Res Commun. 2015;463(4):1278–83. [DOI] [PubMed] [Google Scholar]
- 18.Chae W-J, Bothwell A. Canonical and non-canonical Wnt signaling in immune cells. Trends Immunol. 2018. 10.1016/j.it.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008;10(10):1208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong Z, Zylstra-Diegel CR, Schumacher CA, Baker JJ, Carpenter AC, Rao S, et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci U S A. 2012;109(33):E2197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Z, Ethen NJ, Williams BO. WNT signaling in bone development and homeostasis. Wiley Interdiscip Rev Dev Biol. 2014;3(6):489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leucht P, Lee S, Yim N. Wnt signaling and bone regeneration: can’t have one without the other. Biomaterials. 2018. [DOI] [PubMed] [Google Scholar]
- 23.Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, et al. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol. 2010;12(10):1007–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newton IP, Kenneth NS, Appleton PL, Nathke I, Rocha S. Adenomatous polyposis coli and hypoxia-inducible factor-1{alpha} have an antagonistic connection. Mol Biol Cell. 2010;21(21):3630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9(2):210–7. [DOI] [PubMed] [Google Scholar]
- 26.Iqbal S, Zhang S, Driss A, Liu ZR, Kim HR, Wang Y, et al. PDGF upregulates Mcl-1 through activation of beta-catenin and HIF-1alpha-dependent signaling in human prostate cancer cells. PLoS One. 2012;7(1):e30764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. 2008;111(2):492–503. [DOI] [PubMed] [Google Scholar]
- 28.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–14. [DOI] [PubMed] [Google Scholar]
- 29.Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG, et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7(10):1037–47. [DOI] [PubMed] [Google Scholar]
- 30.Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048–56. [DOI] [PubMed] [Google Scholar]
- 31.Staal FJ, Chhatta A, Mikkers H. Caught in a Wnt storm: complexities of Wnt signaling in hematopoiesis. Exp Hematol. 2016;44(6):451–7. [DOI] [PubMed] [Google Scholar]
- 32.Famili F, Naber BA, Vloemans S, de Haas EF, Tiemessen MM, Staal FJ. Discrete roles of canonical and non-canonical Wnt signaling in hematopoiesis and lymphopoiesis. Cell Death Dis. 2015;6:e1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McClung MR. Sclerostin antibodies in osteoporosis: latest evidence and therapeutic potential. Ther Adv Musculoskelet Dis.2017;9(10):263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9. [DOI] [PubMed] [Google Scholar]
- 35.van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22(1):19–28. [DOI] [PubMed] [Google Scholar]
- 36.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–4. [DOI] [PubMed] [Google Scholar]
- 37.Gori F, Lerner U, Ohlsson C, Baron R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep. 2015;4:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280(29):26770–5. [DOI] [PubMed] [Google Scholar]
- 40.Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, et al. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc Natl Acad Sci U S A. 2015;112(5):E478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouaziz W, Funck-Brentano T, Lin H, Marty C, Ea HK, Hay E, et al. Loss of sclerostin promotes osteoarthritis in mice via beta-catenin-dependent and -independent Wnt pathways. Arthritis Res Ther. 2015;17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.••.Yee CS, Manilay JO, Chang JC, Hum NR, Murugesh DK, Bajwa J, et al. Conditional deletion of Sost in MSC-derived lineages identifies specific cell type contributions to bone mass and B cell development. J Bone Miner Res. 2018;33(10):1748–59 [DOI] [PubMed] [Google Scholar]; This study utilized conditional Sost knockout mice to reveal novel roles of mesenchymal stem cells in control of bone homeostasis and B cell development and revealed that osteocyte specific Sost is not the main driver of these processes.
- 43.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Lierop AH, Hamdy NA, van Egmond ME, Bakker E, Dikkers FG, Papapoulos SE. Van Buchem disease: clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28(4):848–54. [DOI] [PubMed] [Google Scholar]
- 45.Nassar K, Rachidi W, Janani S, Mkinsi O. Van Buchem’s disease. Joint Bone Spine. 2016;83(6):737–8. [DOI] [PubMed] [Google Scholar]
- 46.Kageyama Y, Koshiji M, To KK, Tian YM, Ratcliffe PJ, Huang LE. Leu-574 of human HIF-1alpha is a molecular determinant of prolyl hydroxylation. FASEB J. 2004;18(9):1028–30. [DOI] [PubMed] [Google Scholar]
- 47.Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. [DOI] [PubMed] [Google Scholar]
- 48.Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65(8):3299–306. [DOI] [PubMed] [Google Scholar]
- 49.Liao SH, Zhao XY, Han YH, Zhang J, Wang LS, Xia L, et al. Proteomics-based identification of two novel direct targets of hypoxia-inducible factor-1 and their potential roles in migration/invasion of cancer cells. Proteomics. 2009;9(15):3901–12. [DOI] [PubMed] [Google Scholar]
- 50.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riddle RC, Khatri R, Schipani E, Clemens TL. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J Mol Med (Berl). 2009;87(6):583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu C, Saless N, Wang X, Sinha A, Decker S, Kazakia G, et al. The role of oxygen during fracture healing. Bone. 2013;52(1):220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirao M, Hashimoto J, Yamasaki N, Ando W, Tsuboi H, Myoui A, et al. Oxygen tension is an important mediator of the transformation of osteoblasts to osteocytes. J Bone Miner Metab. 2007;25(5):266–76. [DOI] [PubMed] [Google Scholar]
- 54.Knowles HJ. Hypoxic regulation of osteoclast differentiation and bone resorption activity. Hypoxia (Auckl). 2015;3:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komatsu DE, Hadjiargyrou M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone. 2004;34(4):680–8. [DOI] [PubMed] [Google Scholar]
- 56.Danis A Mechanism of bone lengthening by the Ilizarov technique. Bull Mem Acad R Med Belg. 2001;156(1–2):107–12. [PubMed] [Google Scholar]
- 57.Zhao Q, Shen X, Zhang W, Zhu G, Qi J, Deng L. Mice with increased angiogenesis and osteogenesis due to conditional activation of HIF pathway in osteoblasts are protected from ovariectomy induced bone loss. Bone. 2012;50(3):763–70. [DOI] [PubMed] [Google Scholar]
- 58.Tando T, Sato Y, Miyamoto K, Morita M, Kobayashi T, Funayama A, et al. Hif1alpha is required for osteoclast activation and bone loss in male osteoporosis. Biochem Biophys Res Commun. 2016;470(2):391–6. [DOI] [PubMed] [Google Scholar]
- 59.Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, et al. HIF1alpha is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci U S A. 2013;110(41): 16568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weng T, Xie Y, Huang J, Luo F, Yi L, He Q, et al. Inactivation of Vhl in osteochondral progenitor cells causes high bone mass phenotype and protects against age-related bone loss in adult mice. J Bone Miner Res. 2014;29(4):820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117(6):1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wan C, Gilbert SR, Wang Y, Cao X, Shen X, Ramaswamy G, et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci U S A. 2008;105(2):686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuo GL, Zhang LF, Qi J, Kang H, Jia P, Chen H, et al. Activation of HIFa pathway in mature osteoblasts disrupts the integrity of the osteocyte/canalicular network. PLoS One. 2015;10(3):e0121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.••.Loots GG, Robling AG, Chang JC, Murugesh DK, Bajwa J, Carlisle C, et al. Vhl deficiency in osteocytes produces high bone mass and hematopoietic defects. Bone. 2018;116:307–14 [DOI] [PubMed] [Google Scholar]; The findings in this article highlight the VHL and HIF influences in skeletal development and repair. It has an additional focus on the implications in the immune system in particular of B cell development.
- 65.Chen D, Li Y, Zhou Z, Wu C, Xing Y, Zou X, et al. HIF-1alpha inhibits Wnt signaling pathway by activating Sost expression in osteoblasts. PLoS One. 2013;8(6):e65940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.•.Stegen S, Stockmans I, Moermans K, Thienpont B, Maxwell PH, Carmeliet P, et al. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat Commun. 2018;9(1):2557. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article revealed the role of PHD2 role in the bone homeostasis through epigenetic regulation of sclerostin expression through the Wnt signaling pathway, and a possible strategy to treat osteoporosis.
- 67.Genetos DC, Toupadakis CA, Raheja LF, Wong A, Papanicolaou SE, Fyhrie DP, et al. Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J Cell Biochem. 2010;110(2):457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu JY, Scadden DT, Kronenberg HM. Role of the osteoblast lineage in the bone marrow hematopoietic niches. J Bone Miner Res. 2009;24(5):759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104(13):5431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bader HL, Hsu T. Systemic VHL gene functions and the VHL disease. FEBS Lett. 2012;586(11):1562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537(7619):234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Togo Y, Takahashi K, Saito K, Kiso H, Tsukamoto H, Huang B, et al. Antagonistic functions of USAG-1 and RUNX2 during tooth development. PLoS One. 2016;11(8):e0161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiso H, Takahashi K, Saito K, Togo Y, Tsukamoto H, Huang B, et al. Interactions between BMP-7 and USAG-1 (uterine sensitization-associated gene-1) regulate supernumerary organ formations. PLoS One. 2014;9(5):e96938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collette NM, Yee CS, Murugesh D, Sebastian A, Taher L, Gale NW, et al. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev Biol. 2013;383(1):90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanaka M, Endo S, Okuda T, Economides AN, Valenzuela DM, Murphy AJ, et al. Expression of BMP-7 and USAG-1 (a BMP antagonist) in kidney development and injury. Kidney Int. 2008;73(2):181–91. [DOI] [PubMed] [Google Scholar]
- 77.Manilay JO, Waneck GL, Sykes M. Altered expression of Ly-49 receptors on NK cells developing in mixed allogeneic bone marrow chimeras. Int Immunol. 1998;10(12):1943–55. [DOI] [PubMed] [Google Scholar]
- 78.Tu MM, Mahmoud AB, Makrigiannis AP. Licensed and unlicensed NK cells: differential roles in cancer and viral control. Front Immunol. 2016;7:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136(6):1136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang J, Nguyen-McCarty M, Hexner EO, Danet-Desnoyers G, Klein PS. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012;18(12):1778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476(7360):293–7. [DOI] [PubMed] [Google Scholar]
- 82.Liu D, He XC, Qian P, Barker N, Trainor PA, Clevers H, et al. Leucine-rich repeat-containing G-protein-coupled receptor 5 marks short-term hematopoietic stem and progenitor cells during mouse embryonic development. J Biol Chem. 2014;289(34):23809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150(2):351–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.•.Himburg HA, Doan PL, Quarmyne M, Yan X, Sasine J, Zhao L, et al. Dickkopf-1 promotes hematopoietic regeneration via direct and niche-mediated mechanisms. Nat Med. 2017;23(1):91–9 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that secretion of Dkk1 by BM osteoprogenitors regulates promotes hematopoietic regeneration directly through inhibition of HSC quiescence as well as indirectly through EGF secretion by BM endothelial cells.
- 86.Witcher PC, Miner SE, Horan DJ, Bullock WA, Lim KE, Kang KS, et al. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight. 2018;3(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Florio M, Gunasekaran K, Stolina M, Li X, Liu L, Tipton B, et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat Commun. 2016;7:11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–92 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/beta-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. 2018;39(7):648–58. [DOI] [PubMed] [Google Scholar]
- 90.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol. 2017;1036:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regan JN, Lim J, Shi Y, Joeng KS, Arbeit JM, Shohet RV, et al. Up-regulation of glycolytic metabolism is required for HIF1alpha-driven bone formation. Proc Natl Acad Sci U S A. 2014;111(23): 8673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Z, Frey JL, Wong GW, Faugere MC, Wolfgang MJ, Kim JK, et al. Glucose transporter-4 facilitates insulin-stimulated glucose uptake in osteoblasts. Endocrinology. 2016;157(11):4094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Esen E, Long F. Aerobic glycolysis in osteoblasts. Curr Osteoporos Rep. 2014;12(4):433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.••.Dirckx N, Tower RJ, Mercken EM, Vangoitsenhoven R, Moreau-Triby C, Breugelmans T, et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J Clin Invest. 2018;128(3):1087–105 [DOI] [PMC free article] [PubMed] [Google Scholar]; The findings in this article indicate important metabolic regulation in osteoblasts can extend beyond the bone environment. It showed that deleting VHL in osteoblast cells alters metabolic homeostasis through the VHL/HIF pathway. This highlights the skeleton’s role in global nutrient homeostasis.
- 96.Krzywinska E, Stockmann C. Hypoxia, metabolism and immune cell function. Biomedicines. 2018;6(2):56. [DOI] [PMC free article] [PubMed] [Google Scholar]