

Abstract
Overexpression or activation of AKT is very well known to control cell growth, survival and gene expression in solid tumors. Oridonin, an inflammatory medical and diterpenoid compound isolated from Rabdosia rubescens, has exhibited various pharmacological and physiological properties, including anti-tumor, anti-bacterial and anti-inflammatory effects. In this study, we demonstrated that oridonin is an inhibitor of AKT and suppresses proliferation of esophageal squamous cell carcinoma (ESCC) in vitro and in vivo. The role of AKT in ESCC was studied using immuno- histochemical analysis of a tumor microarray, the effect of AKT knockdown on cell growth, and treatment of cells with MK-2206, an AKT inhibitor. Oridonin blocked AKT kinase activity and interacted with the ATP-binding pocket of AKT. It inhibited growth of KYSE70, KYSE410 and KYSE450 esophageal cancer cells in a time- and concentration-dependent manner. Oridonin induced arrest of cells in the G2/M cell cycle phase, stimulated apoptosis and increased expression of apoptotic biomarkers, including cleaved PARP, caspase-3, caspase-7 and Bims in ESCC cell lines. Mechanistically, we found that oridonin diminished the phosphorylation and activation of AKT signaling. Furthermore, a combination of oridonin and 5-fluorouracil (FU) or cisplatin (clinical chemotherapeutic agents) enhanced the inhibition of ESCC cell growth. The effects of oridonin were verified in patient derived xenograft tumors expressing high levels of AKT. In summary, our results indicate that oridonin acts as an AKT inhibitor to suppress the growth of ESCC by attenuating AKT signaling.
Keywords: oridonin, AKT inhibitor, esophageal squamous cell carcinoma (ESCC)
Introduction
Esophageal cancer is the 6th leading cause of cancer death worldwide and has low survival rates with poor prognosis (1). For men, esophageal cancer is estimated to be the 7th highest cause of cancer-related deaths and, in particular, is the 5th highest cause of cancer death in men aged 40–59 years in the United States (2). Esophageal cancers are classified as esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) based on their histopathological subtypes. The incidence of ESCC is estimated to be 398,000 patients worldwide and about 79% of the total ESCC patients are found in Southeastern and Central Asia (3). Although 5-fluorouracil (5-FU) or cisplatin is consistently used as a chemotherapy agent against ESCC, the 5-year overall survival rate is still limited to about 15% (4).
The inappropriate activation of AKT signaling has been implicated in esophageal carcinomas. AKT, also known as protein kinase B (PKB), is a serine-threonine kinase that activates downstream targets such as glycogen synthase kinase (GSK)-3β, mechanistic target of rapamycin (mTOR), and nuclear factor-kappaB (NF-κB). These cascades of activated signals are involved in enhancing cancer progression and increasing cell proliferation and promoting evasion from apoptosis (5–8). AKT has been validated as an anticancer target for a variety of cancers, including non-small cell lung cancer, gastric cancer, ovarian cancer, leukemia and esophageal cancer (9–13). Thus, targeting AKT appears to be a rational approach for anticancer drug development.
A number of natural products, especially plant constituents, have been reported to deactivate AKT signaling as mechanisms of their anticancer activities (11). Oridonin is an ent-kaurane diterpene compound found in Rabdosia rubescens that is widely grown in China and Japan and has been used in oriental medicine. For example, this compound has antioxidant activities (14) and has been used to treat various disease conditions, such as cancer (15–17), inflammation (18), bacterial infections (19), and also relieves neurodegenerative symptoms (20). The anticancer effects of oridonin involve inhibition of different cellular signaling pathways associated with proliferation, cell cycle and apoptosis (21). The present study aimed to investigate the effects of oridonin on the proliferation and growth of ESCC and to elucidate its underlying mechanisms of action. We found that oridonin is an inhibitor of AKT and induces cell cycle arrest and apoptosis in ESCC cells and attenuates growth of patient derived xenograft (PDX) tumors in vivo by interfering with AKT signaling pathways. Oridonin also potentiated the effects of cisplatin or 5-FU, standard chemotherapy drugs used clinically.
Materials and Methods
Reagents
Oridonin (purity ≥ 97%) was purchased from Xi’an Plant Bio-engineering Co., LTD (CAS: 28957–04-2, Xi’an, Shaanxi, China) and was analyzed and authenticated by high-performance liquid chromatography. 5-Fluorouracil (FU) and cisplatin were bought from Sigma-Aldrich (St. Louis, MO). Active AKT1 and AKT2 were purchased from SignalChem (Richmond, BC, Canada) and the GSK fusion protein for kinase assays was from Cell Signaling Technology (Beverly, MA). Antibodies to detect phosphorylated AKT, total AKT, phosphorylated GSK-3β, total GSK-3β, phosphorylated mTOR, total mTOR and cyclin B1 were also purchased from Cell Signaling Technology. The antibody to detect Bcl-2 was from Santa Cruz Biotechnology (Santa Cruz, CA) and the β-actin antibody was obtained from ZSGB-Bio Company (Beijing, China).
Cell culture
The human esophageal cancer cell lines (KYSE70, KYSE410 and KYSE450) were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI-1640 containing penicillin (100 units/mL), streptomycin (100 μg/mL), and 10% FBS (Biological Industries, Kibbutz Beit-Haemek, Israel). The human immortalized normal esophageal epithelial cell line, SHEE, was donated by Dr. Enmin Li from the Laboratory of Tumor Pathology (Shantou University Medical College, Shantou, Guangdong, China) (22). Cells were maintained at 5% CO2, 37°C in a humidified atmosphere. All cells were cytogenetically tested and authenticated before being frozen. Each vial of frozen cells was thawed and maintained in culture for a maximum of 8 weeks.
Cell proliferation assay
Cells were seeded (6×103 cells/well for KYSE70; 2.5×103 cells/well for KYSE410; 2×103 cells/well for KYSE450) in 96-well plates and incubated for 24 h and then treated with different amounts of oridonin or vehicle. After incubation for 24, 48 or 72 h, cell proliferation was measured by the MTT assay. For anchorage-independent cell growth assessment, cells (8×103 cells/well) suspended in complete medium were added to 0.3% agar with vehicle, 2.5, 5 or 10 μM oridonin in a top layer over a base layer of 0.5% agar with vehicle, 2.5, 5 or 10 μM oridonin. The cultures were maintained at 37°C in a 5% CO2 incubator for 3 weeks and then colonies were visualized under a microscope and counted using the Image-Pro Plus software (v.6.1) program (Media Cybernetics, Rockville, MD).
Cell cycle and apoptosis analyses
Cells (6 × 105 cells for KYSE70; 3 × 105 for KYSE410; 2 × 105 for KYSE450) were seeded in 60-mm plates and treated with 0, 5, 10 or 20 μM oridonin for 48 or 72 h. For cell cycle analysis, cells were then fixed in 70% ethanol and stored at −20°C for 24 h. After staining with annexin-V for apoptosis or propidium iodide for cell cycle assessment, cells were analyzed using a BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA).
Western blot assay
Cell pellets were incubated on ice for 20 min in NP-40 cell lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.5–1% NP-40, protease inhibitor cocktail, dephosphorylate inhibitor tablets, and 1 mM phenylmethylsulfonyl fluoride [PMSF]). After centrifugation at 14,000 rpm for 20 min, the supernatant fractions were harvested as total cellular protein extracts. Determination of protein concentration was performed using the BCA Quantification Kit (Solarbio, Beijing, China). The total cellular protein extracts were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes in transfer buffer. Membranes were blocked with 5% non-fat dry milk in 1× PBST (phosphate buffered saline containing 0.05% Tween-20) and incubated with antibodies against p-AKT1/2, AKT1/2, p-GSK-3β, GSK-3β, p-mTOR, mTOR, cleaved PARP, cleaved caspase-3, cleaved caspase-7, Bax, Bims, cyclin B1 or β-actin. Blots were washed 3 times in 1×PBST buffer, followed by incubation with the appropriate horseradish peroxidase (HRP)-linked IgG. Protein bands were visualized using the enhanced chemiluminescence (ECL) detection reagent (GE Healthcare Life Science, Little Chalfont, HP, UK).
Ex vivo and in vitro pull-down assay
KYSE450 cell lysates (500 μg) and recombinant human active AKT1 or AKT2 (200 ng) were incubated with oridonin-Sepharose 4B (or Sepharose 4B only as a control) beads (50 μL; 50% slurry) in reaction buffer (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol, 0.01% NP-40 and 0.2 mM PMSF, 20× protease inhibitor [1 tablet each]). After incubation with gentle rocking overnight at 4°C, the beads were washed 3 times with buffer (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol, 0.01% NP-40 and 0.2 mM PMSF) and binding was visualized by Western blotting.
Kinase assay
The AKT kinase assay was performed using the Nonradioactive IP-Kinase Assay Kit (Cell Signaling Technology). Twenty μl of the immobilized antibody bead slurry were mixed with 200 μl of the cell lysate (200 μg) and gently rocked overnight at 4°C. After washing twice on day 2, the pellet was suspended in 50 μl of 1× kinase buffer supplemented with 1 μl of 10 mM ATP and 1 μg of kinase substrate. Then, the mixture was incubated for 30 min at 30°C and the reaction was terminated with 25 μl of 3× SDS sample buffer. Binding was assessed by Western blot analysis. For the AKT kinase assay, we used the ADP-Glo™ Kinase Assay Kit (Promega). Active AKT1 or AKT2 kinase and inactive GSK-3β as substrate were mixed by 1x reaction buffer and then added to a white 96-well plate. Pure ATP provided in the kit was serially diluted obtain a final concentration of 0, 1, 10, 50, and 100 μM. Oridonin was added to reach a final concentration of 2.5, 5, 10 or 20 μM and DMSO was used as a control. The mixed solution was incubated at room temperature and luciferase activity was measured using the Luminoskan Ascent plate reader (Thermo-Scientific, Swedesboro, NJ).
Computational modeling
The computer modeling of oridonin with AKT1 and AKT2 was performed using the Schrödinger Suite 2015 software programs (23). The AKT1 and AKT2 crystal structures were downloaded from the Protein Data Bank (PDB) (24) and were prepared under the standard procedures of the Protein Preparation Wizard in Schrödinger. Hydrogen atoms were added consistent with a pH of 7 and all water molecules were removed. The ATP-binding site-based receptor grid was generated for docking. Oridonin was prepared for docking by default parameters using the LigPrep program. Then, the docking of oridonin and AKT1 and AKT2 was accomplished with default parameters under the extra precision (XP) mode using the program Glide. Herein, we could get the best-docked representative structures.
Lentiviral infection and transfections
Each viral vector and packaging vectors (pMD2G, psPAX2, shAKT1 and shAKT2) were transfected using the Simple-Fect Transfection Reagent (Signaling Dawn Biotech, Wuhan, Hubei, China) into 293T cells. The viral particles were harvested by filtration using a 0.22 μm filter and then stored at −20°C. The cultured KYSE70, KYSE410, KYSE450 esophageal cancer cells were infected with virus particles in 8 μg/ml polybrene (Millipore, Billerica, MA) for 24 h. Then, cells were selected with puromycin for 36 h and the selected cells were used for subsequent experiments. For the overexpression of AKT1/2, we used mock vector, pUSE-CA-AKT1 and pUSE-CA-AKT2 plasmids. Each 2.5 μg plasmid and 3-fold Simple-Fect reagent were transfected into 293T cell and the same amount of mock plasmid was transfected at the same time. The medium was changed 12 h after transfection and culture was continued for another 24 h. Subsequent experiments were performed to observe the phenomenon change after AKT1/2 overexpression in 293T cells. For rescue experiments, AKT1 and AKT2 genes were knocked down by lentiviral infection as described above for the KYSE450 cell line. Then 1 μg/ml puromycin was used for selection. At 24 h later, AKT1 or AKT2 was overexpressed in AKT-knock down cells by direct transfection of the pUSE-CA-Akt1/2 plasmid or mock plasmid and cultured for 12 h. At the same time, additional dishes of cells were maintained in 0.1 μg/ml puromycin for 12 h and similar experiments were conducted.
Reporter gene activity assay
Transient transfection was conducted using the Simple-Fect Transfection Reagent (Signaling Dawn Biotech), and the luciferase reporter gene activity assays were performed according to the instructions of the manufacturer (Promega, Madison, WI). Briefly, cells (1ⅹ105) were seeded the day before transfection into 24-well culture plates. Cells were co-transfected with the NF-κB-luc or pGL-3-luc firefly reporter plasmid, and the pRL-SV control Renilla reporter plasmid. Following incubation for 24 h, the cells were treated with vehicle or oridonin for an additional 24 h, and then harvested in lysis buffer. Firefly and Renilla luciferase activities were assessed by using the substrates provided in the reporter assay system (Promega). The Renilla luciferase activity was used for normalization of transfection efficiency. Luciferase activity was measured using the Luminoskan Ascent plate reader (Thermo-Scientific, Swedesboro, NJ).
Patient derived xenograft (PDX) mouse model
Six to eight week-old severe combined immunodeficient (SCID) female mice (Vital River Labs, Beijing, China) were used for these experiments. This study was approved by the Ethics Committee of Zhengzhou University (Zhengzhou, Henan, China). The PDX tumor mass was subcutaneously implanted into the back of SCID mice. When tumors reached an average volume of about 100 mm3, mice were divided into 3 treatment groups for further experimentation as follows: (1) vehicle group (n = 10); (2) 40 mg/kg of oridonin (n = 10); (3) 160 mg/kg of oridonin (n = 10). Oridonin was administered by gavage once a day for 52 days. Tumor volume was calculated from measurements of 3 diameters of the individual tumor base using the following formula: tumor volume (mm3) = (length × width × height × 0.52). Mice were monitored until tumors reached 1.0 cm3 total volume, at which time mice were euthanized and tumors extracted.
Immunohistochemical (IHC) analysis
Paraffin-embedded sections (5 μm) were prepared for IHC analysis. After antigen unmasking, the sections were blocked with 5% goat serum and incubated at 4°C overnight with antibodies to detect Ki-67, phosphorylated AKT (Ser473), mTOR (Ser2448) or GSK-3β (Ser9). After incubation with a rabbit secondary antibody, DAB (3,3’-diaminobenzidine) staining was used to visualize the protein targets according to the manufacturer’s instructions. Sectioned tissues were counterstained with hematoxylin, dehydrated through a graded series of alcohol into xylene, and mounted under glass coverslips.
Statistical Analysis
The student’s t-test was used to perform statistical analysis for single comparisons. A value of p < 0.05 was used as the criterion for statistical significance and the data are shown as mean values ± standard deviation (S.D.) or standard error (S.E.).
Results
AKT is a potential target in ESCC
To evaluate the role of AKT in ESCC, we examined the expression of total and phosphorylated AKT in an ESCC tumor microarray that included 20 pairs of cancer tissues and tissues adjacent to cancer tissues and 4 normal tissues (Fig. 1A). Total AKT was significantly overexpressed in cancer or tissues adjacent to cancer tissues compared with normal tissues (Fig. 1A, left panel). Phosphorylated AKT was significantly overexpressed in cancer tissues, but not in tissues adjacent to cancer tissues, compared with normal tissues (Fig. 1A, right panel). We also evaluated the effects of treatment of ESCC cells with an AKT inhibitor or knock-down of AKT (Fig. 1B–E). The results revealed that treatment with the AKT inhibitor, MK-2206, significantly decreased the growth of KYSE70, KYSE410 or KYSE450 cells (Fig. 1B). Knock-down of AKT was confirmed in KYSE450 cells infected with shRNA-AKT1/2 compared to cells infected with shRNA-mock (Fig. 1C). Depletion of AKT1/2 decreased growth of KYSE450 cells at 48 or 72 h compared with the mock control (Fig. 1D). Furthermore, knock-down of AKT1/2 reduced the number of colonies observed in the anchorage-independent cell growth assay (Fig. 1E).
Figure 1.

AKT is a potential target in ESCC. (A) The expression of total AKT (pan) and phosphorylated AKT (Ser473) was examined by IHC analysis on an ESCC tumor microarray (100X magnification). The top panels show quantitation of all samples. Representative photographs of immunohistochemistry staining, and hematoxylin and eosin (H&E) on an ESCC tumor microarray are shown. AKT and pAKT expression stain a brown color and the nuclei are stained blue with hematoxylin. Two photographs are shown from each group (100X magnification). (B) The effects of an AKT inhibitor, MK-2206, on ESCC cell growth were assessed by MTT assay. The asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant difference between untreated control and MK-2206-treated cells. (C) The expression of AKT in KYSE450 cells expressing shRNA-mock or shRNA-AKT1/2 was evaluated by Western blotting. (D) Growth was assessed in KYSE450 cells expressing shRNA-mock or shRNA-AKT1/2. (E) Anchorage-independent growth was assessed in KYSE450 cells expressing shRNA-mock or shRNA-AKT1/2. Data are shown as means ± S.D. of values from triplicate samples. The asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant difference between shRNA-mock and shRNA-AKT1/2-expressing cells, respectively.
AKT is a direct target of oridonin
We next used binding and kinase profiling assays to identify a direct protein target of oridonin (Fig. 2, Supplementary Fig. 1). First, we performed an in vitro kinase assay with immunoprecipitated AKT in the presence of 5 or 10 μM oridonin and results indicated that the phosphorylation of GSK-3α/β (Ser21/9), an AKT substrate, was decreased by treatment with oridonin compared to an untreated control (Fig. 2A–a). The 50% inhibitory concentration (IC50) of oridonin against AKT1 and AKT2 kinase activity was 8.4 ± 1.34 and 8.9 ± 0.92 μM, respectively (Fig. 2A, b–c). Based on this result, we confirmed the binding and interaction by an ex vivo or in vitro binding assay and a computational docking model between oridonin and AKT1/2 (Fig. 2B–D). Sepharose 4B beads conjugated with oridonin, but not Sepharose 4B beads only, bound with AKT in KYSE450 cell lysates (Fig. 2B). Next, we determined whether the binding is ATP-competitive and results showed that oridonin competed for binding with ATP at the ATP binding pocket of AKT1 or AKT2 (Fig. 2C). To examine how oridonin interacts with AKT1 and AKT2, we docked it in silico to the ATP binding pocket of each protein kinase using several protocols in the Schrödinger Suite 2015 (Fig. 2D). The docking model predicted that oridonin formed two hydrogen bonds each at Asp292 or Lys277/Asp293 in the backbone of AKT1 (Fig. 2D, a–b) and AKT2 (Fig. 2D, c–d), respectively. These results indicated that oridonin might be a potential effecctive inhibitor of AKT1 or AKT2 (images were generated with the UCSF Chimera program) (25).
Figure 2.

AKT1 and AKT2 are potential targets of oridonin. (A) The effect of oridonin on AKT activity was evaluated in an in vitro IP-AKT kinase assay (a), and AKT1 and AKT2 kinase activity assays (b-c). (B) The binding of oridonin to AKT in KYSE450 cell lysates was determined using Sepharose 4B and oridonin-conjugated Sepharose 4B beads. (C) The specificity of the binding of oridonin to active AKT1 or AKT2 in the presence of ATP was evaluated. The values below the respective bands indicate values obtained from densitometry analysis. (D) The interaction between oridonin and AKT1 or AKT2 was predicted using a computational docking model: a) oridonin binding with AKT1; b) ligand interaction diagram of oridonin binding with AKT1; c) oridonin binding with AKT2; d) ligand interaction diagram of oridonin binding with AKT2.
Oridonin inhibits the proliferation of ESCC cells by targeting AKT1/2
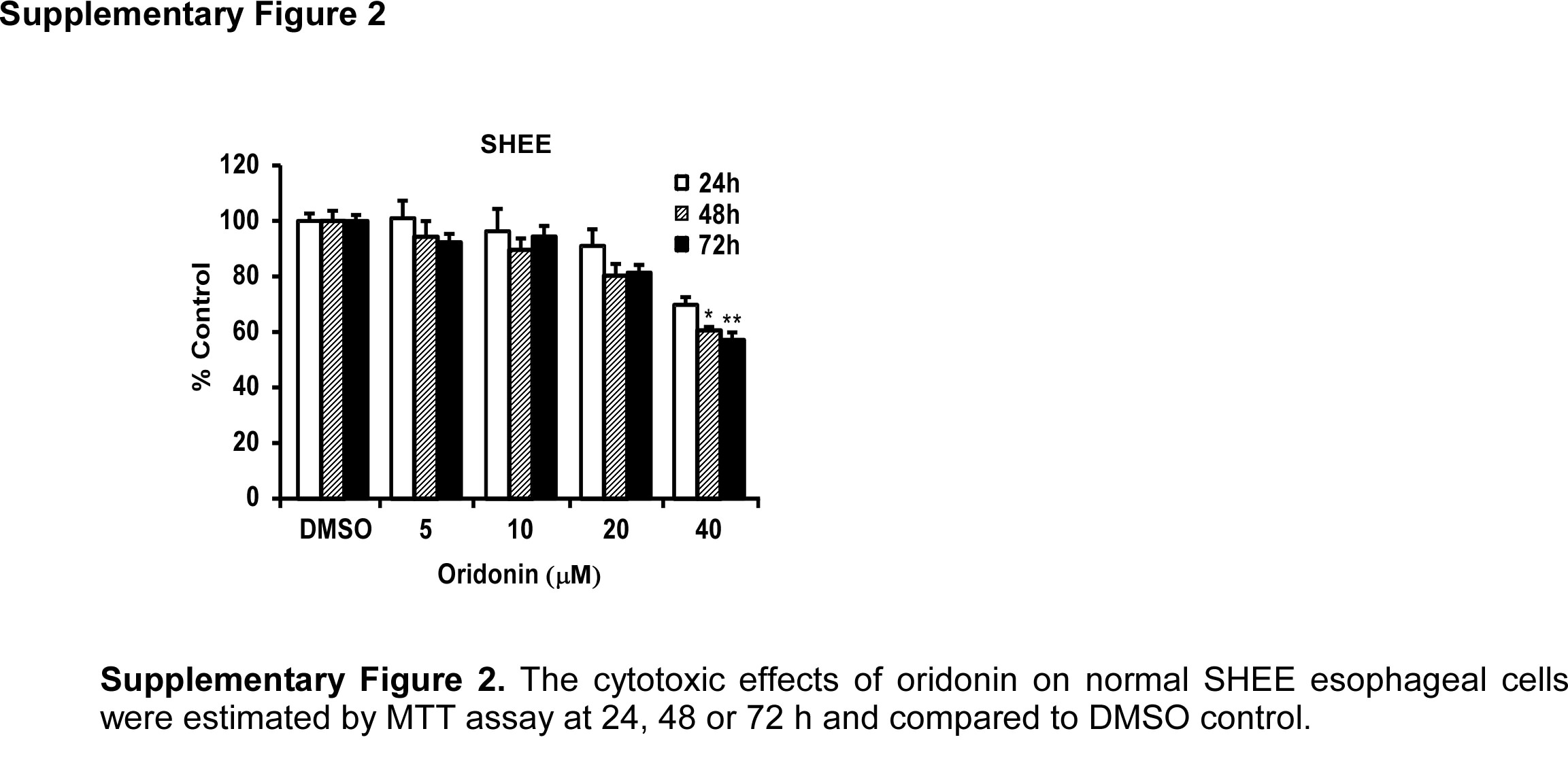
Because an ideal anticancer agent should lack cytotoxicity against normal cells, we first assessed the effect of oridonin on the proliferation of normal human SHEE esophageal cells. Incubation of SHEE cells with oridonin (5, 10, 20 or 40 μM) for 24, 48 or 72 h showed that the compound resulted in approximately 40% toxicity at 40 μM at 72 h (Supplementary Fig. 2). Therefore we selected a maximal nontoxic concentration (20 μM) of oridonin for further experiments. Treatment of KYSE70, KYSE410 and KYSE450 ESCC cells with oridonin (5, 10 or 20 μM) significantly inhibited growth in a time-dependent manner compared to a DMSO control (Fig. 3A). Oridonin also attenuated anchorage-independent growth of these three ESCC cell lines in a concentration-dependent manner compared to an untreated DMSO control (Fig. 3B). Representative images of colonies illustrate the number of colonies (Fig. 3C). We also verified the effects of oridonin on cell proliferation by targeting AKT1/2 (Fig. 4). After knock-down of AKT1 and AKT2 in ESCC cells (Fig. 4A) treatment with oridonin had no effect on anchorage-independent colony growth compared to mock-transfected cells (Fig. 4B). Also the rescue of AKT1 and AKT2 expression in AKT1/2 knock-down cells restored growth of cells as indicated by the MTT and anchorage-independent cell growth assays (Fig. 4C–E). Furthermore, AKT1/2 overexpressing cells showed a greater sensitivity to oridonin compared to mock-transfected cells (Fig. 4F)
Figure 3.

Oridonin inhibits the growth of ESCC cells. (A) Growth effects of oridonin on KYSE70, KYSE410 and KYSE450 ESCC cells were estimated by MTT assay at 24, 48 or 72 h. (B) The effects of oridonin on anchorage-independent growth of ESCC cells were evaluated. (C) Representative photographs of the effects of oridonin on anchorage-independent growth are shown. For A and B, the asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant decrease in proliferation or colony number with oridonin treatment compared with untreated control cells. Data are shown as means ± S.D. of values from triplicate samples and similar results were obtained from 3 independent experiments.
Figure 4.

AKT is a specific target of ESCC cells. (A) Western blot analysis was performed in KYSE70, KYSE410 and KYSE450 cells expressing shRNA-mock or shRNA-AKT1/2. (B) The effect of oridonin treatment on anchorage-independent growth was assessed in KYSE70, KYSE410 and KYSE450 cells expressing shRNA-mock or shRNA-AKT1/2. (C) Western blot analysis was used to examine the protein expression level of shRNA-mock, shRNA-AKT1/2 and overexpressed pUSE-CA-Akt1/2 and corresponding mock control. (D, E) Proliferation of cells expressing shRNA-mock or shRNA-AKT1/2 or overexpressing pUSE-CA-Akt1/2 and corresponding mock control were examined by MTT assay and anchorage-independent cell growth assay. (F) Anchorage-independent cell proliferation was assessed after treatment with serially diluted doses of oridonin in cells expressing mock or AKT1/2. The asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant decrease in proliferation or colony number compared with corresponding control. Data are shown as means ± S.D. of values from triplicate samples and similar results were obtained from 3 independent experiments.
Oridonin induces cell cycle G2/M arrest and apoptosis in ESCC cells
Treatment with oridonin affected cell-cycle distribution and induced apoptosis in ESCC cell lines (Fig. 5A, B, Supplementary Fig. 3, 4). Oridonin (10 or 20 μM) caused significant G2/M phase cell cycle arrest in KYSE70, KYSE410 and KYSE450 cells (Fig. 5A, Supplementary Fig. 4). Oridonin at 20 μM also induced apoptosis in these cell lines (Fig. 5B, Supplementary Fig. 3). Based on the effects induced by oridonin, we examined the expression of proteins associated with the G2/M phase of cell cycle and apoptosis. Oridonin reduced the expression of cyclin B1 in esophageal cancer cells compared with DMSO vehicle control (Fig. 5C). Treatment of ESCC cells with oridonin induced expression of cleaved PARP, caspase-3 and caspase-7 as well as Bax and the short form of Bim (Bims), and also decreased the expression of anti-apoptotic Bcl-2 compared to DMSO controls (Fig. 5D).
Figure 5.

Oridonin induces cell cycle arrest and apoptosis of ESCC cells. The effects of oridonin on cell cycle phase (A) or apoptosis (B) were assessed in ESCC cells. Cells were treated with 0, 5, 10 or 20 μM oridonin and then incubated for 48 h for cell cycle analysis or 72 h for the annexin-V staining (i.e., apoptosis) assay. The asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant difference between untreated control and oridonin-treated cells. Data are shown as means ± S.D. of values from triplicate samples and similar results were obtained from 3 independent experiments. The effects of oridonin on the expression of biomarkers associated with cell cycle (C) and apoptosis (D) are shown. Cells were treated with 0, 5, 10 or 20 μM oridonin and incubated 48 h for examination of cyclin B1 or 72 h for cleaved PARP, caspase-3, and caspase-7, and Bax, Bims (short form of Bim) and Bcl-2.
Oridonin inhibits AKT signaling pathways
Because AKT signaling pathways are involved in cell cycle, apoptosis and cancer cell growth (11), we determined the effects of oridonin on the expression of phosphorylated GSK-3β (Ser9) and phosphorylated mTOR (Ser2448) as well as on the transcriptional activity of NF-κB (Fig. 6). Treatment of KYSE70, KYSE410 or KYSE450 ESCC cells with oridonin had no effect on the expression of phosphorylated AKT or total AKT but decreased the levels of phosphorylated GSK-3β (Ser9) and phosphorylated mTOR (Ser2448) compared to untreated controls (Fig. 6A). Total expression of GSK-3β and mTOR was unchanged compared to untreated controls (Fig. 6A). Furthermore, KYSE70, KYSE410 or KYSE450 ESCC cells transfected with an NF-κB-luciferase plasmid and then treated with oridonin showed significant inhibition of luciferase reporter gene activity (Fig. 6B).
Figure 6. Oridonin attenuates the expression of proteins involved in AKT signaling.

(A) The effects of oridonin on AKT signaling in ESCC cells were assessed by Western blot analysis. Cells were treated with 0, 5, 10 or 20 μM oridonin for 3 h and harvested and then cell lysates were subjected to Western blotting. The values above the bands indicate values obtained from densitometry analysis. (B) The effects of oridonin were evaluated in ESCC cells transfected with an NF-κB luciferase reporter plasmid. Data are shown as means ± S.D. of values from triplicate samples. The asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001) indicate a significant decrease in relative luciferase activity.
A combination of oridonin and cisplatin or 5-FU enhances the suppression of ESCC cell growth
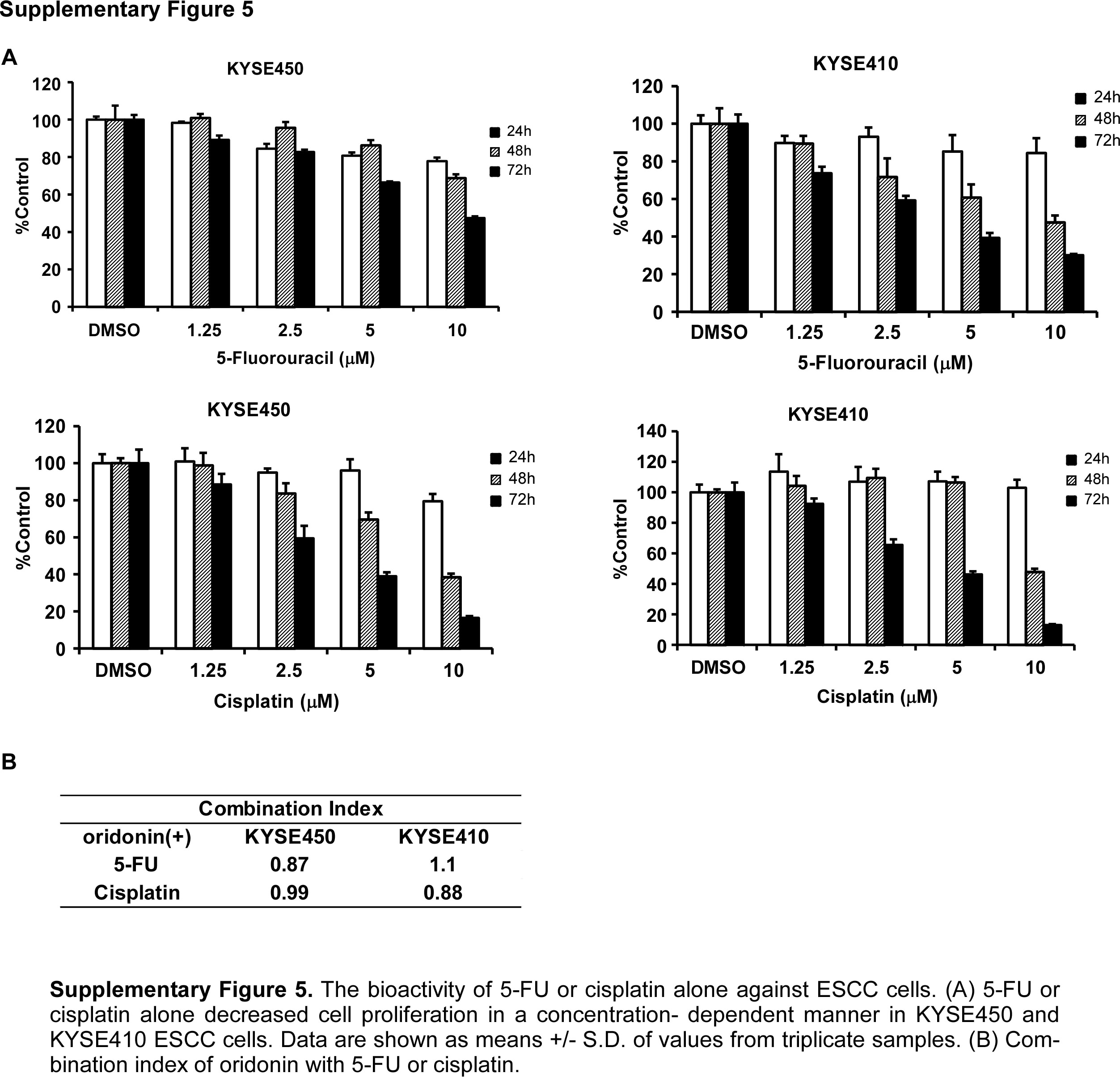
Next, we evaluated whether oridonin could be useful in combination with standard chemotherapy drugs against ESCC. Cells were treated with 1.2 or 1 μM 5-FU or cisplatin, which is the 10% inhibitory concentration (IC10), and vehicle, 5, 10 or 20 μM oridonin for 24, 48 or 72 h (Fig. 7, Supplementary Fig. 5A). The results revealed that growth of KYSE450 and KYSE410 cells was synergistically or additively inhibited by co-treatment with oridonin and 5-FU (Fig. 7A) or cisplatin (Fig. 7B). We calculated the combination index (CI) according to the Chou method (26). The CI values for 1.2 μM 5-FU or 1 μM of cisplatin and oridonin (5–20 μM) were < 0.9 or between 0.9 and 1.1, which was determined to be synergistic or additive between oridonin and 5-FU or cisplatin (Fig. 7A, B, Supplementary Fig. 5B). Colony number formation was also substantially decreased after co-treatment with oridonin and 5-FU (Fig. 7C, E) or cisplatin (Fig. 7D, F).
Figure 7.

A combined treatment of oridonin with 5-FU or cisplatin inhibits cell growth. (A, B) The growth of KYSE410 and KYSE450 cells was determined after co-treatment with various concentrations of oridonin and 5-FU (1.2 μM) or cisplatin (1 μM) for 24, 48 or 72 h. The effect on growth was measured by MTT assay. (C-F) The effect of treatment with a combination of oridonin (5 μM) and 5-FU or cisplatin on anchorage-independent growth of KYSE410 and KYSE450 cells was evaluated. The asterisks (* p < 0.05, ** p < 0.001) indicate a significant decrease in colony number compared to 5 μM oridonin.
Oridonin inhibits PDX growth in vivo in tumors with high expression of AKT
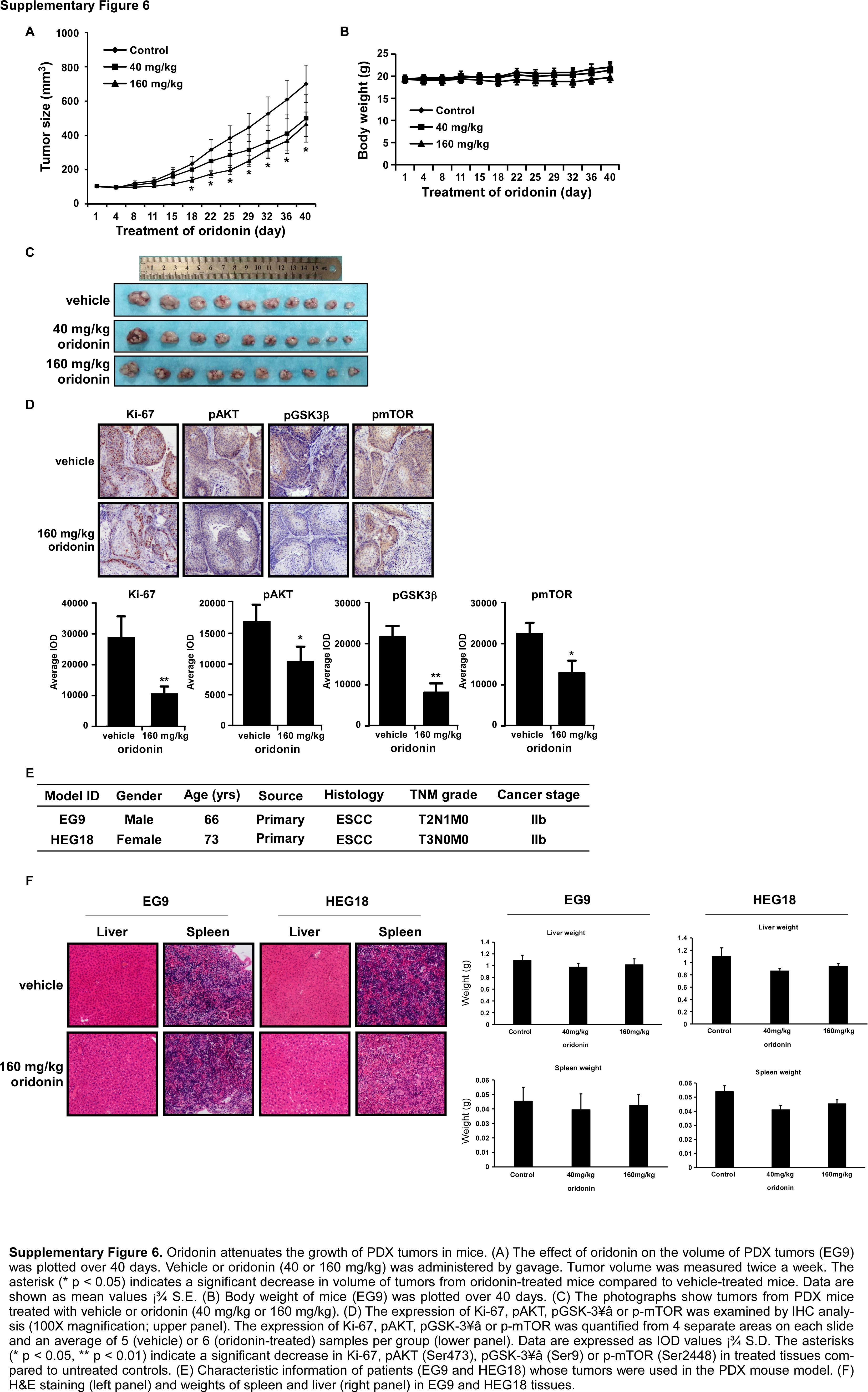
To explore the antitumor activity of oridonin in vivo, we used two PDX models, EG9 and HEG18, which exhibited high expression of AKT (Fig. 8, Supplementary Fig. 6). The PDX tumor mass was implanted into SCID mice and then vehicle or oridonin (40 or 160 mg/kg body weight) was administered by gavage injection, once a day for 40 or 52 days. The results showed that treatment of mice with 160 mg/kg of oridonin significantly reduced tumor growth compared to vehicle-treated group (Fig. 8A, C) with no effect on body weight (Fig. 8B). Tumor tissues were prepared for IHC analysis and the expression of Ki-67, phosphorylated AKT, GSK-3β or mTOR was examined (Fig. 8D, E). Results indicated that all of these protein markers were significantly suppressed by treatment with oridonin compared with vehicle-treated controls (Fig. 8D, E). The characteristics of EG9 and HEG18 are shown in Supplementary Fig. 6E.
Figure 8.

Oridonin attenuates the growth of PDX tumors in mice. (A) The effect of oridonin on the volume of PDX tumors was plotted over 52 days (HEG18). Vehicle or oridonin (40 or 160 mg/kg) was administered by gavage. Tumor volume was measured twice a week. The asterisk (* p < 0.05) indicates a significant decrease in volume of tumors from vehicle or oridonin-treated mice. Data are shown as mean values ± S.E. (B) Body weight of mice was plotted over 52 days. (C) The photographs show tumors from PDX mice treated with vehicle or oridonin (40 mg/kg or 160 mg/kg). (D) The expression of Ki-67, pAKT, pGSK-3β or p-mTOR was examined by IHC analysis (100X magnification). (E) The expression of Ki-67, pAKT, pGSK-3β or p-mTOR was quantified from 4 separate areas on each slide and an average of 5 (vehicle) or 6 (oridonin-treated) samples per group. Data are expressed as IOD values ± S.D. The asterisks (* p < 0.05, ** p < 0.01) indicate a significant decrease in Ki-67, pAKT (Ser473), pGSK-3β (Ser9) or p-mTOR (Ser2448) in treated tissues compared to untreated controls.
Discussion
Because esophageal cancer is a significant welfare and public health issue worldwide, effective chemotherapy/chemoprevention protocols are extremely important. Oridonin is a major diterpenoid component of leaf extracts from Rabdosia rubescens (27) and has been reported to inhibit growth of a variety of cancers, including leukemia, lung, pancreatic, prostate, breast and colon. The compound acts by down-regulating NF-κB activity, Bcl-2 expression, or up-regulating p53, p21, and p38 protein expression (16,28–32). Although oridonin has been reported to inhibit esophageal cancer cell growth (33–35), the molecular mechanisms of its anticancer effects have been poorly elucidated. In this study, we examined the effects of oridonin as a potential AKT inhibitor on the growth of ESCC cell lines and investigated the underlying molecular mechanisms. Results of a tissue microarray showed that phosphorylated AKT and total AKT are highly expressed in ESCC tissues compared to normal tissue (Fig. 1A), suggesting the importance of AKT. In addition, MK-2006, an AKT inhibitor, or AKT gene knock-down inhibited ESCC cell growth (Fig. 1B, D). Nazia Syed, et al reported phosphotyrosine profiling of esophageal cancer cell lines that describes activation status of other kinases including EPHA2, SRC, Src homology 2 domain containing transforming protein 1 (SHC1), mitogen-activated protein kinase 1 (MAPK1), mitogen-activated protein kinase 3 (MAPK3), or cyclin-dependent kinase 5 (CDK5) (36). To identify a direct target for oridonin, we examined the effect of this compound on the kinase activity of AKT, EGFR, Src and FGFR2 by in vitro kinase assay (Fig. 2 A, B, Supplementary Fig. 1A–C). Oridonin clearly inhibited AKT kinase activity (Fig. 2B), but had no effect on EGFR, Src, or FGFR2 kinase activity (Supplementary Fig. 1A–C). We used a kinase-profiling assay to examine the kinase activity of several other kinases associated with AKT signaling. These results indicated that oridonin had no effect on the kinase activity of c-Jun-N-terminal kinase-1/2 (JNK1/2), extracellular signal-regulated kinase 1/2 (ERK1/2), mammalian target of rapamycin (mTOR), phosphoinositide-dependent kinase-1 (PDK1) or phosphoinositide 3-kinase (PI3-K; Supplementary Fig. 1D). Oridonin increased Km values from 2.5 to 4.5 in AKT1 enzyme kinetic assay and from 1.5 to 3 in AKT2 kinetic assay (Supplementary Fig. 1E). Lineweaver-Burke plots indicated that oridonin acts as an ATP-competitive inhibitor in AKT1 and AKT2 kinase assay (Supplementary Fig. 1E). These findings agree with previous reports showing the inhibitory effect of oridonin on AKT phosphorylation in cervical carcinoma, osteosarcoma and prostate cancer (37–39). The novelty of our study is the finding that oridonin interacts with AKT1/2 directly and inhibits AKT1/2 kinase activity (Fig. 2). Our MTT and soft agar assay results also revealed that oridonin significantly inhibited growth of KYSE70, KYSE410 and KYSE450 ESCC cell lines (Fig. 3). Pi, et al. reported that oridonin induced apoptosis in KYSE150 cells at 30 μM (40), whereas, we found that oridonin induced apoptosis and G2/M phase arrest in KYSE70, KYSE410 and KYSE450 ESCC cell lines at 20 μM (Fig. 5, Supplementary Fig. 3, 4). Notably, these cells express very high levels of AKT. These results suggest that AKT is a prominent target of oridonin to inhibit proliferation and induce apoptosis in ESCC cells. AKT mediates several signaling molecules, including GSK-3β, mTOR and NF-κB (8) and oridonin decreased the phosphorylation of GSK-3β and mTOR and NF-κB transcriptional activity (Fig. 6). Furthermore, a combination of oridonin and standard chemotherapy drugs, 5-FU or cisplatin, resulted in synergistic or additive effects against ESCC growth (Fig. 7, Supplementary Fig. 5). Finally we confirmed our findings in in vivo PDX mouse models, which are a useful preclinical model of human tumor growth (41,42). Our results revealed that oral administration of oridonin significantly decreased the growth PDX tumors expressing high levels of AKT (Fig. 8, Supplementary Fig. 6A, C, E) without toxicity based on no loss in body weight, liver or spleen, and H&E staining analysis compared with vehicle-treated control mice (Supplementary Fig. 6B, E). IHC results indicated that oridonin decreased the expression of Ki-67, pAKT, pGSK-3β or p-mTOR in both PDX models (Fig. 8D, E, Supplementary Fig. 6D).
Overall, our study suggests that oridonin can inhibit progression of ESCC tumors, in vitro and in vivo by suppressing AKT signaling through its direct targeting of AKT (Fig. 9). Thus, this study might provide useful information in the clinical application of oridonin for ESCC chemotherapy.
Figure 9.

A proposed scheme illustrating the distinct roles of AKT1/2 in ESCC and their modulation by oridonin. AKT is activated by upstream kinases. Receptor tyrosine kinase (RTK) or phosphatidylinositide 3-kinase (PI3-K) stimulates mTOR, GSK3β or NF-κB thereby promoting cell cycle, preventing apoptosis and increasing cell proliferation. Oridonin targets AKT and blocks the signals of downstream targets, therefore inducing cell cycle arrest and apoptosis as well as inhibiting cell proliferation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We wish to thank Ran Yang and Shen Yang, China-US (Henan) Hormel Cancer Institute for supporting experiments.
Grant Support
This work was supported by grant funding from National Institutes of Health, USA CA187027, CA 166011, CA 196639 and Key program of Henan Province, China
Grant NO.161100510300 (Z.Dong) and the National Natural Science Foundation of China NSFC81672767 (M.Lee), NSFC81372269, NSFC81572812 (K.Liu) and Henan Provincial Government, China.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest
References
- 1.Montgomery E Oesophageal Cancer In Stewart, BW2014 528–43 p.
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 3.Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015;64(3):381–7. [DOI] [PubMed] [Google Scholar]
- 4.Ross P, Nicolson M, Cunningham D, Valle J, Seymour M, Harper P, et al. Prospective randomized trial comparing mitomycin, cisplatin, and protracted venous-infusion fluorouracil (PVI 5-FU) With epirubicin, cisplatin, and PVI 5-FU in advanced esophagogastric cancer. J Clin Oncol 2002;20(8):1996–2004. [DOI] [PubMed] [Google Scholar]
- 5.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 2005;94:29–86. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009;9(8):550–62. [DOI] [PubMed] [Google Scholar]
- 7.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2(7):489–501. [DOI] [PubMed] [Google Scholar]
- 8.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;129(7):1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almhanna K, Strosberg J, Malafa M. Targeting AKT protein kinase in gastric cancer. Anticancer Res 2011;31(12):4387–92. [PubMed] [Google Scholar]
- 10.Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O. The PI3K/Akt pathway as a target in the treatment of hematologic malignancies. Anti-cancer agents in medicinal chemistry 2009;9(5):550–9. [DOI] [PubMed] [Google Scholar]
- 11.Kim MO, Lee MH, Oi N, Kim SH, Bae KB, Huang Z, et al. [6]-shogaol inhibits growth and induces apoptosis of non-small cell lung cancer cells by directly regulating Akt1/2. Carcinogenesis 2014;35(3):683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasano T, Mabuchi S, Kuroda H, Kawano M, Matsumoto Y, Takahashi R, et al. Preclinical Efficacy for AKT Targeting in Clear Cell Carcinoma of the Ovary. Mol Cancer Res 2015;13(4):795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Li J, Xu WW, Guan XY, Qin YR, Zhang LY, et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget 2014;5(22):11576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu ZZ, Fu WB, Jin Z, Guo P, Wang WF, Li JM. Reactive oxygen species mediate oridonin-induced apoptosis through DNA damage response and activation of JNK pathway in diffuse large B cell lymphoma. Leuk Lymphoma 2016;57(4):888–98. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Liu JH, Chai K, Tashiro S, Onodera S, Ikejima T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J Pharm Pharmacol 2013;65(11):1622–42. [DOI] [PubMed] [Google Scholar]
- 16.Xu B, Shen W, Liu X, Zhang T, Ren J, Fan Y, et al. Oridonin inhibits BxPC-3 cell growth through cell apoptosis. Acta Biochim Biophys Sin (Shanghai) 2015;47(3):164–73. [DOI] [PubMed] [Google Scholar]
- 17.Cui Q, Tashiro S, Onodera S, Ikejima T. [Mechanism of downregulation of apoptosis by autophagy induced by oridonin in HeLa cells]. Yao Xue Xue Bao 2007;42(1):35–9. [PubMed] [Google Scholar]
- 18.Wang S, Zhang Y, Saas P, Wang H, Xu Y, Chen K, et al. Oridonin’s therapeutic effect: suppressing Th1/Th17 simultaneously in a mouse model of Crohn’s disease. J Gastroenterol Hepatol 2015;30(3):504–12. [DOI] [PubMed] [Google Scholar]
- 19.Xu S, Li D, Pei L, Yao H, Wang C, Cai H, et al. Design, synthesis and antimycobacterial activity evaluation of natural oridonin derivatives. Bioorg Med Chem Lett 2014;24(13):2811–4. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Yu L, Yang H, Li C, Hui Z, Xu Y, et al. Oridonin Attenuates Synaptic Loss and Cognitive Deficits in an Abeta1–42-Induced Mouse Model of Alzheimer’s Disease. PLoS One 2016;11(3):e0151397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owona BA, Schluesener HJ. Molecular Insight in the Multifunctional Effects of Oridonin. Drugs R D 2015;15(3):233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen ZY, Xu LY, Li EM, Shen J, Zheng RM, Cai WJ, et al. Immortal phenotype of the esophageal epithelial cells in the process of immortalization. Int J Mol Med 2002;10(5):641–6. [PubMed] [Google Scholar]
- 23.Schrödinger. Schrödinger Suite 2015 LLC, New York, NY; 2015. [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004;25(13):1605–12. [DOI] [PubMed] [Google Scholar]
- 26.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 27.Bai N, He K, Zhou Z, Tsai ML, Zhang L, Quan Z, et al. Ent-kaurane diterpenoids from Rabdosia rubescens and their cytotoxic effects on human cancer cell lines. Planta Med 2010;76(2):140–5. [DOI] [PubMed] [Google Scholar]
- 28.Ikezoe T, Yang Y, Bandobashi K, Saito T, Takemoto S, Machida H, et al. Oridonin, a diterpenoid purified from Rabdosia rubescens, inhibits the proliferation of cells from lymphoid malignancies in association with blockade of the NF-kappa B signal pathways. Mol Cancer Ther 2005;4(4):578–86. [DOI] [PubMed] [Google Scholar]
- 29.Wu QX, Yuan SX, Ren CM, Yu Y, Sun WJ, He BC, et al. Oridonin upregulates PTEN through activating p38 MAPK and inhibits proliferation in human colon cancer cells. Oncol Rep 2016;35(6):3341–8. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Li X, Wang J, Ye Z, Li JC. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci 2012;8(6):901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, Zhong Z, Wan J, Tan W, Wu G, Chen M, et al. Oridonin induces apoptosis, inhibits migration and invasion on highly-metastatic human breast cancer cells. Am J Chin Med 2013;41(1):177–96. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Shi QF, Qi M, Tashiro S, Onodera S, Ikejima T. Interruption of hepatocyte growth factor signaling augmented oridonin-induced death in human non-small cell lung cancer A549 cells via c-met-nuclear factor-kappaB-cyclooxygenase-2 and c-Met-Bcl-2-caspase-3 pathways. Biol Pharm Bull 2012;35(7):1150–8. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Jiang L, Wang S, Shi H, Wang J, Wang R, et al. The Antitumor Activity of the Novel Compound Jesridonin on Human Esophageal Carcinoma Cells. PLoS One 2015;10(6):e0130284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Gao J, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. The cytostatic and cytotoxic effects of oridonin (Rubescenin), a diterpenoid from Rabdosia rubescens, on tumor cells of different lineage. Int J Oncol 2005;26(3):579–88. [PubMed] [Google Scholar]
- 35.Li XT, Lin C, Li PY. [Comparison of in vitro assays for the cytotoxic effect of anticancer drugs]. Zhonghua Zhong Liu Za Zhi 1986;8(3):184–6. [PubMed] [Google Scholar]
- 36.Syed N, Barbhuiya MA, Pinto SM, Nirujogi RS, Renuse S, Datta KK, et al. Phosphotyrosine profiling identifies ephrin receptor A2 as a potential therapeutic target in esophageal squamous-cell carcinoma. Proteomics 2015;15(2–3):374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu HZ, Yang YB, Xu XD, Shen HW, Shu YM, Ren Z, et al. Oridonin induces apoptosis via PI3K/Akt pathway in cervical carcinoma HeLa cell line. Acta Pharmacol Sin 2007;28(11):1819–26. [DOI] [PubMed] [Google Scholar]
- 38.Jin S, Shen JN, Wang J, Huang G, Zhou JG. Oridonin induced apoptosis through Akt and MAPKs signaling pathways in human osteosarcoma cells. Cancer Biol Ther 2007;6(2):261–8. [DOI] [PubMed] [Google Scholar]
- 39.Lu J, Chen X, Qu S, Yao B, Xu Y, Wu J, et al. Oridonin induces G2/M cell cycle arrest and apoptosis via the PI3K/Akt signaling pathway in hormone-independent prostate cancer cells. Oncology letters 2017;13(4):2838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pi J, Cai H, Jin H, Yang F, Jiang J, Wu A, et al. Qualitative and Quantitative Analysis of ROS-Mediated Oridonin-Induced Oesophageal Cancer KYSE-150 Cell Apoptosis by Atomic Force Microscopy. PLoS One 2015;10(10):e0140935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9(6):338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunes M, Vrignaud P, Vacher S, Richon S, Lievre A, Cacheux W, et al. Evaluating patient-derived colorectal cancer xenografts as preclinical models by comparison with patient clinical data. Cancer Res 2015;75(8):1560–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.