

Abstract
The synthesis of (–)-salinosporamide A, a proteasome inhibitor, is described. The synthesis highlights the assembly of a densely decorated pyrrolidinone core via an aza-Payne/hydroamination sequence. Central to the success of the synthesis is a late stage C–H insertion reaction to functionalize a sterically encumbered secondary carbon. The latter functionalization leads to an enabling transformation where most of the prototypical strategies failed.
Keywords: salinosporamide A, total synthesis, aza-Payne, hydroamination, C-H insertion
Graphical Abstract
The total synthesis of salinosporamide A is accomplished via a late stage C-H insertion. The core of the molecule was assembled via an aza-Payne/hydroamination sequence of an enantiomerically enriched azidiridine alcohol to yield the pyrrolidinone, with functionalities in place for further elaboration into the target molecule.
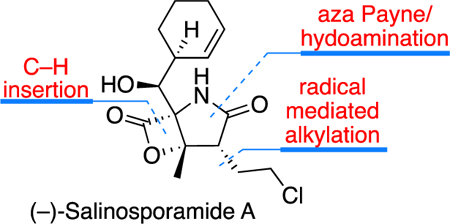
(–)-Salinosporamide A (1), a potent inhibitor of 20S proteasome, was isolated in 2003 by Fenical and co-workers from marine sources found in the ocean sediments.[1] Due to its unique anticancer activity, (–)-salinosporamide A was entered into human clinical trials for treatment of multiple myeloma.[2] Along with its significant biological properties, salinosporamide A contains a highly functionalized skeleton that provides a unique opportunity for reaction discovery. Featuring a bicyclic β-lactone within a highly decorated pyrrolidinone core containing five contiguous stereocenters, salinosporamide A’s structural features has stimulated tremendous activity with diverse strategies for its preparation within the synthetic community.[3]
A year after its isolation, the first total synthesis of salinosporamide A was reported by Corey and coworkers.[3r] A highlight of this ground breaking synthesis is their ingenious endgame strategy that installs the cyclohexenyl moiety. This has become the standard strategy adopted in most of the successful syntheses that followed. Strategically, most of the reported syntheses of salinosporamide A differ in their approach to access the pyrrolidinone core. While Corey and coworkers employed the Baylis-Hillman reaction, the majority of the bioinspired synthetic strategies utilized an intramolecular aldol type reaction that yields the pyrrolidinone. Our approach to the pyrrolidinone core, with the appropriate functionalities in place for completion of the synthesis, differs significantly from previous reports. We aimed to utilize the tandem aza Payne/hydoamination reaction,[4] highlighted in the dashed box in Scheme 1, to create the of pyrolidinone core contained in salinosporamide A.
Scheme 1.
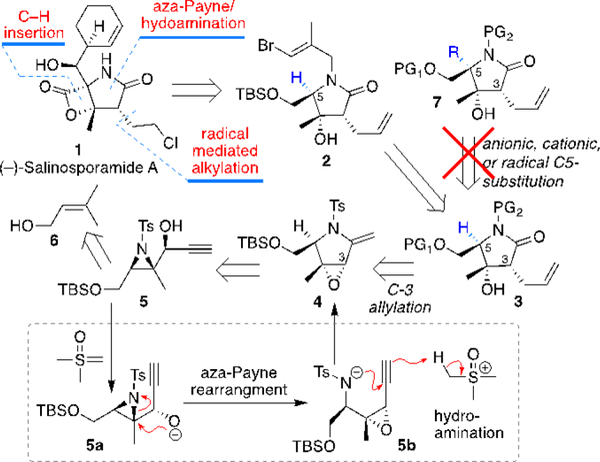
Retrosynthetic strategy towards Salinosporamide A
Initially, our retrosynthetic plan called for C5 acylation of 3 to yield the general structure 7, poised for β-lactone formation with the C4 hydroxyl group (Scheme 1). Pyrrolidinone 3 would be derived from the Lewis acid activated opening of the epoxide in 4, which would also pave the path for intoduction of the C3 allyl group. The aforementioned aza-Payne/hydroamination of 5, obtained from 6 through functional group manipulations and Cordova’s enantioselective aziridination protocol, would provide 4. As will be detailed briefly below, attempts to functionalize C5 via anionic, cationic, and radical routes failed. The C5 substitution was achieved through a C–H insertion reaction from the vinyl bromide 2 via in situ generation of a vinyl carbene, yielding a bicyclic system with an embedded olefin that could be converted to the requisite carbonyl functionality. Functional group interconversions of the latter yield the target compound 1.
Our synthesis commenced with the commercially available alcohol 6, which was protected as a TBS ether, followed by allylic oxidation to furnish the α,β-unsaturated aldehyde 8 in high overall yield (Scheme 2). Asymmetric aziridination of 8 was carried out utilizing Cordova’s protocol,[5] delivering the chiral aziridinal 10 in 88% yield and excellent enantioselectivity. We have previously demonstrated that the chelation-guided addition of ethynyl magnesium bromide to aziridine aldehydes similar to 10 proceeds with high levels of stereocontrol.[6] As such, reaction of ethynylmagnesium bromide with 10 provided the aziridine alcohol 5 as a single diastereomer (structure of 5 was confirmed via x-ray crystallography). The aza-Payne/hydroamination sequence was initiated with the addition of the Corey-Chaykovsky reagent, pretreated with NaH to yield the enamide 4. The crude enamide was readily ozonolyzed to deliver the pyrrolidinone 11 in nearly quantitative yield over two steps. The crude product 11 was subsequently exposed to magnesium bromide in ether, resulting in a highly regio and diastereoselective bromide addition to the epoxide moiety. Bromohydrin 12 was isolated in high overall yield from the propargyl alcohol 5 (86% over three steps), and its structure was further verified by x-ray crystallography.
Scheme 2.
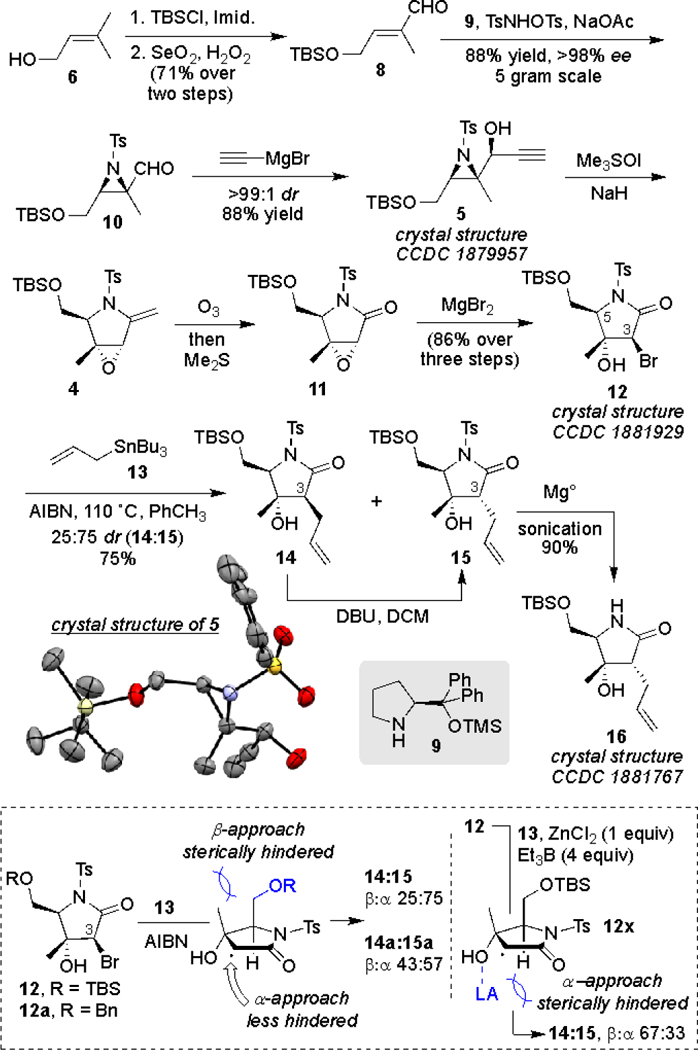
Synthesis of advanced intermediate 16.
Radical mediated allylation of the α-bromo lactam 12 with allyl-tri-butyl stannane (13) installed the three-carbon appendage at C3, favoring the desired diastereomer 15 by a 3:1 ratio. The stereochemical outcome of this reaction is presumably controlled by the large TBS protecting group on the primary alcohol (see dashed box in Scheme 2). Protection with the smaller benzyl group (12a) led to a more equal distribution of diastereomers (14a:15a 43:57), while having a Lewis acid present reversed the selectivity (14:15 67:33, see ZnCl2 mediated allylation of 12, dashed box, Scheme 2). As shown in the proposed complex 12x, the putative coordination of the Lewis acid with the C4-OH blocks the α-approach, and thus favors addition of the allyl group from the opposite face. Since the selectivity attributed to the steric effect of the neighboring groups on the kinetic process of the allylation reaction led to 15 as the major product, we surmised that a similar intrinsic selectivity could apply to the thermodynamics of C3 epimerization. In light of this notion, the mixture of diastereomers was treated with catalytic DBU at room temperature, leading to complete epimerization of C3 to yield 15, exclusively. Removal of the tosyl group with magnesium in methanol with sonication7 provided 16 (structure verified by x-ray crystallography), poised for acylation at C5.
With 15 in hand, installation of the carbonyl or a functionally equivalent moiety on C5 was pursued (Figure 1a). Our initial approach was to deprotonate C5-H for nucleophilic substitution of an activated acyl group (17). Various analogs of compound 15 were prepared to expose an acidic hydrogen on C5. Although with an ester substituted at C5 (see R1=CO2Me in 18) one might argue that the C3 α-H of the lactam moiety might be more acidic, the choice of an aldehyde (R1=CHO in 18) was to ensure effective competition. Nonetheless, under many different conditions, deprotonation/functionalization of C5 failed. Major issues were the kinetic acidity of the C3 proton that would result in the elimination of hydroxyl group (protected and unprotected) from C4 along with acylation of C3. Additionally, leaving the hydroxyl group at C4 unprotected led to significant retro-aldol when C5 carried an electron withdrawing group, thus unraveling the pyrrolidone core. Alternatively, efforts to intramolecularly remove the C5 hydrogen atom were met with disappointing results. Examples 19a and 19b, shown in Figure 1a, utilized radical relay groups to initiate the hydrogen atom removal (see 19a + AIBN), although in most cases the dehalogenated molecules were observed as the major product. Equally disappointing were efforts to use enamine 20 to exploit a cationic based strategy for introducing an acyl equivalent as a nucleophile at C5 (20 + E+); enamine 20 was not stable and underwent rapid loss of water to yield a conjugated diene.
Figure 1.
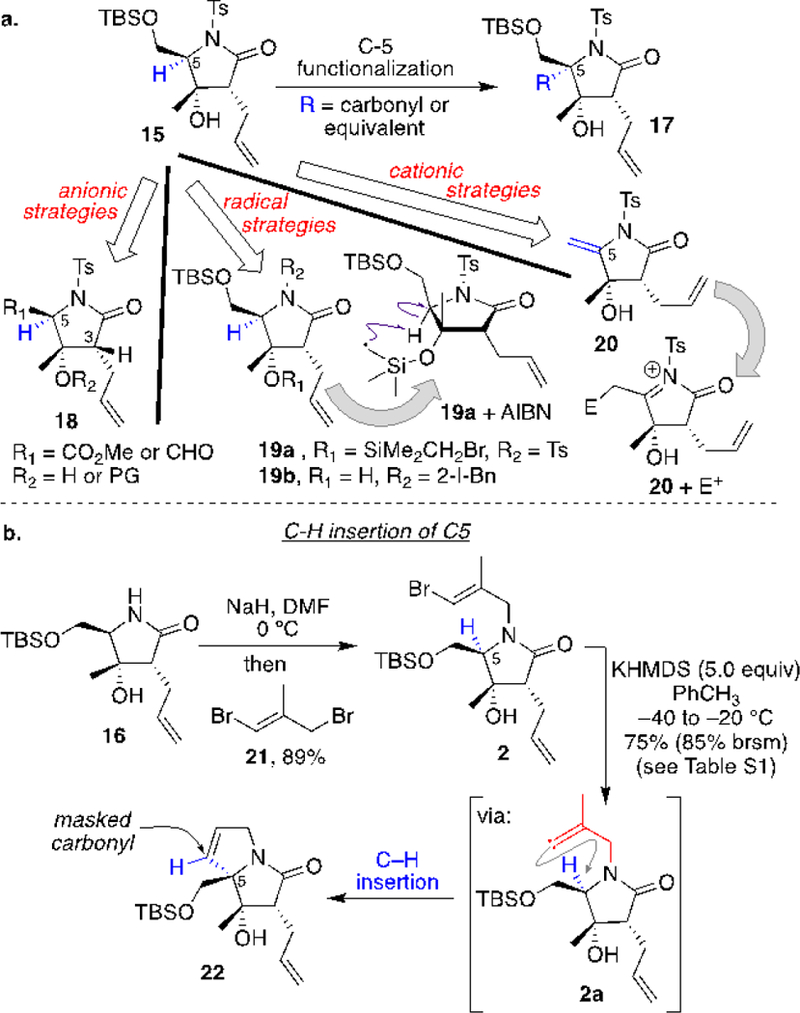
a. Failed strategies to functionalize C5 via anionic, radical, and cationic approaches. b. Intramolecular C-H insertion of the carbene into the C5-H bond.
As described above, polar approaches to C5 functionalization were fraught with complications that led to undesired pathways, while the neutral radical approach seemed to not engage with the C5-H bond, although it minimized degradation via other pathways. Based on these observations, we opted to pursue the neutral activation path, but in lieu of radicals we explored C-H insertion reactions to functionalize C5 (Figure 1b).[8] An in situ generated tethered carbene8e,8h in the vicinity of C5 could lead to the installation of a carbonyl surrogate. The pyrrolidinone nitrogen atom was chosen as a handle to deliver the carbene, thus 16 was reacted with allyl bromide 21 to obtain vinyl bromide 2 in high yield (Figure 1b).[9] Exposure of 2 to basic conditions presumably yields intermediate 2a, leading to the bicyclic product 22. A thorough optimization of the reaction conditions (see Table S1 for details) revealed that employing potassium bis(trimethylsilyl)amide (KHMDS) in toluene at low temperature delivers 22 in high yield. The reaction efficiency was highly dependent on the temperature of the reaction. At higher temperatures the isolated yield of the product and the mass balance of the reaction were significantly lower. This observation is not surprising since the pyrrolidinone core was susceptible to elimination under basic conditions, a process that is retarded at lower temperatures.
Oxidative cleavage of the newly installed double bond in 22 would yield the requisite carbonyl moiety on C5. On the other hand, oxidative cleavage of the terminal olefin on C3 would provide a path to introduce the chloride group on the carbon side chain. Given the concurrent oxidation of both olefins in 22 led to a complex mixture of products, a sequential oxidative cleavage of the two olefins was pursued. Chemoselective cleavage of the monosubstituted alkene was achieved using the Lemieux–Johnson oxidation protocol (Scheme 3). The resulting hemiacetal was directly converted to acetal 23 by reacting with methanol under acidic condition, yielding a separable 1:1 diastereomeric mixture, although the stereochemistry of the newly formed center is inconsequential. For simplifying NMR analyses, the two diastereomers of 23 were carried through the final steps of the synthesis separately, although it should be noted that the yields and selectivities for the products of the reactions that followed were the same (only one diastereomer is shown from 24 to 30).
Scheme 3.
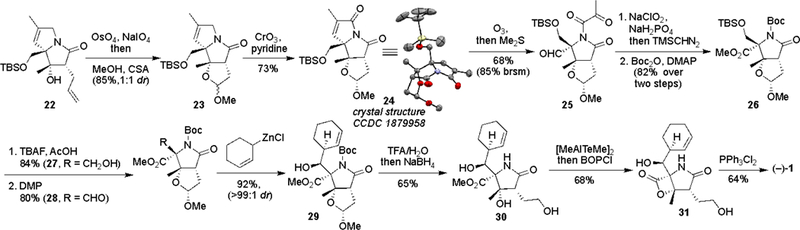
Final steps for the synthesis of (–)-salinosporamide A (1)
The remaining olefin in 23 was ozonolyzed, leading to the installation of the aldehyde group on C5, however, removal of the residual ketone on the pyrrolidinone nitrogen atom proved problematic. Allylic oxidation of the cycloalkene with CrO3[10] led to the α,β-unsaturated imide 24, which was readily cleaved to the dicarbonyl 25 with the desired aldehyde on C5 (see Table S2 for optimization of oxidation conditions). Pinnick-Kraus oxidation of the aldehyde proceeded smoothly, not only delivering the carboxylic acid that was subsequently esterified with (trimethylsilyl)diazomethane, but also hydrolyzing the dicarbonyl group. The resulting lactam was Boc protected, delivering 26 in high yield from aldehyde 25. Removal of the TBS group required care to maintain the Boc group. Tetrabutyl ammonium fluoride buffered with acetic acid[11] provided alcohol 27 in high yield (see Table S3 for details), which upon Dess–Martin periodinane (DMP) oxidation led to the aldehyde 28. Following the strategy reported by Corey and coworkers,[3r] treatment of aldehyde 28 with the cyclohexenyl zinc chloride delivered alcohol 29 in high yield with complete diastereoselectivity. Removal of the Boc group and hydrolysis of the acetal were achieved by treating 29 with aqueous trifluoro acetic acid (TFA).3u The resulting hemiacetal was reduced with sodium borohydride to yield diol 30. Hydrolysis of the methyl ester employing [Me2AlTeMe]2 delivered the corresponding carboxylic acid, which upon treatment with BOPCl led to installation of the β-lactam in 31.[3q] Chlorination of the primary alcohol in 31 with triphenylphosphine dichloride provided the target molecule.
In conclusion, we report the enantioselective synthesis of (–)-salinosporamide A. The synthesis highlights the utility of a key aza-Payne/hydroamination reaction to install the pyrrolidinone core, and an epoxide opening/allylation sequence that decorates the densely functionalized ring. Application of the C–H insertion reaction highlights the use of vinyl carbene chemistry to overcome challenging C-H functionalizations. Although not immune to unwanted side reactions, the power and efficacy of a neutral, but reactive carbon center, in addressing issues that often exists with classical approaches to carbon functionalization in a structurally complex system that could suffer many unproductive pathways is noteworthy.
Supplementary Material
Acknowledgements
We thank Dr. Yu-Sheng Chen at ChemMatCARS, APS, for his assistance with single-crystal data of compound 12. ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant number CHE-0535644. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357. Generous support was provided by the NIH (GM110525).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W, Angew. Chem. Int. Ed. 2003, 42, 355–357. [DOI] [PubMed] [Google Scholar]
- [2].a) Gulder TAM, Moore BS, Angew. Chem. Int. Ed. 2010, 49, 9346–9367; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Groll M, Huber R, Potts BCM, J. Am. Chem. Soc. 2006, 128, 5136–5141. [DOI] [PubMed] [Google Scholar]
- [3].a) Caubert V, Masse J, Retailleau P, Langlois N, Tetrahedron Lett. 2007, 48, 381–384; [Google Scholar]; b) Endo A, Danishefsky SJ, J. Am. Chem. Soc. 2005, 127, 8298–8299; [DOI] [PubMed] [Google Scholar]; c) Fukuda T, Sugiyama K, Arima S, Harigaya Y, Nagamitsu T, Omura S, Org. Lett. 2008, 10, 4239–4242; [DOI] [PubMed] [Google Scholar]; d) Kaiya Y, Hasegawa J, Momose T, Sato T, Chida N, Chem. Asian J. 2011, 6, 209–219; [DOI] [PubMed] [Google Scholar]; e) Ling TT, Macherla VR, Manam RR, McArthur KA, Potts BCM, Org. Lett. 2007, 9, 2289–2292; [DOI] [PubMed] [Google Scholar]; f) Ling TT, Potts BC, Macherla VR, J. Org. Chem. 2010, 75, 3882–3885; [DOI] [PubMed] [Google Scholar]; g) Logan AWJ, Sprague SJ, Foster RW, Marx LB, Garzya V, Hallside MS, Thompson AL, Burton JW, Org. Lett. 2014, 16, 4078–4081; [DOI] [PubMed] [Google Scholar]; h) Ma G, Nguyen H, Romo D, Org. Lett. 2007, 9, 2143–2146; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Marx Léo B, Burton Jonathan W, Chem. Eur. J. 2018, 24, 6747–6754; [DOI] [PubMed] [Google Scholar]; j) Momose T, Kaiya Y, Hasegawa J, Sato T, Chida N, Synthesis 2009, 2983–2991; [Google Scholar]; k) Mosey RA, Tepe JJ, Tetrahedron Lett. 2009, 50, 295–297; [Google Scholar]; l) Mulholland NP, Pattenden G, Walters IAS, Org. Biomol. Chem. 2008, 6, 2782–2789; [DOI] [PubMed] [Google Scholar]; m) Mulholland NP, Pattenden G, Walters LAS, Org. Biomol. Chem. 2006, 4, 2845–2846; [DOI] [PubMed] [Google Scholar]; n) Nguyen H, Ma G, Gladysheva T, Fremgen T, Romo D, J. Org. Chem. 2011, 76, 2–12; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Nguyen H, Ma G, Romo D, Chem. Commun. 2010, 46, 4803–4805; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Reddy LR, Fournier JF, Reddy BVS, Corey EJ, J. Am. Chem. Soc. 2005, 127, 8974–8976; [DOI] [PubMed] [Google Scholar]; q) Reddy LR, Fournier JF, Reddy BVS, Corey EJ, Org. Lett. 2005, 7, 2699–2701; [DOI] [PubMed] [Google Scholar]; r) Reddy LR, Saravanan P, Corey EJ, J. Am. Chem. Soc. 2004, 126, 6230–6231; [DOI] [PubMed] [Google Scholar]; s) Rentsch A, Landsberg D, Brodmann T, Bulow L, Girbig AK, Kalesse M, Angew. Chem. Int. Ed. 2013, 52, 5450–5488; [DOI] [PubMed] [Google Scholar]; t) Sato Y, Fukuda H, Tomizawa M, Masaki T, Shibuya M, Kanoh N, Iwabuchi Y, Heterocycles 2010, 81, 2239–2246; [Google Scholar]; u) Satoh N, Yokoshima S, Fukuyama T, Org. Lett. 2011, 13, 3028–3031; [DOI] [PubMed] [Google Scholar]; v) Shibasaki M, Kanai M, Fukuda N, Chem. Asian J. 2007, 2, 20–38; [DOI] [PubMed] [Google Scholar]; w) Struble JR, Bode JW, Tetrahedron 2009, 65, 4957–4967; [DOI] [PMC free article] [PubMed] [Google Scholar]; x) Takahashi K, Midori M, Kawano K, Ishihara J, Hatakeyama S, Angew. Chem. Int. Ed. 2008, 47, 6244–6246; [DOI] [PubMed] [Google Scholar]; y) Margalef IV, Rupnicki L, Lam HW, Tetrahedron 2008, 64, 7896–7901. [Google Scholar]
- [4].Schomaker JM, Geiser AR, Huang R, Borhan B, J. Am. Chem. Soc. 2007, 129, 3794–3795. [DOI] [PubMed] [Google Scholar]
- [5].Deiana L, Dziedzic P, Zhao GL, Vesely J, Ibrahem I, Rios R, Sun JL, Cordova A, Chem. Eur. J. 2011, 17, 7904–7917. [DOI] [PubMed] [Google Scholar]
- [6].Kulshrestha A, Schomaker JM, Holmes D, Staples RJ, Jackson JE, Borhan B, Chem. Eur. J. 2011, 17, 12326–12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nyasse B, Grehn L, Ragnarsson U, Chem. Commun. 1997, 1017–1018. [Google Scholar]
- [8].a) Davies HML, Angew. Chem. Int. Ed. 2006, 45, 6422–6425; [DOI] [PubMed] [Google Scholar]; b) Davies HML, Beckwith REJ, Chem. Rev. 2003, 103, 2861–2904; [DOI] [PubMed] [Google Scholar]; c) Davies HML, Manning JR, Nature 2008, 451, 417; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Godula K, Sames D, Science 2006, 312, 67–72; [DOI] [PubMed] [Google Scholar]; e) Grainger RS, Owoare RB, Org. Lett. 2004, 6, 2961–2964; [DOI] [PubMed] [Google Scholar]; f) Hinman A, Du Bois J, J. Am. Chem. Soc. 2003, 125, 11510–11511; [DOI] [PubMed] [Google Scholar]; g) Knorr R, Chem. Rev. 2004, 104, 3795–3849; [DOI] [PubMed] [Google Scholar]; h) Taber DF, Christos TE, Neubert TD, Batra D, J. Org. Chem. 1999, 64, 9673–9678; [Google Scholar]; i) Wang D-H, Yu J-Q, J. Am. Chem. Soc. 2011, 133, 5767–5769; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Wardrop DJ, Bowen EG, Chem. Commun. 2005, 5106–5108; [DOI] [PubMed] [Google Scholar]; k) Ye T, Mckervey MA, Chem. Rev. 1994, 94, 1091–1160; [Google Scholar]; l) Jeffrey JL, Sarpong R, Chem. Sci. 2013, 4, 4092–4106; [Google Scholar]; m) Lindsay VN, Viart HM, Sarpong R, J. Am. Chem. Soc. 2015, 137, 8368–8371; [DOI] [PubMed] [Google Scholar]; n) Ye Z, Gettys KE, Dai M, Beilstein J. Org. Chem. 2016, 12, 702–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Attempts to acylate the amide nitrogen atom in 16 with (E)-3-bromo-2-methylacryloyl chloride and (E)-3-bromo-2-methylacrylic anhydride failed, returning the starting material under various forcing conditions. Nonetheless, 21 was capable of alkylating the same nitrogen atom, requiring subsequent oxidation of the methylene to the carbonyl for removal (vida infra)
- [10].Mycock DK, Glossop PA, Lewis W, Hayes CJ, Tetrahedron Lett. 2013, 54, 55–57. [Google Scholar]
- [11].Kawasaki M, Shinada T, Hamada M, Ohfune Y, Org. Lett. 2005, 7, 4165–4167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.