

Abstract
The distribution of telomere length in humans is broad, but it has finite upper and lower boundaries. Growing evidence shows that there are disease processes that are caused by both short and long telomere length extremes. The genetic basis of these short and long telomere syndromes may be linked to mutations in the same genes, such as the telomerase reverse transcriptase (TERT), but through differential effects on telomere length. Short telomere syndromes have a predominant degenerative phenotype marked by organ failure that most commonly manifests as pulmonary fibrosis and are associated with a relatively low cancer incidence. In contrast, insights from studies of cancer-prone families as well as genome-wide association studies (GWAS) have identified both rare and common variants that lengthen telomeres as being strongly associated with cancer risk. We have hypothesized that these cancers represent a long telomere syndrome that is associated with a high penetrance of cutaneous melanoma and chronic lymphocytic leukemia. In this Review, we will synthesize the clinical and human genetic observations with data from mouse models to define the role of telomeres in cancer etiology and biology.
Introduction
Telomeres may at first glance simply appear as geographic boundaries at chromosome ends. However, there is mounting evidence that disturbances in telomere length, at short and, as we will discuss here, long extremes, are linked to disease susceptibility. Telomere DNA is a repetitive hexamer of TTAGGGs that is bound by a specialized protein complex known as shelterin (1–3). Telomerase synthesizes new telomere DNA to offset the shortening that normally occurs during DNA replication (4, 5). Telomerase has two essential components: the telomerase reverse transcriptase (TERT) uses a template within an intrinsic telomerase RNA component, TR (also known as TERC), to add telomere repeats onto the 3′ ends of chromosomes (6–9). Shelterin proteins prevent chromosome ends from fusing and from being recognized as double-strand breaks (3). They also regulate telomerase access to the telomere, and promote telomere repeat addition processivity by allowing TERT to use a relatively short template in TR to iteratively synthesize longer telomere tracks (10–12). As we will discuss here, disorders of telomere length are increasingly appreciated as causing clinically recognizable disease processes (13). The short telomere syndromes are now phenotypically and genetically well characterized (14–19). This knowledge is increasingly integrated into clinical algorithms, especially for patients with lung disease and bone marrow failure (20, 21). Here, we focus on emerging evidence, from cancer-prone families as well as from population-based studies, linking germline variants that promote telomere lengthening to cancer susceptibility. We contrast the genetic basis of the two extreme telomere length phenotypes and highlight how recent human-focused studies provide critical insights into the fundamentals of cancer etiology.
Telomerase is limiting
The foundational understanding of the role of telomeres and telomerase in disease has been rooted in curiosity-driven science, in simple systems and model organisms (22). One major theme that emerges at the intersection between this fundamental science and disease genetics is that relatively small, subtle changes affecting telomerase abundance or function can influence telomere length and, in turn, disease risk (23). The exquisite sensitivity of telomere length to these small changes is related to the fact that telomerase is in very low abundance and its activity is tightly regulated. In yeast, mice, and humans, the number of telomere ends exceeds the number of telomerase molecules (refs. 24, 25, and reviewed in refs. 23, 26). The low levels of telomerase set up a system wherein not all telomeres are elongated during a given cell cycle even when telomerase is normally expressed (27). There are at least three additional limits on telomerase activity. The first is that the essential telomerase components, TERT and TR, are expressed at very low levels relative to other proteins and RNAs (e.g., refs. 24, 28). Even other factors involved in telomerase biogenesis, such as nuclear assembly factor 1 (NAF1), which promotes shuttling of TR to the nucleolus for assembly with TERT, show haploinsufficiency for telomere length (28). Thus, although only 10% of human genes are estimated to show haploinsufficiency, many of the telomerase and related genes that have been heretofore linked to Mendelian disease, including TERT, TR, and NAF1, do so (14, 28–30). A second limit is that telomerase expression is also tightly regulated. After early embryonic development, the TERT promoter is repressed in most somatic cells, likely through promoter hypomethylation (31–33). The repressive effect of this hypomethylation on TERT expression is counter to effects in most other contexts. The timing of TERT silencing leaves a small window during early development for telomeres to be elongated (34), and makes telomere length highly heritable and, in great part, influenced by parental telomere length (27, 35, 36). A third check on telomere elongation is that even when expressed, the timing for telomere repeat addition is cell cycle–regulated and restricted to late S phase (reviewed in ref. 37). For all these reasons, telomeres shorten even in telomerase-expressing somatic cells, such as hematopoietic progenitors and T cells (26). These checks favor a system where telomere shortening is an overall general default, and as we discuss here, evidence linking long telomeres to the risk of multiple cancers underscores the tumor-suppressive advantages of these checks.
Telomere length has definable upper and lower boundaries
One of the major advances in understanding the role of telomere length in human disease has been the standardization of telomere length measurement methods that more robustly define absolute “short” and “long” telomere thresholds (20, 38). Relying on a method that measures telomere length in distinct leukocyte lineages using combined flow cytometry and fluorescence in situ hybridization (flowFISH), there is outstanding concordance and reproducibility across laboratories (20, 38, 39). These flowFISH telomere length measurements show that human telomere length has a definable normal range with discrete upper and lower boundaries (20). This type of telomere length analysis has the advantage of establishing age-, percentile-adjusted values rather than relative comparisons of “longer” and “shorter” descriptors. This advance has made interpretation of telomere length for precision medicine use possible and analogous to other clinical measurements (e.g., white blood cell count) wherein the normal range is broad, but extreme values, relative to healthy controls, may be associated with the risk of certain pathologies. For this Review, “telomere length” refers to the mean length as measured in leukocytes by flowFISH and reported as an age-adjusted percentile. Within each cell, the shortest telomere(s) signal the DNA damage response associated with cellular senescence and apoptosis (40). Remarkably, however, the mean telomere length, in defined and limited clinical contexts, is an outstanding surrogate and can generally distinguish individuals with germline defects in telomere maintenance from their relatives (15, 41). As such, the mean telomere length as measured in leukocyte subsets by flowFISH is used widely as a diagnostic and prognostic tool in patients suspected to have short telomere syndromes (18, 20, 42).
The human short telomere syndrome phenotype
The cancer-prone state associated with telomere lenghtening contrasts with that of short telomere syndromes. To facilitate these comparisons, we will first briefly review the better-described short telomere diseases. Short telomere syndromes encompass a continuum of clinical presentations that manifest from infancy to late adulthood (26). They are caused by mutations in telomerase and telomere maintenance genes. Their onset is determined in great part by the severity of the short telomere defect (20, 43). Short telomere syndromes generally have two primary clinical presentations. A more severe form manifests in infants and children; it causes disease in high-turnover tissues and primarily recognized as immunodeficiency, bone marrow failure, and enteropathy (16, 19, 20, 44). Adult-onset short telomere syndromes are more common and account for at least 90% of presentations (45). They manifest most frequently as idiopathic pulmonary fibrosis (IPF) and other telomere-related lung disease (45). These telomere-related lung disorders in the vast majority show autosomal dominant inheritance and may appear as emphysema in smokers (45, 46). IPF affects 100,000 individuals in the United States alone, and at least 50% of IPF patients have telomere length in the lowest decile of the population (45). In one-third of families with pulmonary fibrosis, a mutation in telomerase or telomere-related genes is detectable (45). The high frequency of telomere defects in IPF and the prevalence of this disease make IPF the most common of the human short telomere phenotypes. A subset of adult IPF patients show extrapulmonary short telomere syndrome features including bone marrow failure, immunodeficiency, and liver disease; their recognition is critical for the diagnosis and management of these patients (18, 21, 41–43, 47).
Cancer is relatively rare in short telomere syndromes
Cancer is an overall relatively rare complication of short telomere syndromes and affects approximately 10% to 15% of patients (48). This rate is far lower than in other common cancer-prone syndromes, such as Li-Fraumeni, which have lifetime risks around 90% (49). The short telomere cancer spectrum is also restricted to mostly hematologic cancers, the most common being myelodysplastic syndrome, an age-associated clonal disease of the bone marrow. This low cancer incidence lies in contrast to predictions from cell-based models, which reported spontaneous immortalization and transformation of cells after short telomere–induced senescence (50). These clinical observations suggest that, in the presence of an intact DNA damage response, as is the case in most patients with short telomere syndromes, degenerative disease is the predominant phenotype and leads to progressive failure of hematopoiesis, T cell immunity, and end-stage lung-liver disease. Below we will highlight how these clinical findings support what has been documented in nearly all tumor-prone mouse models.
The genetic causes of short telomere syndromes
Understanding the genetic mechanisms by which short telomere syndromes arise is particularly relevant for our Review, because mutations in some of the same genes have also been linked to a cancer-prone state, which we hypothesize is long-telomere mediated. Thirteen genes have been implicated to date in Mendelian short telomere syndrome genetics; they explain 50% to 70% of cases (Figure 1A). Two of these genes are also mutated in cancer-prone families. In general, the vast majority of mutations cause telomere shortening by depleting the abundance of telomerase, disturbing its catalytic functions/processivity, or interfering with its recruitment to the telomere. They affect the telomerase holoenzyme itself (TERT, TR, DKC1), adaptors of the dyskerin complex (NHP2, NOP10), genes that affect TR biogenesis and localization (PARN, NAF1, TCAB1), and regulation of telomerase recruitment to the telomere as well as processivity by shelterin (TPP1, also known as ACD, and likely TINF2; ref. 51). There are also mutations in genes that are thought to affect telomere replication (RTEL1) and telomere lagging strand synthesis (CTC1, STN1). The genetic basis of short telomere syndromes has been reviewed elsewhere (13, 28). As further discussed below, for TERT and TPP1, mutations that predict telomere lengthening are also associated with high-penetrance familial cancer syndromes (13).
Figure 1. Schematic of mutant telomerase and telomere genes in Mendelian short and long telomere syndromes and model for TPP1 allele–specific effects on telomere length.

(A) Components with known mutations are shown in color, and their telomere function is indicated above each group. Thirteen genes have been identified, with the short telomere syndrome associations marked by a subscript S. Four genes are associated with long telomere syndrome phenotypes and are marked by a superscript L. Adapted with permission from Current Opinion in Genetics & Development (13). (B) The left panel shows the state of telomere length maintenance normally. The middle panel shows how in-frame deletions in the TEL patch interfere with TERT recruitment and processivity, provoking telomere shortening. The right panel shows a model for how cancer-associated mutations may promote telomere maintenance in cancer-prone families. TPP1 deletions or missense mutations in the POT1-interating domain are predicted to affect POT1’s telomere-binding capacity, allowing TERT to elongate more efficiently. The latter is hypothesized to have a net effect of telomere lengthening and/or telomere maintenance.
Familial cancers caused by telomere-lengthening mutations
Evidence that long telomere length confers a longevity advantage came initially from studies of primary cultured fibroblasts in which cells with longer telomeres had longer replicative potentials (52). Moreover, exogenous TERT expression was sufficient to bypass cellular senescence and immortalize primary cells (53). In humans, evidence that telomerase upregulation confers a risk of familial cancer was first documented in a five-generation autosomal dominant family with cutaneous malignant melanoma (CMM) that was found to carry a mutation in the TERT promoter (54). This gain-of-function mutation upregulates TERT transcription (54). The mutation, located 57 bases upstream of the TERT transcriptional start site, functions similarly to two other common recurrent somatic TERT promoter mutations (54, 55). These promoter mutations create a de novo E26 transformation–specific (ETS) transcription factor family binding site that removes the repressive state on TERT by allowing interaction with an abundant GA-binding protein (GABP) transcription factor to promote TERT transcription (54, 56, 57). A second melanoma family was recently found to carry another TERT promoter mutation (58), but overall, the prevalence of germline TERT promoter mutations in familial melanoma is less than 1% (58). The importance of telomere maintenance to melanoma susceptibility is, however, highlighted in the fact that germline heterozygous mutations in three other telomere genes, POT1, TPP1, and RAP1 (also known as TERF2IP), all shelterin components, have been linked to familial melanoma (Table 1). POT1 mutations are most common, and they account for 2% to 4% of CDKN2A/CDK4-negative CMM families (60% of familial CMM cases fall into this category; refs. 59–61). Mutations in TPP1 and RAP1 account for another 2% of this familial CMM subset (62). Table 1 summarizes these associations.
Table 1. Telomerase and shelterin genes mutated in familial melanoma.
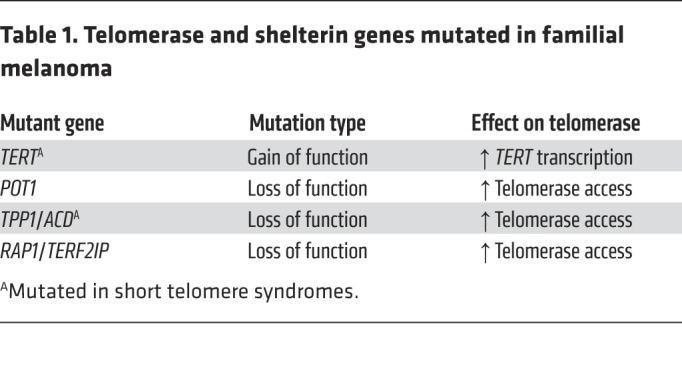
Beyond melanoma, there is also evidence of shelterin gene mutations in other cancer-prone families. Among chronic lymphocytic leukemia (CLL) multigenerational families, mutations in POT1, TPP1, and RAP1 are found in nearly 10% of cases (63). Rare POT1 mutations have also been reported in families with glioma (<1%; ref. 64); cardiac angiosarcoma and Li-Fraumeni–like syndrome (27%, 6 of 22 TP53-negative families; refs. 65, 66); colorectal cancer (0.3%, 3 of 1051 families; ref. 67); and Hodgkin lymphoma (5%, 2 of 41 families; ref. 68). These are generally loss-of-function mutations and are predicted to cause telomere lengthening. While these mutations were identified in familial forms of a single cancer, mutation carriers showed other malignancies, suggesting they confer a broader cancer-prone state (54, 58, 66). In support of this idea, a recent study reported an Ashkenazi founder POT1 mutation that interfered with POT1’s DNA-binding capacity; it simultaneously conferred susceptibility to both melanoma and CLL (69). Because the most prevalent cancers in patients with these TERT and shelterin mutations are melanoma and CLL, we propose that these cancers define core long telomere cancer phenotypes.
Germline mutations in TERT and shelterin are telomere-lengthening
How do mutations in the TERT promoter and shelterin genes promote the risk of melanoma and other cancers? Some studies have suggested that the effect is because of telomere deprotection (65), but this model would not explain the fact that these cancers develop in phenotypically intact adults who show no evidence of genome instability during development. We propose that the single shared consequence of TERT promoter and shelterin mutations is a longer telomere and/or a telomere-lengthening capacity (Table 1). Several pieces of clinical data support this interpretation. The first is that TERT promoter and POT1 mutation carriers have longer telomeres than their unaffected relatives (65, 70). The second is that families with POT1 and TPP1 mutations often show genetic anticipation, for both cancer onset and cancer mortality (13, 59, 60, 65). We have proposed before that this pattern of anticipation is likely because of successive telomere lengthening (13). In one POT1 mutant family, telomere lengthening was observed across generations, although the telomere length measurements were not age-corrected and were performed by nonstandard methods (65). As discussed in detail below, there is an additional independent body of genetic epidemiology showing that long telomere length alone is associated with increased risk of the same cancers (i.e., melanoma, glioma, CLL) that are seen in cancer-prone families with TERT promoter and shelterin mutations.
Different TPP1 mutations cause distinct disease phenotypes
The delicate regulation of telomere length is particularly highlighted in the case of TPP1 mutations, where two distinct types of heterozygous, haploinsufficient mutations show opposing disease phenotypes. Mutations in the TEL patch, which is required for both TERT recruitment and processivity, cause an autosomal dominant short telomere syndrome phenotype (11, 71, 72). By contrast, mutations identified in cancer-prone families fall in the POT1-interacting domain and are predicted to interfere with POT1 binding to the telomere (10, 73). This would have the functional effect of removing the negative regulation on telomere elongation, making the telomere more accessible, and have a net effect of telomere lengthening. A schematic of these TPP1 allele–specific mutations and their putative effects on telomere length is shown in Figure 1B.
Long telomere–associated SNPs increase cancer risk
Although the role of germline telomerase and shelterin mutations in familial cancer may at first appear limited to small subsets of cancer patients, there is epidemiologic evidence supporting long telomere length itself as being associated with cancer risk. This has been shown for melanoma and lung adenocarcinoma as well as other cancers (74–76). Larger genome-wide association studies (GWAS) further assert these associations (77). GWAS are designed to identify common variants that play a role in disease risk (78), and while the associated single nucleotide polymorphisms (SNPs) may not in themselves be pathogenic, they may be in cis with genes that are. GWAS for melanoma, glioma, and CLL risk have all identified SNPs near telomere maintenance genes, including TERT, RTEL1, NAF1, and POT1 (79–82). In a meta-analysis of 372 GWAS data sets, SNPs near TERT were one of the most common recurrent findings in cancer studies (83). One important pattern emerges from examining these cancer GWAS. They show that the cancer-associated risk alleles are also the long-telomere alleles identified in telomere-length GWAS. A Danish study of more than 95,000 individuals found that long telomere–associated SNPs identified in GWAS were also associated with increased risk of cancers, especially melanoma and glioma (84). These data linking genetic variants with long telomere length, along with the data showing that long telomere length itself is cancer-associated, establish that genetically determined long telomere length is a risk factor for a subset of human cancers.
Differential effects of short telomere– and long telomere–associated alleles
Another set of analyses illustrates how differential effects of common alleles affect disease risk. An initial review of the data may show that hits from GWAS for leukocyte telomere length, IPF, and lung cancer converge on hits near telomere-related genes (Figure 2A). To better illustrate this, we will focus on SNPs near TERT, RTEL1, and NAF1, which were identified in studies on telomere length, IPF, and lung adenocarcinoma. For the TERT SNP rs2736100, which is likely the most commonly recurrent hit in cancer GWAS (83, 85), the A short telomere allele, which has a frequency of 0.5, is associated with IPF risk (77, 86), consistent with the known link between short telomeres and IPF risk (41). By contrast, the C allele, which is associated with long telomere length, is a recurrent hit in lung adenocarcinoma (77, 85, 87). SNPs near RTEL1 and NAF1 (rs755017 and rs7675998, respectively) follow similar differential effects, with the short telomere allele associated with IPF and the long telomere allele with lung adenocarcinoma (refs. 86, 87, and Figure 2B).
Figure 2. Shared SNPs identified in GWAS near telomere genes are associated with both telomere length and disease risk, but the directionality of the effect is allele-dependent.

(A) intersection of shared SNPs across GWAS for leukocyte telomere length, lung adenocarcinoma and idiopathic pulmonary fibrosis. The shared SNPs fall near telomere maintenance genes. The alleles for each SNP have differential effects on telomere length with the effect size shown on base pairs. rs2736100 is in intron 2 of TERT. rs755017 is 140 kb downstream of the RTEL1 transcription start site in exon 2. rs7675998 falls 40 kb upstream of the NAF1 transcription start site. (B) Schematic forest plot shows the odds ratio of disease risk with short and long telomere SNPs such as those shown in the table in A. Data in B are adapted with permission from JAMA Oncology (88).
A recent meta-analysis further illustrates the importance of telomere length extremes in disease risk. The study pooled 83 GWAS and collectively included data from 400,000 cases and 1 million controls (88). Among these various phenotypes, the disease that had the strongest association with short telomere SNPs was IPF. The additive effect of these common short telomere SNPs translated to an odds ratio of 10. This finding also underscores the existing literature linking a major subset of IPF risk to short telomere length (14, 15, 41, 89, 90). In contrast, long telomere SNPs were associated with cancers that we have considered here and elsewhere to be part of the long telomere syndrome spectrum, including melanoma and glioma (ref. 88 and Figure 2B).
Limited survival of cancer-prone mice with long telomeres
The evidence that long telomere length is cancer-predisposing has been well documented in vertebrate animal models. When the tumorigenic potential of oncogenes, such as overexpressed Myc or KrasG12D, was compared in short- and long-telomere mice, long-telomere mice invariably had a worse outcome, developing more aggressive tumors and showing decreased survival (refs. 91, 92, and Figure 3A). These adverse outcomes were seen in both telomerase–wild-type and -null long-telomere mice (Figure 3A). Similar patterns have also been seen in cancer-prone models in which cancers are inducible by loss of a tumor suppressor such as ApcMin or Ink4a (93, 94). These models contrast with the exception of Tp53+/– mice, which developed more tumors on the short telomere background (95). Since most humans are germline TP53-intact, and in light of the emerging observations that humans with short telomere syndrome have a relatively low risk of cancer, we believe the current models that are informed primarily on the basis of in vitro data may be overestimating the impact of short telomeres as a driver of genome instability and cancer in humans.
Figure 3. Long telomeres promote cancer-related mortality in mice and proposed mechanism for long-telomere melanomagenesis.
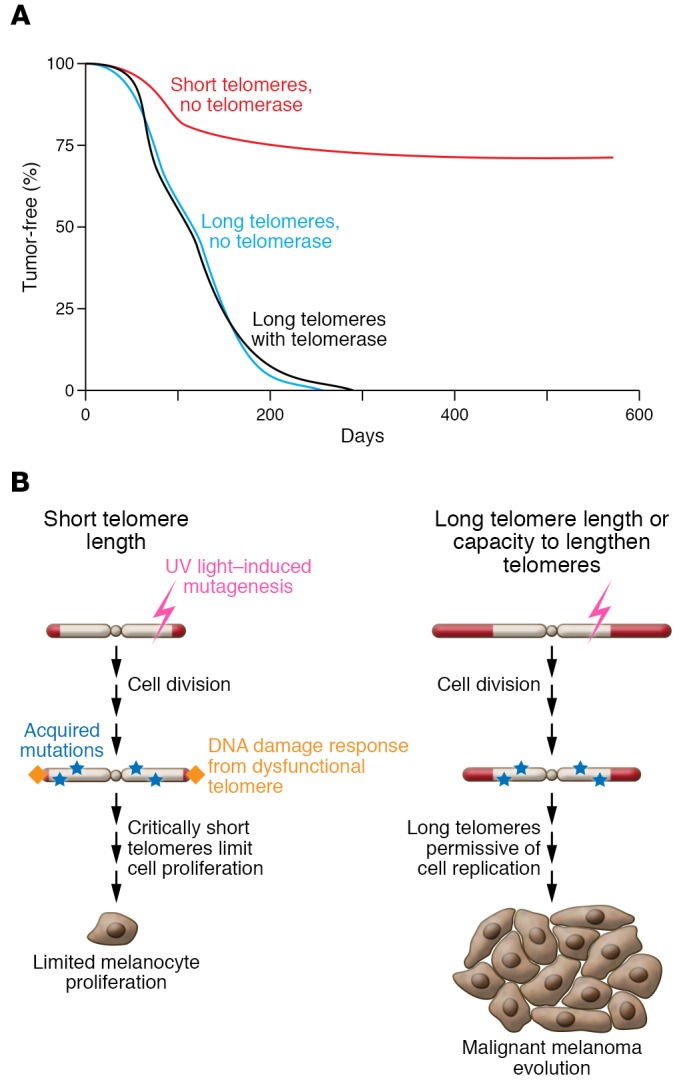
(A) Survival curve summarizing data from mouse models examining the role of telomerase and telomere length in cancer-related survival. It shows a survival advantage for short-telomere mice in a model of Myc-induced lymphoma, according to Feldser et al. (adapted with permission from Cancer Cell; ref. 91). (B) Schematic model for how long-telomere melanoma cells prone to environmentally induced DNA damage may have an advantage in cancer progression.
Longer telomeres in melanocytes may promote a cancer-prone state
The tight association between melanoma risk and long telomere length raises the question of whether there may be some tissue specificity in melanocytes. Melanocytes are highly vulnerable to ultraviolet-induced genotoxic damage. In this context, short telomeres may limit the proliferative potential of mutation-bearing melanocytes, while longer telomere length may be permissive for increased replicative potential. This in turn would allow the acquisition of additional genetic or epigenetic changes that would allow melanomagenesis. This model is clinically supported by the recent observations showing a high penetrance of cutaneous nevi in some POT1 mutation carriers (69). It would also explain the absence of any melanoma cases in patients with short telomere syndrome phenotypes.
Human genetic studies contextualize laboratory-based discoveries
Initial paradigms of the role of telomeres in cancer benefitted from foundational studies in simple organisms and cell-based models. Now, with the advent of new human genetic observations, there is an opportunity to integrate new data to refine the current understanding of the role of telomeres in cancer. Our synthesis of the recent body of work indicates that the risk of cancer susceptibility associated with long telomeres is greater than that associated with genetically determined short telomere length in humans. This observation is also supported by a large body of existing animal model data. The collective overview thus raises the question as to whether current models may be overestimating the role of short telomeres as a driver of human carcinogenesis. The opportunity to study the role of telomere length in human cancer is a prime example of how cancer biology is enriched and challenged by clinical observations. One final note regarding the role of long telomere length in cancer susceptibility relates to the commercial advertising of products that claim to lengthen telomeres for purposes of reversing or preventing aging. This discourse has limitations and does not have a rigorous scientific basis (96). The human genetic observations we reviewed here support the idea that excessively long telomeres do not equate with youth but rather with a capacity for cancer cells to grow unchecked with fewer brakes.
Acknowledgments
We are grateful to Carol Greider, Alexandra Pike, Dustin Gable, and other members of the Armanios laboratory for helpful comments and discussions. Work in the Armanios group is supported by NIH grants CA225027 and HL119476, the Maryland Cigarette Restitution Fund, the Commonwealth Foundation, the Gary Williams Foundation, and an S&R Foundation Kuno Award (to MA). We also acknowledge a gift in the name of P. Godrej. PJL received support from NIH T32CA009071.
Version 1. 08/05/2019
Electronic publication
Version 2. 08/09/2019
Fixed typo in abstract.
Version 3. 09/03/2019
Print issue publication
Footnotes
Conflict of interest: EJM is currently employed by and has stock options in SQZ Biotech, a cell therapy company.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(9):3474–3481.https://doi.org/10.1172/JCI120851.
References
- 1.Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978;120(1):33–53. doi: 10.1016/0022-2836(78)90294-2. [DOI] [PubMed] [Google Scholar]
- 2.Moyzis RK, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988;85(18):6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 4.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 pt 1):405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 5.Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51(6):887–898. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 6.Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59(3):521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 7.Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276(5312):561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- 8.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337(6205):331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 9.Feng J, et al. The RNA component of human telomerase. Science. 1995;269(5228):1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445(7127):506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 11.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012;492(7428):285–289. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol. 2013;14(2):69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley SE, Armanios M. The short and long telomere syndromes: paired paradigms for molecular medicine. Curr Opin Genet Dev. 2015;33:1–9. doi: 10.1016/j.gde.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armanios M, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102(44):15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armanios MY, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 16.Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M. The gastrointestinal manifestations of telomere-mediated disease. Aging Cell. 2013;12(2):319–323. doi: 10.1111/acel.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorgy AI, et al. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest. 2015;148(4):1019–1026. doi: 10.1378/chest.15-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner CL, et al. Short telomere syndromes cause a primary T cell immunodeficiency. J Clin Invest. 2018;128(12):5222–5234. doi: 10.1172/JCI120216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768–779. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 20.Alder JK, et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc Natl Acad Sci U S A. 2018;115(10):E2358–E2365. doi: 10.1073/pnas.1720427115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merck SJ, Armanios M. Shall we call them “telomere-mediated”? Renaming the idiopathic after the cause is found. Eur Respir J. 2016;48(6):1556–1558. doi: 10.1183/13993003.02115-2016. [DOI] [PubMed] [Google Scholar]
- 22.Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12(10):1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- 23.Greider CW. Telomerase RNA levels limit the telomere length equilibrium. Cold Spring Harb Symp Quant Biol. 2006;71:225–229. doi: 10.1101/sqb.2006.71.063. [DOI] [PubMed] [Google Scholar]
- 24.Xi L, Cech TR. Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res. 2014;42(13):8565–8577. doi: 10.1093/nar/gku560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cristofari G, Lingner J. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 2006;25(3):565–574. doi: 10.1038/sj.emboj.7600952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. 2009;85(6):823–832. doi: 10.1016/j.ajhg.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanley SE, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8(351):351ra107. doi: 10.1126/scitranslmed.aaf7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vulliamy T, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 31.Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002;101(4):335–341. doi: 10.1002/ijc.10593. [DOI] [PubMed] [Google Scholar]
- 32.Lee DD, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129(1):223–229. doi: 10.1172/JCI121303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castelo-Branco P, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14(6):534–542. doi: 10.1016/S1470-2045(13)70110-4. [DOI] [PubMed] [Google Scholar]
- 34.Kim NW, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 35.Hao LY, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123(6):1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 36.Aviv A. Genetics of leukocyte telomere length and its role in atherosclerosis. Mutat Res. 2012;730(1–2):68–74. doi: 10.1016/j.mrfmmm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greider CW. Regulating telomere length from the inside out: the replication fork model. Genes Dev. 2016;30(13):1483–1491. doi: 10.1101/gad.280578.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aubert G, Baerlocher GM, Vulto I, Poon SS, Lansdorp PM. Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PLoS Genet. 2012;8(5):e1002696. doi: 10.1371/journal.pgen.1002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1(5):2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 40.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107(1):67–77. doi: 10.1016/S0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 41.Alder JK, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popescu I, et al. Impaired cytomegalovirus immunity in idiopathic pulmonary fibrosis lung transplant recipients with short telomeres. Am J Respir Crit Care Med. 2019;199(3):362–376. doi: 10.1164/rccm.201805-0825OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607–5611. doi: 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170(4):457–471. doi: 10.1111/bjh.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanley SE, Merck SJ, Armanios M. Telomerase and the Genetics of Emphysema Susceptibility. Implications for pathogenesis paradigms and patient care. Ann Am Thorac Soc. 2016;13(suppl 5):S447–S451. doi: 10.1513/AnnalsATS.201609-718AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanley SE, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest. 2015;125(2):563–570. doi: 10.1172/JCI78554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silhan LL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014;44(1):178–187. doi: 10.1183/09031936.00060014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mai PL, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122(23):3673–3681. doi: 10.1002/cncr.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5(7):577–584. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- 51. doi: 10.1101/435958. Posted on bioRxiv October 5, 2018. [DOI]
- 52.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 53.Bodnar AG, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 54.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 55.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mancini A, et al. Disruption of the β1L isoform of GABP reverses glioblastoma replicative immortality in a TERT promoter mutation-dependent manner. Cancer Cell. 2018;34(3):513–528.e8. doi: 10.1016/j.ccell.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell RJ, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348(6238):1036–1039. doi: 10.1126/science.aab0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harland M, et al. Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer. 2016;15(1):139–144. doi: 10.1007/s10689-015-9841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robles-Espinoza CD, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet. 2014;46(5):478–481. doi: 10.1038/ng.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi J, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46(5):482–486. doi: 10.1038/ng.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Potrony M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. doi: 10.1111/bjd.17443. doi: 10.1111/bjd.17443. [published online ahead of print November 19, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aoude LG, et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst. 2015;107(2):dju408. doi: 10.1093/jnci/dju408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Speedy HE, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood. 2016;128(19):2319–2326. doi: 10.1182/blood-2016-01-695692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bainbridge MN, et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2015;107(1):384. doi: 10.1093/jnci/dju384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calvete O, et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat Commun. 2015;6:8383. doi: 10.1038/ncomms9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calvete O, et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur J Hum Genet. 2017;25(11):1278–1281. doi: 10.1038/ejhg.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chubb D, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. 2016;7:11883. doi: 10.1038/ncomms11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMaster ML, et al. Germline mutations in protection of telomeres 1 in two families with Hodgkin lymphoma. Br J Haematol. 2018;181(3):372–377. doi: 10.1111/bjh.15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong K, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. doi: 10.1001/jamadermatol.2018.3662. doi: 10.1001/jamadermatol.2018.3662. [published online ahead of print December 26, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rachakonda S, et al. Telomere length, telomerase reverse transcriptase promoter mutations, and melanoma risk. Genes Chromosomes Cancer. 2018;57(11):564–572. doi: 10.1002/gcc.22669. [DOI] [PubMed] [Google Scholar]
- 71.Kocak H, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014;28(19):2090–2102. doi: 10.1101/gad.248567.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo Y, et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124(18):2767–2774. doi: 10.1182/blood-2014-08-596445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ. The telomerase database. Nucleic Acids Res. 2008;36(Database issue):D339–D343. doi: 10.1093/nar/gkm700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seow WJ, et al. Telomere length in white blood cell DNA and lung cancer: a pooled analysis of three prospective cohorts. Cancer Res. 2014;74(15):4090–4098. doi: 10.1158/0008-5472.CAN-14-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lan Q, et al. Longer telomere length in peripheral white blood cells is associated with risk of lung cancer and the rs2736100 (CLPTM1L-TERT) polymorphism in a prospective cohort study among women in China. PLoS One. 2013;8(3):e59230. doi: 10.1371/journal.pone.0059230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han J, et al. A prospective study of telomere length and the risk of skin cancer. J Invest Dermatol. 2009;129(2):415–421. doi: 10.1038/jid.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Codd V, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45(4):422–427e1. doi: 10.1038/ng.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Visscher PM, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ptaszek LM, Saldana F, Palacios IF, Wu SM. Platypnea-orthodeoxia syndrome in two previously healthy adults: a case-based review. Clin Med Cardiol. 2009;3:37–43. doi: 10.4137/cmc.s2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Speedy HE, et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2014;46(1):56–60. doi: 10.1038/ng.2843. [DOI] [PubMed] [Google Scholar]
- 81.Barrett JH, et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43(11):1108–1113. doi: 10.1038/ng.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Law MH, et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet. 2015;47(9):987–995. doi: 10.1038/ng.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell. 2012;11(5):727–731. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rode L, Nordestgaard BG, Bojesen SE. Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol. 2016;45(5):1634–1643. doi: 10.1093/ije/dyw179. [DOI] [PubMed] [Google Scholar]
- 85.Landi MT, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85(5):679–691. doi: 10.1016/j.ajhg.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fingerlin TE, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McKay JD, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126–1132. doi: 10.1038/ng.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Telomeres Mendelian Randomization Collaboration, et al. Association between telomere length and risk of cancer and non-neoplastic diseases: a mendelian randomization study. JAMA Oncol. 2017;3(5):636–651. doi: 10.1001/jamaoncol.2016.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alder JK, et al. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat. 2013;34(11):1481–1485. doi: 10.1002/humu.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147(5):1361–1368. doi: 10.1378/chest.14-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11(5):461–469. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perera SA, et al. Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis. 2008;29(4):747–753. doi: 10.1093/carcin/bgn050. [DOI] [PubMed] [Google Scholar]
- 93.Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28(2):155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 94.Greenberg RA, et al. Short dysfunctional telomeres impair tumorigenesis in the INK4a(δ2/3) cancer-prone mouse. Cell. 1999;97(4):515–525. doi: 10.1016/S0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 95.Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406(6796):641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 96.Armanios M. Telomeres in the clinic, not on TV. Mayo Clin Proc. 2018;93(7):815–817. doi: 10.1016/j.mayocp.2018.05.024. [DOI] [PubMed] [Google Scholar]