

Abstract
Cyclooxygenase 2 (Cox2) total knockout and myeloid knockout (MKO) mice develop Crohn’s-like intestinal inflammation when fed cholate-containing high-fat diet (CCHF). We demonstrated that CCHF impaired intestinal barrier function and increased translocation of endotoxin, initiating TLR/MyD88-dependent inflammation in Cox2-KO but not WT mice. Cox2-MKO increased proinflammatory mediators in LPS-activated macrophages, and in the intestinal tissue and plasma upon CCHF challenge. Cox2-MKO also reduced inflammation resolving lipoxin A4 (LXA4) in intestinal tissue, whereas administration of an LXA4 analog rescued disease in Cox2-MKO mice fed CCHF. The apolipoprotein A-I (APOA1) mimetic 4F mitigated disease in both the Cox2-MKO/CCHF and piroxicam-accelerated Il10–/– models of inflammatory bowel disease (IBD) and reduced elevated levels of proinflammatory mediators in tissue and plasma. APOA1 mimetic Tg6F therapy was also effective in reducing intestinal inflammation in the Cox2-MKO/CCHF model. We further demonstrated that APOA1 mimetic peptides (a) inhibited LPS and oxidized 1-palmitoyl-2-arachidonoyl-sn-phosphatidylcholine–dependent (oxPAPC-dependent) proinflammatory responses in human macrophages and intestinal epithelium, and (b) directly cleared proinflammatory lipids from mouse intestinal tissue and plasma. Our results support a causal role for proinflammatory and inflammation-resolving lipids in IBD pathology and a translational potential for APOA1 mimetic peptides for the treatment of IBD.
Keywords: Gastroenterology
Keywords: Eicosanoids, Inflammatory bowel disease, Lipoproteins
Introduction
Inflammatory bowel disease (IBD), inclusive of Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic and relapsing inflammatory disorder of the gastrointestinal tract. IBD consists of a dysregulated mucosal immune response to gut microbiota, brought about by host and/or environmental triggers in genetically susceptible individuals (1). However, there exists no single pathogenic mechanism for IBD. Several genetic susceptibility loci have been identified for IBD (2). The candidate genes broadly implicate several distinct functional categories, including epithelial barrier function, innate immunity, T cell signaling, and autophagy; but even within a category, the underlying pathways are largely distinct (2, 3). The range of environmental factors associated with the development of IBD is also complex (4). In the absence of a single pathogenic mechanism, investigation into the etiology and pathogenesis of IBD requires careful elucidation of the disparate causal contributions of the many susceptibility-linked genes and disease-associated environmental factors.
IBD exhibits high morbidity, but there are currently few effective pharmacological interventions. For example, between 70% and 90% of all patients with CD eventually undergo surgery within their lifetime, and approximately 40% will require repeated surgery (5). Anti-TNF therapies including both infliximab and adalimumab offer a treatment option for IBD that is refractory to conventional therapies, including corticosteroids and immunomodulators (6). However, treatment failures with anti-TNF therapies are common (7). There exists a pressing need for new IBD therapies.
Cyclooxygenase 2 (COX2) is the inducible form of cyclooxygenase and is responsible for the rate-limiting step in the conversion of arachidonic acid to prostanoids (prostacyclins, prostaglandins, and thromboxanes). Prostanoids have been well-characterized as proinflammatory mediators, and their biosynthesis is blocked by nonsteroidal antiinflammatory drugs, including selective COX2 inhibitors. However, COX2 can play a role in the resolution of inflammation, partially through its induction of the proresolving eicosanoid lipoxin A4 (LXA4) (8, 9).
Multiple genetic susceptibility studies have linked COX2 to an altered risk of developing IBD (2, 10, 11), but the exact nature of the link is not clear due to conflicting associations when utilizing data from COX2 polymorphisms. Nevertheless, it has been shown that COX2 and COX2-dependent prostaglandin E2 (PGE2) help maintain adaptive immune tolerance to dietary antigens in the proximal small intestine (12, 13). Consistent with the observations of Newberry et al., we previously reported that both Cox2 total knockout (Cox2-TKO) and Cox2 myeloid-specific knockout (Cox2-MKO) mice or COX2-inhibited (COX2i) mice develop severe CD-like inflammation in their ileo-ceco-colic junctions when challenged with a cholate-containing or cholate-containing high fat diet (CCHF) (14, 15). Our prior findings suggested that COX2 plays an antiinflammatory role at the ileo-ceco-colic junction in mice. In the present study, we investigated the molecular mechanisms by which COX2 deficiency can contribute to the development of IBD.
Apolipoprotein A-I (APOA1) mimetic peptides, including both 4F and 6F, are short (18 amino acids) sequences that recapitulate the secondary structure and partial function of APOA1, the main structural protein of high density lipoprotein (HDL) (16). 4F and transgenic 6F (Tg6F; extract from tomatoes expressing the 6F peptide) are protective in numerous animal models of inflammatory diseases, including atherosclerosis (16). We previously reported that circulating 4F selectively targets the small intestine, where it is transported into the intestinal lumen, reabsorbed by the intestinal mucosa, and mediates the trans-intestinal efflux of cholesterol (17). We also demonstrated that both orally and subcutaneously administered 4F reduce the levels of proinflammatory fatty acid metabolites in enterocytes of low-density lipoprotein receptor–null (LDLR-null) mice on Western diet (18). We thus sought further to determine whether orally administered 4F and Tg6F could inhibit the development of intestinal inflammation in animal models of IBD including the Cox2-KO/CCHF model.
Our results indicate that (a) myeloid COX2 generates an antiinflammatory/proresolving lipid and proinflammatory check on TLR-dependent mural inflammation that can drive intestinal inflammation in IBD when compromised; (b) proinflammatory lipids play a causal role in IBD pathology, while antiinflammatory/proresolving lipids play a protective role; and (c) APOA1 mimetic peptides hold promise as therapies for IBD, in part through their capacities to inhibit both LPS and oxidized 1-palmitoyl-2-arachidonoyl-sn-phosphatidylcholine (oxPAPC) signaling and to promote luminal clearance of mural proinflammatory lipids.
Results
Dysregulated response to TLR-dependent signaling drives intestinal inflammation in the Cox2-KO/CCHF models of IBD.
Consistent with our prior reports (15), Cox2-TKO and COX2i but not control mice developed severe chronic inflammation in their ileo-ceco-colic junctions when fed CCHF for 2 weeks (Figure 1A). Disease was characterized by inflammatory lesions in both the submucosa and muscularis propria, sporadic foci of mucosal ulceration, and thickening of the intestinal wall.
Figure 1. Dysregulated response to TLR-dependent signaling drives intestinal inflammation in the COX2-KO/CCHF models of IBD.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A) Mice treated with Cox2-TKO (upper panel) and BL6 + celecoxib (CX) (lower panel) develop transmural and sporadically ulcerating inflammation in their ileo-ceco-colic junctions when fed CCHF for 2 weeks (representative images). Scale bars: 250 μm. (B) C57BL/6J mice fed CCHF diet with or without CX were assessed at multiple time points (n = 4 mice/group/time point). CCHF significantly increased whole intestinal barrier permeability over the course of 0–7 days independently of CX (upper left panel). Endotoxin in portal vein serum significantly increased in both groups by 7 days (lower left panel). Inflammation significantly increased in CX-treated mice by 11 days (upper right panel), whereas epithelial damage did not significantly increase until day 14 (lower right panel) (histological assessments, 0–4 point scale). (C) Mice were pretreated with vancomycin, ampicillin, neomycin, and metroniadazole for 7 days and continued on antibiotics (AB) for 14 days of CCHF or CCHF + CX (n = 4/group). AB abrogated intestinal inflammation in mice treated with COX2-TKO (upper panel) and mice treated with C57BL6 + CX (BL6 + CX) (lower panel), as assessed by ileo-ceco-colic thickness. (D) Cox2-TKO and WT mice were treated 3 times per week i.p. with the MyD88 inhibitor T6167923 (0.25 mg/injection) over the course of 2 weeks on CCHF (n = 5/group). MyD88 inhibition (MyD88i) significantly inhibited intestinal inflammation in Cox2-TKO mice. (E) Cox2-MKO significantly enhanced RNA expression of Il1b (upper panel) and Tnf (lower panel) in LPS-activated BMDMs compared with FLOX controls (n = 3/group) (fold change vs. FLOX, 0 hours) (statistics shown only for FLOX vs. MKO). Two-way ANOVA and Tukey’s multiple comparisons test with adjusted P values were used for statistical analyses.
Bile acids pose a risk of cellular toxicity (19), while disrupted barrier function (20) or epithelial damage itself (21) can cause intestinal inflammation. We thus sought to determine if altered barrier function or epithelial damage preceded the development of inflammation in the COX2 models. C57BL/6J (BL6) mice were fed CCHF or CCHF with the COX2 inhibitor celecoxib (CX). We assessed intestinal epithelial damage (22), changes in whole intestinal barrier permeability (23), translocation of endotoxin into portal vein serum, and ileo-ceco-colic inflammation over 2 weeks (Figure 1B). CCHF significantly increased whole intestinal permeability equally in both CX-treated and control mice, as assessed by urinary excretion of sucralose (Figure 1B). We also observed that CCHF comparably increased intestinal permeability in wild-type (WT) and Cox2-TKO mice (Supplemental Figure 1), indicating that off-target effects of celecoxib had not confounded our prior result (24).
The increase in permeability was accompanied by a significant increase in endotoxin translocated into portal vein serum in CX-treated and control mice (Figure 1B). The increases in both permeability and endotoxin preceded histological evidence of inflammation in the ileo-ceco-colic junctions of CX-treated mice (Figure 1B). Nonetheless, despite similar increases in both permeability and endotoxin levels, CX-treated but not control mice developed severe intestinal inflammation. Epithelial ulceration was observed in CX-treated mice at day 14 only (Figure 1B), after the initiation of inflammation in these mice. Since ulceration followed the development of inflammation, ulceration itself was likely a consequence rather than a cause of the inflammation.
We pursued the hypothesis that CCHF initiates intestinal inflammation by causing loss of barrier function and translocation of pathogen-associated molecular patterns (PAMPs), including lipopolysaccharide (LPS). We thus determined the dependence of the intestinal inflammation on gut microbiota. We pretreated Cox2-TKO and BL6 mice for 1 week with antibiotics (AB) to eliminate intestinal bacteria (25). We challenged the mice with CCHF or CCHF + CX for 2 weeks, while continuing AB. AB treatment abrogated inflammation in the ileo-ceco-colic junctions of Cox2-TKO and CX-treated mice, as assessed by gross pathology (Figure 1C) and histological disease score (Supplemental Figure 2). (For this and subsequent histological measures of disease, we employed a 0–12 point scoring system that incorporated assessments of both inflammation and epithelial damage/altered mucosal architecture; see Supplemental Methods.) To exclude the possibility that AB treatment inhibited inflammation by blocking the bacterial conversion of cholate to the more cytotoxic deoxycholate (26), we determined the effects of cholate and deoxycholate on barrier permeability in AB-treated TKO mice, which treatment prevented the conversion of cholate to deoxycholate. Cholate and deoxycholate significantly increased barrier permeability, but we observed no difference between their effects (Supplemental Figure 3).
LPS induces the production of proinflammatory mediators by stimulating the LBP/CD14/MD-2/TLR4 cell surface formation. This complex then signals through the adaptor protein MyD88 to trigger the nuclear translocation of NFκB and the transcription of proinflammatory target genes (27). We inhibited MyD88 with the small molecule T6167923 (28) in Cox2-TKO mice fed CCHF. As with AB treatment, MyD88 inhibition abrogated gross pathological (Figure 1D) and histological (Supplemental Figure 4) evidence of ileo-ceco-colic inflammation.
If LPS/PAMP translocation and TLR/MyD88 signaling initiate disease in our models, there must exist a differential response to bacteria-dependent TLR ligands in COX2-deficient versus control mice. Cox2 myeloid conditional knockout (MKO) mice (Cox2fl/fl LysMcre/+) developed ileo-ceco-colic inflammation comparable to that of Cox2-TKO and COX2i mice, upon 7- or 10-week CCHF (Supplemental Figure 5). Elevated levels of neutrophils and macrophages but not CD4+ or CD8+ T cells characterized the inflammatory lesions (Supplemental Figure 6) (15). CCHF significantly increased intestinal barrier permeability through 3 weeks in Cox2-MKO and floxed control (FLOX) mice as assessed by FITC-dextran 4 kD (29) (Supplemental Figure 7). The LysM promoter of these mice is expressed in almost 100% of nonsplenic macrophages and approximately 60% of neutrophils (30). We conducted a pilot study using neutrophil-specific Cox2-KO mice (Cox2fl/fl Mrp8Cre/+) whose promotor is unexpressed in macrophages (30). Following 7 weeks of CCHF, we did not observe any evidence of ileo-ceco-colic inflammation (data not shown). As a result, we restricted our investigation into differential responses to LPS/PAMP to the macrophage compartment. We generated bone marrow–derived macrophages (BMDMs) from Cox2-MKO and FLOX mice (Supplemental Figure 8). We activated the macrophages with LPS and determined the expression of NFκB proinflammatory target genes using quantitative PCR (qPCR) for 24 hours. We had validated the loss of COX2 activity by measuring COX2 metabolites, including PGE2, PGD2, PGJ2, and 15dPGJ2, via liquid chromatography with tandem mass spectrometry (LC-MS/MS) (Supplemental Figure 9). We observed that the expressions of proinflammatory genes including Tnf and Il1b were significantly higher in Cox2-MKO BMDMs (Figure 1E). Dysregulation of TLR/MyD88-dependent proinflammatory signaling within the macrophage compartment could thus partially explain the differential response to CCHF between control and COX2-deficient mice.
Cox2-MKO mice fed CCHF exhibit elevated levels of proinflammatory lipid mediators in intestine and plasma, while the loss of inflammation-resolving LXA4 in intestine appears partially causal for disease.
Cox2-MKO enhanced the acute inflammatory response and inhibited its resolution in the carrageenan hind paw model (Supplemental Figure 10). COX2 can modulate the production of proresolving lipid mediators including LXA4 (8), an eicosanoid that promotes resolution in part by stimulating efferocytosis (31). Macrophage COX2 can directly mediate intracellular production of LXA4 through LPS-dependent generation of 15HETE (32). Loss of macrophage COX2 can also shunt arachidonic acid (AA) through the 5-lipoxygenase (5LOX) and 12- or 15-lipoxygenase (12/15LOX in mice) pathways, leading to the production of proinflammatory eicosanoids (32). We thus hypothesized that Cox2-MKO mice fed CCHF would exhibit higher proinflammatory and lower proresolving lipid mediator levels compared with FLOX controls.
We developed a multiple reaction monitoring LC-MS/MS method to measure a panel of proinflammatory, antiinflammatory, and proresolving lipid mediators (see Supplemental Methods for details). The method quantitatively evaluates most of the bioactive signaling metabolites of AA, DHA, and EPA in the COX and LOX pathways, together with pathway markers and stable end products (Table 1 and Supplemental Table 1). We validated the method for intestine, plasma, and cell culture lysate and media (Supplemental Tables 2–5 and Supplemental Figure 11).
Table 1. Bioactive lipid inflammatory mediators measured by LC-MS/MS either directly or via stable products.
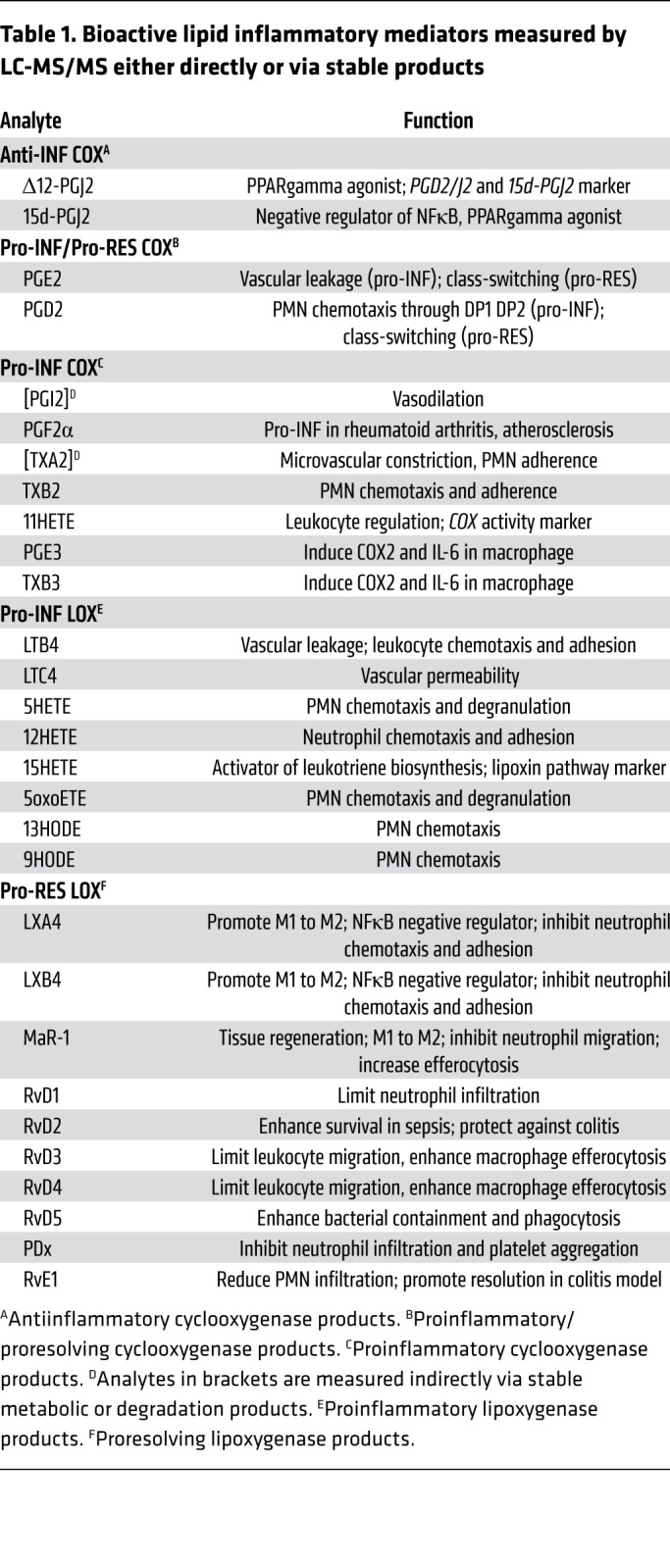
We fed Cox2-MKO and FLOX mice either chow or CCHF for 8.5 weeks then determined the levels of the above mediators in the ileo-ceco-colic junction and plasma (Figure 2, A and B, and Supplemental Tables 6 and 7). See Supplemental Tables 8–10 for application of Benjamini-Hochberg procedure (33) to all lipidomic data sets for the control of false discovery rate (FDR) at level α = 0.05. On CCHF, Cox2-MKO mice had significantly increased intestinal levels of several proinflammatory signals compared with FLOX controls (Figure 2C), including the proinflammatory LOX mediators 12HETE, 15HETE, and 13HODE, and the proinflammatory COX-dependent PGI2 degradation product 6ketoPGF1α. Cox2-MKO fed CCHF had significantly increased plasma levels of the proinflammatory LOX signals 12HETE, 5HETE, and 15HETE, and the proinflammatory COX signal PGF2α together with its degradation product 13,14-dihydro-15-PGF2α, and TXB2 (Figure 2D). In tissue and plasma, Cox2-MKO fed CCHF also had a significantly increased total level of all LOX and COX proinflammatory mediators. In plasma, PGE2 was also increased (Figure 2E).
Figure 2. Cox2-MKO (MKO) mice challenged with CCHF exhibit elevated levels of lipid proinflammatory mediators in intestine and plasma, whereas the loss of inflammation-resolving LXA4 in intestine appears partially causal for disease in MKO mice fed CCHF.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A–F) Lipid inflammatory mediators were determined by LC-MS/MS in the ileo-ceco-colic junctions (A) and plasmas (B) of MKO and FLOX mice fed CCHF or chow for 8.5 weeks (n = 5–7/group). All significant differences between MKO and FLOX on CCHF are presented: proinflammatory (Pro-INF) LOX and COX mediators in the intestines (C) and plasmas (D); PGE2, PGD2, and associated metabolites in both intestine (Int) and plasma (Pl) (E). Tt, total. In intestine, Cox2-MKO significantly reduced the levels of both the specialized proresolving mediator (SPM) LXA4 and total 5LOX SPM, together with the ratio of total 5LOX SPM to LTB4 + 6trans12epiLTB4 (Tt LTB4) (F). (G) A stable analog of LXA4 (BML111) administered twice weekly i.p. significantly inhibited markers of disease including ileo-ceco-colic thickness (n = 10/group) (left) and H&E score (n = 3–5/group) (right). For A–F, we used the Benjamini-Hochberg procedure applied to 1-way ANOVA for each lipidomic analyte with FDR at level α = 0.05, followed by Tukey’s multiple comparisons test with adjusted P values for statistical analyses. For G, we used a 1-way ANOVA with Tukey’s multiple comparisons test and adjusted P values.
We investigated whether these elevated proinflammatory signals in plasma might constitute biomarkers for IBD. In an independent experiment, we determined the levels of proinflammatory LOX mediators in plasma of Cox2-MKO and FLOX mice fed chow, and those fed CCHF for 3 or 8.5 weeks (Supplemental Figure 12). Proinflammatory LOX signals, including 12HETE and 5HETE, were significantly elevated in Cox2-MKO mice with 8.5 weeks CCHF. The differential plasma levels of these signals correlated with the degree of intestinal inflammation in these mice. We also observed that a significant increase in the systemic inflammation marker serum amyloid A (SAA) in FLOX mice did not associate with elevated signals (Supplemental Figure 13). These results suggest that the plasma levels of these proinflammatory LOX products possess at least prima facie specificity with respect to IBD. The significant (P < 0.0001) but modest increase in SAA in FLOX mice, which can be induced by IL-1 and IL-6 but also by LPS (34), provides further evidence of the otherwise modest effect of CCHF in normal mice.
Cox2-MKO also significantly reduced proresolving LOX signals in the ileo-ceco junctions of CCHF-fed mice (Figure 2F). Both LXA4 and the total of 5LOX specialized proresolving mediators (SPMs) (LX + RvD series) were significantly decreased. Moreover, the ratio of 5LOX-associated SPM to proinflammatory LTB4 and its product 6trans12epiLTB4 was significantly decreased (see 5LOX SPM/Tt LTB4 in Figure 2F).
We had observed the copresence of neutrophils and macrophages in the inflammatory lesions of Cox2-MKO mice fed CCHF (Supplemental Figure 6). This copresence suggested that the resolution of inflammation, which involves the efferocytosis of neutrophils by macrophages, failed (35). LXA4 can block neutrophil migration and stimulate efferocytosis (36). We thus hypothesized that the significant reduction (P < 0.05) of LXA4 in the ileo-ceco-colic junctions of Cox2-MKO + CCHF mice was partially causal for the chronic inflammation in these mice. We administered a stable analog of LXA4 (BML111) to Cox2-MKO mice across the course of an 8.5-week CCHF (37). BML111 treatment significantly improved several measures of intestinal inflammation, including gross pathology and histological disease score (Figure 2G). (See Supplemental Figure 14 for additional measures and representative images.)
The APOA1 mimetic peptides 4F and Tg6F inhibit the development of intestinal inflammation in the Cox2-MKO/CCHF model of IBD.
Gene expression of APOA1 is reduced in the ilea of patients with ileal or colonic CD (38). Colonic APOA1 is also protective in mouse models of UC (39). We determined both APOA1 protein and gene expression in the proximal colons and terminal ilea of MKO and FLOX mice fed CCHF or chow for 8.5 weeks (Supplemental Figure 15). Gene expression was significantly reduced in the proximal colons of Cox2-MKO mice fed CCHF.
We cotreated Cox2-MKO mice fed CCHF with the APOA1 mimetic D-4F (500 μg/mL drinking water) for 7 and 10 weeks. 4F protected against ileo-ceco-colic inflammation in Cox2-MKO mice at both time points. 4F treatment blocked the increase in ileo-ceco-colic thickness (Figure 3A), significantly improved disease score (Figure 3B), and blocked the increase in muscularis thickness (Figure 3C). 4F treatment significantly inhibited the influx of F4/80+ macrophages and Ly6G+ neutrophils into the ileo-ceco-colic junctions (Figure 3, D and E). 4F-treated mice also expressed significantly less Tnf but more Il10 in their ileo-ceco-colic junctions (Figure 3F).
Figure 3. APOA1 mimetics inhibit the development of intestinal inflammation in the Cox2-MKO and CCHF model of IBD.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A–F) Cox2-MKO (MKO) mice fed chow or CCHF for 7 and 10 weeks and treated with oral D-4F (500 μg/mL drinking water). (A) 4F significantly inhibited thickening of the ileo-ceco-colic junctions. (B) Representative images of MKO mice fed CHOW, CCHF, or CCHF + 4F for 10 weeks (left) (scale bars, 250 μm); 4F significantly inhibited the H&E disease score (0–12 points) (right). (C) 4F significantly reduced CCHF-dependent cecal muscularis thickening. (D–E) 4F treatment significantly reduced infiltration of macrophages (F4/80+) (D) and neutrophils (Ly6G+) (E) into the ileo-ceco-junctions of MKO mice on CCHF diet (left panels, representative images at 7 weeks; scale bars, 200 μm). (F) 4F significantly altered expression of Tnf and Il10 in the ileo-ceco-colic junctions of MKO mice fed CCHF for 7 weeks. (G–H) MKO mice fed CCHF for 10 weeks were treated with Tg6F for the full 10 weeks or the last 5 weeks. Both treatments significantly suppressed ileo-ceco-colic thickening (G) while also improving histopathology (H; representative images; scale bars, 200 μm). For A–C, and G we used 1-way ANOVA with Tukey’s multiple comparisons test and adjusted P values for statistical analyses. For D–F, we used Student’s t tests with Holm-Sidak correction for multiple tests and adjusted P values.
To determine whether the protective effect of 4F in this model extended to other APOA1 mimetic peptides, we cotreated Cox2-MKO mice fed CCHF with 0.06% Tg6F (wt/wt) for 10 weeks (40). To assess whether Tg6F could inhibit the progression of existing disease, we delayed the start of treatment with Tg6F in another group until the mice had been fed CCHF for 5 weeks. Both early and late onset Tg6F therapy rescued gross pathology and improved histology (Figure 3, G and H and Supplemental Figure 16). These results suggest that APOA1 mimetic peptides as a class may be protective against intestinal inflammation in the Cox2-KO/CCHF model of IBD even after the disease has been well established.
4F treatment improves the balance of proinflammatory and inflammation-resolving lipid mediators in Cox2-MKO mice fed CCHF, while suppressing the proinflammatory phenotype of LPS-treated macrophages.
We determined the effect of oral D-4F therapy on the levels of lipid inflammatory mediators in the ileo-ceco-colic junctions and plasmas of Cox2-MKO mice fed CCHF for 8.5 weeks (Figure 4, A and B; Supplemental Table 6 and Supplemental Table 7). 4F treatment rescued the intestinal levels of total proinflammatory LOX products as well as 12HETE and 13HODE (Figure 4C). In plasma, 4F significantly reduced the level of every proinflammatory LOX mediator elevated by Cox2-MKO together with most additional mediators in this class (Figure 4D). 4F also significantly reduced in plasma most proinflammatory COX mediators as well as PGE2 (Figure 4E).
Figure 4. 4F treatment improves the inflammatory mediator profile of mouse intestinal tissue and plasma.

*P < 0.05; **P < 0.01; ***P < 0.001. (A–E) The effect of 4F on the intestinal and plasma lipid mediator profile of Cox2-MKO (MKO) and FLOX mice fed chow or CCHF for 8.5 weeks was determined by LC-MS/MS (n = 5–7/group). Heatmaps of lipid mediator changes in intestine (A) and plasma (B) are shown. Significant reductions of intestinal proinflammatory LOX (C) and plasma proinflammatory LOX (D) and COX (D) mediators. Tt, total. Statistical analyses were conducted using the Benjamini-Hochberg procedure applied to 1-way ANOVA for each lipidomic analyte with FDR at level α = 0.05, followed by Tukey’s multiple comparisons test with adjusted P values.
We determined the effect of 4F on plasma paraoxonase-1 (PON1) activity as a measure of HDL and on plasma total cholesterol (TC) (Supplemental Figure 17) (41). 4F significantly reduced TC, consistent with our prior reports that 4F can enhance the trans-intestinal efflux of cholesterol (17). 4F also significantly improved PON1 activity. We last determined the effect of 4F on broad classes of plasma lipids in Cox2-MKO mice fed CCHF. We employed ion mobility MS/MS shotgun lipidomics (42) through the SCIEX Lipidyzer platform to determine the concentrations of 1103 lipid species from 13 lipid classes. We observed significant reductions in total triacylglycerols, diacylglycerols, cholesterol esters, and lyso-phosphatidylethanolamines (Supplemental Figure 18).
Last, we investigated the protective mechanism of 4F by asking whether 4F could inhibit the proinflammatory effects of LPS on mouse macrophages. We cotreated LPS-activated BMDMs with 4F (15 μg/mL) and assessed the expression of NFκB proinflammatory target genes. 4F significantly inhibited expression of Il1b (Figure 5A) and Tnf (Figure 5B) in Cox2-MKO and FLOX BMDMs for 24 hours. We also assessed the effect of 4F on lipid inflammatory mediators in BMDM cell lysates. 4F significantly reduced the levels of LPS-induced COX metabolites including PGE2 (Figure 5C) and PGD2 (Figure 5D). Comparing the mono-hydroxylated COX and LOX AA-metabolites 11HETE and 15HETE, respectively, we observed that 4F treatment significantly lowered the levels of 11HETE but not 15HETE (Figure 5E). This differential effect on structurally comparable lipids suggests that 4F did not lower the levels of COX metabolites by binding and clearing these lipid species. Rather, these results altogether suggest that 4F inhibits the effects of LPS by directly interfering with LPS-dependent signaling.
Figure 5. 4F treatment improves the inflammatory mediator profile of mouse macrophages.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A–C) LPS-activated (25 ng/mL) BMDMs from FLOX and MKO mice were cotreated with D-4F (15 mg/mL) for 24 hours (n = 3/group). 4F significantly suppressed Il1b (A) and Tnf (B) expression in both FLOX and MKO BMDMs (fold change vs. 0 hours). LPS significantly increased prostanoid production in FLOX but not MKO mice, whereas 4F significantly reduced the levels of PGE2 (C) and PGD2 (D) in FLOX lysate from 10 hours. In FLOX lysate, 4F also selectively suppressed the COX pathway marker 11HETE without reducing the lipoxygenase product 15HETE (E). Statistical analyses were conducted as follows: (A, B) For each of the FLOX or MKO mice with or without 4F, 2-way ANOVA with Tukey’s multiple comparisons test and adjusted P values were used. (C, D) Three-way ANOVA with Tukey’s multiple comparisons test and adjusted P values were used. (E) For each of the 2 analytes, 2-way ANOVA with Tukey’s multiple comparisons test and adjusted P values were used.
4F inhibits the LPS-mediated proinflammatory response of human macrophages and intestinal epithelium.
We stimulated human THP-1 macrophages with LPS and cotreated with 4F for 24 hours. As with mouse macrophages (Figure 5), we observed that 4F significantly inhibited the induction of PGE2 (Figure 6A) and other prostanoids (Supplemental Figure 19). We examined by Western blot the LPS-dependent degradation of the NFκB inhibitor IκBα over the course of 60 minutes. 4F significantly prevented the degradation of IκBα at both 20 and 30 minutes (Figure 6B and Supplemental Figure 20; see complete unedited blots in the supplemental material). We obtained comparable results using Cox2-MKO peritoneal macrophages (data not shown). Macrophages do not internalize 4F (GM Anantharamaiah, unpublished observations), and 4F can bind to oxidized and nonoxidized fatty acids with high affinity (43). We hypothesized that 4F inhibits the LPS-dependent activation of the NFκB pathway by binding to the lipid A moiety of LPS, thereby preventing lipid A from activating LBP/CD14/MD-2/TLR4. We employed surface plasmon resonance analysis to determine the binding affinity between 4F and both LPS and lipid A (Figure 6C). 4F bound to both with high affinity (KD = approximately 1 nM and 17 nM, respectively), supporting our hypothesis.
Figure 6. 4F inhibits the LPS-mediated proinflammatory response of human macrophages and intestinal epithelium.

(*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A–B) Human THP1 macrophages were treated with 4F (15 μg/mL), LPS (20 ng/mL), or LPS + 4F. (A) 4F significantly inhibited total PGE2 in cell lysates and media for 24 hours (n = 3/group). (B) 4F (4) significantly inhibited LPS dependent (L) IκBα degradation at 30 minutes, as determined by Western blot (NT, no treatment) (left: representative blot; right: densitometric analysis of 3 experiments) (noncontiguous sections of same blots). (C) 4F binds both LPS (left; KD = 0.86 nM) and lipid A (right; KD = 17.5 nM) with high affinity, as determined by surface plasmon resonance analysis. (D) Crypts were isolated from human small intestine and grown into enteroids in matrigel (representative image; scale bar: 200 μm). (E–I) Conditioned media from THP-1 cells treated with LPS or LPS + 4F for 4 hours (LPS 4h; LPS + 4F 4h) or 12 hours (LPS 12h; LPS + 4F 12h) was added to the enteroids, and proinflammatory gene expression at 12 hours was determined by qPCR (n = 3/group; Media indicates THP1 media only; fold change vs. NT). For statistical analyses, 1-way (B, E) or 2-way (A) ANOVA with Tukey’s multiple comparisons test and adjusted P values were used.
We next investigated the effect of 4F on the LPS-dependent proinflammatory response of human intestinal epithelium ex vivo. We isolated small intestinal crypts from human surgical samples and cultured them in Matrigel (see Supplemental Methods). Under these conditions, the crypts expand into small intestinal epithelial structures known as enteroids (Figure 6D) that recapitulate intestinal epithelial physiology (44). We examined the way that small intestinal epithelium might amplify the proinflammatory response of macrophages. We collected conditioned media (CM) from THP-1 macrophages that had been treated with LPS or LPS + 4F for either 4 or 12 hours. We exposed the human enteroids to CM for 12 hours and determined proinflammatory gene expression by qPCR. CM from LPS-activated macrophages significantly induced gene expression of TLR4, COX2, IL1B, IL6, and TNF, with 12-hour CM significantly increasing expression over 4-hour CM (Figure 6, E–I). By contrast, CM from macrophages treated with LPS + 4F did not increase expression of these genes. The magnitude of the proinflammatory response of the enteroids to CM from LPS-activated macrophages highlights the potential amplifying effect of proinflammatory stimuli in the lamina propria by intestinal epithelium. The degree of the inhibition of this epithelial response by 4F suggests a protective mechanism by which 4F not only inhibits the primary proinflammatory response of resident macrophages but also interrupts the positive feedback loops that propagate and amplify this response within the intestinal mucosa.
OxPAPC is elevated in the plasma and ceca of Cox2-MKO + CCHF mice, while 4F reduces oxPAPC in vivo and inhibits the oxPAPC-dependent proinflammatory response of human macrophages.
Lipid peroxidation markers are elevated in plasma (45) and affected intestinal tissue (46) of patients with CD, and lipid peroxides may be pathogenic for CD (47). POVPC, PGPC, and KOdiAPC are all phospholipids in which the polyunsaturated arachidonoyl group at the sn2 position of PAPC oxidatively truncates following a peroxidation step at one of its double bonds (48). Because PAPC is a common and representative phospholipid, these oxPAPC species are considered biomarkers of lipid peroxidation (49).
We determined the levels of POVPC, PGPC, and KOdiAPC (Supplemental Methods) in the ileo-ceco-colic junction (Figure 7A) and plasma (Figure 7B) of Cox2-MKO and FLOX mice fed chow or 8.5 wk CCHF. We observed that these species were significantly elevated in both compartments of Cox2-MKO mice fed CCHF. We further determined oxPAPC levels in the proximal colon and terminal ileum of these mice, as well as in the ileo-ceco-colic junctions of FLOX and Cox2-MKO mice fed CCHF for 3 weeks. Elevated oxPAPC levels correlated both spatially and temporally with advanced intestinal inflammation (Supplemental Figure 21). In contrast to human CD, the increase in oxPAPC species did not associate with an increase in expression of dual oxidase 2 (Duox2) in the terminal ileum or proximal colon (Supplemental Figure 22) (38).
Figure 7. Oxidized PAPC is elevated in the plasma and ceca of Cox2-MKO and CCHF-fed mice, whereas 4F both reduces oxidized PAPC in vivo and inhibits oxidized PAPC-dependent proinflammatory response in human macrophages.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Levels of oxPAPC products (POVPC, PGPC, and KOdiAPC) were determined in the ceca (n = 7) (A) and plasmas (n = 10) (B) of Cox2-MKO (MKO) and FLOX mice fed CCHF or chow for 8.5 weeks. Independently, the effect of 4F on oxPAPC in the ceca (C) and plasma (D) of MKO and FLOX mice fed CCHF for 3 weeks was determined. Last, human THP-1 macrophages were treated with 1 μM POVPC for 3 hours with and without the apoA-I mimetic peptides 4F and 3F(14); and expression of IL1B was determined by qPCR (n = 3–6) (fold change vs. NT) (E) After 8.5 weeks, MKO + CCHF diet significantly increased oxPAPC species in both ceca and plasma. 4F significantly lowered levels of oxPAPC in vivo, and 4F but not 3F(14) significantly inhibited the proinflammatory effect of POVPC on macrophages. For each analyte in A–D, 2-way ANOVA with Tukey’s multiple comparisons test and adjusted P values was used. A 1-way ANOVA with Tukey’s multiple comparisons test and adjusted P values was used for E.
We investigated the effect of 4F on these oxPAPC species in both Cox2-MKO and FLOX mice fed CCHF. After only 3 weeks of therapy, oral D-4F had significantly reduced oxPAPC in the cecum and plasma (Figure 7, C and D) of both groups.
Oxidized PAPC species are themselves biologically active and can be proinflammatory (48, 50). We considered the hypotheses that oxPAPC helps amplify intestinal inflammation, while 4F inhibits this effect. POVPC significantly increased IL1B gene expression in THP-1 macrophages by 3 hours (Figure 7E). 4F cotreatment blocked this induction, while 3F(14) — an 18 amino acid peptide that lacks the affinity of 4F for oxidized lipids (43) — did not.
4F can directly clear lipid proinflammatory mediators from intestinal tissue and plasma.
4F reduced the levels of proinflammatory LOX but not COX mediators in Cox2-MKO mice fed CCHF (Figure 8A and Figure 4C). This selective reduction of disease-elevated mediators weighs against the possibility that 4F only indirectly lowers these mediators by otherwise inhibiting disease. We hypothesized that 4F could directly clear proinflammatory LOX mediators from intestinal tissue. We previously employed Ussing chambers to study the effect of 4F on trans-intestinal cholesterol efflux (TICE) ex vivo (17). We now mounted in Ussing chambers small intestinal explants from Cox2-MKO mice fed CCHF for 8.5 weeks. Mucosal/luminal side D-4F significantly increased the clearance into the mucosal media of most proinflammatory LOX mediators (Figure 8B).
Figure 8. 4F can directly clear lipid proinflammatory mediators from intestinal tissue and plasma.

*P < 0.05. (A) 4F fails to significantly suppress the disease-elevated inflammatory COX mediators in the intestinal tissue of Cox2-MKO (MKO) mice challenged for 8.5 weeks with CCHF, in contrast to its differential effect on disease-elevated inflammatory LOX mediators (see also Figure 4D). (B) Pairs of matching intestinal explants from MKO mice challenged for 8.5 weeks with CCHF (1 pair/mouse) were mounted in Ussing chambers. As determined by LC-MS/MS, lumen-side D-4F (50 mg/mL) significantly increased the pair-wise clearance of proinflammatory LOX mediators from the intestinal tissue into mucosal media. (C–D) Deuterated proinflammatory LOX mediators (15HETE-d, 12HETE-d, 13HODE-d) were combined with HDL and LDL (1 mg oxFA-d/[2.5 mg HDL + 5 mg LDL]) and added to the serosal sides of pairs of matching BL6 intestinal explants mounted in Ussing chambers. (C) There existed a baseline trans-intestinal transport of these tagged lipids (representative LC-MS/MS chromatogram). (D) Mucosal-side 4F (50 mg/mL) significantly increased the pair-wise trans-intestinal transport of both 13HODE-d and 15HETE-d (n = 3–6/group) For A, we used the Benjamini-Hochberg procedure applied to 1-way ANOVA for each lipidomic analyte with FDR at level α = 0.05, followed by Tukey’s multiple comparisons test and adjusted P values (see also Figure 2). For B and D, we used Student’s t tests with Holm-Sidak correction for multiple tests and adjusted P values.
We previously reported that proinflammatory LOX mediators associate with lipoproteins, including HDL (51, 52). We also demonstrated that 4F enhances TICE, while showing ex vivo that luminal 4F can increase the trans-intestinal efflux of lipoprotein-associated cholesterol (17). We now asked whether luminal 4F could also affect the trans-intestinal transport of lipoprotein-associated proinflammatory mediators. We combined deuterated 15HETE, 12HETE, and 13HODE with HDL and LDL and loaded the mixture into the serosal media of Ussing chambers, in which we mounted explants from the proximal small intestine. We determined by LC-MS/MS that trans-intestinal transport of these tagged lipids occurred even at baseline (Figure 8C). There thus may exist trans-intestinal transport of lipid species besides cholesterol in vivo. Further, mucosal-side 4F significantly increased the trans-intestinal transport of 15HETE-d and 12HETE-d (Figure 8D). Luminal 4F might directly enhance trans-intestinal lipid transport, and thereby possible clearance, of lipoprotein-associated inflammatory mediators.
4F treatment inhibits development of colitis in the piroxicam-accelerated Il10–/– model.
4F binds to cholesterol with high affinity (KD = approximately 11 nM) (43). We considered the possibility that 4F is protective in the Cox2-KO/CCHF models by binding and inactivating dietary cholate. We determined the binding affinity between D-4F and cholate, observing only low-affinity binding (Supplemental Figure 23). 4F also did not inhibit the early effect of CCHF on barrier permeability (Supplemental Figure 24), further indicating that 4F does not inactivate CCHF.
We nonetheless assessed the efficacy of 4F therapy in a second mouse model of IBD, the piroxicam accelerated (PAC) Il10–/– model (53). Il10–/– mice that are fed the NSAID piroxicam (PX) for 9 days develop colitis that is characterized by hyperplasia and focal transmural inflammation reminiscent of human CD. In the PAC Il10–/– model, disease is assessed at day 14 (5 days after PX) by determining colon length, colitis severity, and histopathological score (53).
Oral D-4F (500 μg/mL drinking) improved several markers of disease in PAC Il10–/– mice. PX significantly decreased colon length while 4F therapy rescued this effect (Figure 9A). Colitis severity was determined on a 4-point scale by assessing the thickening of the mucosa, vascularity, granularity, and feces consistency (53). 4F therapy significantly improved colitis disease score (Figure 9A). 4F therapy also significantly improved histological disease score, as assessed by the degree of lesions, hyperplasia, ulceration, and percentage of area involved (53) (Figure 9B).
Figure 9. 4F treatment inhibits the development of colitis in the piroxicam-accelerated Il10–/– model of IBD.

*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (A–B) Il10–/– mice were treated with and without piroxicam (PX) and 4F (n = 4–7/group). (A) 4F partially rescued the effect of PX on colon length and colitis score. (B) 4F also improved disease histopathology: representative H&E images (scale bars, 200 μm) and H&E disease score are shown. Green bars indicate thickness of mucosa in Il10–/– + PX. Red arrows indicate inflammatory foci. (C) PX increased both whole intestinal barrier permeability of Il10–/– mice in a biphasic manner, as determined by LC-MS/MS of urinary excretion of sucralose (left), as well as the translocation of endotoxin into portal vein serum at day 9 (right). (D) 4F inhibited the production of prostanoids including PGE2 in LPS-activated Il10–/– BMDMs with or without PX (n = 3/group). (E–F) The lipid inflammatory mediator profile in the colons of the mice shown in A and B was determined by LC-MS/MS (see Supplemental Table 8) (n = 4–8 mice/group). (E) Heatmap of changes. (F) All significant differences are represented. 4F significantly inhibited the disease-dependent increase of proinflammatory LOX mediators. Tt, total. Statistical analyses were as follows: 1-way (A, B), repeated measures 1-way (C, left), or 3-way (D) ANOVA with Tukey’s multiple comparisons test and adjusted P values; Student’s t test (C, right). For E–F, we used the Benjamini-Hochberg procedure applied to 1-way ANOVA for each lipidomic analyte with FDR at level α = 0.05, followed by Tukey’s multiple comparisons test and adjusted P values.
The PAC Il10–/– model depends on the presence of gut microbiota to initiate disease (53). Given the off-target epithelial toxicity of PX, it has been conjectured but not demonstrated that PX accelerates disease by impairing barrier function (54). We thus assessed the effect of PX on whole intestinal permeability in Il10–/– mice over the course of 9 days of PX challenge. We observed a biphasic response with an early increase in permeability (day 0–3) followed by a late increase (day 7–9) that coincides with increased disease (Figure 9C, left). We also observed increased endotoxin in portal vein serum at day 9 (Figure 9C, right).
4F inhibited the effect of LPS on Il10–/– peritoneal macrophages treated with PX (Figure 9D). We also determined the levels of lipid inflammatory mediators in colon from Il10–/– and PAC Il10–/– mice with or without 4F. Multiple proinflammatory mediators were increased in the colon of PAC Il10–/–mice (Figure 9E and Supplemental Table 11). 4F therapy rescued the increase of all proinflammatory LOX products, without inhibiting the significant increase in the sole elevated COX product TXB2 (Figure 9F).
Finally, we determined the effect of 4F on plasma lipid inflammatory mediators in these mice (Supplemental Table 12). 4F therapy significantly reduced the levels of most proinflammatory LOX and COX mediators. The broad nature of the effects of 4F on plasma lipids suggests a broad clearance mechanism capable of affecting all plasma-associated mediators together.
Discussion
We investigated the pathogenic mechanisms of the Cox2-KO/CCHF models of IBD together with the protective mechanisms of APOA1 mimetic peptides (Figure 10, A and B). CCHF increased barrier permeability and initiated TLR/MyD88-dependent intestinal inflammation, while the loss of myeloid COX2 enhanced the proinflammatory response of LPS-activated macrophages, elevated the levels of lipid proinflammatory mediators in intestinal tissue and plasma, and reduced the level of proresolving LXA4 in intestinal tissue. An LXA4 analog (BML111) rescued disease in the Cox2-MKO/CCHF model. 4F therapy inhibited the proinflammatory effects of LPS and oxPAPC on macrophages (and intestinal epithelium), and it reversed the disease-dependent increase of lipid proinflammatory mediators while enhancing luminal clearance of mural proinflammatory lipids. These observations support the correlate hypotheses that dysregulated levels of inflammatory mediators are pathogenic for disease in the Cox2-MKO model, and that rescue of these levels is protective.
Figure 10. Proposed pathogenic mechanisms of Cox2-MKO and CCHF-dependent intestinal inflammation together with the protective mechanisms of APOA1 mimetic peptides.

(A) Cholate increases barrier permeability, initiating both bacteria and MyD88-dependent inflammation in Cox2-KO mice. Cox2-KO macrophages in the lamina propria exhibit an increased proinflammatory response to PAMP/LPS activation in response to PAMP/LPS activation, perhaps through loss of negative feedback control of NFκB. In turn, intestinal epithelium amplifies the proinflammatory stimuli of macrophages, resulting in a dysregulated proinflammatory positive feedback loop. Elevated oxPAPC levels further amplify the proinflammatory response. Loss of LXA4 production in Cox2-KO macrophages impairs the resolution of inflammation. The APOA1 mimetic peptide 4F interrupts the positive feedback loops by inhibiting the LPS-dependent activation of macrophages, blocking proinflammatory signal amplification by intestinal epithelium, and reducing the levels of oxPAPC while directly blocking their effects on macrophages. (B) Upon CCHF challenge, Cox2-MKO elevates lipid proinflammatory mediators in intestinal tissue and plasma while reducing proresolving mediators in tissue. 4F reduces the levels of proinflammatory LOX mediators in the cecum (right), possibly through direct clearance. 4F also reduces the levels of proinflammatory lipid mediators in plasma (left), possibly through enhancement of the TILT of lipoprotein-associated lipid inflammatory mediators into the lumen of the proximal small intestine (SI), in a manner comparable to the effect of 4F on the TICE of free cholesterol (FC) (17).
Disease in the Cox2-MKO/CCHF model overlaps with human CD in several respects. The inflammation is localized to the ileo-colic junction and is transmural in nature (Figure 1A). Apoa1 expression is inhibited in this region (Supplemental Figure 14), as occurs in human CD (38). Lipid peroxidation markers are elevated in plasma and inflamed intestine (Figure 6), as in human CD (55, 56). However, unlike human CD (38), expression of Duox2 was not increased in the terminal ilea of diseased mice (Supplemental Figure 22). Furthermore, the reduction of Apoa1 expression in the colon was offset by lack of change in the ileal and plasma compartments.
Increased intestinal permeability has been proposed as a primary etiologic factor in IBD (57). However, the inflammatory process itself can impair barrier function (58). There remains debate whether changes in barrier permeability are early events in pathogenesis or mere secondary phenomena (59). Our results indicate that alteration in barrier function is an important initiating event in both the Cox2-KO/CCHF and PAC Il10–/– models (Figure 1 and Figure 8C). Nonetheless, we did observe delayed increases in barrier permeability in both the Cox2-MKO/CCHF and PAC Il10–/– models, indicative of an inflammation-dependent secondary effect (Supplemental Figure 7 and Figure 9C).
Loss of barrier function alone was not sufficient to cause intestinal inflammation in Cox2-MKO mice fed CCHF. Barrier permeability increased along the whole intestine (Figure 1B and Supplemental Figure 1), but inflammation was localized to the ileo-ceco-colic junction. We originally considered the possibility that disease depended on the conversion of cholate to deoxycholate within this compartment, but we observed no difference between the functional effects of these bile salts (Supplemental Figure 3). However, disease in this model did depend on TLR/MyD88-specific ligands (Figure 1), whose concentrations are greatest in the ceca of mice.
Cox2-MKO enhanced the proinflammatory phenotype of LPS-activated BMDMs (Figure 1E) (compare our results with those in refs. 60, 61). Cox2-MKO also prevented production of PGJ2 and 15d-PGJ2 in these macrophages (Supplemental Figure 9). The cyclopentenone prostaglandins PGJ2 and 15d-PGJ2 can act as negative regulators of NFκB (36). It is possible that a loss of negative feedback control of NFκB partially explains the proinflammatory effects of Cox2-MKO in LPS-activated macrophages. Because additional PAMPs including lipoteichoic acid (LTA) and peptidoglycan (PG) also can signal through NFκB (27), we speculate that Cox2-MKO also enhances the proinflammatory effects of these additional PAMPs.
The proinflammatory LOX and COX mediators 15HETE, 12HETE, 5HETE, 11HETE, PGE2, and TXB2 are elevated in the colons of human patients with UC (62). We observed comparable increases in proinflammatory LOX and COX mediators in the intestinal tissues of both the Cox2-MKO and PAC Il10–/– models (Figure 2 and Figure 9). The overlap between the proinflammatory eicosanoid signatures of these mouse models with that of human UC suggests that the common elevations may constitute a more general signature for IBD. Intestinal inflammation also correlated with elevated plasma lipid proinflammatory mediators including both LOX metabolites and oxPAPC (Figure 2, Figure 7, and Figure 9; Supplemental Figure 12 and Supplemental Figure 21). These plasma-associated inflammatory mediators may thus be of interest as possible IBD biomarkers.
Proinflammatory lipid mediators may be causative for disease in both IBD models. 4F therapy rescued the increase of proinflammatory LOX mediators in the intestinal tissue without affecting the levels of proinflammatory COX mediators (Figure 4C, Figure 8A, and Figure 9F). 4F can also directly clear proinflammatory LOX mediators from intestinal tissue (Figure 8). Together, these observations support a causal role for the LOX mediators. Of note, 12HETE enhances vascular permeability and neutrophil recruitment (63), and elevated 12HETE has been associated with human IBD (62). Furthermore, disease correlated with elevated oxPAPC in intestine, and oxPAPC induced a proinflammatory response in macrophages. 4F lowered intestinal oxPAPC and inhibited the direct effect of oxPAPC on macrophages (Figure 7). These further observations support a role for oxPAPC in amplifying disease in Cox2-MKO mice fed CCHF.
Loss of proresolving lipid mediators may be causative for disease in the Cox2-MKO/CCHF model. Macrophage COX2 can mediate intracellular production of LXA4 (9), and Cox2-MKO lowered the level of LXA4 compared with FLOX controls (Figure 2F). LXA4 can trigger antiinflammatory and proresolving functions, including blocking neutrophil migration and stimulating efferocytosis (36). LXA4 formation is lower in colonic mucosal biopsies from patients with active UC versus healthy controls (64), while stable analogs of LXA4 were protective in dextran sodium sulfate (DSS) and trinitrobenzene sulphonic acid models of UC (65, 66). The stable LXA4 analog BML111 inhibited disease in Cox2-MKO mice fed CCHF (Figure 2G), supporting the hypothesis that COX2-dependent loss of LXA4 is partially causative of disease in this model. By contrast, LTB4 is a potent proinflammatory mediator involved in neutrophil recruitment (67) whose production can be enhanced by loss of COX2 (9). Altered balance of 5LOX-associated resolving mediators to LTB4 has been associated with plaque instability in atherosclerosis (68) and could further explain the chronic inflammation of this model.
We concluded that myeloid COX2 constitutes a check on TLR-dependent intestinal inflammation in the ileo-colic junction. Our account thus extends earlier work that also demonstrated a role for COX2 in intestinal immune tolerance. Newberry et al. showed that COX2 and PGE2 help maintain adaptive immune tolerance to dietary antigens in the proximal small bowel (12), which function they further attributed to constitutive Cox2 expression and PGE2 production by stromal cells in the lamina propria (13). Despite the general overlap, however, our current account differs from this prior work in at least one respect. Contrary to Newberry et al., we did not observe an expansion of the T lymphocyte population in the inflammatory lesions of the Cox2-MKO/CCHF model (Supplemental Figure 5). Rather, the immunosuppressive effect of myeloid COX2 in our model appears to bear primarily on innate immunity. However, Newberry et al. identified stromal-derived PGE2 as the essential mediator of COX2-dependent adaptive immune suppression. By contrast, myeloid Cox2-KO did not result in a decrease in PGE2 within the ileo-ceco-colic junctions of Cox2-MKO mice fed CCHF (Figure 2E), indicating complimentary production of PGE2 by other cell types. Our results thus appear consistent with the earlier work of Newberry et al. on COX2 and intestinal immune tolerance.
The APOA1 mimetic peptides 4F and Tg6F inhibited intestinal inflammation in our mouse models. In the Cox2-MKO/CCHF model, 4F directly suppressed LPS-dependent signaling and appeared to interrupt many of the amplifying proinflammatory feedback loops present in disease. In particular, 4F blocked the proinflammatory effect of LPS-activated macrophages on intestinal epithelium, lowered oxPAPC while inhibiting its proinflammatory effects, and cleared mural proinflammatory lipids. These results thus extend earlier reports on the efficacy of mimetic peptides against DSS-induced colitis (39, 69). APOA1 mimetic peptides offer several advantages over existing IBD therapies. These drugs are not immune suppressive, and unlike current biologics, they can be taken orally and are not antigenic (70–72).
4F therapy reduced the plasma levels of lipid proinflammatory mediators (Figure 4, Figure 7, and Figure 8). We demonstrated ex vivo that lipid inflammatory mediators can be transported across intestinal explants from lipoproteins into mucosal media, indicating that there may exist trans-intestinal lipid transport (TILT) of lipid species over and above cholesterol. 4F can enhance this trans-intestinal transport (Figure 8D). It is thus possible that TILT mediates the effect of 4F on plasma lipid inflammatory mediators in vivo. We previously demonstrated that 4F can enhance TICE (17), and that 4F lowered total cholesterol in the plasmas of Cox2-MKO/CCHF mice (Supplemental Figure 17). 4F may mediate the reduction of plasma cholesterol and plasma prolipid inflammatory mediators through comparable trans-intestinal mechanisms (Figure 10). It is also possible that 4F may act like APOA1 to increase macrophage expression of the antioxidant APOE (73), which effect could further account for the antioxidant properties of 4F.
The current study contains several limitations. First, inflammation is a dynamic process but we sampled lipid inflammatory mediators at only a few time points. Second, we do not fully elucidate the mechanisms by which loss of myeloid COX2 results in the dysregulated lipid mediator levels that we observe. Third, we do not fully establish the causal significance of most of the dysregulated lipid inflammatory mediators.
Nonetheless, the present study identifies a role for myeloid COX2 in limiting TLR-dependent intestinal inflammation. It highlights the potential importance of proinflammatory and proresolving lipids as modulators of IBD. Finally, we offer further evidence of the possible translational value of APOA1 mimetics as therapy for IBD, while adding to our understanding of their protective mechanisms.
Methods
Mice.
Cox2-MKO (Cox2fl/flLysMCre/+), Cox2 FLOX (Cox2fl/fl), Cox2-TKO (Cox2luc/luc), Cox2fl/fl Mrp8Cre/+, and Il10–/– mice were all generated, bred, and maintained as described (15, 30, 53, 74). C57BL/6J mice were from The Jackson Laboratory. Equal numbers of male and female mice were used in all in vivo studies.
APOA1 mimetic peptides.
D-4F and 3F(14) peptides were synthesized as previously described (17, 75). For in vivo studies, D-4F was supplied as 500 μg/mL in drinking water, and the mice were allowed to drink ad libitum. For cell culture studies, D-4F or 3F(14) were added at 15 μg/mL to the cell culture media. Tg6F extracts were prepared as described previously (40, 76). For studies involving Tg6F, the final diets contained 0.06% of Tg6F extract by weight (40).
Study approval.
All human tissues used in this study were obtained from de-identified and discarded surgical specimens following clinical surgical pathology evaluation. The UCLA Institutional Review Board approved procurement and use of surgical samples. The requirement for informed consent for tissues obtained from the UCLA Translational Pathology Core Laboratory (IRB 11-002504) was waived. The Animal Research Committee at UCLA approved the animal protocols used in this study and the methods were carried out in accordance with the approved guidelines.
Statistics.
For lipidomic analyses (Figure 2, Figure 4, and Figure 9), FDR corrections were performed by application of the Benjamini-Hochberg procedure to P values determined for each analyte by 1-way ANOVA, with FDR controlled at level α = 0.05. For each analyte within a data set deemed a discovery/significant, post hoc analyses consisted of Tukey’s multiple comparisons test. For all other multiple comparisons, 1-, 2-, or 3-way ANOVA was followed by Tukey’s or Dunnett’s multiple comparisons test. For comparisons involving 1 independent variable with 2 groups, we performed Student’s t tests, and for studies consisting of more than 1 Student’s t test, we performed Holm-Sidak’s correction for multiple tests. In all cases of multiple comparisons/tests, we adjusted P values in accord with the appropriate correction. Statistical significance was set at P less than 0.05 with respect to final (adjusted) P values. Variation is reported as SEM. Additional information about Materials and Methods can be found in the Supplemental Materials.
Author contributions
DM designed studies, conducted experiments, analyzed data, and wrote the manuscript. DS conducted the Ussing chamber experiments. CV, AD, and HT helped conduct the animal studies and maintain the animal colonies. VG performed macrophage studies. ND did Western blots and lipid extractions. RSSV and JW helped perform the human enteroid studies. EO and JP helped develop the LC-MS/MS lipid analysis methods. ML provided the Ussing chambers and training for their use. MNP and GMA synthesized the APOA1 mimetic peptides D-4F and 3F. HRH generated and provided the Cox2-TKO and neutrophil-specific knockout mice and reviewed the experimental protocols and results. MGM and AMF provided guidance, feedback, supervision, and financial support. STR provided guidance and feedback regarding the overall study and individual experiments, edited the manuscript, and provided laboratory and financial support.
Supplementary Material
Acknowledgments
This work was supported in part by NIH National Heart, Lung, and Blood Institute grant 2P01 HL-30568 (to AMF and STR); the Castera, Laubisch, and Milt Grey funds at UCLA; F31 DK108592 and T32 2T32HL007895-16 (to DM); NIH P30 DK 41301 (to ML); and R01 HL071776 (to STR). Research reported in this publication was also supported by the National Institute of Diabetes and Digestive and Kidney Disorders under grant DK41301 (to MGM) and the National Institute of Allergy and Infectious Diseases under grant U01DK085535 (to MGM). The content is solely the responsibility of all the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Dr. Martin Philips and Chun-Ling Jung for their contributions to our SPR analyses.
Version 1. 06/11/2019
In-Press Preview
Version 2. 08/05/2019
Electronic publication
Version 3. 09/03/2019
Print issue publication
Footnotes
HT’s present address is: 1711 Granville Avenue #5, Los Angeles, California, 90025, USA.
Conflict of interest: GMA, AMF, and STR are principals in Bruin Pharma, and AMF is an officer in Bruin Pharma.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(9):3670–3685.https://doi.org/10.1172/JCI123700.
Contributor Information
David Meriwether, Email: dmeriwether@mednet.ucla.edu.
Dawoud Sulaiman, Email: dsulaiman@g.ucla.edu.
Carmen Volpe, Email: cvolpe@mednet.ucla.edu.
Anna Dorfman, Email: adorfman@mednet.ucla.edu.
Victor Grijalva, Email: vgrijalva@mednet.ucla.edu.
Nasrin Dorreh, Email: ndorreh@mednet.ucla.edu.
R. Sergio Solorzano-Vargas, Email: ssolorzano@mednet.ucla.edu.
Ellen O’Connor, Email: eioconnor@ucla.edu.
Jeremy Papesh, Email: jpapesh@mednet.ucla.edu.
Muriel Larauche, Email: MLarauche@mednet.ucla.edu.
Hannah Trost, Email: hktrost@ucla.edu.
Mayakonda N. Palgunachari, Email: palgun@uab.edu.
G.M. Anantharamaiah, Email: ananth@uab.edu.
Harvey R. Herschman, Email: hherschman@mednet.ucla.edu.
Martin G. Martin, Email: mmartin@mednet.ucla.edu.
Alan M. Fogelman, Email: afogelman@mednet.ucla.edu.
Srinivasa T. Reddy, Email: sreddy@mednet.ucla.edu.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Liu JZ, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JC, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat Genet. 2017;49(2):262–268. doi: 10.1038/ng.3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ananthakrishnan AN, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol. 2018;15(1):39–49. doi: 10.1038/nrgastro.2017.136. [DOI] [PubMed] [Google Scholar]
- 5.Lewis RT, Maron DJ. Efficacy and complications of surgery for Crohn’s disease. Gastroenterol Hepatol (NY) 2010;6(9):587–596. [PMC free article] [PubMed] [Google Scholar]
- 6.Peyrin-Biroulet L. Anti-TNF therapy in inflammatory bowel diseases: a huge review. Minerva Gastroenterol Dietol. 2010;56(2):233–243. [PubMed] [Google Scholar]
- 7.Kopylov U, Seidman E. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Therap Adv Gastroenterol. 2016;9(4):513–526. doi: 10.1177/1756283X16638833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174(8):5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 9.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci U S A. 2014;111(35):12746–12751. doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox DG, Crusius JB, Peeters PH, Bueno-de-Mesquita HB, Pena AS, Canzian F. Haplotype of prostaglandin synthase 2/cyclooxygenase 2 is involved in the susceptibility to inflammatory bowel disease. World J Gastroenterol. 2005;11(38):6003–6008. doi: 10.3748/wjg.v11.i38.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Vries HS, te Morsche RH, van Oijen MG, Nagtegaal ID, Peters WH, de Jong DJ. The functional -765G→C polymorphism of the COX-2 gene may reduce the risk of developing crohn’s disease. PLoS ONE. 2010;5(11):e15011. doi: 10.1371/journal.pone.0015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-dependent arachidonic acid metabolites are essential modulators of the intestinal immune response to dietary antigen. Nat Med. 1999;5(8):900–906. doi: 10.1038/11341. [DOI] [PubMed] [Google Scholar]
- 13.Newberry RD, McDonough JS, Stenson WF, Lorenz RG. Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria: directing the tone of the intestinal immune response. J Immunol. 2001;166(7):4465–4472. doi: 10.4049/jimmunol.166.7.4465. [DOI] [PubMed] [Google Scholar]
- 14.Lin JA, et al. Atherogenic diet causes lethal ileo-ceco-colitis in cyclooxygenase-2 deficient mice. Prostaglandins Other Lipid Mediat. 2007;84(3-4):98–107. doi: 10.1016/j.prostaglandins.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe J, et al. Novel anti-inflammatory functions for endothelial and myeloid cyclooxygenase-2 in a new mouse model of Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G842–G850. doi: 10.1152/ajpgi.00468.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Navab M, Reddy ST, Meriwether D, Fogelman SI, Fogelman AM.ApoA-I Mimetic Peptides: A review of the present status. In: Anantharamaiah GM, Goldberg D eds. Apolipoprotein mimetics in the management of human disease. Cham, Switzerland: Springer International Publishing; 2015:15–27. [Google Scholar]
- 17.Meriwether D, et al. Transintestinal transport of the anti-inflammatory drug 4F and the modulation of transintestinal cholesterol efflux. J Lipid Res. 2016;57(7):1175–1193. doi: 10.1194/jlr.M067025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navab M, et al. D-4F-mediated reduction in metabolites of arachidonic and linoleic acids in the small intestine is associated with decreased inflammation in low-density lipoprotein receptor-null mice. J Lipid Res. 2012;53(3):437–445. doi: 10.1194/jlr.M023523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann AF. Bile Acids: The good, the bad, and the ugly. News Physiol Sci. 1999;14:24–29. doi: 10.1152/physiologyonline.1999.14.1.24. [DOI] [PubMed] [Google Scholar]
- 20.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117(3):514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyapati RK, Rossi AG, Satsangi J, Ho GT. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 2016;9(3):567–582. doi: 10.1038/mi.2016.14. [DOI] [PubMed] [Google Scholar]
- 22.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res. 2004;55(4):622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 23.Meddings JB, Gibbons I. Discrimination of site-specific alterations in gastrointestinal permeability in the rat. Gastroenterology. 1998;114(1):83–92. doi: 10.1016/S0016-5085(98)70636-5. [DOI] [PubMed] [Google Scholar]
- 24.Assefnia S, et al. Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: common target, common therapies. Oncotarget. 2014;5(6):1458–1474. doi: 10.18632/oncotarget.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4(4):337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vyvoda OS, Coleman R, Holdsworth G. Effects of different bile salts upon the composition and morphology of a liver plasma membrane preparation. Deoxycholate is more membrane damaging than cholate and its conjugates. Biochim Biophys Acta. 1977;465(1):68–76. doi: 10.1016/0005-2736(77)90356-x. [DOI] [PubMed] [Google Scholar]
- 27.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Olson MA, Lee MS, Kissner TL, Alam S, Waugh DS, Saikh KU. Discovery of small molecule inhibitors of MyD88-dependent signaling pathways using a computational screen. Sci Rep. 2015;5:14246. doi: 10.1038/srep14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta J, Nebreda AR. Analysis of intestinal permeability in mice. Bio-protocol. 2014;4(22):e1289. doi: 10.21769/BioProtoc.1289. [DOI] [Google Scholar]
- 30.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 32.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci U S A. 2014;111(35):12746–12751. doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 34.Migita K, et al. Lipopolysaccharide signaling induces serum amyloid A (SAA) synthesis in human hepatocytes in vitro. FEBS Lett. 2004;569(1-3):235–239. doi: 10.1016/j.febslet.2004.05.072. [DOI] [PubMed] [Google Scholar]
- 35.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2(10):787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, et al. Lipoxin A4 and its analog suppress inflammation by modulating HMGB1 translocation and expression in psoriasis. Sci Rep. 2017;7(1):7100. doi: 10.1038/s41598-017-07485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haberman Y, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124(8):3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gkouskou KK, et al. Apolipoprotein A-I inhibits experimental colitis and colitis-propelled carcinogenesis. Oncogene. 2016;35(19):2496–2505. doi: 10.1038/onc.2015.307. [DOI] [PubMed] [Google Scholar]
- 40.Chattopadhyay A, et al. Efficacy of tomato concentrates in mouse models of dyslipidemia and cancer. Pharmacol Res Perspect. 2015;3(4):e00154. doi: 10.1002/prp2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vakili L, et al. Systemic inflammation, intestine, and paraoxonase-1. In: Camps J, ed. Oxidative stress and inflammation in non-communicable diseases - molecular mechanisms and perspectives in therapeutics. Cham, Switzerland: Springer International Publishing; 2014:83–88. [Google Scholar]
- 42.Lintonen TP, et al. Differential mobility spectrometry-driven shotgun lipidomics. Anal Chem. 2014;86(19):9662–9669. doi: 10.1021/ac5021744. [DOI] [PubMed] [Google Scholar]
- 43.Van Lenten BJ, et al. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49(11):2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zachos NC, et al. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem. 2016;291(8):3759–3766. doi: 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alzoghaibi MA, Al Mofleh IA, Al-Jebreen AM. Lipid peroxides in patients with inflammatory bowel disease. Saudi J Gastroenterol. 2007;13(4):187–190. doi: 10.4103/1319-3767.36750. [DOI] [PubMed] [Google Scholar]
- 46.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201(1):28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 47.Alzoghaibi MA. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J Gastroenterol. 2013;19(39):6540–6547. doi: 10.3748/wjg.v19.i39.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies SS, Guo L. Lipid peroxidation generates biologically active phospholipids including oxidatively N-modified phospholipids. Chem Phys Lipids. 2014;181:1–33. doi: 10.1016/j.chemphyslip.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egea J, et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS) Redox Biol. 2017;13:94–162. doi: 10.1016/j.redox.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vladykovskaya E, et al. Reductive metabolism increases the proinflammatory activity of aldehyde phospholipids. J Lipid Res. 2011;52(12):2209–2225. doi: 10.1194/jlr.M013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meriwether D, et al. Enhancement by LDL of transfer of L-4F and oxidized lipids to HDL in C57BL/6J mice and human plasma. J Lipid Res. 2011;52(10):1795–1809. doi: 10.1194/jlr.M016741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgantini C, et al. HDL lipid composition is profoundly altered in patients with type 2 diabetes and atherosclerotic vascular disease. Nutr Metab Cardiovasc Dis. 2014;24(6):594–599. doi: 10.1016/j.numecd.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holgersen K, Kvist PH, Markholst H, Hansen AK, Holm TL. Characterisation of enterocolitis in the piroxicam-accelerated interleukin-10 knock out mouse--a model mimicking inflammatory bowel disease. J Crohns Colitis. 2014;8(2):147–160. doi: 10.1016/j.crohns.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Hale LP, Gottfried MR, Swidsinski A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflamm Bowel Dis. 2005;11(12):1060–1069. doi: 10.1097/01.MIB.0000187582.90423.bc. [DOI] [PubMed] [Google Scholar]
- 55.Wendland BE, et al. Lipid peroxidation and plasma antioxidant micronutrients in Crohn disease. Am J Clin Nutr. 2001;74(2):259–264. doi: 10.1093/ajcn/74.2.259. [DOI] [PubMed] [Google Scholar]
- 56.Bouzid D, et al. Oxidative stress markers in intestinal mucosa of Tunisian inflammatory bowel disease patients. Saudi J Gastroenterol. 2013;19(3):131–135. doi: 10.4103/1319-3767.111956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munkholm P, et al. Intestinal permeability in patients with Crohn’s disease and ulcerative colitis and their first degree relatives. Gut. 1994;35(1):68–72. doi: 10.1136/gut.35.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bruewer M, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171(11):6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 59.Cobrin GM, Abreu MT. Defects in mucosal immunity leading to Crohn’s disease. Immunol Rev. 2005;206:277–295. doi: 10.1111/j.0105-2896.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 60.Na YR, Yoon YN, Son D, Jung D, Gu GJ, Seok SH. Consistent inhibition of cyclooxygenase drives macrophages towards the inflammatory phenotype. PLoS ONE. 2015;10(2):e0118203. doi: 10.1371/journal.pone.0118203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, et al. Macrophage cyclooxygenase-2 protects against development of diabetic nephropathy. Diabetes. 2017;66(2):494–504. doi: 10.2337/db16-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masoodi M, et al. Altered colonic mucosal polyunsaturated fatty acid (PUFA) derived lipid mediators in ulcerative colitis: new insight into relationship with disease activity and pathophysiology. PLoS ONE. 2013;8(10):e76532. doi: 10.1371/journal.pone.0076532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rossaint J, Zarbock A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun. 2013;5(4):348–357. doi: 10.1159/000345943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mangino MJ, Brounts L, Harms B, Heise C. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2006;79(1–2):84–92. doi: 10.1016/j.prostaglandins.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 65.Gewirtz AT, et al. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J Immunol. 2002;168(10):5260–5267. doi: 10.4049/jimmunol.168.10.5260. [DOI] [PubMed] [Google Scholar]
- 66.Fiorucci S, et al. A beta-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc Natl Acad Sci U S A. 2004;101(44):15736–15741. doi: 10.1073/pnas.0404722101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lämmermann T, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498(7454):371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fredman G, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nowacki TM, et al. The 5A apolipoprotein A-I (apoA-I) mimetic peptide ameliorates experimental colitis by regulating monocyte infiltration. Br J Pharmacol. 2016;173(18):2780–2792. doi: 10.1111/bph.13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bloedon LT, et al. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. 2008;49(6):1344–1352. doi: 10.1194/jlr.P800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dunbar RL, et al. Oral apolipoprotein A-I mimetic D-4F lowers HDL-inflammatory index in high-risk patients: a first-in-human multiple-dose, randomized controlled trial. Clin Transl Sci. 2017;10(6):455–469. doi: 10.1111/cts.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lau JL, Dunn MK. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26(10):2700–2707. doi: 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 73.Rees D, Sloane T, Jessup W, Dean RT, Kritharides L. Apolipoprotein A-I stimulates secretion of apolipoprotein E by foam cell macrophages. J Biol Chem. 1999;274(39):27925–27933. doi: 10.1074/jbc.274.39.27925. [DOI] [PubMed] [Google Scholar]
- 74.Ishikawa TO, Jain NK, Taketo MM, Herschman HR. Imaging cyclooxygenase-2 (Cox-2) gene expression in living animals with a luciferase knock-in reporter gene. Mol Imaging Biol. 2006;8(3):171–187. doi: 10.1007/s11307-006-0034-7. [DOI] [PubMed] [Google Scholar]
- 75.Handattu SP, et al. ApoA-I mimetic peptides with differing ability to inhibit atherosclerosis also exhibit differences in their interactions with membrane bilayers. J Biol Chem. 2007;282(3):1980–1988. doi: 10.1074/jbc.M606231200. [DOI] [PubMed] [Google Scholar]
- 76.Mukherjee P, et al. Transgenic tomatoes expressing the 6F peptide and ezetimibe prevent diet-induced increases of IFN-β and cholesterol 25-hydroxylase in jejunum. J Lipid Res. 2017;58(8):1636–1647. doi: 10.1194/jlr.M076554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.