

Abstract
We previously generated 32 rotavirus-specific (RV-specific) recombinant monoclonal antibodies (mAbs) derived from B cells isolated from human intestinal resections. Twenty-four of these mAbs were specific for the VP8* fragment of RV VP4, and most (20 of 24) were non-neutralizing when tested in the conventional MA104 cell–based assay. We reexamined the ability of these mAbs to neutralize RVs in human intestinal epithelial cells, including ileal enteroids and HT-29 cells. Most (18 of 20) of the “non-neutralizing” VP8* mAbs efficiently neutralized human RV in HT-29 cells or enteroids. Serum RV neutralization titers in adults and infants were significantly higher in HT-29 than MA104 cells and adsorption of these sera with recombinant VP8* lowered the neutralization titers in HT-29 but not MA104 cells. VP8* mAbs also protected suckling mice from diarrhea in an in vivo challenge model. X-ray crystallographic analysis of one VP8* mAb (mAb9) in complex with human RV VP8* revealed that the mAb interaction site was distinct from the human histo-blood group antigen binding site. Since MA104 cells are the most commonly used cell line to detect anti-RV neutralization activity, these findings suggest that prior vaccine and other studies of human RV neutralization responses may have underestimated the contribution of VP8* antibodies to the overall neutralization titer.
Keywords: Vaccines, Virology
Keywords: Adaptive immunity, B cells
Introduction
Group A rotaviruses (RVs) are non-enveloped, double-stranded, segmented RNA viruses with a triple-layered particle (TLP) composed of inner layer (VP2), intermedia layer (VP6), and outer layer (VP7, VP4) proteins (1). The RV surface spike protein, VP4, is proteolytically cleaved into 2 non–covalently bonded subunits, a head subunit (VP8*) and a stem subunit (VP5*). The VP4 cleavage is necessary for efficient RV cell entry and infection (2). VP8* is thought to be responsible for initial target cell binding, while VP5* is likely involved in cell membrane penetration and cell entry (3). RVs replicate primarily, but not exclusively, in mature enterocytes of the small intestine and RV infection is highly species specific, such that RV strains isolated from one animal species have significantly reduced infectivity and ability to spread among heterologous animal species (1). RV infection induces a specific antibody response that provides substantial protection from subsequent severe RV reinfection and more moderate protection from mild or asymptomatic reinfection (4). Results primarily from monoclonal antibody (mAb) studies demonstrate that antibodies specific for VP7, VP5*, or VP8* can all neutralize RV in cell culture and confer protection against RV infection in animal models in either a monotypic or heterotypic manner, depending on the particular antibody’s serologic specificity (1). In some animal studies, antibodies against RV proteins VP6 or NSP4 have also been implicated in inhibiting RV infection or RV-induced diarrhea. However, these mAbs do not have in vitro neutralization activity and their relevance to protection in vivo in humans or animals is not well established (5, 6).
Despite the availability of several safe and effective RV vaccines, RVs remain the single most important cause of severe diarrheal disease in infants and young children around the world. RV infection results in approximately 200,000 annual deaths, predominantly in low-income countries (7). There are 2 main vaccine strategies that are currently used for all licensed live attenuated human RV vaccines. The RotaTeq (Merck Inc.) and Rotasiil (Serum Institute of India) vaccines are both based on a so-called modified Jennerian strategy in which animal-origin (bovine in these cases) RVs, which are attenuated due to their host-range restriction, were used to generate a multivalent set of reassortant RVs containing human-origin VP7 G1, G2, G3, and G4 and a human-origin VP4. The serotypic diversity of these vaccines is postulated to enhance the breadth of their protective efficacy (8). On the other hand, Rotarix (GSK) and Rotavac (Bharat) both consist of single, multiple tissue culture passaged and hence attenuated human RV strains. They are postulated to generate protective immunity based on epidemiologic data demonstrating substantial protective immunity following a single natural RV infection (9). Both types of RV vaccines have been shown to provide high levels of effective protection against severe RV diarrhea in various high- or middle-income countries despite the serotypic heterogeneity of circulating human RV strains, suggesting that vaccination by one serotype appears to efficiently induce heterotypic protective immunity (9, 10).
The mechanism by which live attenuated RV vaccines induce protection, as well as the RV protein targets of that protective immune response, are still not fully understood. Vaccine efficacy correlates poorly with serum neutralization responses (11–14). RV vaccine efficacy is significantly reduced in low-income countries and the reason for this lower efficacy is also not clear (9, 10). Higher levels of preexisting maternal antibody, poor nutrition, higher incidence of concurrent enteric infection, and differences in the gut microbiome at the time of vaccination may be some of the contributors to the reduced RV vaccine efficacy in less developed countries (9, 10).
In vitro, RVs replicate in many human and animal epithelial cell lines with varying efficiencies. However, there does not appear to be a clear enteric tissue tropism or species specificity for RV replication in cell lines comparable to the replication restriction encountered in vivo. A monkey kidney cell line, MA104, was found to be highly permissive for RV replication over 40 years ago and it has been very widely used for RV propagation, infectivity, and neutralization studies ever since (15, 16). In fact, the great majority of neutralization assays undertaken over the past 40 or more years of RV research have been performed using MA104 cells.
Recently, we generated 32 recombinant human RV–specific mAbs based on immunoglobulin (Ig) sequences derived from selected antibody-secreting B cells isolated from resected small intestinal tissues of adult bariatric surgery patients (17). We identified 3 VP5*-specific and 4 VP7-specific mAbs and all but one VP7 mAb effectively neutralized RV in either a homotypic or heterotypic manner. These mAbs were also able to prevent RV-induced diarrhea in suckling mice in a passive protection assay (17). Interestingly, the great majority (24 of 32) of the isolated, human small bowel–derived, recombinant mAbs were VP8* specific. Surprisingly, only 4 of these were found to neutralize RV when assayed in a conventional MA104 cell–based neutralization assay. This observation was noted as unusual at the time since multiple prior mouse-based mAb studies had identified many neutralizing VP8*-directed mAbs (18).
In this study, we provide data based on the reexamination of the neutralization activity of these “non-neutralizing” VP8*-directed human mAbs examined in human small intestinal enteroids and in the human HT-29 colon cancer–derived cell line as opposed to MA104 cells. We find that most of the previously “non-neutralizing” VP8* mAbs efficiently neutralize RV in the human-origin intestinal cells but not in monkey kidney cell lines (MA104 or CV1) or a human kidney cell line (HEK293).
Results
Neutralization of human RVs in human small bowel enteroid cultures.
We previously reported that among 32 recombinant mAbs generated from Ig sequences isolated from individual human small intestinal B cells, 3 of 3 VP5*-directed and 3 of 4 VP7-directed mAbs effectively neutralized selected RVs in a conventional MA104 cell–based neutralization assay. On the other hand, only 4 of 24 anti-VP8* mAbs demonstrated neutralizing activity (17). In the current study, we reassessed our prior neutralization results using several additional cell substrates, but focusing on human small intestinal enteroid cultures and 2 human-origin colon cancer cell lines with small intestinal characteristics (19). We observed that in a human ileal enteroid monolayer culture, most of the previously non-neutralizing VP8* mAbs effectively inhibited human RV replication by more than 95% (Figure 1 and Table 1). On the other hand, VP5* (mAb2 and mAb41), VP8* (mAb30), and VP7 (mAb27), which were identified as neutralizing in MA104 (17), remained neutralizing in the enteroids versus human strain RVs WI61 (G9, P[8]) and Wa (G1, P[8]) (Figure 1 and Table 1). As expected from prior MA104-based neutralization data, negative control VP6-specific human mAbs did not neutralize RV in the enteroid assay (Figure 1 and Table 1). Interestingly, in preliminary studies we observed that RVs infected the basolateral side of the enteroid monolayer cultures more efficiently than the apical surface, an observation made previously by others in both intestinal cell lines and enteroids (ref. 20 and S. Blutt, S. Crawford, and M. Estes, Baylor College of Medicine, personal communication). Therefore, we carried out the bulk of our enteroid monolayer neutralization assays and all the results found in both Figure 1 and Table 1 using a basolateral infection route in the enteroid monolayer cultures. However, we did observe that our VP8* mAbs also neutralized RV efficiently when an apical route of infection was used, even though RV infectivity itself, from the apical side of the monolayers, was much less efficient (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI128382DS1). Inhibition of RV infectivity by VP8* mAbs was also observed in 3D enteroid cultures where enteroids are embedded and grown in the basement membrane matrix, Matrigel (Figure 2, A and B).
Figure 1. Neutralization of human RV WI61 (G9, P[8]) by recombinant RV-specific human mAbs in human ileal enteroid monolayer cultures.

WI61 RV was incubated with the indicated human recombinant mAbs (100 ng/mL) for 1 hour at 37°C, and then added to the basolateral side of the enteroid monolayer culture. Cells were fixed 16 hours after infection and then stained with FITC-labeled polyclonal rabbit anti-RV antibody (green), Texas red–phalloidin (red), and DAPI (blue). (A) No mAb, (B) mAb30 (anti-VP8*, neutralizing in MA104 cells), (C) mAb2 (anti-VP5*), (D) mAb27 (anti-VP7) [against Wa (G1, P[8])], (E) mAb11 (anti-VP8*, non-neutralizing in MA104 cells), and (F) mAb72 (anti-VP6, non-neutralizing in MA104 cells). Original magnification, ×10.
Table 1. Neutralization of selected RV-specific human mAbs versus human rotavirus, [WI61 (G9, P[8]) or Wa (G1.P[8])], in human ileal enteroid monolayer cultures.
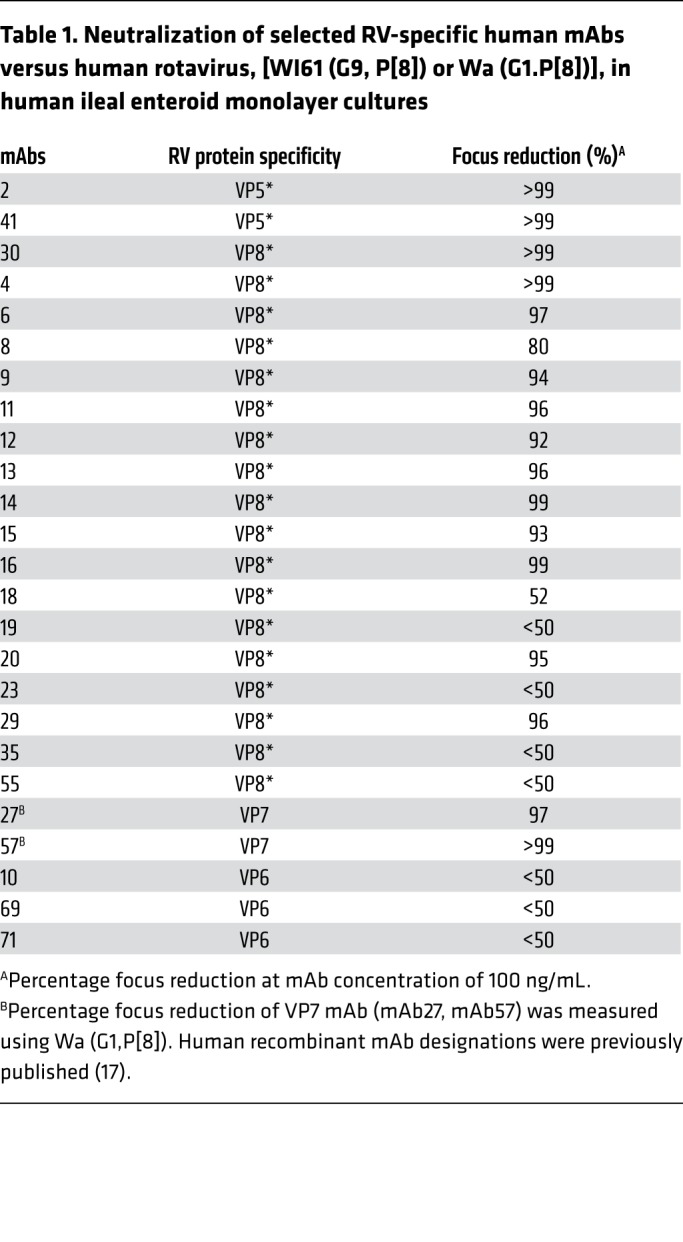
Figure 2. Comparison of neutralization activity of human RV VP8*–specific mAb11 against WI61 (G9, P[8]) RV in ileal enteroids and HT-29, MA104, CV-1, and HEK293 cells.

Indicated cell types were infected with WI61 in the presence or absence of human VP8* mAb11 (100 ng/mL) for 16 hours. Cells were then fixed and stained with FITC-labeled polyclonal rabbit anti-RV antibody (green), Texas red–phalloidin (red), and DAPI (blue). Ileal enteroid 3D Matrigel culture with (A) or without (B) mAb. Ileal enteroid monolayer culture, basolateral infection with (C) or without (D) mAb. HT-29 cells with (E) or without (F) mAb. MA104 cells with (G) or without (H) mAb. CV-1 cells with (I) or without (J) mAb. HEK293 cells with (K) or without (L) mAb. Percentages of RV focus reduction by mAb11: ileal enteroid monolayer culture (97%), HT-29 cells (94%), MA104 cells (0%), CV-1 cells (11%), and HEK293 cells (0%). Original magnification, ×10.
Human VP8* mAbs neutralize RV in human intestinal epithelial cell lines but not in human or monkey kidney cell lines.
We next tested the neutralizing capacity of VP8*-specific mAbs in several other commonly used cell lines including the HT-29 human colonic cell line, the HEK293 human kidney cell line, and the CV-1 monkey kidney cell line in addition to the MA104 monkey kidney cell line. As shown in Table 1 and Figure 2, VP8* mAb11, which neutralized human RV WI61 (G9, P[8]) efficiently in enteroids, also neutralized RV efficiently in HT-29, but not in MA104, CV-1, or HEK293 cells. The percentages of focus reduction of mAb11 in enteroid monolayer culture and HT-29 cells were 97% and 94%, respectively. The percentages of focus reduction of mAb11 in MA104, CV-1, and HEK293 were 0%, 11%, and 0%, respectively. Based on these initial findings, we retested all our previously characterized human RV–specific mAbs in MA104 and HT-29 cells by neutralization versus 3 strains of human RVs including WI61 (G9, P[8]), Wa (G1, P[8]), and DS1 (G2, P[4]). We found that neutralization titers were similar between MA104 and HT-29 cells for all the VP5*, VP8*, and VP7 mAbs that previously were shown to neutralize RVs in MA014 cells (Table 2 and ref. 17). However, the great majority of VP8*-specific mAbs, which had little or no neutralizing activity in MA104 cells, now neutralized one or two of the indicated human RV strains efficiently in HT-29 cells. Additionally, several VP8* mAbs, including mAb9 and mAb11, efficiently inhibited WI61 replication by at least 10-fold in HT-29 cells but not in MA104 cells, as measured by a quantitative RT-PCR–based assay (data not shown). Only 4 of 24 VP8* mAbs (mAb31, -35, -44, and -55; Table 2) had no detectable neutralizing activity in any of the cell lines examined (Table 2). Interestingly, the VP8* mAbs in our study tended to neutralize human WI61 RV more effectively than the Wa RV strain, even though both strains share the same VP4 P[8] type. This suggests that, as has been shown previously with RV VP7–neutralizing mAbs (21), the VP8* protein can demonstrate some level of P[8] subtype neutralization specificity when assayed with mAbs.
Table 2. Comparison of neutralization activity of RV-specific human mAbs against indicated human RV strains in HT-29, CaCo-2, and MA104 cells.

Most (19 of 24) of the VP8* mAbs were monotypic, neutralizing only P[8] VP8* RVs. However, 5 VP8* mAbs (mAb6, -9, -23, -30, and -47) also neutralize P[4] RV strains. Only one of the 24 examined VP8* mAbs (mAb30), neutralized a P[6] RV strain (17). In summary, when tested in the HT-29 cell line (or in enteroid monolayer cultures) the percentage of neutralizing VP8* mAbs changed from 17% in the MA104 cell–based assay to 83% in the HT-29 cell–based assay. On the other hand, the percentages of neutralizing VP5* and VP7 mAbs remained unchanged in the 2 cell lines.
We next expanded our examination of cell lines that might potentially be useful for detecting human anti-VP8* neutralizing activity by examining another frequently used human-origin intestinal epithelial cell line, CaCo-2 cells (16). Neutralization titers were generally similar between CaCo-2 and MA104 cells for those VP7, VP5*, and the few VP8* mAbs that neutralized RV in MA104 cells. However, for most of the VP8* mAbs that neutralized only in HT-29 cells and enteroids but not in MA104 cells, both the number and efficiency of neutralization in CaCo-2 cells were significantly lower than in HT-29 cells, but higher than in MA104 cells (Table 2).
VP8*-specific mAbs protect suckling mice from RV-induced diarrhea.
We previously demonstrated that RV-neutralizing VP5* and VP7 mAbs could protect suckling mice from either homotypic or heterotypic heterologous human RV–induced diarrhea following experimental challenge (17). Here we examined whether human VP8*-specific mAbs with neutralizing activity in enteroids and HT-29 cells, but not in MA104 cells, could also protect suckling mice from diarrhea following heterologous human RV challenge in a passive protection assay. Human RV WI61 was preincubated with anti-VP8* mAb11 or mAb14 (250 ng/mL) and then administered by oral gavage to 5-day-old 129sv suckling mice. Both VP8* mAbs significantly reduced RV-associated diarrhea despite the fact that these mAbs were non-neutralizing when assayed in MA104 cells (Figure 3).
Figure 3. Passive protection assay of human RV–specific VP8* mAb in suckling mice.

Five-day-old suckling 129sv mice were orally inoculated with WI61 human RV with or without indicated mAbs (250 ng/mL). At indicated days after infection mice were observed for diarrhea. The figure represents the combination of 2 independent experiments. Number of mice per group: no Ab, 19; mAb11, 12; and mAb14, 14. *P < 0.001 compared with mAb groups by Fisher’s exact test.
VP8*-specific neutralizing activity can be detected in normal human adult sera using HT-29 but not MA104 cells.
We next examined whether the HT-29 cell–based neutralization assay could be used to directly detect and quantify RV anti-VP8* neutralizing activity in human sera that might be undetected by the conventional MA104 assay. We first compared neutralization titers of 10 normal adult serum samples against Wa (a prototypic G1 human RV strain) in HT-29 and MA104 cells (Figure 4A). Neutralization titers in HT-29 cells were significantly higher than in MA104 cells in 8 of 10 subjects. The median neutralization titer in the HT-29 cell assay was 384 versus 96 in the MA104 cell assay (P = 0.04 by t test of means of log2-transformed titers). The mean fold increase of titers in the HT-29 cell assay was 4.4 (±2.6 [SD]) (Figure 4A). To test whether these increased titers resulted from the detection of anti-VP8* antibodies in the HT-29 cell–based assay, we preincubated the adult serum samples with the indicated soluble recombinant P[8], P[4], or P[6] VP8* (10 μg/mL). The increased neutralization titers in HT-29 versus MA104 cells were completely eliminated by incubation with recombinant P[8] or P[4] VP8* (Figure 4, B and C). Interestingly, despite the fact that none of our HT-29–specific VP8*-neutralizing mAbs actually neutralized P[6] RV strains, the antigenically distinct P[6] peptide efficiently adsorbed out anti-VP8* activity from 4 of the serum samples (Figure 4D). To confirm if the addition of soluble recombinant VP8* specifically blocked anti-VP8* antibody neutralization activity in the serum specimens, we incubated recombinant P[8] or P[6] VP8* with human mAbs against VP7 (mAb27), VP5* (mAb41), or VP8* (mAb9) prior to carrying out neutralization assays against Wa in MA104 or HT-29 cells (Supplemental Figure 2). We found that VP8* mAb’s (mAb9) neutralizing activity was only eliminated by incubation with recombinant P[8], but not recombinant P[6] VP8*. VP7 or VP5* mAb neutralizing activities were not affected by the addition of recombinant VP8* (Supplemental Figure 2). Taken together, these results strongly suggested that the HT-29 cell–based assay detects human VP8*-specific neutralizing antibody responses that are underestimated or not detected at all by the conventional MA104 cell neutralization assay.
Figure 4. Effect of soluble VP8* on RV neutralization titers of normal adult human sera in HT-29 and MA104 cells.

Diluted normal adult serum samples (in duplicate) were preincubated with or without soluble recombinant VP8*s (10 μg/mL) for 1 hour at 37°C and incubated with Wa (G1, P[8]) for an additional hour. The mixtures were then added to MA104 or HT-29 cells for infection (1-hour adsorption at 37°C and 16-hour incubation). Focus-forming units (FFUs) of RV were measured by immunostaining using rabbit polyclonal anti-RV antibody 16 hours after infection. Focus reduction titer was defined as the maximum serum dilution that resulted in a 50% or more focus reduction. The data shown are representative of 2 independent experiments of similar results. (A) Without soluble VP8* preincubation. The neutralization titer difference between MA104 and HT-29 cells was statistically significant (P = 0.04 by Student’s t test of means of log2-transformed titers. (B) With P[8] VP8* preincubation. (C) With P[4] VP8* preincubation. (D) With P[6] VP8* preincubation.
Comparative MA104 cell and HT-29 cell neutralization titers in infant sera from R1 RV vaccine studies in India and the US.
Most published vaccine studies have used MA104 cell–based neutralization assays to evaluate vaccine immunogenicity and to look for correlates of protection (11, 22, 23). Our findings strongly suggested that the MA104 cell–based assay significantly underestimates neutralization activity by failing to detect neutralizing anti-VP8* antibodies in adults. Therefore, we next examined if HT-29 cells can also be used to detect anti-VP8* antibody neutralizing activity in infant serum samples collected after R1 RV (Rotarix) vaccination. Neutralization titers in infant serum samples collected from Indian and US R1 RV vaccine studies were examined in MA104 and HT-29 cells (Figure 5). In the Indian serum samples (Figure 5A), the median MA104 neutralization titer against the Wa RV strain was 20, while it was 40 in the HT-29 cell–based assay (P = 0.01 by t test of means of log2-transformed titers). The average fold neutralization titer increase in HT-29 versus MA104 cells was 2.8 (±2.2). In the US infant samples (Figure 5B), the median MA104 neutralization titer versus Wa was 160, while it was 480 in the HT-29 cell–based assay (P = 0.01 by t test of means of log2-transformed titers). The average fold increase in neutralization titer in the HT-29 cell assay was 4.3 (±5.8).
Figure 5. Neutralization titers of post–RV1 vaccination serum samples from Indian or US infants measured in MA104 or HT-29 cells.

Diluted infant serum samples (in duplicate) and Wa (G1P[8]) mixtures were added to MA104 or HT-29 cells and incubated at 37°C. At 16 hours after infection, RV FFUs were determined by immunostaining using a rabbit polyclonal anti-RV antibody. Focus reduction titers were defined as the maximum serum dilution that resulted in a 50% or greater focus reduction. (A) Infant serum samples from India RV1 vaccine study (45). (B) Infant serum samples from US RV vaccine study (46, 47). Infant serum samples from the US were tested 2 times with similar results and infant serum samples from India were tested only once due to small sample volumes. The neutralization titer differences between MA104 and HT-29 cells were statistically significant (P = 0.01 by t test of means of log2-transformed titers for both Indian and US samples).
Crystal structure of scFv9 in complex with P[4] VP8*.
To better understand the structural basis of human VP8* mAb interaction with VP8* and to potentially gain insight into the mechanism by which these human VP8* mAbs inhibit RV replication, we generated a single-chain fragment variable (scFv) construct of P[8] and P[4] VP8*–reactive mAb9 by connecting the variable domains of the heavy (VH) and light chains (VL) with a flexible linker for structural studies (24). We determined the cocrystal structure of P[4] VP8* in complex with scFv9 to a resolution of 2.4 Å (Supplemental Table 1). Three VP8* and 3 scFv9 molecules were found in the crystallographic asymmetric unit, with each scFv9 binding to 1 VP8*. No density for the flexible linker was observed. P[4] VP8* displays a galectin-like fold and the VH and VL exhibit a typical Ig fold (Figure 6A). The hypervariable complementarity-determining regions (CDRs) of both the heavy and light chains recognize P[4] VP8*, with the heavy chain contributing the majority of the interactions. The epitope on the P[4] VP8* recognized by scFv9 is formed by residues from 4 regions: β-strands C and L; the C–D and K–L loops (residues 94–99 and 195–199); the G–H loop (residues 145–148); and the β-hairpin (residues 122–124) (Figure 6, A and B). The paratope of scFv9 consists of all CDRs: (a) Y33 and K34 of CDRL1; (b) V58 and G59 of CDRL2; (c) D98 and S101 of CDRL3; (d) S33 and T35 of CDRH1; (e) F54, S56, and S58 of CDRH2; and (f) residues 100–105 of CDRH3 (Figure 6, C–E, and Supplemental Figure 3). These CDRs of scFv9 bind to P[4] VP8* via an extensive network of hydrogen bonding, electrostatic, and hydrophobic interactions with a buried surface area of 371.9 Å2 and 545.6 Å2 contributed by VL and VH, respectively. Some of the P[4] VP8* residues in this extensive network of interactions include Q148 that forms both direct hydrogen bonds and hydrophobic interactions with K34 of CDRL1 and D98 of CDRL3 (Figure 6C) and residue N195 interacting with F54, S56, and S58 of CDRH2 (Figure 6D). In addition, 7 of the VP8* residues interact with Y101, Y102, and Y103 of CDRH3 either through direct hydrogen bonding or hydrophobic contacts (Figure 6E).
Figure 6. Crystal structure of scFv9 in complex with P[4] VP8*.

(A) The overall structure of scFv9 in complex with P[4] VP8*. The scFv9 heavy chain (VH) and light chain (VL) are colored in blue and light blue, respectively. P[4] VP8* is shown in gold. The β-sheets and C-terminal α-helix of VP8* are labeled. (B) The footprint of scFv9 on P[4] VP8*. The surface of VP8 is shown with 60% transparency, and the antibody binding residues are shown as stick models. (C–E) Molecular interactions between P[4] VP8* and CDRs of scFv9. The proteins are colored as in A. The interacting residues of P[4] VP8* and scFv9 are represented with stick models and are labeled. The hydrogen bonds and electrostatic interactions are shown using dashed black lines.
Structural basis of the genotypic specificity of human mAb9.
To investigate the molecular basis of the genotypic specificity of mAb9, we carried out structure-based sequence alignment with VP8*s of several genotypes using the program Chimera (Figure 7A). Sequence analyses show that the scFv9 binding residues are conserved in the prevalent P[4] and P[8] human RV genotypes, consistent with its genotype-specific binding and neutralizing activities (17). These residues are not conserved in other human RVs, such as P[6], P[11], and P[14], or animal RVs, including P[3] and P[7]. Because P[6] human RVs are phylogenetically closely related to P[4] and P[8] human RVs, we sought to understand why scFv9 does not recognize P[6] VP8*. Structural superimposition of P[6] VP8* with the P[4] VP8*/scFv9 complex reveals that there are several critical sequence changes in P[6] VP8* that lead to the loss of antibody binding (Figure 7, B and C). The residue Q148 in P[4] VP8*, which interacts with CDRL1, is changed to S148 in P[6] (Figure 7B), and residue N98, which interacts with 3 of the CDRs via hydrogen bonding and hydrophobic interactions, is mutated to K98 in P[6] VP8* (Figure 7C). Furthermore, P[6] VP8* contains several mutations in the amino acid sequence 195–199 that also contribute to the loss of binding to scFv9.
Figure 7. Structural basis of the genotypic specificity of human mAb9.

(A) Structure-based sequence alignment of VP8*s. The scFv9 and the known glycan binding sites in VP8*s are denoted in different colors: the scFv9 binding residues are shown in blue, the H-type 1 HBGA binding site in gray, the precursor HBGA binding residues in red, and the A-type HBGA-interacting residues in P[14] and the sialic acid binding residues in P[3] and P[7] in green. The PDB ID is given for each structure. (B and C) Superposition of scFv9/P[4] VP8* structure with that of P[6] RV3 VP8* (PDB ID: 5VX8) showing how the sequence changes in P[6] VP8* abrogates its binding to scFv9. The P[4] and P[6] VP8*s are colored in green and yellow, respectively; the VL and VH of scFv9 are shown in light and dark blue, respectively.
ScFv9 does not bind to the glycan receptor binding site on VP8*.
Recent biochemical, epidemiological, and structural studies have shown that P[4] VP8* binds to H-type 1 human histo-blood group antigen (HBGA) that may act as a cell attachment factor during RV entry (25). To investigate whether mAb9 neutralizes RV infections by blocking the HBGA binding site, we superimposed the P[4] VP8*/scFv9 structure onto the structure of the P[4] VP8*/H-type 1 HBGA complex (Figure 8A). The structural comparison shows that scFv9 binds to a site distant from the HBGA binding site, indicating that the mechanism by which mAb9 neutralizes the virus is not by blocking the glycan binding. Further structure-based sequence comparisons with VP8*s of other genotypes reveal that scFv9 epitope is also distinct from other known glycan binding sites on VP8*, such as the precursor HBGA binding site on P[11] VP8* and the sialic acid binding site on P[3] and P[7] VP8*s (Figure 8A). We modeled the binding of mAb9 in the context of an RV TLP (Figure 8B). Consistent with X-ray crystallography studies, mAb9 did not block the binding of P[8] VP8* to H-type I HBGA (data not shown).
Figure 8. Structural basis for how mAb9 may interact with VP8*.

(A) Superimposition of scFv9/P[4] VP8* structure onto the VP8* domain of the VP4 spike in the P[3] RRV structure (PDB ID: 4V7Q) along with the superposition of the structure of P[4] VP8* in complex with H-type 1 HBGA (PDB ID: 5VX5) to show the HBGA binding site. The glycan molecules are presented as ball-and-stick models in green. The inset shows a close-up view of the VP8* domain. (B) Modeling of binding of an IgG to VP4 spike on an RV TLP based on the scFv9/P[4] VP8* structure. The crystal structure of an IgG (PDB ID: 1IGT) molecule is docked near the VP4 spikes to show that 1 mAb9 molecule can bind to adjacent VP4 spikes separated by approximately 150 Å, around the icosahedral 5-fold axis on the TLP.
Discussion
Numerous prior studies, using both hyperimmune and/or monoclonal anti-RV antibodies generated in various animal species, demonstrated that antibodies against both VP7 and VP4 were able to neutralize RVs (1, 26–28). Depending on the specific antibody, the neutralization specificity could be either highly strain (serotype) specific or heterotypic to a greater or lesser degree (29). For VP4-directed antibodies, both VP8* and VP5* were identified as targets of neutralization, with VP8* generally being a more type-specific target and VP5*-directed antibodies being more heterotypic in their neutralization specificity (18). It has been more difficult to directly examine the protein specificity of neutralizing responses in humans, but experiments employing reassortant RVs, and one case of an experiment in which 3 human mAbs obtained through a phage display library were examined, generally confirmed the results derived from animal studies (30, 31). Recently we were able to isolate a modest-sized library of human-derived anti–RV TLP mAbs from small intestinal B cells of adults undergoing bariatric surgery (17). A very surprising observation from that study was the finding that most (20 of 24) of the VP8*-directed mAbs isolated did not neutralize RVs when assayed in the standard MA104 monkey kidney cell assay.
In the current study, we took advantage of the recent availability of a human small intestinal enteroid culture system to reexamine our library of human anti-RV mAbs and to compare enteroid culture neutralization results with those of a traditional MA104 cell assay. While the mAbs against VP7 and VP5* neutralized RV efficiently in both cell substrates, we found that most of the previously “non-neutralizing” human VP8*-specific mAbs now efficiently neutralized RVs in the enteroids, either cultivated as monolayers or as 3D spherules (Figures 1 and 2 and Table 1). As previously noted by others (ref. 20 and S. Blutt, S. Crawford, and M. Estes, personal communication), when polarized enteroid monolayers are infected with RV, infection occurs much more efficiently from the basolateral than the apical surface. However, infection at both surfaces was neutralized with similar efficiency by mAbs directed against VP8*, as well those against VP5* and VP7. Whether the basolateral specificity for infection found in the enteroid system accurately recapitulates what takes place in vivo remains to be determined. However, it does appear likely that whatever the mechanism is by which the VP8* mAbs inhibit RV infection, it is similar on both cell surfaces.
The human enteroid culture system proved very sensitive for detecting previously undetectable neutralizing activity in our human VP8* mAb collection. However, the enteroid system was not easily adapted to large numbers of serologic assays and their repetition. We therefore examined several other commonly used cell lines as possible substitutes for the enteroid monolayer system (Figure 2 and Table 2). Two other commonly used kidney cell lines (CV-1 and MA104) of monkey origin were similar to human HEK293 cells, in that we could not detect neutralizing activity in any of our previously negative VP8*-specific mAbs. Interestingly, we did detect neutralizing activity in some of the VP8*-directed mAbs using the human intestine–derived CaCo2 cell line, but the activity was much less efficient than that observed with the HT-29 cells (Table 2). Given these findings, we concluded that HT-29 cells were the best cell substrate available to more fully examine the role of human anti-VP8* immunity in RV infection and immunization.
As reported for the binding specificity of the VP8* mAbs (17), the neutralization specificity was also found to be primarily P-genotype specific, and in this case, directed primarily at P[8] human RV strains. This specificity is not unexpected considering the fact that the labeled RV TLP used to initially select the RV-directed enteric B cells for this study was a P[8] strain (17). However 4 of 22 VP8* mAbs also neutralized the prototypic DS1 P[4] strain, while none neutralized a P[6] RV (Table 2 and data not shown). These findings are consistent with, and would be supportive of, the recent change to a multivalent formulation of the candidate VP8* recombinant RV vaccine currently undergoing clinical trials (32, 33). Of interest, the VP8* mAbs did not neutralize P[6] strains efficiently and neutralization activity was not affected by preincubation with recombinant P[6] VP8* (Supplemental Figure 2). However, the recombinant P[6] VP8* was able to adsorb out VP8*-specific neutralizing activity from several adult sera (Figure 4D). A plausible explanation for this finding is that the serum specimens we studied almost certainly contained a much wider VP8* serotypic repertoire than our limited sampling of human VP8* mAbs. We hypothesize that found within some serum samples there are neutralizing antibodies with [P]6 binding ability that can also neutralize [P]8 or [P]4 RVs and these antibodies are adsorbed by the added recombinant P[6]. The isolation of an expanded library of human anti-VP8* mAbs will be needed to test this hypothesis.
A variety of animal studies indicated that VP8*-directed antibodies can protect animals from experimental RV challenge (29). It has been more difficult to directly evaluate the role of VP8*-specific immunity in people. We tested the in vivo activity of human anti-VP8* antibodies using a murine RV model of heterologous human RV–induced diarrhea (17). We found that 2 representative human VP8* mAbs significantly suppressed human P[8] RV–associated diarrhea following challenge, indicating that the in vitro ability to suppress human RV replication of these mAbs correlated with an in vivo suppression of diarrhea in a small animal model system (Figure 3) and providing experimental support for the rationale of testing a recombinant RV VP8* vaccine in humans.
We next evaluated the utility of the HT-29 cell neutralization test for measuring the overall neutralization potency of anti-RV antibodies in normal adult sera (Figure 4). Our findings demonstrate that a substantial fraction of overall neutralization capacity in these normal US adult sera specimens (50% to 88%, average 69%) appeared to be contributed by antibodies against VP8* and, therefore, could be adsorbed out of specific serum specimens using bacterially expressed fragments of either a P[8] or a P[4] VP8*. Following VP8* adsorption, neutralization titers in HT-29 cells and MA104 cells were equalized, suggesting that the inequality in titers prior to adsorption was likely to be specifically accounted for by antibodies against VP8*. Interestingly, although recombinant P[4] quite efficiently removed VP8*-neutralizing activity from adult sera, our human VP8* mAbs did not neutralize a prototype P[4] RV strain very efficiently (Table 2). The precise reason for this difference is currently unknown but likely relates to the affinity of the mAbs for P[8] versus P[4] or P[6] proteins. Of interest, all the human mAbs in our library were initially selected for their ability to bind to a P[8] TLP. Therefore, these VP8* mAbs might represent a biased selection sample of what is actually present in the total normal adult VP8* repertoire.
We also thought it useful to examine the role of anti-VP8* antibodies in mediating neutralization in 2 quite different sets of sera from children in the US or India after R1 RV (G1, P[8]) vaccination (Figure 5). If we assume, as with the adult sera, that the majority of the difference in neutralization titer between HT-29 cells and MA104 cells can be accounted for by anti-VP8* antibody detection present only in the HT-29 cells, then in the Indian samples (0% and 88%, average 31%) and in the US samples (0% and 94%, average 59%) all of the neutralization activity measured in the post–R1 vaccination HT-29 cell assays was due to anti-VP8* reactivity. This observation, if correct, has some potentially important implications. We examined 20 RV vaccine trials published between 1996 and 2017 that contained neutralization-testing data using a variety of neutralization assay protocols. All carried out their neutralization assays using an MA104 cell system. Fifty percent of these studies attempted to correlate neutralization titer responses with vaccine efficacy. Our preliminary findings suggest that the neutralization-response data previously obtained using the MA104-based assay may have significantly underestimated actual responses. Perhaps some of these past vaccine studies can now be reexamined using an HT-29 or other neutralization assay that more reliably detects anti-VP8* responses. It will then be interesting to determine if HT-29–based neutralization data correlate better with protection outcomes than the current MA104-based neutralization data.
It is challenging to speculate as to the mechanistic basis for the difference between the neutralization results obtained in the MA104 and HT-29 cell–based assays. Prior studies using murine anti-VP8* mAbs and MA104 cells indicated that these anti-VP8* mouse mAbs functioned primarily by inhibiting binding of the simian rhesus RV (RRV) strain to an MA104 cell monolayer. VP8* is the RV hemagglutinin and is responsible for RBC binding (34, 35). If our current library of VP8* mAbs are also mediating neutralization by inhibiting human RV binding to HT-29 cells, then it seems reasonable to postulate that MA104 cells have an RV receptor that efficiently mediates infection, is not blocked by the current set of human mAbs, and that this receptor is not present on HT-29 cells. Studies are currently underway to explore this and other mechanistic hypotheses that might account for the RV VP8* neutralization differences observed between MA104 and HT-29 cells.
Recently, the non-sialylated human HBGAs have been found to bind to VP8* and proposed as cellular receptors for human RVs (36). Status of HBGA expression (secretor or nonsecretor status) has been clearly associated with RV susceptibility (37–40) and vaccine “take” (41, 42) in several recent studies. The sites of VP8*/HBGA interactions have recently been identified (25, 35). However, neutralizing antibodies that function by blocking VP8* interactions with HBGA were not well documented in people after natural infection or vaccination, unlike what has been observed for norovirus (43). In this study we find that, for at least 1 neutralizing human VP8* mAb (mAb9), the HBGA binding site and the VP8* mAb binding sites do not overlap (Figure 8) and this mAb does not block VP8* binding to HBGA, suggesting that the RV-neutralizing effects of this VP8*-directed antibody are unlikely to be due to inhibition of the HBGA/RV interaction. Detailed analysis of HBGA binding to VP8* and the potential interference of VP8* mAbs with RV/HBGA interactions, along with delineation of the mechanisms of neutralization mediated by VP8* mAbs are important issues that will require additional studies.
Methods
Cells, RV strains, human RV–specific mAbs, human serum samples, and recombinant VP8* reagents.
The MA104 monkey kidney cell line was maintained in medium 199 (M199) (Gibco). The CV1 monkey kidney cell line, human kidney HEK293 cell line, and human colonic HT-29 epithelial cell line were maintained in Advanced DMEM/F12 (Adv DMEM) (Gibco). The human colonic cell line CaCo-2 was maintained in DMEM (Gibco). All these cell lines were originally obtained from ATCC. All the above media were supplemented with L-glutamine, penicillin and streptomycin (Corning), and 10% fetal bovine serum (FBS) (Gibco). Propagation and titer determination of human rotaviruses Wa (G1, P[8]), WI61 (G9, P[8]), and DS1 (G2, P[4]) were as described previously (44). The RV protein specificity and neutralization activity in MA104 cells of the recombinant human anti-RV mAbs in this study were previously described (17). Normal adult serum samples were obtained from the Stanford Blood Center. Antibody levels in these adult sera in general reflect antibody responses after natural RV infection since the current RV vaccines were not licensed in the US until 2006 to 2008. Indian infant post–RV vaccination serum specimens were collected at 14 weeks of age (4 weeks after vaccination) and US infant post–RV vaccination serum specimens were collected at 24–38 weeks of age (4–6 weeks after vaccination) as part of prior RV (G1, P[8]) vaccine studies in India and the US (45–47). All adult and infant sera used in this study were deidentified. The human vaccine studies in which these samples were originally collected were approved by the relevant institutional review boards. The recombinant P[8], P[4], and P[6] VP8*s were derived from Wa (G1, P[8]), DS1 (G2, P[4]), and 1076 (G2, P[6]), respectively, as previously described (48) and were a gift from the Program for Appropriate Technology in Health (PATH) (Seattle, Washington, USA). These bacterially expressed VP8*s were originally developed as part of an alternative parenterally administered RV vaccine program (48).
Ileal enteroid culture.
Ileal enteroids were derived from deidentified intestinal tissues from secretor patients collected after pediatric endoscopy and were generated as part of an NIH-funded U19 grant program at Stanford. Ileal enteroids were grown as 3D cultures in Matrigel matrix (BD Bioscience) using enteroid growth media (49). Methods for making enteroid monolayer cultures were previously described (50). Briefly, Matrigel was dissolved using 0.5 mM EDTA in Ca++- and Mg++-free PBS at 4°C for 1 hour and enteroids were isolated. Enteroids were digested with trypsin-EDTA (Gibco) for 10 minutes to obtain a single-cell suspension. After washing, 5 × 105 cells were added to 33 μg/mL collagen IV–coated (MilliporeSigma) 0.33 cm2, 0.4-μm polycarbonate transwell plates (Costar). The enteroid monolayer cultures were grown in enteroid differentiation media consisting of enteroid growth media without WNT and R-spondin at 37°C and 10% CO2. RV infection and neutralization assays were performed when the transepithelial electrical resistance (TEER) of the culture exceeded 3000 Ω/cm2.
Neutralization assay.
General methods for RV neutralization assays have been previously described and the method used here was similar for MA104, HT-29, CaCo-2, CV-1, and HEK293 cells (44). Briefly, serially diluted human RV–specific mAbs or serum specimens from adults or infants were incubated with indicated RV strains at 37°C for 1 hour. The mixtures were then added to cells in 96-well plates (Costar) for a 1-hour adsorption at 37°C. Cells were washed and medium without FBS was added back to the cells for an additional 16-hour incubation. The cells were fixed with 10% formalin and stained with rabbit polyclonal anti-RV antibody and followed by peroxidase-conjugated goat anti–rabbit IgG (γ chain specific [MilliporeSigma]). A color reaction was developed using the 3-amino-9-ethylcarbozole (AEC) substrate (Vector). Neutralization titers were expressed as the minimum mAb concentration (ng/mL) or maximum dilution of sera that resulted in at least a 50% reduction in the number of foci as compared with a no-antibody control. Because RV infectivity was highest in MA104 cells and significantly lower in the other cell lines used in this study, using the same dose of RV as used for MA104 cells for neutralization assays would have resulted in too few foci to evaluate in the other cell lines. We generally added 5 to 10 times more virus to HT-29, CaCo-2, CV-1, and HEK293 cells to reach a similar infected-cell density as in the MA104 cells so that neutralization titers could be calculated accurately and consistently.
RV neutralization assays in 3D enteroid cultures were performed by first isolating enteroids from Matrigel using 0.5 mM EDTA, as described above. Isolated enteroids were washed and then incubated with an RV and mAb (100 ng/mL) mixture in Adv DMEM without FBS for 1 hour at 37°C. Enteroids were then washed again and re-embedded in Matrigel for an additional 16-hour incubation at 37°C in enteroid growth media. RV neutralization using enteroid monolayer cultures was performed in transwell plates after TEER was above 3000 Ω/cm2. RV and mAb mixtures were added to the lower chamber of the transwells for 1 hour. Cells were washed and differentiation media was added back for an additional 16-hour incubation at 37°C and 10% CO2. Enteroid cells in both 3D or monolayer cultures and the cell lines listed in Figures 1 and 2 were then fixed and stained with FITC-labeled rabbit polyclonal anti-RV antibody (green), Texas red–labeled phalloidin (red), and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Cells were observed and images acquired using a Keyence BZ-X710 All-in-one fluorescence microscope.
Soluble recombinant VP8* competition assay.
To investigate whether the quantity of human serum neutralization activity detected in HT-29 cells differed from that detected in MA104 cells and whether the difference was mediated by anti-VP8* antibodies, we first preincubated serially diluted human sera specimens with soluble recombinant P[8], P[4], or P[6] VP8* (10 μg/mL) for 1 hour at 37°C. Wa RV was then added to the mixture for 1 hour at 37°C. The Wa/serum/recombinant VP8* mixtures were added to MA104 or HT-29 cell monolayers in 96-well plates for a 1-hour adsorption at 37°C and then incubated for an additional 16 hours, as described above. Cells were then immunostained and neutralization titers determined as described above. For confirmation of the specificity of recombinant VP8* to inhibit VP8*-directed neutralization activity, we preincubated serially diluted human mAbs against VP7 (mAb27), VP5* (mAb41), or VP8* (mAb9) with soluble P[8] or P[6] VP8* (10 μg/mL) and performed neutralization assays against Wa in MA104 and HT-29 cells, as described above.
VP8* human mAb passive protection studies.
Human recombinant VP8* mAb passive protection studies were done as described previously (17). Briefly, VP8*-specific mAb11 or mAb14 (250 ng/mL) was incubated with human RV WI61 (1 × 106 PFU per mouse) for 1 hour at 37°C. The mixtures were then orally administered by gavage to 5-day-old suckling 129sv mice. At indicated days after infection mice were observed for diarrhea as previously described (51).
Expression and purification scFv9 and VP8* proteins.
The cDNA of scFv9 was synthesized with a 15–amino acid linker, (GGGGS)3, connecting the VH and VL domains of mAb9 (24). The synthesized gene was subcloned into the bacterial expression vector pET28 with an N-terminal His tag followed by a TEV protease cleavage site (Epoch Life Science). Escherichia coli Shuffle cells (NEB) were transformed with the scFv9/pET28 plasmid and induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (MilliporeSigma) when the optical density at 600 nm reached 0.6. Cells were resuspended in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 10 mM imidazole, supplemented with protease inhibitor cocktail (Roche). Cells were lysed using a microfluidizer (Microfluidics), followed by removal of cell debris by centrifugation at 39,000 g for 30 minutes at 4°C. His-tagged scFv9 was bound to Ni-NTA Agarose (Qiagen) and eluted using a gradient of 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 250 mM imidazole. The eluted His-tagged scFv9 was concentrated using a 10-kDa centrifugal filter unit (Millipore) and dialyzed into 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 10 mM imidazole for His-TEV protease cleavage overnight at 4°C. The cleaved protein mixture was reloaded onto the Ni-NTA column to remove the His-TEV protease and uncleaved His-scFv9. The human rotavirus G2P[4] VP8* was expressed in E. coli BL21(DE3) cells (Novagen) and purified with a Glutathione Sepharose 4 Fast Flow (GE Healthcare) affinity column as previously described (25).
Production of scFv9/P[4] VP8* complex and crystallization.
The scFv9/P[4] VP8* complex was formed by incubation at a molar ratio of 1:1.2 in a buffer containing 10 mM HEPES (pH 8.0) and 150 mM NaCl at 4°C overnight before loading onto a HiLoad 16/60 Superdex 75 prep-grade column (GE Healthcare). The fractions with the scFv9/P[4] VP8* complex were collected and concentrated to 8 mg/mL in 10 mM HEPES (pH 8.0) and 150 mM NaCl for crystallization. The scFv9/P[4] VP8* crystals were grown at 20°C by hanging-drop vapor diffusion using the Mosquito crystallization robot (TTP LabTech) and imaged using a Rock Imager (Formulatrix). Each drop contained 0.2 μL of scFv9/P[4] VP8* and 0.2 μL of crystallization buffer containing 100 mM HEPES (pH 7.0) and 20% w/v PEG 8000. The crystals were transferred into cryoprotectant solution with 20% glycerol and flash-frozen in liquid nitrogen.
Data processing, structure determination, and refinement.
X-ray diffraction data for scFv9 complexed with P[4] VP8* were collected on the Beamline 5.0.1 at Advanced Light Source, Lawrence Berkeley National Laboratory. Diffraction data were processed using the program HKL2000 (52). The native structure of human rotavirus G2P[4] VP8* (PDB ID: 5VX4) and the predicted structure of scFv9 as generated by the antibody modeling software ABodyBuilder (53) were used as search models for molecular replacement using PHASER (54). Iterative cycles of refinement were carried out using PHENIX (55) and further model building using COOT based on the difference maps (56). Data collection and refinement statistics following the final refinement cycle are given in Supplemental Table 1. The interactions between P[4] VP8* and scFv9 were analyzed using the program LigPlot+ v.2.1 (57). The figures were prepared with Chimera (58).
Statistics.
Statistical analysis was performed using SPSS Statistics 21 (IBM). Neutralization titers of human sera in MA104 or HT-29 cells were log2 transformed and analyzed using a 2-tailed Student’s t test. Percentages of diarrheal disease in suckling mice in the passive protection study were analyzed using Fisher’s exact test. A P value less than 0.05 was considered significant.
Study approval.
129sv mice were bred in-house at the Veterinary Medical Unit of the Palo Alto VA Health Care System (PAVAHCS). The animal study was approved by the Institutional Animal Care and Use Committee (IACUC) at PAVAHCS. Infant sera in this study were obtained as deidentified samples from vaccine studies of India (45) and the US (46, 47). The human vaccine studies were approved by the IRBs of the respective institutions.
Author contributions
Functional studies on VP8* mAb and human serum intestinal cell–specific neutralization were primarily performed by NF with significant contributions from SD. LLY and YS provided important assistance. The structural studies of VP8* interaction with HBGA and VP8* mAb were primarily performed by LH, BZ, and BS. MS managed the recombinant human mAb stocks. SR and MM provided critical serum samples from vaccine studies in India and the US and valuable advice for this study. BVVP and HBG as senior authors provided guidance for the experimental design and analysis of this study. NF, LH, and HBG wrote the manuscript. All authors read and revised the manuscript.
Supplementary Material
Acknowledgments
We thank Mary Estes, Sarah Blutt, and Sue Crawford (Baylor College of Medicine, Houston, Texas, USA) for their invaluable advice on human enteroid culture. This work was supported by NIH grants R01 AI125249 and U19 AI116484 and by VA Merit grant GRH0022 awarded to HBG, and by NIH R01 AI36040 and the Robert Welch Foundation Q1279 to BVVP. SD is supported by an Early Career Award from the Thrasher Research Fund and NIH grant K99 AI135031.
Version 1. 08/12/2019
Electronic publication
Version 2. 08/13/2019
Corrected B.V. Venkataram Prasad's name.
Version 3. 09/03/2019
Print issue publication
Footnotes
Conflict of interest: HBG consults for and is on the advisory board of Aridis, a biotech company that has done preclinical research on novel delivery systems for rotavirus vaccines. He has provided educational lectures for Abbot, a pharmaceutical company that markets a licensed rotavirus vaccine in India.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(9):3839–3851.https://doi.org/10.1172/JCI128382.
Contributor Information
Ningguo Feng, Email: ngf@stanford.edu.
Liya Hu, Email: lhu@bcm.edu.
Siyuan Ding, Email: syding@stanford.edu.
Mrinmoy Sanyal, Email: msanyal@stanford.edu.
Boyang Zhao, Email: boyangz@bcm.edu.
Banumathi Sankaran, Email: BSankaran@lbl.gov.
Sasirekha Ramani, Email: ramani@bcm.edu.
Monica McNeal, Email: monica.mcneal@cchmc.org.
Linda L. Yasukawa, Email: llew@stanford.edu.
Yanhua Song, Email: syh85@stanford.edu.
References
- 1. Estes MK, Greenberg HB. Rotaviruses. In: Fields Virology. Knipe DM, Howley HM, eds. Philadelphia, USA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013:1347–401. [Google Scholar]
- 2.Estes MK, Graham DY, Mason BB. Proteolytic enhancement of rotavirus infectivity: molecular mechanisms. J Virol. 1981;39(3):879–888. doi: 10.1128/jvi.39.3.879-888.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arias CF, Silva-Ayala D, López S. Rotavirus entry: a deep journey into the cell with several exits. J Virol. 2015;89(2):890–893. doi: 10.1128/JVI.01787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velázquez FR, et al. Rotavirus infection in infants as protection against subsequent infections. N Engl J Med. 1996;335(14):1022–1028. doi: 10.1056/NEJM199610033351404. [DOI] [PubMed] [Google Scholar]
- 5.Choi NW, Estes MK, Langridge WH. Oral immunization with a shiga toxin B subunit: rotavirus NSP4(90) fusion protein protects mice against gastroenteritis. Vaccine. 2005;23(44):5168–5176. doi: 10.1016/j.vaccine.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Feng N, et al. Inhibition of rotavirus replication by a non-neutralizing, rotavirus VP6-specific IgA mAb. J Clin Invest. 2002;109(9):1203–1213. doi: 10.1172/JCI14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization–Coordinated Global Rotavirus Surveillance Network Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis. 2016;62(Suppl 2):S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward RL, Clark HF, Offit PA. Influence of potential protective mechanisms on the development of live rotavirus vaccines. J Infect Dis. 2010;202(Suppl):S72–S79. doi: 10.1086/653549. [DOI] [PubMed] [Google Scholar]
- 9.Velasquez DE, Parashar U, Jiang B. Decreased performance of live attenuated, oral rotavirus vaccines in low-income settings: causes and contributing factors. Expert Rev Vaccines. 2018;17(2):145–161. doi: 10.1080/14760584.2018.1418665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desselberger U. Differences of rotavirus vaccine effectiveness by country: likely causes and contributing factors. Pathogens. 2017;6(4):E65. doi: 10.3390/pathogens6040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angel J, Franco MA, Greenberg HB. Rotavirus vaccines: recent developments and future considerations. Nat Rev Microbiol. 2007;5(7):529–539. doi: 10.1038/nrmicro1692. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein DI, Glass RI, Rodgers G, Davidson BL, Sack DA. Evaluation of rhesus rotavirus monovalent and tetravalent reassortant vaccines in US children. US Rotavirus Vaccine Efficacy Group. JAMA. 1995;273(15):1191–1196. [PubMed] [Google Scholar]
- 13.Ward RL, Bernstein DI. Lack of correlation between serum rotavirus antibody titers and protection following vaccination with reassortant RRV vaccines. US Rotavirus Vaccine Efficacy Group. Vaccine. 1995;13(13):1226–1232. doi: 10.1016/0264-410X(95)00060-E. [DOI] [PubMed] [Google Scholar]
- 14.Ward RL, et al. Evidence that protection against rotavirus diarrhea after natural infection is not dependent on serotype-specific neutralizing antibody. J Infect Dis. 1992;166(6):1251–1257. doi: 10.1093/infdis/166.6.1251. [DOI] [PubMed] [Google Scholar]
- 15.Estes MK, Graham DY, Gerba CP, Smith EM. Simian rotavirus SA11 replication in cell cultures. J Virol. 1979;31(3):810–815. doi: 10.1128/jvi.31.3.810-815.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitamoto N, Ramig RF, Matson DO, Estes MK. Comparative growth of different rotavirus strains in differentiated cells (MA104, HepG2, and CaCo-2) Virology. 1991;184(2):729–737. doi: 10.1016/0042-6822(91)90443-F. [DOI] [PubMed] [Google Scholar]
- 17.Nair N, et al. VP4- and VP7-specific antibodies mediate heterotypic immunity to rotavirus in humans. Sci Transl Med. 2017;9(395):eaam5434. doi: 10.1126/scitranslmed.aam5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackow ER, Shaw RD, Matsui SM, Vo PT, Dang MN, Greenberg HB. The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutralization and identification of a putative fusion region. Proc Natl Acad Sci U S A. 1988;85(3):645–649. doi: 10.1073/pnas.85.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96(3):736–749. doi: 10.1016/0016-5085(89)90897-4. [DOI] [PubMed] [Google Scholar]
- 20.Cevallos Porta D, López S, Arias CF, Isa P. Polarized rotavirus entry and release from differentiated small intestinal cells. Virology. 2016;499:65–71. doi: 10.1016/j.virol.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Coulson BS, Kirkwood C. Relation of VP7 amino acid sequence to monoclonal antibody neutralization of rotavirus and rotavirus monotype. J Virol. 1991;65(11):5968–5974. doi: 10.1128/jvi.65.11.5968-5974.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franco MA, Angel J, Greenberg HB. Immunity and correlates of protection for rotavirus vaccines. Vaccine. 2006;24(15):2718–2731. doi: 10.1016/j.vaccine.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 23.Angel J, Franco MA, Greenberg HB. Rotavirus immune responses and correlates of protection. Curr Opin Virol. 2012;2(4):419–425. doi: 10.1016/j.coviro.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yusakul G, Sakamoto S, Pongkitwitoon B, Tanaka H, Morimoto S. Effect of linker length between variable domains of single chain variable fragment antibody against daidzin on its reactivity. Biosci Biotechnol Biochem. 2016;80(7):1306–1312. doi: 10.1080/09168451.2016.1156482. [DOI] [PubMed] [Google Scholar]
- 25.Hu L, et al. Glycan recognition in globally dominant human rotaviruses. Nat Commun. 2018;9(1):2631. doi: 10.1038/s41467-018-05098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoshino Y, Saif LJ, Sereno MM, Chanock RM, Kapikian AZ. Infection immunity of piglets to either VP3 or VP7 outer capsid protein confers resistance to challenge with a virulent rotavirus bearing the corresponding antigen. J Virol. 1988;62(3):744–748. doi: 10.1128/jvi.62.3.744-748.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Offit PA, Clark HF, Blavat G, Greenberg HB. Reassortant rotaviruses containing structural proteins vp3 and vp7 from different parents induce antibodies protective against each parental serotype. J Virol. 1986;60(2):491–496. doi: 10.1128/jvi.60.2.491-496.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Offit PA, Shaw RD, Greenberg HB. Passive protection against rotavirus-induced diarrhea by monoclonal antibodies to surface proteins vp3 and vp7. J Virol. 1986;58(2):700–703. doi: 10.1128/jvi.58.2.700-703.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui SM, et al. Passive protection against rotavirus-induced diarrhea by monoclonal antibodies to the heterotypic neutralization domain of VP7 and the VP8 fragment of VP4. J Clin Microbiol. 1989;27(4):780–782. doi: 10.1128/jcm.27.4.780-782.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higo-Moriguchi K, Akahori Y, Iba Y, Kurosawa Y, Taniguchi K. Isolation of human monoclonal antibodies that neutralize human rotavirus. J Virol. 2004;78(7):3325–3332. doi: 10.1128/JVI.78.7.3325-3332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward RL, McNeal MM, Sander DS, Greenberg HB, Bernstein DI. Immunodominance of the VP4 neutralization protein of rotavirus in protective natural infections of young children. J Virol. 1993;67(1):464–468. doi: 10.1128/jvi.67.1.464-468.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fix AD, et al. Safety and immunogenicity of a parenterally administered rotavirus VP8 subunit vaccine in healthy adults. Vaccine. 2015;33(31):3766–3772. doi: 10.1016/j.vaccine.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Groome MJ, et al. Safety and immunogenicity of a parenteral P2-VP8-P[8] subunit rotavirus vaccine in toddlers and infants in South Africa: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2017;17(8):843–853. doi: 10.1016/S1473-3099(17)30242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiore L, Greenberg HB, Mackow ER. The VP8 fragment of VP4 is the rhesus rotavirus hemagglutinin. Virology. 1991;181(2):553–563. doi: 10.1016/0042-6822(91)90888-I. [DOI] [PubMed] [Google Scholar]
- 35.Hu L, et al. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature. 2012;485(7397):256–259. doi: 10.1038/nature10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramani S, Hu L, Venkataram Prasad BV, Estes MK. Diversity in rotavirus-host glycan interactions: a “sweet” spectrum. Cell Mol Gastroenterol Hepatol. 2016;2(3):263–273. doi: 10.1016/j.jcmgh.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imbert-Marcille BM, et al. A FUT2 gene common polymorphism determines resistance to rotavirus A of the P[8] genotype. J Infect Dis. 2014;209(8):1227–1230. doi: 10.1093/infdis/jit655. [DOI] [PubMed] [Google Scholar]
- 38.Nordgren J, et al. Both Lewis and secretor status mediate susceptibility to rotavirus infections in a rotavirus genotype-dependent manner. Clin Infect Dis. 2014;59(11):1567–1573. doi: 10.1093/cid/ciu633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne DC, et al. Epidemiologic association between FUT2 secretor status and severe rotavirus gastroenteritis in children in the United States. JAMA Pediatr. 2015;169(11):1040–1045. doi: 10.1001/jamapediatrics.2015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Trang N, Vu HT, Le NT, Huang P, Jiang X, Anh DD. Association between norovirus and rotavirus infection and histo-blood group antigen types in Vietnamese children. J Clin Microbiol. 2014;52(5):1366–1374. doi: 10.1128/JCM.02927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee B, et al. Histo-blood group antigen phenotype determines susceptibility to genotype-specific rotavirus infections and impacts measures of rotavirus vaccine efficacy. J Infect Dis. 2018;217(9):1399–1407. doi: 10.1093/infdis/jiy054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kazi AM, et al. Secretor and salivary ABO blood group antigen status predict rotavirus vaccine take in infants. J Infect Dis. 2017;215(5):786–789. doi: 10.1093/infdis/jix028. [DOI] [PubMed] [Google Scholar]
- 43.Blazevic V, Malm M, Vesikari T. Induction of homologous and cross-reactive GII.4-specific blocking antibodies in children after GII.4 New Orleans norovirus infection. J Med Virol. 2015;87(10):1656–1661. doi: 10.1002/jmv.24237. [DOI] [PubMed] [Google Scholar]
- 44.Hoshino Y, Wyatt RG, Greenberg HB, Flores J, Kapikian AZ. Serotypic similarity and diversity of rotaviruses of mammalian and avian origin as studied by plaque-reduction neutralization. J Infect Dis. 1984;149(5):694–702. doi: 10.1093/infdis/149.5.694. [DOI] [PubMed] [Google Scholar]
- 45.Lazarus RP, et al. The effect of probiotics and zinc supplementation on the immune response to oral rotavirus vaccine: A randomized, factorial design, placebo-controlled study among Indian infants. Vaccine. 2018;36(2):273–279. doi: 10.1016/j.vaccine.2017.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein DI, et al. Efficacy of live, attenuated, human rotavirus vaccine 89-12 in infants: a randomised placebo-controlled trial. Lancet. 1999;354(9175):287–290. doi: 10.1016/S0140-6736(98)12106-2. [DOI] [PubMed] [Google Scholar]
- 47.Bernstein DI, et al. Safety and immunogenicity of live, attenuated human rotavirus vaccine 89-12. Vaccine. 1998;16(4):381–387. doi: 10.1016/S0264-410X(97)00210-7. [DOI] [PubMed] [Google Scholar]
- 48.Wen X, Cao D, Jones RW, Li J, Szu S, Hoshino Y. Construction and characterization of human rotavirus recombinant VP8* subunit parenteral vaccine candidates. Vaccine. 2012;30(43):6121–6126. doi: 10.1016/j.vaccine.2012.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saxena K, et al. Human intestinal enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol. 2016;90(1):43–56. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou WY, et al. Human intestinal enteroids: new models to study gastrointestinal virus infections. Methods Mol Biol. doi: 10.1007/7651_2017_1. [published online ahead of print March 31, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burns JW, Krishnaney AA, Vo PT, Rouse RV, Anderson LJ, Greenberg HB. Analyses of homologous rotavirus infection in the mouse model. Virology. 1995;207(1):143–153. doi: 10.1006/viro.1995.1060. [DOI] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 53.Leem J, Dunbar J, Georges G, Shi J, Deane CM. ABodyBuilder: Automated antibody structure prediction with data-driven accuracy estimation. MAbs. 2016;8(7):1259–1268. doi: 10.1080/19420862.2016.1205773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 58.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.