

Abstract
Sialyl Lewis A (sLeA, also known as CA19-9), a tetrasaccharide selectively and highly expressed on advanced adenocarcinomas including colon, stomach, and pancreatic cancers, has long been considered as an attractive target for active and passive vaccination. While progress in antibodies targeting tumor-associated protein antigens resulted in an impressive array of therapeutics for cancer treatment, similar progress in exploiting tumor-associated carbohydrate antigens, such as sLeA, has been hampered by the lack of a detailed understanding of the singular characteristics of these antigens. We have addressed this issue by analyzing antibodies derived from patients immunized with an sLeA/KLH vaccine. These antibodies were engineered to mediate tumor clearance in vivo in preclinical models through Fc-FcγR interactions. However, in contrast to protein antigens in which hFcγRIIIA engagement was both necessary and sufficient to mediate tumor clearance in both preclinical and clinical settings, a similar selective dependence was not seen for anti-sLeA antibodies. Thus, re-engineering the Fc portion of sLeA-targeting antibodies to broadly enhance their affinity for activating FcγRs led to an enhanced therapeutic effect. These findings will facilitate the development of more efficient anticancer therapies and further advance this promising class of therapeutic antibodies into clinical use.
Keywords: Immunology
Keywords: Cancer immunotherapy, Immunoglobulins
Introduction
Antibody-based therapies are now routinely used in the clinic to treat various malignancies (1). These antibodies target either the tumor directly, activating intrinsic or extrinsic cytotoxic pathways, or target the immune system to induce its activation with resulting tumor clearance. Antibodies of this first class target tumor-associated antigens (TAAs), which are expressed preferentially (but not exclusively) by tumor cells and can potentiate immune-mediated tumor cell killing (2). TAAs can be further subdivided based on their structure: tumor-associated proteins, carbohydrates, and glycolipids (3). Altered carbohydrate expression and aberrant glycan modifications are one of the hallmarks of cancer (4, 5) and many tumor-associated carbohydrate antigens (TACAs) have been identified and routinely used in the clinic to monitor tumor progression and response to treatment. While antibodies and vaccines targeting these antigens have been proposed as potential therapeutic interventions (6), very few TACA-targeting antibodies have been successfully developed and reached clinical trials due to low affinity and specificity, coupled with toxicity and adverse effects (7). To date, only one TACA-targeting antibody has been FDA approved (dinutuximab, which targets the ganglioside GD2; ref. 8), in contrast to the growing number of therapeutic antibodies that are directed against tumor-associated proteins.
Previous studies have focused on antibodies targeting tumor-associated proteins, such as CD20 (9) and Her2/Neu (10), as well as antibodies targeting the checkpoint targets CTLA4 (11), CD40 (12), and PD-1/PD-L1 (13), establishing the mechanistic role by which such antibodies confer their therapeutic activity in vivo through engagement of Fcγ receptors (FcγRs). Whether those same mechanisms apply to antibodies targeting TACAs has not been previously examined. We have focused on the TACA sialyl Lewis A (sLeA), a carbohydrate that belongs to the family of Lewis antigens (14). Patient data revealed that sLeA is expressed on various types of tumor cells (including gastrointestinal, breast, and pancreatic malignancies; refs. 15–17) and that high expression of sLeA correlates with metastasis and poor survival (16, 18). Moreover, sLeA has been shown to promote cancer development and progression by facilitating tumor cell adhesion, angiogenesis, and rolling of tumor cells on endothelial cells resulting in extravasation and metastasis formation (19, 20). Owing to these observations, sLeA has been considered as an attractive therapeutic target for cancer therapy (14). In this study, we analyzed antibodies that were derived from patients immunized with an sLeA/KLH vaccine, and utilized antibody engineering techniques in order to enhance their ability to perform tumor clearance in preclinical models.
Efficient elimination of tumor cells by the immune system requires not only binding of the TAA-targeting antibodies to their antigens on the malignant cells via their Fab, but necessitates activation of effector functions by the antibody Fc portion (21). Activation of effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), relies on binding of the antibody Fc portion to FcγRs, a complex family of predominately low-affinity IgG receptors, differentially expressed on the surface of leukocytes and capable of mediating both activating and inhibitory signals (22) upon cross-linking by immune complexes or IgG-coated target cells. The IgG Fc portion is subject to significant structural variation, resulting from differences both in the IgG subclasses and the heterogeneity of the N-linked biantennary glycan on the CH2 domain. These structures affect the binding affinity for different FcγRs (23) and thus impact the ability of an antibody to mediate cytotoxicity. A key determinant of the cytotoxic capacity of an IgG Fc is the ratio of binding to activating versus inhibitory FcγRs (A/I ratio), which thereby determines the level of ADCC or ADCP induction, impacting the nature and extent of the elicited antitumor response (24). Understanding the FcγR requirements necessary for eliciting effective antitumor cytotoxicity has led to development of specific engineered Fc sequences and glycoforms with selective enhancement for specific activating FcγRs and consequently improved in vivo activity. For example, although the following 2 CD20-targeting antibodies recognize different epitopes, the Fc-glycoengineered antibody obinutuzumab, which is enhanced for binding to the activating receptor FcγRIIIA, displays improved therapeutic activity in chronic lymphocytic leukemia when compared with rituximab (25, 26). Thus, re-engineering the Fc of tumor protein–targeting antibodies has been proven as a successful strategy for developing more efficient cancer therapeutics. Nevertheless, to date, the FcγR-engagement requirements of carbohydrate-targeting antibodies have not been characterized, and Fc engineering of such antibodies has not been attempted.
Using murine tumor cell lines stably expressing surface sLeA (sLeA+) inoculated into immunocompetent mice expressing human FcγRs we have established that engagement of either of the low-affinity activating receptors, hFcγRIIA or hFcγRIIIA, is necessary and sufficient for generation of an effective antitumor immune response and prevention of tumor cell colonization in the lungs. Overall, our results suggest that carbohydrate-targeting antibodies (targeting sLeA) demonstrate different FcγR-engagement requirements compared with those of protein-targeting antibodies (targeting gp75 or CD20), which only require engagement of hFcγRIIIA (9).
Results
Modeling sLeA-expressing murine tumor cell lines.
In order to study the TACA sLeA in an immunocompetent environment, we established murine tumor cell lines that stably express sLeA. This glycan is synthesized by 2 sequential reactions, catalyzed by the enzymes β-galactoside α-2,3-sialyltransferase 3 (ST3GAL3) and fucosyltransferase III (FUT3). Because sLeA is not expressed in murine tissues (due to the absence of a murine homolog of FUT3) we transduced murine tumor cell lines to express human FUT3. We chose to transduce B16 melanoma and EL4 lymphoma murine tumor cell lines, as these are well-established model systems for studying the antitumor activities of TAA-targeting antibodies, and have been used extensively by many research groups (27–30). FUT3 transduction led to surface expression of sLeA in both cell lines, as determined by flow cytometry (Figure 1A). sLeA expression levels were comparable to those reported for some human tumor cell lines, such as Colo-205 colorectal cells (31). We also performed a direct comparison of the sLeA expression levels between our engineered cell lines (EL4-FUT3 and B16-FUT3) and several human tumor cell lines. Supplemental Figure 1 (supplemental material available online with this article; https://doi.org/10.1172/JCI128437DS1) demonstrates the heterogeneous expression levels within HT-29 colorectal carcinoma cells (showing high sLeA expression within a subpopulation of the tumor cells) and OVCAR3 ovarian carcinoma cells (showing moderate expression). Nevertheless, the levels of sLeA expression within the sLeA+ fraction of these cells were comparable to those of our engineered murine cell lines (Supplemental Figure 1). Consistent with the clinical observation that sLeA is shed by tumor cells and can be used as a clinical serum biomarker in several human cancers (32, 33), we also observed shedding of sLeA in the supernatant of FUT3-transduced cells by sandwich ELISA (Figure 1B).
Figure 1. Modeling sLeA-expressing murine tumor cell lines.

B16 melanoma cells and EL4 lymphoma cells were transduced to stably express the human enzyme fucosyltransferase III (FUT3), which synthesizes sLeA. (A) Surface expression of sLeA. B16 and EL4 tumor cells were labeled with an anti-sLeA primary Ab (5B1-hIgG1) followed by Alexa Fluor 488–conjugated goat anti–human IgG antibody. The panel shows a representative experiment (n > 3), all showing similar results. (B) Secretion of sLeA. Supernatants were collected from tumor cells 72 hours after seeding, filtered, and analyzed by sandwich ELISA for detection of extracellular sLeA. Data were pooled from n = 3 experiments and presented as mean ± SEM. (C) Lung colonization of sLeA+ B16 cells. WT C57BL/6 mice were inoculated i.v. with 5 × 105 B16 or B16-FUT3 tumor cells. Fourteen days after inoculation, mice were euthanized, lungs were excised and fixed, and metastatic foci were counted. Data were pooled from n = 3 experiments, n ≥ 20/group. ***P < 0.005 (unpaired 2-tailed t test). The box extends from the 25th to 75th percentile, the line within the box represents the median value, and the whiskers correspond to the 5th to 95th percentile. (D) Tumor growth of B16 cells. WT C57BL/6 mice were inoculated subcutaneously with 5 × 105 B16 or B16-FUT3 tumor cells. Average sizes of primary tumors ± SEM are presented in mm3, measured biweekly by caliper. Data were pooled from n = 2 experiments, n > 18/group. (E) Survival of mice inoculated with EL4 cells. WT C57BL/6 mice were inoculated i.v. with 5 × 105 EL4 or EL4-FUT3 tumor cells. Survival was followed daily. Data were pooled from n = 3 experiments, n = 28/group.
Although several clinical studies demonstrate that high expression levels of sLeA correlates with metastatic disease and poor survival (16, 18), the specific molecular mechanisms by which sLeA promotes disease progression remain elusive. Proposed mechanisms include angiogenesis potentiation (34) and enhancement of sLeA-expressing tumor cell adhesion to E-selectin–expressing endothelial cells (35), facilitating metastatic colonization (20). We validated that the transduced tumor cell lines retained their tumorigenic ability upon transduction with FUT3 and that they were not rejected by the immune system of immunocompetent mice. WT C57BL/6 immunocompetent mice were inoculated i.v. (Figure 1C) or subcutaneously (Figure 1D) with B16 tumor cells (parental cells that do not express sLeA) or B16-FUT3 tumor cells (which express sLeA). Both inoculation methods suggested that the B16-FUT3 tumor cells show comparable engraftment capabilities to those of B16 cells, giving rise to comparable primary tumors, and slightly higher numbers of lung metastatic foci. In the systemic EL4 lymphoma tumor model, expression of sLeA did not substantially impact the overall survival of mice inoculated with EL4 versus EL4-FUT3 cells (Figure 1E). As a whole, these findings suggest that FUT3-expressing cells can serve as a model system to study the activity of anti-sLeA antibodies in an immunocompetent syngeneic tumor setting.
sLeA-targeting antibodies derived from sLeA/KLH–immunized patients protect mice from sLeA+ tumor challenge.
Several human clones of sLeA-specific antibodies have been derived from patients immunized with an sLeA/KLH vaccine, including clones 5B1 (originally human IgG1) and 7E3 (originally human IgM) (31). The affinity of the 2 antibody clones for sLeA was comparable, as determined by surface plasmon resonance (SPR) (31) and ELISA of synthetic sLeA, as well as by cell surface immunostaining of sLeA+ tumor cells (Supplemental Figure 1). These antibodies have been shown be to be highly specific for the carbohydrate sLeA, and do not cross-react with similar carbohydrates (31). First, we used the variable region sequences of these 2 patient-derived antibodies (patent US9475874B2) to generate chimeric recombinant antibodies with murine Fc portions.
The Fab regions of clones 5B1 and 7E3 were expressed either as murine IgG1, IgG2a, or with a point mutation D265A that severely hampers binding to murine FcγRs (36) in order to evaluate the contribution of specific FcγRs to the antitumor efficacy of these antibodies in immunocompetent mice endogenously expressing murine FcγRs. Administration of either 5B1-mIgG2a or 7E3-mIgG2a antibodies to immunocompetent C57BL/6 mice inoculated i.v. with sLeA-expressing B16 cells dramatically reduced metastatic colonization, while the antibodies showed no therapeutic activity when the mice were inoculated with parental (sLeA-negative) B16 cells (Figure 2A). The sLeA-targeting antibodies exhibited comparable therapeutic effect to that of the TA99-mIgG2a that targets the melanoma-associated protein antigen gp75 (Supplemental Figure 3A). Similarly, when mice were inoculated with EL4-FUT3 (sLeA+) tumor cells, treatment with either of the 2 antibodies (5B1-mIgG2a or 7E3-mIgG2a) rescued the majority of mice, but could not rescue any of the animals inoculated with parental EL4 cells (Figure 2B). Overall, our findings suggest that these carbohydrate-targeting antibodies were capable of triggering tumor clearance in an immunocompetent environment, similarly to protein-targeting antibodies.
Figure 2. sLeA-targeting Abs protect mice from sLeA+ tumor challenge.

(A) Anti-sLeA Abs inhibit lung colonization of sLeA+ B16 cells. WT C57BL/6 mice were inoculated i.v. with 5 × 105 B16 or B16-FUT3 tumor cells. One hundred micrograms of anti-sLeA Abs (5B1-mIgG2a or 7E3-mIgG2a) or isotype-matched control Abs was administered i.p. on days 1, 4, 7, and 11. Fourteen days after inoculation, mice were euthanized, lungs were excised and fixed, and metastatic foci were counted. The panel summarizes data pooled from n = 3 experiments, and shows representative images of 3 excised lungs from mice inoculated with B16-FUT3 cells. n > 20 for all groups, except 7E3-mIgG2a (n = 6–8/group). **P < 0.01, ****P < 0.0001 (1-way ANOVA with Bonferroni’s post hoc test). The box extends from the 25th to 75th percentile, the line within the box represents the median value, and the whiskers correspond to the 5th to 95th percentile. (B) Anti-sLeA Abs rescue mice inoculated with sLeA+ EL4 cells. WT C57BL/6 mice were inoculated i.v. with 5 × 105 EL4 or EL4-FUT3 tumor cells. One hundred micrograms of anti-sLeA Abs (B1: 5B1-mIgG2a; B2: 7E3-mIgG2a) or isotype-matched control Abs was administered i.p. on days 1, 4, 7, and 11. Survival was assessed daily. For 5B1, data were pooled from n = 2 experiments, n > 20/group. For 7E3, n = 10–20/group. ****P < 0.0001 (log-rank test). NS, not significant.
Antibody-mediated antitumor cytotoxicity is mediated by engagement of activating FcγRs and is dictated by the tumor.
It has been established that effective antibody-mediated tumor elimination requires not only binding of the antibody to tumor-expressed antigens via its Fab, but also induction of effector functions (e.g., ADCC and ADCP) via interaction of its Fc portion with leukocyte-expressed FcγRs (23). In order to explore whether the antitumor activity of anti-sLeA monoclonal antibodies is FcγR dependent, we employed 2 complementing approaches: (a) comparing the antitumor activity of parental nonengineered antibodies in WT mice (expressing murine FcγRs) versus activating FcγR–null mice (carrying a deletion of the murine γ chain, resulting in the absence of murine activating FcγRs); and (b) comparing the therapeutic activity of parental antibodies versus Fc re-engineered variants, in which FcγR-binding ability was eliminated.
B16-FUT3 cells were inoculated into WT C57BL/6 mice and into activating FcγR–null mice (Fcγ chain–KO mouse strain, lacking mFcγRI, mFcγRIII, and mFcγRIV) and treated with anti-sLeA antibody. Whereas 5B1-mIgG2a led to a significant decrease in tumor burden in WT mice, this effect was completely abolished in mice lacking activating FcγRs (Figure 3A). Similarly, when WT mice were treated with a 5B1-mIgG1-D265A variant that is nearly unable to engage FcγRs a similar diminution of antitumor efficacy was observed (Figure 3A). Differences in the antitumor activity among the 5B1 Fc variants were not attributed to antigen binding, as all 5B1 Fc variants exhibited comparable binding to sLeA+ tumor cells (Supplemental Figure 2A). Overall, these findings indicate that in the B16 melanoma metastasis model, the antitumor effect of antibodies targeting the carbohydrate sLeA rely fully on engagement of activating FcγRs by the administered antibodies.
Figure 3. Ab-induced antitumor effect is mediated by engagement of Fcγ receptors (FcγRs), and is dictated by the tumor.

(A) The anti-sLeA effect is fully mediated by FcγR engagement in an sLeA+ B16 tumor model. WT C57BL/6 or activating FcγR-null (aFcγR-null, γ chain–KO) mice (see Methods for detailed mouse strain information) were inoculated i.v. with 5 × 105 B16-FUT3 tumor cells. One hundred micrograms of anti-sLeA Abs (5B1-mIgG2a or 5B1-mIgG1-D265A variants) or isotype-matched control Abs was administered i.p. on days 1, 4, 7, and 11. Fourteen days after inoculation, mice were euthanized, lungs were excised and fixed, and metastatic foci were counted. The panel summarizes the data pooled from n = 2 experiments, n ≥ 11/group, and shows representative images of 3 excised lungs. *P < 0.05, **P < 0.01 (1-way ANOVA with Bonferroni’s post hoc test). The box extends from the 25th to 75th percentile, the line within the box represents the median value, and the whiskers correspond to the 5th to 95th percentile. Data for groups WT mice – isotype, and WT – 5B1-mIgG2a also appear in Figure 2A. (B) The anti-sLeA effect is partially mediated by FcγR engagement in an sLeA+ EL4 tumor model. WT C57BL/6 or aFcγR–null mice (see Methods for detailed mouse strain information) were inoculated i.v. with 5 × 105 EL4-FUT3 tumor cells. One hundred micrograms of anti-sLeA Abs (5B1-mIgG2a or 5B1-mIgG1-D265A) or isotype-matched control Abs was administered i.p. on days 1, 4, 7, and 11. Survival was assessed daily. n = 8/group. *P < 0.05, **P < 0.01 (log-rank test).
In contrast to the B16 metastasis model, the role of Fc-FcγR interactions in the EL4 tumor model revealed differential Fc requirements. While mIgG2a sLeA-targeting antibodies were able to rescue all WT mice challenged with EL4-FUT3 tumor cells, it could only rescue approximately half of the mice in the activating FcγR–null group, implicating a non–Fc-mediated protection (Figure 3B). Similarly, engineered 5B1-mIgG1-D265A antibodies (which are nearly unable to engage FcγRs) also displayed partial therapeutic effects. Thus, in contrast to the B16 metastasis model, the antitumor effect of sLeA-targeting antibodies against EL4 lymphoma cells is only partially mediated by FcγR engagement, suggesting a Fab-mediated protective effect by the antibody. These findings are in contrast to antibodies targeting protein antigens such as CD20 in the EL4 tumor model that confer protection in a fully FcγR-dependent mechanism (9). Targeting sLeA in EL4 cells likely mediates disruption of tumor-stromal cell interactions or signaling of the EL4 cells in the context of the in vivo setting that are required for tumor cell survival or proliferation. It is likely that the difference in the necessity of FcγR engagement stems from the inherent differences between the 2 tumor models, since other parameters (such as the mode of tumor inoculation and treatment regimen) were identical between the 2 sets of experiments.
Although it differs in the extent of the contribution of FcγR engagement to the antibody-mediated antitumor activity in these 2 models, it is clear from these data that full protection required FcγR engagement regardless of the tumor model. To determine which Fc receptors mediated the antitumor effect we compared the antitumor activity of the 5B1 clone Fab, expressed either as a murine IgG2a or IgG1. These 2 isotypes differ in their binding affinity for activating versus inhibitory murine FcγRs, which can be described using the A/I ratio. While the mIgG2a antibody preferentially engages the activating murine receptor mFcγRIV and is thus characterized by a high A/I ratio (A/I = 69), mIgG1 preferentially engages the inhibitory murine receptor mFcγRIIB and is characterized by a low A/I ratio (A/I = 0.1) (37). This concept remains similar for carbohydrate-targeting antibodies, as the 5B1-mIgG2a variant (high A/I ratio) outperformed the same antibody as a mIgG1 subclass variant (low A/I ratio), leading to a more pronounced tumor clearance (Supplemental Figure 3B), highlighting the importance of engaging activating FcγRs by tumor-antigen–targeting antibodies, in vivo.
Human IgG1 sLeA-targeting antibodies promote tumor clearance and their antitumor potency is enhanced by engaging activating human FcγRs.
In order to recapitulate the interactions of antibodies designed for clinical use with a human Fc with human FcγRs, B16-FUT3 cells were inoculated into FcγR-humanized mice, a strain that lacks all murine FcγRs while carrying transgenes of all human FcγRs (38), resulting in the recapitulation of the cellular expression pattern of human FcγRs in a fully immunocompetent murine background. B16 tumor–bearing mice were treated with sLeA-targeting antibodies, clones 5B1 and 7E3, expressed in the hIgG1 subclass. Both 5B1 and 7E3 clones exhibited comparable therapeutic efficacy (Figure 4A), leading to a significant reduction in the number of metastatic foci in the lungs. As observed with the chimeric human-mouse antibodies (Figure 3A), engineering 5B1-hIgG1 with an Fc mutation (N297A) that severely hampers its ability to engage human FcγRs results in the loss of the therapeutic effect of sLeA-targeting antibodies (Supplemental Figure 3C).
Figure 4. Engagement of either hFcγRIIA or hFcγRIIIA is necessary and sufficient for tumor clearance, mediated by sLeA-targeting Abs with an hIgG1 Fc.

Mice were inoculated i.v. with 5 × 105 B16-FUT3 tumor cells. One hundred micrograms of anti-sLeA Abs or isotype-matched control Abs was administered i.p. on days 1, 4, 7, and 11. Fourteen days after inoculation, mice were euthanized, lungs were excised and fixed, and metastatic foci were counted. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (1-way ANOVA with Bonferroni’s post hoc test). For all panels, the box extends from the 25th to 75th percentile, the line within the box represents the median value, and the whiskers correspond to the 5th to 95th percentile. (A) Fc-engineered anti-sLeA Ab variants demonstrate superior antitumor efficacy. FcγR-humanized mice were treated with clones 5B1 or 7E3, hIgG1 or hIgG1-GAALIE with G236A/A330L/I332E mutations. Data were pooled from n = 2–3 experiments, n ≥ 13/group, except for 7E3-hIgG1-GAALIE (n = 7). (B) 5B1-hIgG1 Abs with enhanced binding affinity for hFcγRIIA, or hFcγRIIIA, or both demonstrate a superior antitumor effect. FcγR-humanized mice were treated with Fc variants 5B1-hIgG1, 5B1-hIgG1-GA with a G236A mutation, 5B1-hIgG1-ALIE with A330L/I332E mutations, or 5B1-hIgG1-GAALIE with G236A/A330L/I332E mutations. Data were pooled from n = 2–4 experiments. n ≥ 12/group. (C) hFcγRIIA or hFcγRIIIA engagement is essential for efficient tumor clearance of sLeA+ tumors. Activating FcγR-null (aFcγR-null, γ chain–KO), FcγR-humanized, hFcγRIIA/IIB–transgenic, and hFcγRIIIA/IIIB–transgenic mice were treated with the Ab 5B1-hIgG1-GAALIE with G236A/A330L/I332E mutations. Data were pooled from n ≥ 2 experiments for aFcγR-null and FcγR-humanized mice, n ≥ 12/group. For hFcγRIIA/IIB–transgenic and hFcγRIIIA/IIIB–transgenic, n = 5–8/group.
In light of the above-described role of activating FcγRs in mediating antibody-induced tumor clearance we sought to increase the therapeutic potency of sLeA-targeting antibodies by increasing their affinity for activating FcγRs. To do this, we re-engineered hIgG1 sLeA-targeting antibodies by introducing 3 point mutations (G236A/A330L/I332E, herein termed “GAALIE”), substantially enhancing the affinity of sLeA-targeting antibodies for 2 activating human FcγRs, hFcγRIIA and hFcγRIIIA, while reducing the binding to the inhibitory receptor, hFcγRIIB, without interfering with their binding affinity for sLeA (Supplemental Figure 2B). The re-engineered 5B1 and 7E3 antibody variants demonstrated superior antitumor activity compared with the parental antibody with a WT hIgG1 Fc portion (Figure 4A). These results reinforce our previous findings that engagement of activating FcγRs is a crucial step in the process of efficient antibody-mediated tumor clearance.
Engagement of either hFcγRIIA or hFcγRIIIA is necessary and sufficient for antibody-mediated tumor clearance.
We previously reported that engagement of hFcγRIIIA alone is both necessary and sufficient for antibody-mediated tumor clearance in several tumor models, while the engagement of the activating receptor hFcγRIIA was insufficient to mediate tumor clearance (9). Since our previous study focused on CD20-targeting antibodies (protein-targeting antibodies), we aimed to determine whether these findings also hold true for sLeA-targeting antibodies (carbohydrate-targeting antibodies). Thus, we compared the antitumor activity of 3 Fc variants with enhanced affinities for hFcγRIIA (GA), hFcγRIIIA (ALIE), or both (GAALIE) in FcγR-humanized tumor-bearing mice (Table 1). The affinity of the GA and ALIE hIgG1 Fc variants for different human FcγRs has been reported (9, 39, 40); the GAALIE Fc variant exhibits a higher affinity for hFcγRIIA and hFcγRIIIA, with reduced affinity for hFcγRIIB (Supplemental Figure 4 and Table 1), and an in vivo half-life comparable to that of hIgG1, while demonstrating a superior ADCC capability compared with the parental hIgG1 (Supplemental Figure 5).
Table 1. Binding affinity of hIgG1 Fc variants for human FcγRs.
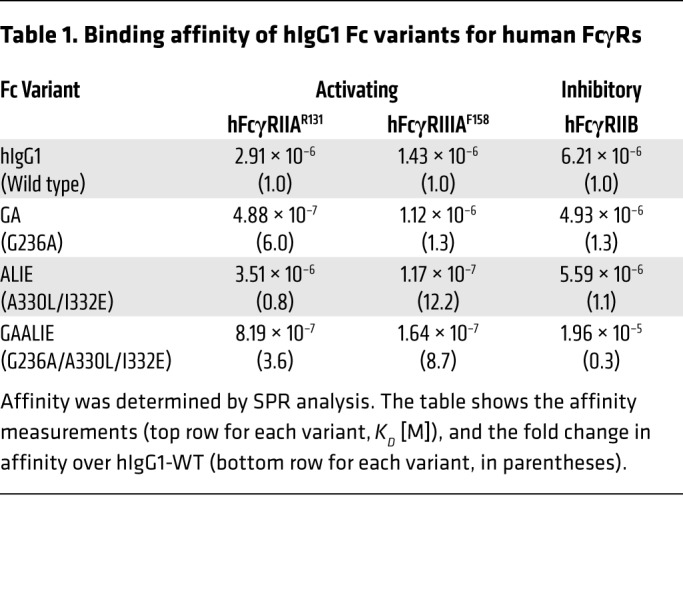
All 3 Fc variants exhibited a comparable antitumor potential, which was significantly higher than that of the WT parental human IgG1 antibody (Figure 4B). To confirm these findings, we compared the antitumor activity of the Fc variant 5B1-hIgG1-GAALIE (with enhanced affinity for both activating FcγRs) in several transgenic mouse strains expressing human FcγRs. Figure 4C indicates that the 5B1-hIgG1-GAALIE variant demonstrates a pronounced, yet comparable, antitumor activity not only in FcγR-humanized mice (which express all human FcγRs, including hFcγRIIA, hFcγRIIB, and hFcγRIIIA), but also in hFcγRIIA/IIB–only mice and hFcγRIIIA/IIIB–only mice. As expected, tumor clearance was not observed in activating FcγR–null mice. NK depletion did not substantially hamper the antitumor activity of this sLeA-targeting antibody (Supplemental Figure 6), suggesting that tumor cell depletion is primarily mediated by effector cells expressing hFcγRIIIA and hFcγRIIA, such as macrophages, consistent with what we have previously reported (9).
Overall, our results suggest that engagement of hFcγRIIA or hFcγRIIIA by sLeA-targeting antibodies is necessary for efficient tumor clearance; however, in this context of lung colonization, the receptors are redundant, and activation of either one suffices. These findings differ from those of gp75-targeting antibodies (clone TA99) for which enhancing the affinity for hFcγRIIIA (ALIE variant) improves ADCC activity, while enhancing the affinity for hFcγRIIA does not improve upon that of the parental hIgG1 variant (Supplemental Figure 7). Thus, re-engineering the Fc portion of sLeA-targeting antibodies, to enhance their affinity for activating human FcγRs, may dramatically increase the therapeutic potential of antibodies designed for clinical use.
Discussion
In the 2 decades since the first approval of a therapeutic antibody targeting a tumor-associated protein antigen it has become generally accepted that the in vivo therapeutic efficacy of this class of antibodies requires Fc-mediated effector activity, exerted through engagement of FcγRs expressed on leukocytes (41). Recruitment of effector cells, such as macrophages, expressing these receptors to tumors results in tumor cell elimination. We have shown in several preclinical models the dependence of antibodies targeting tumor-associated protein antigens on hFcγRIIIA or its murine homolog, mFcγRIV, to confer antitumor activity. DiLillo et al. (9) demonstrated that hCD20-targeting antibodies were able to clear tumors in mice expressing only hFcγRIIIA/IIIB. The study by Nimmerjahn et al. (42) demonstrated that in mFcγRIV-knockout mice, the antitumor effect of gp75-targeting antibodies (clone TA99) is completely abolished, suggesting complete dependence on mFcγRIV. These observations were later confirmed by large-scale genetic association clinical studies that correlated the expression of the high-affinity hFcγRIIIA allele (V158) with improved response rates of lymphoma patients treated with rituximab (43) and breast cancer patients treated with trastuzumab (44). Although some studies did not find a correlation between FcγR polymorphism and response to antibody therapy (reviewed in ref. 45), it has become routine to consider both Fab and Fc interactions when developing antitumor therapeutic antibodies. This is evidenced by the FDA-approved therapeutic antibodies obinutuzumab (targeting CD20) and mogamulizumab (targeting CCR4), which are both glycoengineered for enhanced hFcγRIIIA binding and thus improved for ADCC (25, 46).
However, despite this understanding of the role of effector cells in antitumor responses, the precise contribution of specific cellular populations and individual FcγRs remains incompletely understood. Human FcγRs comprise a complex family of cell surface receptors with distinct cellular expression patterns, signaling motifs, and allelic heterogeneity (24). FcγRs capable of mediating cellular activation through immunoreceptor tyrosine–based activation motif (ITAM) signaling include the high-affinity hFcγRI and low-affinity hFcγRIIA, hFcγRIIC, and hFcγRIIIA receptors. Inhibitory signaling is mediated by the low-affinity hFcγRIIB receptor through an immunoreceptor tyrosine-based inhibition motif (ITIM) found in its cytoplasmic domain. In general, effector cells express both activating and inhibitory FcγRs, thus setting a threshold for activation by IgG bound to a target or as an immune complex. This diversity of FcγR structure is paired with the diversity found in the IgG Fc domain, dictated by individual subclasses that differ in their affinity for specific FcγRs and the structure of the N-linked, complex biantennary glycan linked to Asn 297 in the CH2 domain of all IgGs (47).
Thus, while the general features of antibody-mediated tumor clearance through FcγR engagement are known, the impact of tumor type and targeted antigen has not been systematically studied. Despite the identification of many carbohydrate antigens, developing and characterizing antibodies against carbohydrate tumor antigens have been challenging, resulting in a near neglect of this entire category of tumor antigens. In this study, we have begun to address these issues by focusing on antibodies against sLeA, a TACA overexpressed on a wide variety of human tumors (15–17). Antibodies against this antigen originated in a patient participating in a cancer vaccine trial, and are now in clinical development for both diagnostic and therapeutic indications (48–50); however, their mode of action remains unknown. Using several syngeneic tumor models, we demonstrated some mechanisms shared with protein-targeting antibodies, yet revealed key differences in FcγR-engagement patterns. Thus, while antibodies against both classes of TAAs display a requirement for FcγR engagement in the B16 metastatic melanoma model, the specific FcγR requirements for each class of antibodies differ. Furthermore, targeting of EL4 lymphoma cells revealed that antibodies against a protein antigen had an absolute FcγR requirement for protection against lethal disease, while antibodies against a carbohydrate antigen showed only a partial FcγR requirement. These differences suggest that the precise tumor microenvironment is contributing to the FcγR-dependent mechanism of action of these antibodies, targeting different classes of TAAs. Finally, we demonstrate that precise Fc engineering to enhance specific FcγR engagement results in significant enhancement of in vivo potency and therapeutic efficacy of several clones of sLeA-targeting antibodies. Translating Fc-optimized carbohydrate-targeting antibodies to the clinic could not only enhance the therapeutic effect, but could also assist with reducing toxicities and adverse events, since it may allow decreasing the administered dose.
The antibodies that we have studied here that target the TACA sLeA display an FcγR-dependent phenotype not seen for antibodies that target protein antigens. In the B16 metastasis model (Figure 4), these antibodies mediate clearance of metastatic nodules by engaging either hFcγRIIIA or hFcγRIIA, displaying a redundancy not seen for antibodies targeting protein antigens (9). Similarly, the protective effect of these antibodies in the lethal EL4 lymphoma model is only partially FcγR dependent, in contrast to antibodies that target protein antigens on these tumors (9). The mechanistic basis for these differences remains to be determined but cannot be attributed to differences in the Fc binding characteristics of these 2 classes of antibodies nor their half-life in vivo. It suggests that differences in the interactions of the tumor cell with its tumor microenvironment may be responsible. Since sLeA has the capacity to bind to selectins on endothelial cells and to Siglecs on immune effector cells, among other adhesion receptors, they may be responsible for altering the microenvironment and thereby modifying the effector cell pathways that are engaged.
Previous studies have indicated that both in the B16 melanoma model and in the EL4 lymphoma model, antibody-dependent tumor clearance is mediated by macrophages, which are activated by engagement of their FcγRs (9). Treating WT tumor-bearing mice with an antibody with a murine IgG2a Fc results in potent tumor clearance in a variety of syngeneic tumor models, despite the inability of this subclass to engage murine NK cells through its mFcγRIII receptor. Therefore, the relevant effector cell for mIgG2a-mediated tumor clearance and protection expresses mFcγRIV, thus implicating macrophages and potentially other myeloid cells (37). This observation has also been supported by our NK cell depletion experiments in the present study (Supplemental Figure 5). The collective conclusion from these studies suggests that in the mouse, NK cells, despite their in vitro ability to kill tumor targets by ADCC, do not contribute to the in vivo protection by antibodies targeting tumor antigens. Whether the same is true for human NK cells has yet to be determined.
Our research provides what we believe is the first characterization of FcγR requirements for carbohydrate-targeting antibodies. While it is tempting to speculate that other carbohydrate-targeting antibodies will display the same mechanisms of action and share the same FcγR requirements as the sLeA-targeting antibodies, further research on antibodies targeting other TACAs is indicated.
Overall, we believe that our observations will facilitate development of antibodies against this somewhat neglected class of targets, promoting the introduction of novel and effective immunotherapeutics. As indicated above, sLeA-targeting antibodies are currently in phase I clinical trials (NCT02672917) and have been reported to be well tolerated, suggesting that an Fc-optimized variant could potentially be translated to the clinic, offering a higher therapeutic benefit to the patients.
Methods
Transgenic mouse models
WT C57BL/6 mice were purchased from The Jackson Laboratories. Activating FcγR–null mice have a deletion of the murine γ chain (36). FcγR-humanized mice (murine α chain KO, hFcγRI+, hFcγRIIAR131+, hFcγRIIB+, hFcγRIIIAF158+, and hFcγRIIIB+) were generated in our laboratory and extensively characterized (38). While FcγR-humanized mice do not express hFcγRIIC, this gene is expressed only by approximately 20% of the human population, and thus this strain represents 80% of the population. hFcγRIIA/IIB–only mice and hFcγRIIIA/IIIB–only mice express a single activating human FcγR on a background of α chain KO (lacking murine FcγRs, as previously described in ref. 9). All mice were maintained in the Rockefeller University Comparative Bioscience Center. Tumor studies were performed on age- and sex-matched females and male mice, 9 to 13 weeks old.
Cell lines and sLeA expression
Murine B16 melanoma and EL4 lymphoma tumor cells were obtained from ATCC and maintained in Dulbecco’s minimum essential medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Life Technologies). Tumor cells were transduced with constructs encoding the human gene FUT3, selected in 500 μg/mL Geneticin (Life Technologies) to generate stable cell lines, and sorted using anti-sLeA antibodies, to obtain cells that homogeneously expressed surface sLeA.
Antibody engineering and expression
The variable heavy and light regions of anti-sLeA antibody clones 5B1 and 7E3 (described in patent US9475874B2) were synthesized (IDT) and subcloned into mammalian expression vectors with human IgG1, mouse IgG2, mouse IgG1 heavy chains, or human κ or mouse κ light chains, as previously described (51). For the generation of Fc-domain variants of human IgG1 (N297A, G236A, A330L/I332E, and G236A/A330L/I332E) and mouse IgG1 (D265A), site-directed mutagenesis using specific primers was performed based on the QuikChange site-directed mutagenesis Kit II (Agilent Technologies) according to the manufacturer’s instructions. Mutated plasmid sequences were validated by direct sequencing (Genewiz).
Antibodies were generated by transient cotransfection of Expi293F cells with heavy-chain and light-chain constructs. Expi293F cells were maintained in serum-free Expi293 Expression Medium, and transfected using an ExpiFectamine 293 Transfection Kit (all from Thermo Fischer Scientific). Supernatants were collected 7 days after transection, centrifuged, and filtered (0.22 μm). Antibodies were purified from clarified supernatants using Protein G Sepharose 4 Fast Flow (GE Healthcare), dialyzed in PBS, and sterile filtered (0.22 μm) as previously described (52).
Flow cytometry
Surface expression of sLeA on murine tumor cell lines.
Expression was assessed using recombinantly expressed 5B1 and 7E3 antibodies, generated in our laboratory. Tumor cells (5 × 105) were incubated with 0.5 μg antibody, followed by Alexa Fluor 488–conjugated secondary antibodies against mouse IgG (catalog A11029, Invitrogen) or human IgG (catalog A11013, Invitrogen). Baseline staining was obtained using isotype-matched antibodies as controls.
NK cell staining.
Μurine lungs were perfused with HBSS/1 mM EDTA and dissociated using a mouse lung dissociation kit (Miltenyi Biotec), according to the manufacturer’s protocol. Erythrocytes were lysed using Hybri-Max buffer (MilliporeSigma), and remaining cells were washed in PBS and labeled using the following antibodies (all from BioLegend): Alexa Fluor 594–conjugated against CD45 (catalog 103144), PerCP-Cy5.5–conjugated against NKp46 (catalog 137609), BV510-conjugated against CD3 (catalog 100353), BV510-conjugated against B220 (catalog 103247), BV510-conjugated against CD19 (catalog 115546), and BUV395-conjugated against CD49b (catalog 740250, BD Bioscience). Live/Dead Aqua (catalog L34966, Thermo Fisher Scientific) was used to assess cell viability. NK cells were gated as follows: live CD3–CD19–B220–CD45+NKp46+CD49b+.
ELISA
sLeA secretion from transduced tumor cells was detected by sandwich ELISA. High-binding 96-well microtiter plates (Nunc) were coated overnight at 4°C with 7E3-mIgG2a (1 μg/mL in PBS). All sequential steps were performed at room temperature. Plates were blocked for 1 hour with PBS/2% BSA and were subsequently incubated for 1 hour with serially diluted cell supernatants from 72-hour cultures. CA19-9 was used as a standard curve and for determining the linear range (BioRbyt; 1:3 consecutive dilutions in PBS/2% BSA, starting at 100 U/mL). Plates were incubated for 1 hour with 5B1-hIgG1 (2.5 μg/mL), followed by a 1-hour incubation with horseradish peroxidase–conjugated anti–human IgG antibodies (Jackson ImmunoResearch).
Binding of sLeA-targeting antibodies to sLeA was determined by indirect ELISA. High-binding 96-well microtiter plates (Nunc) were coated overnight at 4°C with recombinant CA19-9 (BioRbyt; 1,000 U/mL in PBS). All sequential steps were performed at room temperature. Plates were blocked for 1 hour with PBS/2% BSA and were subsequently incubated for 1 hour with serially diluted 5B1-hIgG1 and 7E3-hIgG1 antibodies (1:3 consecutive dilutions in PBS/2% BSA, starting at 2 μg/mL). Plates were incubated for 1 hour with horseradish peroxidase–conjugated anti–human IgG (catalog 709-035-149, Jackson ImmunoResearch).
IgG levels of antibodies in murine sera (antibody pharmacokinetics) were quantified by ELISA as previously described (53). Briefly, FcγR-humanized mice were injected i.v. with 100 μg of hIgG1 Fc variants (clone 3BNC117). High-binding 96-well microtiter plates (Nunc) were coated overnight at 4°C with Neutravidin (2 μg/mL in PBS; Thermo Fisher Scientific). All sequential steps were performed at room temperature. Plates were blocked for 1 hour with PBS/2% BSA and incubated with biotinylated goat anti–human IgG antibodies for 1 hour (5 μg/mL; catalog 109-066-170, Jackson ImmunoResearch). Serum samples were serially diluted and incubated for 1 hour, followed by incubation with horseradish peroxidase–conjugated anti–human IgG.
In all ELISA experiments, detection was performed using a 3,3′,5,5′-tetramethylbenzidine (TMB) 2-Component Peroxidase Substrate Kit (KPL) and reactions stopped with the addition of 2 M phosphoric acid. Αbsorbance at 450 nm was immediately recorded using a SpectraMax Plus spectrophotometer (Molecular Devices), background absorbance from negative control samples was subtracted, and duplicate wells were averaged.
Tumor challenge, antibody treatments, and NK cell depletion
B16, B16-FUT3, EL4, or EL4-FUT3 (5 × 105 cells/mouse) cells were inoculated i.v. into the lateral tail vein in 200 μL PBS. Mice were randomized and received intraperitoneal injections of 100 μg of sLeA-targeting antibodies (clones 5B1 or 7E3) or isotype-matched IgGs serving as control, on days 1, 4, 7, and 11 after inoculation. For the B16 lung colonization model, the lungs were harvested on day 14 and analyzed for the presence of surface metastatic foci using a dissecting microscope. For the B16 solid tumor model, 5 × 105 cells/mouse were inoculated subcutaneously and tumor volumes were measured biweekly with an electronic caliper and reported as volume (mm3) using the formula (L12 × L2)/2, where L1 is the shortest diameter and L2 is the longest diameter. For the EL4 model, overall survival was assessed daily.
NK cell depletion was performed by intranasal administration of antibodies against NK1.1 (300 μg/mouse; catalog BE0036, Bio X Cell) or isotype-matched controls on days –3, –1, 1, 4, 7, and 11 after tumor cell inoculation.
SPR
Binding of hIgG1 Fc variants to hFcγRs was measured by SPR using a Biacore T200 instrument (GE Healthcare). Protein G was immobilized at 2,000 resonance units (RU) on a CM5 biosensor chip (GE Healthcare) using amine coupling chemistry at pH 4.5. Human IgG Fc variants were captured on the Protein G–coated sensor surface at a density of 500–800 RU and serial dilutions of FcγRs were injected as analytes. The FcγRs tested were FcγRIIIA (F158), FcγRIIA (R131), and FcγRIIB. Recombinant soluble FcγR ectodomains (2-fold serial dilutions; 2 μM to 7.8 nM) were injected at 30 μL/min for 180 seconds, followed by a 300-second dissociation. After each cycle, the surface was regenerated by injecting a glycine HCl buffer (10 mM, pH 2.0; 50 seconds). Background binding to blank immobilized flow cells was subtracted and affinity constants were calculated using BIAcore T100 evaluation software (GE Healthcare) using the 1:1 Langmuir binding model.
In vivo ADCC
The ADCC activity of different hIgG1 Fc variants was assessed in vivo using the platelet depletion assay, as previously described (38). FcγR-humanized mice were injected intravenously with 10 μg of hIgG1 Fc variants (clone 6A6, targeting a platelet-associated antigen, produced in-house). Mice were bled at the indicated time points before and after antibody injections, and platelet counts were measured using an Advia 120 hematology system (Bayer Healthcare).
Statistics.
One-way ANOVA with Bonferroni’s post hoc test was used to compare groups in lung colonization and flow cytometry (NK cell staining) experiments. An unpaired 2-tailed t test was used when 2 groups were compared. Statistical differences between survival rates were analyzed by comparing Kaplan-Meier curves using the log-rank test. In box-and-whisker plots, the box extends from the 25th to 75th percentile, the line within the box represents the median value, and the whiskers correspond to the 5th to 95th percentile. Data were analyzed with GraphPad Prism software and P values of ≤ 0.05 were considered to be statistically significant, indicated as *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ***P ≤ 0.0001 in the figures.
Study approval
All animal experiments were performed in compliance with federal laws and institutional guidelines and had been approved by the Rockefeller University IACUC.
Author contributions
PW and JVR conceptualized the work and are responsible for the methodology. PW and SB performed experiments and analyzed data. PW wrote the original draft of the manuscript. All authors contributed to reviewing and editing the manuscript. PW and JVR secured funding. JVR supervised the work.
Supplementary Material
Acknowledgments
We would like to thank Holly Smith and Ming Ye Pearly for excellent technical assistance. Research reported in this publication was supported in part by the National Cancer Institute of the NIH under award numbers R35CA196620 and P01CA190174, and by the National Institute of Allergy and Infectious Diseases under award number R01AI129795. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. PW is supported by a postdoctoral fellowship from the National Cancer Center.
Version 1. 08/19/2019
Electronic publication
Version 2. 09/03/2019
Print issue publication
Funding Statement
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Footnotes
Conflict of interest: JVR was a paid consultant and member of the board of directors of MabVax Therapeutics (2014 to 2017). SB and JVR are inventors on a patent application (WO2019125846A1) describing the GAALIE variant and its use in immunotherapy.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(9):3952–3962.https://doi.org/10.1172/JCI128437.
Contributor Information
Polina Weitzenfeld, Email: pvaitsenfe@rockefeller.edu.
Stylianos Bournazos, Email: sbournazos@rockefeller.edu.
References
- 1.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 2.Wurz GT, Kao CJ, DeGregorio MW. Novel cancer antigens for personalized immunotherapies: latest evidence and clinical potential. Ther Adv Med Oncol. 2016;8(1):4–31. doi: 10.1177/1758834015615514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seledtsov VI, Goncharov AG, Seledtsova GV. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum Vaccin Immunother. 2015;11(4):851–869. doi: 10.1080/21645515.2015.1009814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamczyk B, Tharmalingam T, Rudd PM. Glycans as cancer biomarkers. Biochim Biophys Acta. 2012;1820(9):1347–1353. doi: 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15(9):540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda M. Possible roles of tumor-associated carbohydrate antigens. Cancer Res. 1996;56(10):2237–2244. [PubMed] [Google Scholar]
- 7.Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014;588(2):288–297. doi: 10.1016/j.febslet.2013.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Dhillon S. Dinutuximab: first global approval. Drugs. 2015;75(8):923–927. doi: 10.1007/s40265-015-0399-5. [DOI] [PubMed] [Google Scholar]
- 9.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 11.Simpson TR, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell. 2016;29(6):820–831. doi: 10.1016/j.ccell.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell. 2015;28(3):285–295. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Ugorski M, Laskowska A. Sialyl Lewis(a): a tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells. Acta Biochim Pol. 2002;49(2):303–311. [PubMed] [Google Scholar]
- 15.Itai S, et al. Significance of 2-3 and 2-6 sialylation of Lewis A antigen in pancreas cancer. Cancer. 1988;61(4):775–787. doi: 10.1002/1097-0142(19880215)61:4<775::AID-CNCR2820610423>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Jeschke U, et al. Expression of sialyl Lewis X, sialyl Lewis A, E-cadherin and cathepsin-D in human breast cancer: immunohistochemical analysis in mammary carcinoma in situ, invasive carcinomas and their lymph node metastasis. Anticancer Res. 2005;25(3A):1615–1622. [PubMed] [Google Scholar]
- 17.Nakayama T, Watanabe M, Katsumata T, Teramoto T, Kitajima M. Expression of sialyl Lewis(a) as a new prognostic factor for patients with advanced colorectal carcinoma. Cancer. 1995;75(8):2051–2056. doi: 10.1002/1097-0142(19950415)75:8<2051::AID-CNCR2820750804>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Matsui T, et al. Sialyl Lewisa expression as a predictor of the prognosis of colon carcinoma patients in a prospective randomized clinical trial. Jpn J Clin Oncol. 2004;34(10):588–593. doi: 10.1093/jjco/hyh110. [DOI] [PubMed] [Google Scholar]
- 19.Takada A, et al. Adhesion of human cancer cells to vascular endothelium mediated by a carbohydrate antigen, sialyl Lewis A. Biochem Biophys Res Commun. 1991;179(2):713–719. doi: 10.1016/0006-291X(91)91875-D. [DOI] [PubMed] [Google Scholar]
- 20.Kannagi R. Carbohydrate antigen sialyl Lewis a--its pathophysiological significance and induction mechanism in cancer progression. Chang Gung Med J. 2007;30(3):189–209. [PubMed] [Google Scholar]
- 21.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19(2):239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119(24):5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 23.DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. 2015;3(7):704–713. doi: 10.1158/2326-6066.CIR-15-0120. [DOI] [PubMed] [Google Scholar]
- 24.Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15(8):707–716. doi: 10.1038/ni.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illidge T, Klein C, Sehn LH, Davies A, Salles G, Cartron G. Obinutuzumab in hematologic malignancies: lessons learned to date. Cancer Treat Rev. 2015;41(9):784–792. doi: 10.1016/j.ctrv.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Goede V, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–1110. doi: 10.1056/NEJMoa1313984. [DOI] [PubMed] [Google Scholar]
- 27.Kao D, et al. A monosaccharide residue is sufficient to maintain mouse and human IgG subclass activity and directs IgG effector functions to cellular Fc receptors. Cell Rep. 2015;13(11):2376–2385. doi: 10.1016/j.celrep.2015.11.027. [DOI] [PubMed] [Google Scholar]
- 28.Patel D, et al. Enhanced suppression of melanoma tumor growth and metastasis by combined therapy with anti-VEGF receptor and anti-TYRP-1/gp75 monoclonal antibodies. Anticancer Res. 2008;28(5A):2679–2686. [PubMed] [Google Scholar]
- 29.Abès R, Gélizé E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116(6):926–934. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 30.Kohrt HE, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123(5):678–686. doi: 10.1182/blood-2013-08-519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawada R, et al. Human monoclonal antibodies to sialyl-Lewis (CA19.9) with potent CDC, ADCC, and antitumor activity. Clin Cancer Res. 2011;17(5):1024–1032. doi: 10.1158/1078-0432.CCR-10-2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song YX, et al. Clinicopathologic and prognostic value of serum carbohydrate antigen 19-9 in gastric cancer: a meta-analysis. Dis Markers. 2015;2015:549843. doi: 10.1155/2015/549843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poruk KE, et al. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: diagnostic and prognostic updates. Curr Mol Med. 2013;13(3):340–351. doi: 10.2174/1566524011313030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terraneo L, et al. Expression of carbohydrate-antigen sialyl-Lewis a on colon cancer cells promotes xenograft growth and angiogenesis in nude mice. Int J Biochem Cell Biol. 2013;45(12):2796–2800. doi: 10.1016/j.biocel.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Takada A, et al. Contribution of carbohydrate antigens sialyl Lewis A and sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res. 1993;53(2):354–361. [PubMed] [Google Scholar]
- 36.Baudino L, et al. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181(9):6664–6669. doi: 10.4049/jimmunol.181.9.6664. [DOI] [PubMed] [Google Scholar]
- 37.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science. 2005;310(5753):1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 38.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci U S A. 2012;109(16):6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. 2008;7(8):2517–2527. doi: 10.1158/1535-7163.MCT-08-0201. [DOI] [PubMed] [Google Scholar]
- 40.Lazar GA, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103(11):4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012;12:13. [PMC free article] [PubMed] [Google Scholar]
- 42.Nimmerjahn F, et al. FcγRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci U S A. 2010;107(45):19396–19401. doi: 10.1073/pnas.1014515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cartron G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 44.Musolino A, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 45.Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013;6:1. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. 2012;4(4):419–425. doi: 10.4161/mabs.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bournazos S, Ravetch JV. Diversification of IgG effector functions. Int Immunol. 2017;29(7):303–310. doi: 10.1093/intimm/dxx025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Houghton JL, et al. Establishment of the in vivo efficacy of pretargeted radioimmunotherapy utilizing inverse electron demand Diels-Alder click chemistry. Mol Cancer Ther. 2017;16(1):124–133. doi: 10.1158/1535-7163.MCT-16-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houghton JL, Abdel-Atti D, Scholz WW, Lewis JS. Preloading with unlabeled CA19.9 targeted human monoclonal antibody leads to improved PET imaging with 89Zr-5B1. Mol Pharm. 2017;14(3):908–915. doi: 10.1021/acs.molpharmaceut.6b01130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poty S, et al. Leveraging bioorthogonal click chemistry to improve 225Ac-radioimmunotherapy of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2019;25(2):868–880. doi: 10.1158/1078-0432.CCR-18-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333(6045):1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23(1):41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 53.Bournazos S, Gazumyan A, Seaman MS, Nussenzweig MC, Ravetch JV. Bispecific anti-HIV-1 antibodies with enhanced breadth and potency. Cell. 2016;165(7):1609–1620. doi: 10.1016/j.cell.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.