

Abstract
Rationale
The ventricular conduction system (VCS) rapidly propagates electrical impulses through the working myocardium of the ventricles to coordinate chamber contraction. Genome-wide association studies (GWAS) have associated nucleotide polymorphisms, most are located within regulatory intergenic or intronic sequences, with variation in VCS function. Two highly correlated polymorphisms (r2>0.99) associated with VCS functional variation (rs13165478 and rs13185595) occur 5’ to the gene encoding the bHLH transcription factor HAND1.
Objective
Here, we test the hypothesis that these polymorphisms influence HAND1 transcription thereby influencing VCS development and function.
Methods and Results
We employed transgenic mouse models to identify an enhancer that is sufficient for left ventricle (LV) cis-regulatory activity. Two evolutionarily conserved GATA transcription factor cis-binding elements within this enhancer are bound by GATA4 and are necessary for cis-regulatory activity, as shown by in vitro DNA binding assays. CRISPR/Cas9-mediated deletion of this enhancer dramatically reduces Hand1 expression solely within the LV but does not phenocopy previously published mouse models of cardiac Hand1 loss-of-function. Electrophysiological and morphological analyses reveals that mice homozygous for this deleted enhancer display a morphologically abnormal VCS, and a conduction system phenotype consistent with right bundle branch block. Using 1000 Genomes Project data, we identify three additional SNPs, located within the Hand1 LV enhancer, that compose a haplotype with rs13165478 and rs13185595. One of these SNPs, rs10054375, overlaps with a critical GATA cis-regulatory element within the Hand1 LV enhancer. This SNP, when tested in electrophoretic mobility shift assays (EMSA), disrupts GATA4 DNA-binding. Modeling two of these SNPs in mice causes diminished Hand1 expression and mice present with abnormal VCS function.
Conclusions
Together, these findings reveal that SNP rs10054375, which is located within a necessary and sufficient LV-specific Hand1 enhancer, exhibits reduces GATA DNA-binding in EMSA and this enhancer in total, is required for VCS development and function in mice and perhaps humans.
Keywords: Arrhythmia, conduction system, cardiac development, transcription factors, genetics, Arrhythmias, Animal Models of Human Disease, Developmental Biology, Gene Expression and Regulation, Genetically Altered and Transgenic Models
Graphical Abstract
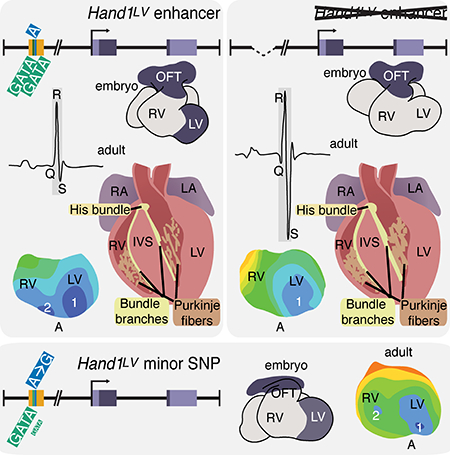
INTRODUCTION
The cardiomyocytes that initiate and transmit the electrical impulses that drive coordinated heart contraction compose the cardiac conduction system (CCS). Within the ventricles, the ventricular conduction system (VCS) rapidly propagates these electrical impulses from the atrioventricular node to the ventricular apex to depolarize the working cardiomyocytes.1 The VCS, which consists of the His bundle, the left bundle branch (LBB), right bundle branch (RBB), and the Purkinje fiber (PF) network, is derived from trabecular myocardium and septum crest,.2 VCS disorders often manifest as arrhythmias.3 The genetic and developmental mechanisms that underlie arrhythmias are poorly understood. In humans, the QRS complex in a surface electrocardiogram (ECG) depicts VCS-mediated depolarization of the ventricles.4 GWAS analyses have identified numerous QRS duration-associated single nucleotide polymorphisms (SNPs).5–8 The majority of these SNPs occur within intergenic or intronic sequences that are hypothesized to lie within regulatory DNA;9, 10 thereby impacting transcription and deleteriously influencing CCS development and/or function.10, 11
Here, we identify a 336 bp evolutionarily conserved murine enhancer (Hand1LV), located ~15Kb 5’ to the transcriptional start site of the gene encoding the bHLH transcription factor HAND1, that is necessary and sufficient to drive reporter gene expression specifically within the LV. In mice, HAND1 functions within the embryonic LV as the heart undergoes trabeculation.12 Conditional cardiac Hand1 ablation during embryogenesis leads to ventricular septal defects (VSDs) and hyperplastic atrioventricular valves.13 Although CRISPR/Cas9-mediated deletion of the Hand1LV enhancer dramatically reduces LV Hand1 expression, it does not phenocopy the Hand1 cardiomyocyte-specific knockout phenotype. In humans, two highly correlated SNPs (r2>0.99) associated with QRS duration (rs13165478 and rs13185595) flank the region syntenic to the Hand1LV enhancer.7 Individuals carrying the minor variant of rs13165478 exhibit a QRS interval shortening of approximately 1 ms.7, 8, 14, 15 In mice, transgenic Hand1 overexpression within the adult myocardium causes QRS prolongation, a predisposition to ventricular arrhythmia, and elevated cardiac expression levels of the gap junction subunit CONNEXIN40 (encoded by Gja5).16 Given the proximity of these SNPs to the Hand1LV enhancer, and the potentially novel role for HAND1 in the cardiac conduction system, we hypothesized that these SNPs may influence the function of one or more essential Hand1 cis-regulatory elements within the Hand1LV enhancer. Mice homozygous for the Hand1LV enhancer deletion present with decreased LV Hand1 expression, a prolonged QRS interval, shortened PR interval, dysregulated LV gene expression, aberrant VCS development, and impaired CCS function. The Hand1LV enhancer contains two evolutionarily conserved GATA cis-regulatory elements, which are capable of GATA4 DNA-binding and are necessary for Hand1LV enhancer transcriptional activity. Using 1000 Genomes Project data, 17 we identified three additional SNPs located within the evolutionarily conserved Hand1LV enhancer, which are highly correlated with the QRS-associated SNP rs13165478 (all r2>0.95 in both European and African ancestry populations). One of these novel SNPs, rs10054375, directly overlaps and alters sequence of the 3’ critical GATA cis-regulatory element within the Hand1LV enhancer resulting in a reduction of GATA4 DNA-binding affinity. Modeling two of the three novel SNPs, rs10076436 and rs10054375 in mice results in modest yet significant reduction of Hand1 LV expression accompanied by abnormal VCS function. Together, these findings reveal that SNP rs10054375 impedes efficient GATA DNA-binding within an LV-specific Hand1 enhancer which is necessary and sufficient for normal VCS development and function in mice and potentially in humans.
METHODS
Data availability statement
All datasets featured within this manuscript will be made available from the corresponding author upon request.
Bioinformatics
All sequences were obtained from Ensembl (http://www.ensembl.org) via BLASTN analyses using the mouse Hand1 sequence as a point of reference. Synteny to the Hand1 locus was validated for all conserved non-coding sequences. mVISTA alignment, PATTERNMATCH and CLUSTALW analyses were performed on the following web sites: (http://genome.lbl.gov/vista/index.shtml and http://workbench.sdsc.edu).
Cloning
To generate Constructs #22, the putative Hand1 enhancer element was isolated from C57Bl/6 genomic DNA using the primers BoxA(F) TATCACGTGCCTAGCTGAGGGGACTCT and BoxA(R) CATATGTCGACTTGTCTCAATCCCCCT and cloned into the hsp68-lacZ expression construct.18 This sequence was subcloned using primers BoxAEMSA_01 and BoxAEMSA_04 RC (sequence below) to generate Constructs #23. All constructs were sequence validated.
To generate Construct #22.G1,2, point mutagenesis was performed using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer’s instructions with the following primers and their reverse complements: H1BoxA[GATAmut1], 5’-ACCCTTGTCTCTTCCACCTTCGCGAAGCCTCACACGTTTGGGG-3’; H1BoxA[GATAmut2], 5’-AAACAGAGGCGGGTGGCATGCCACCATAGCCCCTTCTTGCC-3’. Mutagenized base pairs are in bold font.
Experimental mice
All transgenic mice were generated either by the Indiana University Transgenic and Knock-Out Mouse Core or the University of Michigan Transgenic Animal Model Core. Genotyping of the Hand1tm1Eno, Cntn23’UTR-IRES-Cre-EGFP and Gt(ROSA)26Sortm1Sor alleles have been previously described.13, 19, 20 Yolk sac DNA was used to assess transgene integration using the primer pairs: lacZ(F), 5’-GATCCGCGCTGGCTACCGGC-3’ and lacZ(R), 5’-GGATACTGACGAAACGCCTGCC-3’.
CRISPR/Cas9 deletion of the Hand1LV enhancer was performed by SAGE Labs, a Horizon Discovery Group Company. CRISPR/Cas9 generation of the Hand136,75 allele was performed by the Genome Engineering and iPSC Center at Washington University. Both alleles were generated and maintained on an FVB genetic background. For the Hand1ΔLV allele, the upstream and downstream gRNAs used were 5’-ACTACGTATAGCTGAGACTATGG-3’ and 5’-GTGGAGAGTCCCCTCAGCTAGGG-3’, respectively. Pups and embryos were genotyped using the primers Hand1LV114(F), 5’-GCTGTGTATTGTGATGGAGAGG-3’ and Hand1LV114(R), 5’-CTTTTGACACAAAGGCTCCTG-3’, which generated an 854 bp wild-type amplicon, and a 104 bp Hand1ΔLV amplicon.
For the Hand136,75 allele, the upstream and downstream gRNAs used were MS644.Hand1.sp22 5’-CCACCCGCCTCTGTTTCGTTNGG-3’ and MS644.Hand1.sp8 5’-CCAAACGAAACAGAGGCGGGNGG-3’. Pups and embryos were genotyped using the primers Hand1LV_dual_mod(F) 5’-ATCCCCACCCTTGTCTCTTC-3’, and Hand1LV_dual_mod(R), 5’-GGTGCAGAAACACGAGATCA-3’. Amplicons were digested with FokI, which generated an 351 bp wild-type amplicon, and 246 bp and 105 bp Hand136,75 amplicons. For both the Hand1ΔLV and Hand136,75 alleles, founder mice were bred with wild-type FVB mice to generate F1 mice. These F1 mice and intercrossed F2 mice were then intercrossed to generate F2 and F3 mutant mice and embryos for analysis. Both male and female embryos and pups were used for all analyses. All animal maintenance and procedures were performed in accordance with the Indiana University School of Medicine Animal Care and use committee.
X-gal staining
X-gal staining was performed as previously described. 21 If stained embryos were to be sectioned and immunohistochemically stained, they were fixed both pre- and post-staining in 10% neutral buffered formalin, rather than, respectively, 2% paraformaldehyde-0.2%glutaraldehyde and 4% paraformaldehyde. P7 hearts were dehydrated in ethanol and cleared in a 1:1 mixture of methyl salicylate:benzyl benzoate.
Immunohistochemistry
Immunohistochemistry was performed as previously described.22 α-CNTN2 (R&D Systems, #AF4439) antibody was used at a dilution of 1:50. α-GJA5 (Boster Bio: PA1368) antibody was used at a dilution of 1:100. Primary antibodies were bound with biotinylated α-goat secondary antibody and SA-HRP (Vector Laboratories). HRP color reaction was developed using the DAB Peroxidase Substrate kit (Vector Laboratories) according to the manufacturer’s instructions. α-HCN4 (Merck Millipore: AB5808) rabbit polyclonal and α-ACTC1 (Sigma: A9357) mouse monoclonal antibodies were diluted 1:200 and 1:400, respectively. Control immunohistochemistry lacking the addition of the primary antibody fail to produce immunodetection within CCS tissues (CNTN2, HCN4, CX40) or cardiomyocytes (ACTC1).
In situ hybridization
Whole mount in situ hybridization was performed as described.23 Antisense digoxygenin-labeled Hand1 riboprobe was synthesized using T7, T3 or SP6 polymerases (Promega) and DIG-Labeling Mix (Roche) using a linearized plasmid template.
Immunohistochemical 3D reconstruction
3D reconstruction of HCN4 expression was performed as previously described.24 Briefly, P0 hearts were sectioned at 10 μm, and each third section was immunohistochemically stained with antibodies against both HCN4 and ACTC1. Images of immunohistochemically stained sections were then captured with a Leica DM 6000 fluorescence microscope. The stacks of images were aligned in Amira and then labeled in the same program to render a 3D reconstruction.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as previously described.25, 26 In vitro transcription and translation was performed using a pCS2-FLAG+Gata4 expression plasmids and the TnT rabbit reticulocyte lysate in vitro transcription system (Promega) as per manufacturer’s instructions. To generate whole cell lysates, HeLa cells were transfected using X-tremeGENE HP transfection reagent (Roche) according to manufacturer’s instructions. Cells were harvested and lysed via sonication. The following oligos, which were annealed to their complements, were used to generate radiolabelled probes;
GATA 5’, 5’-ACCCTTGTCTCTTCCACCTTTATCAAGCCTCACACGTTTGGGG-3’; GATA 3’, 5’-AAACAGAGGCGGGTGGCAGATAACCATAGCCCCTTCTTGCC-3’; GATAmut 5’, 5’-ACCCTTGTCTCTTCCACCTTCGCGAAGCCTCACACGTTTGGGG-3’; GATAmut 3’, 5’-AAACAGAGGCGGGTGGCATGCCACCATAGCCCCTTCTTGCC-3’; WT, 5’-CAAACGAAACAGAGGCGGGTGGCAGATAACCATAGCC-3’; rs10076436, 5’-CAAACGAAACAGACGCGGGTGGCAGATAACCATAGCC-3’; rs10054375, 5’-CAAACGAAACAGAGGCGGGTGGCGGATAACCATAGCC-3’; rs10076436/rs10054375, 5’-CAAACGAAACAGACGCGGGTGGCGGATAACCATAGCC-3’
Statistical analyses
Significance of QRTPCR results was determined by two tailed students T-test followed by post hoc Benjamini-Hochberg FDR correction that is automatically calculated by QuantStudio 3 qRTPCR thermal cycler software analysis package. ECG measurements were assessed under blinded conditions. For analyses of ECG parameters, the mean ± standard error was determined for the indicated number of mice assayed. Normal distribution of the data was determined by Shapiro-Wilk test. Significant variation among data sets fitting a normal distribution was then assessed using one-way ANOVA followed by Tukey’s test, for post hoc comparisons, as specified. For non-parametric comparison between 2 groups, the Mann-Whitney U test was employed. F0 transgenic reporter sets were compared via Fisher Exact Test (p ≤ 0.05). Box plots were generated using BoxPlotR (http://shiny.chemgrid.org/boxplotr/). Center lines within the boxes indicate medians. The box limits represent the 25th and 75th percentiles. Whiskers represent 1.5X the interquartile range from the box limits and outliers are represented by open dots. Outliers are defined according to Tukey’s rule. Mann-Whitney U tests were applied to all Optical mapping analysis.
Surface ECG
All mice in ECG studies were assayed at 5-weeks of age. Mice were anesthetized with isoflurane (2.5% vol vapor) and placed in a supine position on a heating pad. Body temperature was continuously monitored via a rectal thermistor and maintained between 37° and 38° Celsius. Platinum needle electrodes (Model F-E2, Grass Technologies, West Warwick, RI) were placed subcutaneously in the right and left axilla and in the left groin. Bipolar leads I, II, and III were sequentially recorded for 1 min each, using an isolated biological amplifier (ISO-Dam, World Precision Instruments, Inc. Sarasota, FL) with a high-frequency filter of 1 kHz and 1,000-fold gain. The signal was fed into an analog-to-digital converter (DAQCard Al-16XE-50; National Instruments, Austin, TX), sampled with 2 kHz at 16-bit resolution, and displayed in real-time on the screen of a laptop computer by using a custom-written data acquisition system implemented in Matlab (The Math Works, Natick, MA). ECG recordings were analyzed using the Clampfit module of the pClamp11 software (Molecular Devices, Sunnyvale, CA). A signal-averaged ECG was generated by overlaying all the cycles in a recording, using QRS maximum or QRS minimum for alignment. The PR-interval was measured from the beginning of the P-wave to the beginning of the QRS complex. QRS duration was measured from the first deflection of the Q-wave (or R-wave in the absence of a Q-wave) to the nadir of the S-wave (defined as the point of minimum voltage in the terminal portion of the QRS complex; method I), and to the onset of the J wave (method II)27. The QT interval was defined as the interval from the beginning of the QRS complex to the end of the T-wave (defined as the point where the T-wave merges with the isoelectric line). The R-R interval was obtained as the average R-R interval over the sampling period.
Epicardial optical voltage mapping of Langendorff-perfused hearts
High-resolution optical mapping experiments were performed on 7- to 50-week-old Hand1LV mice, 7- to 15-week-old Hand136;75/36/75 mice, wild-type littermates, and 5 47-to 70-week-old Hand136;75/36/75 mice and assayed as described.28 Hearts were isolated and retrogradely perfused in Langendorff-mode with temperature-controlled (37°C) Krebs-Henseleit solution (pH 7.4 when gassed with a mixture of 95% O2 and 5% CO2) at an aortic pressure of 70 cm H2O. A volume-conducted ECG was monitored continuously throughout the experiment. After 10 minutes of stabilization, the hearts were stained with the voltage-sensitive dye Di-4-ANEPPS (2 μL of a 2-mmol/L stock solution). The heart was then washed with dye-free solution for 5 min followed by the addition of (±)-blebbistatin to uncouple contraction from excitation (10 μmol/L; Tocris Bioscience, Minneapolis, MN). The stained hearts were illuminated with a laser at a 532 nm wavelength and the fluorescence was collected by a MiCAMUltima-L CMOS camera (SciMedia, Costa Mesa, CA) through a 715-nm long-pass filter. The fluorescence was recorded at a 1 ms/frame rate in a 100 ×100 pixel grid with a spatial resolution of 0.35×0.35 mm2 per pixel. Optical signals were processed with both spatial (3×3 pixels Gaussian filter) and temporal (3 frames moving average) filtering. Hearts were paced at the right atrium at a cycle length of 120 ms. Three 1-s recordings were captured sequentially while the right atrium was paced at cycle lengths of 120, 150 and 200 ms. This protocol was repeated once. Finally, three 1-s recordings were acquired while the hearts were paced from the apex at cycle lengths of 120, 150 and 200 ms. Activation time was measured as described previously.28
Quantitative RT-PCR
Total RNA was isolated from E11.5 ventricles using the High Pure RNA Isolation Kit (Roche). This RNA was then used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For qRT-PCR, cDNA was amplified using TaqMan Probe-Based Gene Expression Assays (Applied Biosystems) and the QuantStudio 3 Real-Time PCR System (ThermoFisher). Normalization to GAPDH was used to determine relative gene expression and statistical analysis automatically applied by the instrument software.
RESULTS
A GATA-dependent distal Hand1 enhancer is necessary and sufficient for gene expression within the LV
To identify potential cis-regulatory elements controlling HAND1 LV transcription, we interrogated a 744 bp murine conserved non-coding sequence (CNS) that displays > 75% conservation between mammals and reptiles over 100 bp (Fig. 1A; red). We isolated this sequence, located at chr11:57660605–57661348 (mm9), and generated a β-galactosidase (lacZ) reporter construct (Fig. 1A; Construct #22). Of 12 transgenic E10.5 F0 embryos, 8 displayed robust X-gal staining within the LV myocardium similar to Hand1lacZ knock-in mice21 (Fig. 1, compare 1B, E with C, F). We further refined the Hand1LV enhancer to the 5’ most 336 bp of this CNS (Fig. 1A; Construct #23), observing LV-specific X-gal staining, consistent with Hand1lacZ mice, in all 8 transgene-positive F0 embryos (Fig. 1D, G).
Figure 1. A GATA-dependent Hand1 enhancer is sufficient to drive LV-specific gene expression.

A) Schematic representation of the mouse Hand1 locus, including the two conserved GATA cis-elements (green), and hsp68-lacZ reporter constructs. Exons and conserved non-coding sequence are highlighted in grey and red, respectively. Position, in Kb, relative to the Hand1 transcription start site is noted. Positive transgenic expressers within the LV, and the total number of transgenic embryos examined are indicated. B-G) Whole mount (B-D) and transverse sections (E-G) of X-gal-stained E10.5 F0 embryos showing robust transgene expression in the LV (arrowhead), but not the neural crest cells (white arrow) or myocardial cuff (black arrow) of the outflow tract, of transient transgenics expressing Constructs #22 (C, F) and #23 (D, G). An E10.5 Hand1lacZ/+ embryo is included for comparison (B, E). LV – left ventricle, RV – right ventricle. H) EMSA using lysate programmed with FLAG+GATA4 demonstrates GATA4 binding (arrowhead) to radiolabeled oligonucleotides mimicking the two GATA consensus sites (GATA 5’ and GATA 3’) in the Hand1 LV enhancer. Western blots for α-FLAG (inset) verifies synthesis of FLAG-GATA4 that was added equally to each EMSA where indicated. I, J) A representative X-gal-stained E10.5 F0 embryo positive for Construct #22.G1,2 showing a lack of transgene expression in the LV (arrowheads). All Scale bars = 200 μM. n reflects biological replicates in transgenic analysis.
To identity evolutionarily conserved cis-elements that regulate Hand1LV enhancer activity, we employed phylogenetic footprinting. Comparison of the orthologous Hand1LV enhancer sequences in different species revealed two GATA cis-regulatory elements that are evolutionarily conserved. The 5’ GATA cis-element is conserved among mammals, and 3’ GATA cis-element is conserved through reptiles (Fig. 1A). Gene regulation within the developing ventricular myocardium involves GATA transcription factors.29, 30 Publicly available ChIP-Seq data sets indicate that GATA4 occupies the Hand1LV enhancer within cardiomyocyte progenitor cells (Online Fig. I). Electro Mobility Shift Assay (EMSA) confirms specific GATA4 DNA-binding to both GATA cis-element sequences (Fig. 1H, arrowhead). Additionally, a 10-fold excess of unlabeled competitor oligo (WT) reduces binding to the labeled probe; however, addition of an unlabeled competitor oligo in which the GATA cis-regulatory element is mutagenized (mut) does not effectively compete for GATA4 DNA binding (Fig. 1H).
We next assessed whether these GATA cis-elements are necessary for Hand1LV activity in vivo. We generated point mutants in the GATA elements within Construct #22 (Construct #22.G1,2; Fig. 1A). None of the 5 transgenic E10.5 F0 embryos generated with Construct #22.G1,2 exhibited LV X-gal staining (Fig. 1I, J; p = 0.029 compared to Construct #22 transgenics, Fisher Exact Test). We conclude that the conserved GATA cis-binding elements within the minimal Hand1 enhancer are necessary for LV transcription.
To validate that the Hand1LV enhancer is necessary for myocardial Hand1 expression and LV development, we employed CRISPR/Cas9 to delete the genomic region containing this enhancer (Fig. 2A). Two independent F0 founders were identified. The line containing the smaller 750 bp deletion of Hand1LV, we termed Hand1ΔLV. This deletion encompasses all but 4 bp on the 5’ end of the 744 bp fragment included in Construct #22, as well as an additional 10 bp on the 3’ end. We generated Hand1ΔLV/ΔLV mutants at E10.5 to test whether in vivo Hand1LV deletion disrupts Hand1 LV gene expression. Whole mount Hand1 in situ hybridization reveals a marked reduction of LV Hand1 expression within Hand1ΔLV/ΔLV embryonic hearts (compare Fig. 2B, C, arrowheads). Hand1 expression within the cardiac outflow tract (OFT) and lateral mesoderm (LM) is unaffected. Next, E11.5 ventricles isolated from Hand1ΔLV/ΔLV and Hand1+/+ littermates were subjected to qRT-PCR using Taqman primers specific for Hand1 (Fig. 2D). Consistent with in situ hybridization findings, Hand1 RNA levels are markedly reduced to roughly 20% of its normal levels in Hand1ΔLV/ΔLV mutants. Expression of the putative upstream Hand1 transcriptional regulator, Gata4, was unchanged (Fig. 2D). Additionally, the expression of evolutionarily conserved genes located within 0.5 Mb upstream and downstream of Hand1 are unaltered in E11.5 ventricles of Hand1ΔLV/ΔLV mutants (Online Fig. II). These findings demonstrate that deletion of Hand1LV specifically reduces LV Hand1 expression and does not impact the expression of neighboring genes or any Hand1 expression domains outside the LV at mid-gestation.
Figure 2. The Hand1LV enhancer is necessary for LV gene expression of Hand1 and its downstream targets.

A) Schematic representation of the mouse Hand1 locus, and the CRISPR/Cas9-generated Hand1ΔLV allele. Exons and conserved non-coding sequence are highlighted in grey and orange, respectively. gRNA locations are shown in green. Arrowheads depict genotyping primers. B, C) Whole mount in situ hybridization shows a marked reduction in Hand1 expression within the LV of Hand1ΔLV/ΔLV mutant embryos (white arrowheads). LM, lateral mesoderm; OFT, outflow tract. D) qRT-PCR of isolated E11.5 ventricles reveal diminished Hand1 (# p=6.4X10−06) and Hand1 downstream target genes Cited1 (# p=2.02X10−04) and Gaj5 (# p=2.02X10−04) between Hand1ΔLV/ΔLV mutants (n=6) and wild-type controls (n = 4). Gata4 expression is unaffected (P=0.142). Significance changes in mRNA levels were calculated using a two-tailed students T-test with a post hoc Benjamini-Hochberg false discovery rate (FDR). Scale bars = 200 μM. Sample sizes indicates biological replicates.
Hand1ΔLV/ΔLV mice display abnormal VCS gene regulation, function, and development
We then assessed whether Hand1ΔLV/ΔLV mutants display changes in cardiac gene expression similar to what is observed in HAND1 loss-of-function.13 Conditional inactivation of Hand1 within the embryonic heart downregulates LV expression of the transcriptional co-activator Cited1.13 qRT-PCR reveals that Cited1 expression is likewise significantly downregulated in Hand1ΔLV/ΔLV E11.5 mutant ventricles (Fig. 2D). Hand1 loss- or gain-of-function is associated with altered expression of the VCS marker Gja5.13, 16 qRT-PCR shows that expression of Gja5 is significantly downregulated within E11.5 Hand1ΔLV/ΔLV ventricles (Fig. 2D). Together, these findings demonstrate that deletion of the Hand1LV alters normal E11.5 expression of HAND1 downstream targets.
In contrast to the high penetrance neonatal lethality observed in cardiomyocyte HAND1 loss-of-function,13 we observed that Hand1ΔLV/ΔLV mice are viable and fertile (Online Table I). Although histological analyses at E17.5 reveal that roughly 1 of 6 Hand1ΔLV/ΔLV mutant hearts present with a VSD, we observe no neonatal or embryonic lethality in Hand1ΔLV/ΔLV mice. The presence of VSDs is consistent with both the conditional myocardial Hand1 13 and conditional LV Hand1 knockouts.12 The penetrance; however is markedly reduced within Hand1ΔLV/ΔLV mutants. Also in contrast to both the myocardial and LV Hand1 conditional knockouts, the LV free wall of Hand1ΔLV/ΔLV mutants is not grossly dysmorphic. Thus, homozygosity of the Hand1ΔLV allele does not impart significant lethality.
In humans, the conserved HAND1LV enhancer is located in between two QRS interval–associated SNPs, rs13165478 and rs13185595. We therefore examined whether reduced embryonic Hand1 LV expression alters postnatal cardiac electrical function. ECG recordings in anesthetized 5-week-old Hand1ΔLV/ΔLV mice reveal significantly shortened PR intervals and prolonged QRS intervals when compared to both Hand1+/+ and Hand1+/ΔLV littermates (Fig. 3A–C and Online Table II). RR and QT intervals were not significantly affected (Fig. 3D and Online Table II). These results are largely consistent with observations in humans where SNP rs13165478 has been associated with a shorter PR interval, in addition to its association with QRS interval, although this association is not at genome-wide significance31 (P=0.003).
Figure 3. VCS function is abnormal in Hand1ΔLV/ΔLV mutants.

A) Representative lead II ECG tracings show prolonged QRS in Hand1ΔLV/ΔLV mutants (blue) compared to Hand1+/+ controls (red) at P35. B-D) Lead II ECG analyses confirm that PR interval (B) is shortened (p=0.041) and QRS duration (C) is prolonged in Hand1ΔLV/ΔLV mutants compared with Hand1+/+ (p=0.01) and Hand1ΔLV/+ littermates (p=0.021). QT interval (D) is unaltered. Center lines - medians; box limits - 25th and 75th percentiles; whiskers - 1.5X the interquartile range from the box limits. Individual data points are represented by open circles outliers represented by closed circles. n = 18 wild-type, 19 Hand1ΔLV/+, and 25 Hand1ΔLV/ΔLV mutants. E-G) Ventricular epicardial activation mapping in Langendorff-perfused hearts during right atrial pacing at a cycle length of 120 ms. Schematized atrial pacing (E) illustrates sites (blue dots 1 and 2) corresponding to the endocardial insertions of the Purkinje network branches. (F, G) Activation maps obtained from the anterior epicardial surfaces, with color-coded isochronal lines 1 ms apart. Time scales presented in ms, are to the right of the optical maps: dark blue, 0–5.7 ms; light blue to light green, 5.7 to 8.55 ms; green to light yellow 8.55 to 11.4 ms; light yellow to light orange 11.4 to 14.25 ms; light orange to red 14.25 to 17.1 ms; red to dark red 17.1 to 20 ms. H) Differences in epicardial activation times (p=2X10−6) during atrial pacing between Hand1+/+ (white) and Hand1ΔLV/ΔLV (grey) hearts. I) RV epicardial breakthroughs, relative to the LV, in Hand1+/+ (n=8) and Hand1ΔLV/ΔLV (n=7) hearts (p=0.029). J) Schematized apical pacing showing to point of initial epicardial activation (blue). K and L) Isochronal activation maps obtained during direct ventricular pacing at a cycle length of 120 ms in the hearts respectively shown in (F) and (G) time scale identical to that of F and G. M) Epicardial activation times during apical pacing for the same hearts as in (H). p values calculated for B and C employed one-way ANOVA followed by Tukey’s test for post hoc analyses. P values in H and I were calculated using Mann-Whitney Rank Sum test. All sample sizes indicate biological replicates.
In contrast to human ECGs, in which the QRS complex represents the depolarization of the ventricles, the mouse QRS complex represents both ventricular depolarization and repolarization.27 QRS prolongation in 5-week-old Hand1ΔLV/ΔLV mice indicates impaired electrical activation of the ventricles, but extrapolating this to human data is a challenge. To determine ventricular activation patterns, we next performed optical voltage mapping of isolated perfused hearts from Hand1+/+ and Hand1ΔLV/ΔLV mice between 7 and 50 weeks of age. Representative colored activation maps obtained from the anterior ventricular surface are shown in Figure 3. During atrial pacing (Fig. 3E), Hand1+/+ hearts show two quasi-simultaneous epicardial breakthroughs close to the apices of the LV and right ventricle (RV; denoted as 1 and 2 in Fig. 3F, Online Movie 1). These breakthroughs form wave fronts that fully activate the field of view within an average of 4.8 ms (Fig. 3H). Within roughly half (7/15) of atrial paced Hand1ΔLV/ΔLV hearts, singular breakthroughs near the LV apex are observed at all three pacing cycle lengths tested (120, 150 and 200 ms) and this is observed in both young and old adults (Fig. 3G; Online Movie 2). An additional four Hand1ΔLV/ΔLV hearts, rather than exhibiting quasi-synchronous LV and RV epicardial breakthroughs, displayed a second, delayed (>2 ms later) epicardial breakthrough proximal to the RV apex (Online Movie 3). This delayed breakthrough phenotype is observed in both young and old adults (Online Fig. III). We also observe two Hand1ΔLV/ΔLV hearts that exhibited a second, ectopic breakthrough near the base of the LV (Online Movie 4), and one heart that presented with a total of three breakthroughs – two proximal to the apex of the LV, and one near the apex of the RV (Online Movie 5). Given the rarity of these observations, we cannot account for any possible age correlations (Online Fig. III). The ventricular activation sequence of the one atrial paced Hand1ΔLV/ΔLV heart is indistinguishable from activation sequences observed in Hand1+/+ hearts (Online Movie 5; summarized in Table 1). Wave fronts emanating from breakthroughs in Hand1ΔLV/ΔLV hearts activated the field of view within 7.2 ms on average; significantly longer than Hand1+/+ hearts (Fig. 3H). Given that the PR interval is shortened in Hand1ΔLV/ΔLV mutants, we hypothesized that conduction from the atria to the ventricles is affected in these mutants. We measured the time elapsed from the epicardial activation of the right atrium (RA) to the LV breakthrough, observing similar durations at all cycle lengths tested (Online Fig. IV A–D). Epicardial breakthrough in the RV, relative to the LV, is significantly delayed in Hand1ΔLV/ΔLV mutants at cycle lengths of 120 and 150 ms (Fig. 3I, p=0.029 120 ms; Online Fig. IV: p=0.029, 120 ms; p=0.0122, 150 ms). Apical pacing (Fig. 3J), which propagates an electrical signal through both the working myocardium and the Purkinje network, resulted in similar epicardial activation patterns and activation times in both Hand1+/+ and Hand1ΔLV/ΔLV hearts (Fig. 3K–M; Online Movies 6 and 7). Optical mapping of mutants homozygous for a second, independent Hand1ΔLV allele, in which 823 bp overlapping Hand1LV has been deleted (termed Hand1ΔLV106), also reveals similar aberrant activation patterns when atrial paced (n = 3, Table 1). No functional evidence for the existence of atrioventricular accessory pathways could be demonstrated with decremental atrial or ventricular pacing (Online. Fig. IV B–D). Collectively, these findings indicate that conduction across the right VCS is impeded in Hand1ΔLV/ΔLV mutants, despite the fact that Hand1LV drives expression only within the LV. Further, the results suggest that the modest PR interval shortening seen in Hand1ΔLV/ΔLV mutants in vivo is due to enhanced atrioventricular nodal conduction rather than an atrioventricular bypass tract (Online Fig. IV B–D).
Table 1.
Epicardial activation phenotypes in Hand1LV enhancer mutants.
| Phenotype | Hand1+/+ (n=13) | Hand1ΔLV/ΔLV (n=15) | Hand1ΔLV106/ΔLV106 (n=3) | Hand136;75/36 75 (n=11) |
|---|---|---|---|---|
| Two breakthrough points in the LV | 0 (0%) | 0 (0%) | 0 (0%) | 1 (9.1%) |
| Single breakthrough point in the LV | 0 (0%) | 7 (46.7%) | 0 (0%) | 0 (0%) |
| Late (> 2 ms) breakthrough point in the RV | 0 (0%) | 4 (26.7%) | 1 (33.3%) | 2 (18.2%) |
| Single breakthrough point in the RV | 0 (0%) | 0 (0%) | 0 (0%) | 1 (9.1%) |
| Diffuse RV breakthrough misplaced toward base | 0 (0%) | 0 (0%) | 0 (0%) | 1 (9.1%) |
| Ectopic breakthrough proximal to the base of the LV | 0 (0%) | 2 (13.3%) | 0 (0%) | 0 (0%) |
| Two breakthroughs in the RV - one proximal to the apex and one diffuse and displaced toward the base | 0 (0%) | 0 (0%) | 0 (0%) | 1 (9.1%) |
| Two breakthroughs proximal to the apex of the LV, one in the RV | 0 (0%) | 1 (6.7%) | 1 (33.3%) | 1 (9.1%) |
| Late, ectopic breakthrough at the mid RV | 0 (0%) | 0 (0%) | 1 (33.3%) | 0 (0%) |
| Phenotypically Normal* | 13 (100%) | 1 (6.7%) | 0 (0%) | 4 (36.4%) |
Two quasi-synchronous epicardial breakthroughs proximal to the apices of the LV and RV
Suspecting VCS phenotypes congenitally derived from embryonic Hand1 function,21, 32 we assessed conduction system morphology in Hand1ΔLV/ΔLV mutants. Whole mount X-gal staining from the Hand1lacZ allele21 shows that Hand1lacZ expression is detectable within the postnatal (P42) VCS. Immunohistochemistry showed clear overlap between Hand1-driven X-gal stain and GJA5 immunoreactivity (Online Fig. V) confirming Hand1 expression within the adult VCS. Breeding the Hand1ΔLV allele onto the Hand1lacZ knockout allele (Hand1ΔLV/lacZ) did not decrease postnatal survival (Online Table I). However, comparison of X-gal stained Hand1+/lacZ and Hand1ΔLV/lacZ hearts revealed an increase in staining along the LV septal wall of Hand1ΔLV/lacZ hearts (Fig. 4A, B arrows), suggesting that the Hand1ΔLV/ΔLV LV VCS may be hyperplastic. To test this, Hand1+/lacZ and Hand1ΔLV/lacZ hearts were stained for both X-gal and immunoreactivity of the CCS marker CONTACTIN2 (CNTN2).33 Hand1lacZ and CNTN2 double-positive cells are observed within the left His bundle (Fig. 4C, D, arrowheads) and the LBB (arrow; Fig. 4D). Hand1lacZ-positive cell localization further reveals that, in Hand1ΔLV/lacZ hearts, the cells on the left side of the His bundle appear abnormally dispersed (arrowheads, Fig. 4D), and the LBB appears hyperplastic (Fig. 4D, arrows).
Figure 4. The Hand1 VCS expression domain is expanded in Hand1ΔLV mutants.

A, B) Whole mount X-gal staining of the Hand1lacZ allele shows X-gal expression in the postnatal (P42) LV septal wall (A, arrow). This staining is expanded in Hand1lacZ/ΔLV mutants (B). C, D) Coronal sections of Hand1lacZ/+ hearts, stained with X-gal (blue) in which the CCS has been labeled immunohistochemically with α-CNTN2 (brown), show partial co-localization of Hand1-reporter-positive cells to the His bundle (arrowhead) and left bundle branch (arrow).
To confirm the dysmorphic VCS phenotype, Hand1ΔLV/ΔLV mutants were bred onto mice heterozygous for the postnatal CCS-specific Cre driver, Cntn2IRES-Cre19 and the R26RlacZ reporter allele. P7 hearts were X-gal stained and cleared. Compared to Hand1+/+ and Hand1+/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ controls, Hand1ΔLV/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ visual observation of hearts reveals a hyperplastic His bundle, and a significantly broader more robust X-gal-positive staining domain overlapping with the LBB (Fig. 5A–C, arrowhead and arrow, respectively; n = 6). These data support that the left side of the Hand1ΔLV/ΔLV VCS is dysmorphic and hyperplastic. Additional staining of P42 hearts support that, not only is the left side of the VCS hyperplastic, (observed in 4 out of 5 hearts), Fig. 5D, E), the peripheral portions of the RBB network (white arrowheads) appear more compacted, and the proximal portions (white arrows) more diffuse, with more observed x-gal stained fascicles than Hand1+/+ controls (observed in 3out of 5 hearts), Fig. 5F, G). To examine the His bundle and LBB in greater detail, immunohistochemistry for the potassium/sodium channel HCN4 was performed upon sections of P0 Hand1+/+ and Hand1ΔLV/ΔLV hearts, which were then reconstructed in three dimensions (Fig. 5H–K). In 2 of 3 Hand1ΔLV/ΔLV hearts examined, the left sided ventricular septum is malformed, with 1 mutant displaying a membranous VSD and 1 displaying an aberrant canal that invades the membranous septum (Fig. 5I). All three Hand1ΔLV/ΔLV mutants displayed abnormally dispersed, hyperplastic His bundles and LBBs when compared to Hand1+/+ controls (Fig. 5I, K).
Figure 5. VCS hyperplasia in Hand1ΔLV/ΔLV mutants.

A-C) P7 hearts of Hand1ΔLV/ΔLV mutants bred onto a Cntn2IRES-Cre; R26RlacZ CCS reporter. The atria and great vessels have been removed, and the hearts have been stained in whole mount with X-gal and cleared. Hand1ΔLV/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ hearts (C; n = 7) displayed an increased number of X-gal-positive His bundle (arrowhead) and left bundle branch (arrow) cells, relative to Hand1+/+;Cntn2+/IRES-Cre;R26R+/lacZ (A) and Hand1+/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ hearts (B). D-G) P42, X-gal-stained Hand1ΔLV/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ hearts (E) display a hyperplastic His bundle (arrowhead), and a significantly broader X-gal-positive staining domain overlapping with the LBB (arrow; n = 4/5), than Hand1+/+ controls (D). The peripheral portions of the RBB network (white arrowheads) in Hand1ΔLV/ΔLV;Cntn2+/IRES-Cre;R26R+/lacZ hearts (G) appear more compacted, and the proximal portions (white arrow) more spread out, and with more fascicles than Hand1+/+ controls (F; n = 3/5). Scale bars = 200 μM. H-K) 3D-reconstructions of HCN4 immunohistochemistry (red) upon sections of P0 hearts. Hand1ΔLV/ΔLV mutants (I, K) display abnormally dispersed, hyperplastic His bundles (white arrowheads) and left bundle branches (white arrows) compared with wild-type controls (H, J). Ventral views of the heart showing HCN4-positive cells with (H, I) and without (J, K) the surrounding myocardial cells (gray) are shown. All Scale bars = 200 μM. Sample sizes indicates biological replicates.
Polymorphisms within the human HAND1LV enhancer are associated with QRS duration
The Hand1ΔLV phenotype supports that functional variation within the syntenic HAND1 enhancer may contribute to altered QRS duration in humans. However, SNPs rs13165478 and rs13185595 are located 5’ and 3’ respectively of the putative human HAND1LV (schematized in Fig. 6A). Moreover, rs13165478 and rs13185595 are not conserved in mouse. We therefore looked for additional genomic variants occurring between rs13165478 and rs13185595, ideally within Hand1LV, that are also associated with altered QRS duration. Three additional HAND1 SNPs occur in virtually perfect LD (r2>0.99) with rs13165478 (Table 2). Two SNPs within this haplotype, rs10076436 and rs10054375, are directly located within the Hand1LV sequence (Fig. 6A). Intriguingly, SNP rs10054375 alters the sequence of the necessary 3’- GATA cis-element binding site. We employed EMSA to assess whether these two SNPs influence GATA-factor DNA binding to HAND1LV. Oligos mimicking the minor variant of SNP rs10054375 are not bound as robustly by GATA4 (Fig. 6B) and, when used as an unlabeled DNA competitor, do not compete with GATA4 binding to the major variant as efficiently (Online Fig. VI). These data suggest that the minor variant of SNP rs10054375 is a weak GATA-factor binding site, and this mutation may decrease transcriptional output of HAND1LV.
Figure 6. Single Nucleotide Polymorphisms within the Human HAND1LV enhancer affect GATA4 binding, Hand1 expression, and VCS function.

A) Schematic representation of the human HAND1 locus illustrating the location of the five QRS-associated SNPs (orange) relative to the two critical GATA binding sites (green) within the conserved non-coding sequence (red). B) EMSA using lysate programmed with FLAG-GATA4 demonstrates GATA4 binding (arrowhead) to radiolabeled oligonucleotides mimicking the major SNP variants of the human HAND1 sequence (WT). GATA4 does not bind as strongly to probes featuring the minor variant of rs10054375. The minor variant of rs10076436 does not impede GATA4 binding. Western blot for α-FLAG (inset) verifies synthesis of FLAG-GATA4 added equally to each EMSA where indicated. + indicates the TnT lysate and the DNA probe represented in the gel shift lane. C) Schematic illustration of the minor SNP variants introduced into the Hand1+/36;75 mouse lines (highlighted in orange). A third, intervening nucleotide (denoted in charcoal) was mutated from G>A to block insertion-deletions. GATA binding site is highlighted in green. D) qRT-PCR of isolated E11.5 ventricles show reduced Hand1 gene expression in Hand136;75/36;75 hearts (n = 14) compared to wild-type controls (n = 18; p=0.039, according to a two-tailed students T-Test and accompanying Benjamini-Hochberg false discovery rate). E-G, K-O) Epicardial activation maps obtained from Langendorff-perfused Hand136;75/36; 75 hearts (7–15 weeks; n=6) during right atrial pacing (pacing cycle length = 120 ms) displayed one (F) or three (G) epicardial breakthroughs, compared to 2 focused epicardial breakthroughs on the anterior walls of the right and left ventricle, characteristic of wild-type controls (E). Additional hearts (47–70 weeks; n=5) displayed two breakthrough points in the LV (K), a late (> 2 ms) breakthrough point in the RV (L, M), Two breakthroughs in the RV, one proximal to the apex and one diffuse and displaced toward the base (N), and a diffuse RV break-through misplaced toward base (O). H-J, P-T) Activation patterns are unaltered (Mann-Whitney U test) during direct ventricular pacing (cycle length = 120 ms) in Hand136;75/36;75 hearts compared with wild-type control. (Time scales presented in milliseconds (ms), are to the right of the optical maps: dark blue, 0–5.7 ms; light blue to light green, 5.7 to 8.55 ms; green to light yellow 8.55 to 11.4 ms; light yellow to light orange 11.4 to 14.25 ms; light orange to red 14.25 to 17.1 ms; red to dark red 17.1 to 20 ms. Sample sizes indicates biological replicates.
Table 2.
HAND1 SNPs associated with altered QRS duration.
| chr5 bp (hg37) | Hispanics15 | Europeans7 | African Americans8 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP ID | CAF | A1/A2 | Beta | SE | P-value | CAF | A1/A2 | Beta | SE | P-value | CAF | A1/A2 | Beta | SE | P-value | |
| rs13185595 | 153872170 | 0.33 | A/G | −0.7002 | 0.1053 | 2.88E-11 | 0.37 | A/G | −0.5587 | 0.0728 | 1.72E-14 | 0.53 | A/G | −0.4359 | 0.1239 | 0.0004356 |
| rs10076436 | 153871841 | 0.33 | G/C | −0.6993 | 0.1052 | 3.02E-11 | ||||||||||
| rs10054375 | 153871832 | 0.33 | C/T | −0.6992 | 0.1052 | 3.00E-11 | ||||||||||
| rs62379942 | 153871070 | 0.33 | T/C | −0.6992 | 0.1051 | 2.87E-11 | ||||||||||
| rs13165478 | 153869040 | 0.34 | A/G | −0.6842 | 0.0978 | 2.69E-11 | 0.36 | A/G | −0.5451 | 0.0708 | 1.38E-14 | 0.53 | A/G | −0.4531 | 0.1354 | 0.0008583 |
To test this hypothesis, we employed CRISPR/Cas9 to generate mice harboring the minor variants of the QRS duration-associated SNPs rs10076436 and rs10054375 (the Hand136;75 allele; Fig. 6C). qRT-PCR of E11.5 Hand136;75/36;75 mutant ventricles revealed a modest but significant (~20%) reduction in Hand1 expression (Fig. 6D). ECGs performed on 5-week-old anesthetized Hand136;75/36;75 mice revealed no significant changes in PR, QRS, or QT interval when compared to Hand1+/+ or Hand1+/36;75 mice (Online Table II). However, epicardial activation maps obtained from 6 atrial paced hearts ranging between 7- and 15-weeks of age reveal a single breakthrough site on the anterior RV free wall in one Hand136;75/36;75 heart (Fig. 6F; Online Movie 8), and three epicardial breakthroughs, one proximal to the apex of the RV and two proximal to the apex of the LV, in another heart (Fig. 6G; Online Movie 9). Identical activation patterns were observed at cycle lengths of 120, 150 and 200 ms. The remaining four Hand136;75/36;75 mutant hearts display activation patterns indistinguishable from Hand1+/+ controls (Fig. 6E; Online Movie 10). Suspecting that age could be a contributing factor given the low penetrance observed between 7 and 15 weeks, we next assayed five hearts isolated from older Hand136;75/36;75 mutants – between 46- and 70-weeks of age. All hearts assayed displayed aberrant activation maps. (Fig 6K–T) One Hand136;75/36;75 heart exhibited two breakthrough points in the LV (Fig. 6K; Online Movie 13). Two others exhibited a late (> 2 ms) breakthrough point within the RV (Fig. 6L, M; Online Movies 14 and 15), phenotypes consistent with those observed in Hand1ΔLV/ΔLV mutant hearts. The remaining two Hand136;75/36;75 hearts displayed, respectively, two breakthroughs within the RV, one proximal to the apex and one diffuse and displaced toward the base (Fig. 6N; Online Movies 16) and a diffuse RV breakthrough misplaced toward the base (Fig. 6O; Online Movies 17). The average time elapsed from the initiation of the wave front at the breakthrough site to activation of the ventricular base was not different among atrial paced Hand1+/+ and Hand136;75/36;75 hearts, nor did we find a significant difference in the epicardial conduction time during ventricular pacing from the apex (Fig. 6H–J, P-T; compare Online Movies 11 and 12). To look for morphological differences within the Hand136;75/36;75 VCS, we X-gal stained P42 Hand136;75/36;75;Cntn2+/IRES-Cre;R26R+/lacZ hearts and compared them to Hand1+/+;Cntn2+/IRES-Cre;R26R+/lacZ controls (n = 5, Online Fig. VII). We observe no differences in VCS morphology. To validate that the Hand136;75/36;75 heart phenotypes are significant when compared to controls, a p-value of 0.001 was obtained using a Fisher exact test. We therefore conclude that the minor variants of SNPs rs10076436 and rs10054375 significantly affect Hand1LV transcriptional output. Although this modest level of regulation is not sufficient to alter CCS morphology, Hand136;75/36;75 hearts display defects in VCS function.
DISCUSSION
Abnormal cardiac conduction can lead to sudden death and is a challenging clinical problem. The origin of conduction defects is broad and GWAS studies have identified hundreds of loci associated with cardiac conduction.7, 8, 14, 15 The majority of conduction-associated SNPs occur within intronic or intergenic sequences suggesting a transcriptional regulation mechanism for these non-coding SNPs. Refined GWAS analyses has localized QRS- and QT-associated SNPs within putative enhancers.10 The goal of these human genetics studies is to functionally characterize these SNPs within the context of gene regulatory elements, and to define how these SNPs impact development and/or physiology, and thereby contribute to complex human traits. Here, we identify an evolutionarily conserved LV-specific HAND1 enhancer located between two intergenic SNPs that are associated with altered QRS duration.7, 8, 14, 15 In mice, Hand1LV is necessary (Fig. 2) and sufficient (Fig. 1) to drive LV-specific gene expression. Two GATA cis-elements directly regulate this enhancer. Hand1ΔLV/ΔLV mice are viable but exhibit both altered LV gene expression and display a morphologically abnormal VCS (Fig. 5). Hand1ΔLV/ΔLV mice also show a significantly broadened QRS interval, shortened PR interval, and 14 out of 15 Hand1ΔLV/ΔLV mutant hearts assayed display altered ventricular activation patterns (Fig. 3). Humans carrying the minor variant of rs13165478 exhibit a roughly 1 ms shortening of the QRS interval, as well as a shortened PR interval that is not significant genome-wide.7, 8, 14, 15 The additional SNP rs10054375 located within the HAND1LV directly changes a necessary GATA cis-element sequence and reduces GATA4 DNA-binding (Fig. 5). This reduced GATA DNA binding within the HAND1LV lowers Hand1 transcriptional activity, reducing Hand1 expression levels.
Variations within the HAND1LV enhancer are associated with QRS interval shortening in humans. Here we show that deletion of the Hand1LV enhancer results in QRS interval broadening in mice. Indeed, recapitulating the human SNPs in mice results in a QRS pattern indistinguishable from controls. The lack of observable ECG phenotype in the Hand136;75/36;75 mice may reflect subtle differences in GATA regulation of Hand1 expression between mice and humans as well as the differences in the physiology of the two species. For example, in humans, QRS represents the depolarization phase of the ventricles whereas in mice it represents both ventricular repolarization and depolarization making understanding of changes to QRS complex between mice and humans a challenge.27 Moreover, the Hand1ΔLV/ΔLV mice delete the entire Hand1LV enhancer, and are therefore likely not an appropriate comparison to humans, in which the majority of the HAND1LV enhancer is unaltered. These data, taken in total, do provide evidence that human subjects that carry the minor rs10054375 SNP, likely exhibit reduced GATA DNA binding to the 3’ most GATA site, and by consequence, reduced HAND1 expression. Subjects that carry this HAND1 minor allele may be susceptible to variation in VCS development in utero that could in part explain the shortened QRS duration with which this allele is associated.
Although we did not identify the etiology of the PR interval shortening observed in Hand1ΔLV/ΔLV mice, the results of the present study are most consistent with enhanced atrio-ventricular conduction along the physiological anatomical pathway, most notably the defective His bundle morphology. The absence of accelerated atrioventricular conduction in ex vivo Hand1ΔLV/ΔLV hearts (Online Fig. IV B–D) suggests that the PR shortening in vivo results from functional (e.g., autonomic influences), rather than structural, differences between genotypes. However, whether and how His bundle hyperplasia as seen in Hand1ΔLV/ΔLV hearts also contributes to enhanced atrioventricular condition remains to be determined.
The congenital origins of the VCS are found within the complex cell specification, differentiation, and morphological patterning gene regulatory networks that drive cardiogenesis. In the mouse, Hand1 LV expression is first detectable at E8.5 34 peaking between 9.5–10.5.35–37 Hand1 expression subsequently restricts and becomes undetectable by in situ hybridization by E13.5. Interestingly, Gja5 and Hcn4, two genes mis-regulated in Hand1ΔLV/ΔLV hearts, are essential for normal VCS function. Both Gja5 and Hcn4 show expression within the entire LV at E10.5 and, as the heart develops, restrict to the maturing VCS. Previous reports have shown Hand1 expression in the adult heart in rodents by PCR38 and humans (GTEx Analysis Release V7, dbGaP Accession phs000424.v7.p2). Our X-gal staining shows that Hand1 transcriptional elements mark the His bundle, LBB, and left PF networks, co-localized with GJA5 and CNTN2 (Online Fig. V and Fig. 4C, D). Postnatally, Gja5 expression marks the VCS; however, at E9.5, Gja5 is expressed transmurally, and at E11.5 Gja5-negative cardiomyocytes expand the compact myocardium of the free wall. After birth, the trabecular zone remodels into the definitive VCS.4 In addition to its role in VCS development, reported here, HAND1 has been shown to play a role in ventricular cardiomyocyte identity.12 Our data supports a model in which the Hand1LV enhancer drives gene expression within embryonic trabecular myocytes. Function of this enhancer is affected by a SNP associated with an overlapping GATA cis-binding element. HAND1 appears to suppress the differentiation of embryonic myocytes into the conduction lineage. Additionally, HAND1 may transcriptionally regulate factors that control the intrinsic electrophysiological properties of conduction cells or their electrical continuity with working cardiomyocytes and describing these gene regulatory networks will be the subject of ongoing studies.
The mature VCS is morphologically asymmetrical, indicating that chamber-specific gene regulatory networks are integral to its normal development.1 Hand1 expression is restricted to the developing LV. Unexpectedly, Hand1LV deletion generates a cardiac phenotype consistent with RBB block. Given that the RV breakthroughs in Hand1ΔLV/ΔLV hearts are always focused and concentric, the RBB phenotype in these mutants most likely results from localized defects that derive from the malformed His bundle disrupting patent connection with the proximal portion of right VCS. We feel this is the most likely mechanism rather than one in which a diffuse deceleration within the RBB and/or right peripheral Purkinje cells that would impact Purkinje – myocyte coupling for two reasons. First, Hand1 expression is detectable at the base of the RV septal wall (Online Fig. V B), and within the His bundle, which is malformed in Hand1ΔLV/ΔLV hearts (Fig. 5). Neither Hand1 expression nor Hand1-lineage 34 marks the right VCS. Second, an apically paced action potential propagates through both the working myocardium and the conduction system (Fig. 3K; Fig. 6H; Online Movies 6 and 12). If Purkinje fiber coupling to the working myocardium were asymmetrically impaired in Hand1 mutants, then this action potential would not propagate at the same rate across both the left and right ventricles, as we have observed in all Hand1 mutant models assayed (Fig. 3L; Fig. 6I, J; Online Movies 7 and 11). Therefore, a model in which variation in Purkinje cell number and/or diminished Purkinje cell coupling to working myocardium causes variation in QRS interval is less likely although possible. Nevertheless, a causal link between VCS gene expression, morphology, and function in Hand1ΔLV/ΔLV hearts remains obscure, and will be the subject of further study.
Hand136;75/36;75 mutants only show a 20% reduction in Hand1 mRNA. Yet, surprisingly, this modest reduction may be sufficient to cause VCS phenotypes as two of six mutants exhibit VCS abnormalities (Fig. 6E–J); however, in older Hand136;75/36;75 mutant hearts VCS phenotypes are observed in all five mice tested (Fig. 6K–T). This may indicate that aging plays an important role in the SNP mediated defects and further studies of this are planned. The variations in the VCS phenotypes observed between the Hand136;75/36;75 and Hand1ΔLV/ΔLV mutants is likely influenced by the level of Hand1 message expressed within each model. Transcription factors that can bind and influence expression of the Hand1 minor variant, such as the 5’ GATA cis-element still present within the Hand136;75/36;75 mice, likely account for the variation in severity between the Hand1 mutant VCS phenotypes. Indeed, HAND1 SNPs associated with altered QRS duration do show evidence of genetic heterogeneity7, 8, 14, 15 and identifying modifier genes that influence HAND1-mediated VCS morphogenesis will be of importance to better define the gene regulatory networks that drive VCS formation in mammals.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Ventricular Conduction System (VCS) disorders often manifest as arrhythmias; however, the genetic and developmental mechanisms that underlie VCS disfunction are not well understood.
A large number of Single Nucleotide Polymorphisms (SNPs) are associated with VCS variation but SNPs located outside of protein coding domains often provide no mechanistic insight.
SNPs located 5’ to the gene encoding the basic Helix-loop-Helix (bHLH) transcription factor HAND1 are associated with variation in ventricular conduction in Hispanic, African American and European populations.
What New Information Does This Article Contribute?
Located between these HAND1 SNPs, we identify a GATA-dependent Hand1 left ventricular enhancer (LV) that is necessary and sufficient for Hand1 LV expression.
We identify additional SNPs associated with VCS function, one within a necessary GATA DNA-binding site.
Deletion of the Hand1 LV enhancer or incorporation of the SNP sequences that alter this GATA DNA binding site in mice results in decreased Hand1 expression, altered VCS morphology, and defective VCS function.
Arrhythmias are a predictor of cardiac arrest and death. Two SNPs associated with VCS functional variation are located 5’ of the coding region of HAND1. These SNPs flank an evolutionarily-conserved non-coding sequence that is necessary and sufficient for Hand1 expression within the embryonic LV. Two highly conserved GATA DNA binding sites are required for this LV-specific expression. Further analysis shows that an additional unreported SNP in near perfect linkage disequilibrium with the flanking SNPs alters one of the GATA DNA binding sites in a way that diminishes GATA factor DNA binding. Deletion of this LV-specific Hand1 enhancer or incorporation of the minor human SNP variants into the murine Hand1 locus produce adult mice that exhibit hypertrophic VCS structures as well as abnormal VCS function. These findings show a rare and important validation of genome wide association analysis as an approach to identify causes of cardiac disease and identify HAND1 as a critical transcription factor necessary for the formation and function of the cardiac conduction system. This new understanding of HAND1 during VCS formation and function allows for specific evaluation of the HAND1 dependent VCS gene regulatory network which will provide mechanistic insight into the etiology of arrythmias.
ACKNOWLEDGEMENTS
We thank Danny Carney, for technical assistance. We thank Marco Osterwalder at the Lawrence Berkeley National Laboratory for enhancer analyses. We thank Nikhil Munshi at UT Southwestern Medical Center for sharing the Cntn23’UTR-IRES-Cre-EGFP mice. We thank Stacey Rentschler at the Washington University School of Medicine for technical insight and assistance with data interpretation. We thank the Herman B Wells Center Cardiac Developmental Biology Group for helpful discussions. Infrastructural support at the Herman B Wells Center is partially supported by the Riley Children’s Foundation and the Carleton Buehl McCulloch Chair of Pediatrics.
SOURCES OF FUNDING
This work is in part is supported by the NIH 1R01HL122123–04 P01HL134599–03, R01HL145060–01 (to A.B.F.), NIH R01HL111089, R01HL116747, R01HL141989 (to N.S.) by an award from the American Heart Association and The Children’s Heart Foundation (16SDG27260072 to J.W.V.), and by the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative, foundation Leducq (14CVD01) and from CVON ConcorGenes to V.M.C.
Nonstandard Abbreviations and Acronyms
- bHLH
basic helix–loop–helix
- CCS
cardiac conduction system
- CNS
conserved non-coding sequence
- E
embryonic day
- ECG
electrocardiogram
- EMSA
electrophoretic mobility shift assay
- GWAS
genome-wide association study
- LBB
left bundle branch
- LD
linkage disequilibrium
- LV
left ventricle
- P
postnatal day
- PF
Purkinje fiber
- RBB
right bundle branch
- RV
right ventricle
- SNP
single nucleotide polymorphism
- VCS
ventricular conduction system
- VSD
ventricular septal defect
Footnotes
DISCLOSURES
The authors declare no competing interests.
REFERENCES
- 1.Miquerol L, Beyer S, Kelly RG. Establishment of the mouse ventricular conduction system. Cardiovascular research. 2011;91:232–242 [DOI] [PubMed] [Google Scholar]
- 2.Christoffels VM, Moorman AF. Development of the cardiac conduction system: Why are some regions of the heart more arrhythmogenic than others? Circ Arrhythm Electrophysiol. 2009;2:195–207 [DOI] [PubMed] [Google Scholar]
- 3.Sizarov A, Ya J, de Boer BA, Lamers WH, Christoffels VM, Moorman AF. Formation of the building plan of the human heart: Morphogenesis, growth, and differentiation. Circulation. 2011;123:1125–1135 [DOI] [PubMed] [Google Scholar]
- 4.van Weerd JH, Christoffels VM. The formation and function of the cardiac conduction system. Development. 2016;143:197–210 [DOI] [PubMed] [Google Scholar]
- 5.Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, pr interval and qrs duration. Nature genetics. 2010;42:117–122 [DOI] [PubMed] [Google Scholar]
- 6.Chambers JC, Zhao J, Terracciano CM, et al. Genetic variation in scn10a influences cardiac conduction. Nature genetics. 2010;42:149–152 [DOI] [PubMed] [Google Scholar]
- 7.Sotoodehnia N, Isaacs A, de Bakker PI, et al. Common variants in 22 loci are associated with qrs duration and cardiac ventricular conduction. Nature genetics. 2010;42:1068–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans DS, Avery CL, Nalls MA, et al. Fine-mapping, novel loci identification, and snp association transferability in a genome-wide association study of qrs duration in african americans. Human molecular genetics. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Tucker NR, Rizki G, et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Boogaard M, Smemo S, Burnicka-Turek O, et al. A common genetic variant within scn10a modulates cardiac scn5a expression. J Clin Invest. 2014;124:1844–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincentz JW, Toolan KP, Zhang W, Firulli AB. Hand factor ablation causes defective left ventricular chamber development and compromised adult cardiac function. PLoS Genet. 2017;13:e1006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN. The hand1 and hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development. 2005;132:189–201 [DOI] [PubMed] [Google Scholar]
- 14.Prins BP, Mead TJ, Brody JA, et al. Exome-chip meta-analysis identifies novel loci associated with cardiac conduction, including adamts6. Genome Biol. 2018;19:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swenson BR, Louie T, Lin HJ, et al. Gwas of qrs duration identifies new loci specific to hispanic/latino populations. PLoS One. 2019;14:e0217796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breckenridge RA, Zuberi Z, Gomes J, Orford R, Dupays L, Felkin LE, Clark JE, Magee AI, Ehler E, Birks EJ, Barton PJ, Tinker A, Mohun TJ. Overexpression of the transcription factor hand1 causes predisposition towards arrhythmia in mice. J Mol Cell Cardiol. 2009;47:133–141 [DOI] [PubMed] [Google Scholar]
- 17.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, Rossant J. Inducible expression of an hsp68-lacz hybrid gene in transgenic mice. Development. 1989;105:707–714 [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharyya S, Bhakta M, Munshi NV. Phenotypically silent cre recombination within the postnatal ventricular conduction system. PLoS One. 2017;12:e0174517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soriano P Generalized lacz expression with the rosa26 cre reporter strain. Nature genetics. 1999;21:70–71 [DOI] [PubMed] [Google Scholar]
- 21.Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bhlh transcription factor hand1. Nature genetics. 1998;18:266–270 [DOI] [PubMed] [Google Scholar]
- 22.Vincentz JW, VanDusen NJ, Fleming AB, Rubart M, Firulli BA, Howard MJ, Firulli AB. A phox2- and hand2-dependent hand1 cis-regulatory element reveals a unique gene dosage requirement for hand2 during sympathetic neurogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:2110–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vincentz JW, McWhirter JR, Murre C, Baldini A, Furuta Y. Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis. 2005;41:192–201 [DOI] [PubMed] [Google Scholar]
- 24.Soufan AT, Ruijter JM, van den Hoff MJ, de Boer PA, Hagoort J, Moorman AF. Three-dimensional reconstruction of gene expression patterns during cardiac development. Physiol Genomics. 2003;13:187–195 [DOI] [PubMed] [Google Scholar]
- 25.Ghosh TK, Packham EA, Bonser AJ, Robinson TE, Cross SJ, Brook JD. Characterization of the tbx5 binding site and analysis of mutations that cause holt-oram syndrome. Human molecular genetics. 2001;10:1983–1994 [DOI] [PubMed] [Google Scholar]
- 26.Durocher D, Chen CY, Ardati A, Schwartz RJ, Nemer M. The atrial natriuretic factor promoter is a downstream target for nkx-2.5 in the myocardium. Mol Cell Biol. 1996;16:4648–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boukens BJ, Rivaud MR, Rentschler S, Coronel R . Misinterpretation of the mouse ecg: ‘Musing the waves of mus musculus’. J Physiol. 2014;592:4613–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maruyama M, Li BY, Chen H, et al. Fkbp12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ Res. 2011;108:1042–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stefanovic S, Christoffels VM. Gata-dependent transcriptional and epigenetic control of cardiac lineage specification and differentiation. Cell Mol Life Sci. 2015;72:3871–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14:529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Setten J, Brody JA, Jamshidi Y, et al. Pr interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat Commun. 2018;9:2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riley P, Anson-Cartwright L, Cross JC. The hand1 bhlh transcription factor is essential for placentation and cardiac morphogenesis. Nature genetics. 1998;18:271–275 [DOI] [PubMed] [Google Scholar]
- 33.Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI. Contactin-2 expression in the cardiac purkinje fiber network. Circ Arrhythm Electrophysiol. 2010;3:186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnes RM, Firulli BA, Conway SJ, Vincentz JW, Firulli AB. Analysis of the hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Developmental dynamics : an official publication of the American Association of Anatomists. 2010;239:3086–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cserjesi P, Brown D, Lyons GE, Olson EN. Expression of the novel basic helix-loop-helix gene ehand in neural crest derivatives and extraembryonic membranes during mouse development. Developmental biology. 1995;170:664–678 [DOI] [PubMed] [Google Scholar]
- 36.Firulli BA, McConville DP, Byers JS, 3rd, Vincentz JW, Barnes RM, Firulli AB Analysis of a hand1 hypomorphic allele reveals a critical threshold for embryonic viability. Developmental dynamics : an official publication of the American Association of Anatomists. 2010;239:2748–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H. Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol Cell Biol. 1995;15:3813–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thattaliyath BD, Livi CB, Steinhelper ME, Toney GM, Firulli AB. Hand1 and hand2 are expressed in the adult-rodent heart and are modulated during cardiac hypertrophy. Biochem Biophys Res Commun. 2002;297:870–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets featured within this manuscript will be made available from the corresponding author upon request.