

Abstract
Inhibition of β‐secretase 1 (BACE‐1; also known as β‐site amyloid precursor protein‐cleaving enzyme‐1) is a current approach to fight the amyloid‐β (Aβ) deposition in the brains of patients with Alzheimer's disease, and a number of BACE‐1 inhibitors are being tested in clinical trials. The BACE‐1 inhibitor NB‐360, although not a clinical compound, turned out to be a valuable pharmacological tool to investigate the effects of BACE‐1 inhibition on the deposition of different Aβ species in amyloid precursor protein (APP) transgenic mice. Furthermore, chronic animal studies with NB‐360 revealed relationships between BACE‐1 inhibition, Aβ deposition, and Aβ‐related downstream effects on neuroinflammation, neuronal function, and markers of neurodegeneration. NB‐360 effects on the processing of physiological BACE‐1 substrates as well as on nonenzymatic BACE‐1 functions have been investigated, complementing studies in BACE‐1 knockout mice. Because NB‐360 is also an inhibitor for BACE‐2, nonclinical studies in adult animals revealed physiological effects of BACE‐2 inhibition.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- Arc
Arctic
- Aβ
amyloid‐β
- BACE
β‐secretase
- NfL
neurofilament light chain
- pKa
logarithm of the acid dissociation constant
- PS‐1
presenilin‐1
- S1, S2, S3
protease subsites that bind substrate side chains of the first, second, third amino acid C‐terminal of the scissile bond
- SEZ‐6
seizure‐6 protein
- tg
transgenic
1. AMYLOID PRECURSOR PROTEIN, β‐SECRETASE, and AMYLOID PRECURSOR PROTEIN‐CLEAVING ENZYME INHIBITORS
In 1987, amyloid precursor protein (APP) was identified as the precursor of the amyloid‐β (Aβ) peptide found in amyloid plaques and vascular deposits in Alzheimer's disease (AD) brains (Kang et al., 1987). Soon thereafter, an enzymatic activity that cleaved APP at the Met596–Asn597 bond and created the N‐terminus of the Aβ peptide (Asn1–Ala2–Glu3–Phe4 …) was identified in neuronal and nonneuronal cells and called β‐secretase (Haass et al., 1993). In a major parallel effort in the late 1990s of the last century, the membrane‐bound aspartic protease β‐site APP‐cleaving enzyme‐1 (BACE‐1) was discovered as the predominant β‐secretase (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999). An X‐ray structure of the catalytic domain complexed with the peptide inhibitor OM99‐2 identified key enzyme–ligand interactions, mostly hydrogen bonds of the backbone amides, and was thought to be a starting point for inhibitor design (Hong et al., 2000).
Because standard high‐throughput screening campaigns on large compound collections failed to deliver potent hits for further optimization, industrial and academic researchers applied approaches similar to those developed for inhibitors of other aspartyl proteases like renin and human immunodeficiency virus (HIV) protease (Ghosh, Osswald, & Prato, 2016; Nguyen, Hamada, Kimura, & Kiso, 2008). In the coming years, these resource‐intense drug discovery programmes provided a large number of compounds of reasonable MW, good in vitro potency, and Aβ reduction in animals (Huang et al., 2012; Hussain et al., 2007; Lerchner et al., 2010; Rueeger et al., 2011; Rueeger et al., 2013). However, overall, the results were disappointing, because applying the strategies for the inhibition of peripheral/systemic proteases regularly led to peptidic or peptide‐mimetic structures. Those molecules did not provide the required combination of potency, selectivity, metabolic stability, and absence of cytochrome P450 inhibition together with acceptable brain penetration necessary for the BACE‐1 inhibitors. Furthermore, in parallel to the drug discovery efforts, our understanding of the treatment needs in AD increased substantially. It was realized that a potential therapy with a BACE‐1 inhibitor would need to be long term, but the bar was raised even higher when the concepts of early or even preventive treatment were put forward. Such treatments clearly require compounds with an excellent overall profile, not available from peptidomimetics. The initial hype turned into frustration, BACE‐1 was sometimes classified as an “undruggable” target, and investments into BACE‐1 inhibitor programmes were reduced or discontinued in many research organizations. The situation only changed with the application of fragment screening approaches for hit finding and the subsequent optimization of the hits (Jennings et al., 2008; Mandal et al., 2016; Stamford et al., 2012; Wang et al., 2010; Zhu et al., 2010). This novel approach delivered chemical starting points for inhibitor design in completely nonpeptidic chemical space, not previously associated with inhibition of aspartyl proteases. These new binding motifs generally shared an acyl‐guanidine or amidine structure embedded in a 5‐ or 6‐membered ring, allowing two nitrogens to interact with the catalytic aspartates Asp32 and Asp228 in the enzyme active site of BACE‐1 (Figure 1). In subsequent years, most of the BACE‐1 inhibitor research turned towards these new substance classes, which were quickly proven to hold great promise for in vitro potency and selectivity. They also appeared to suffer less from the metabolism and brain penetration problems encountered before with the peptidomimetics.
Figure 1.

Structures of BACE inhibitors from peptidomimetics to NB‐360. Fully peptidic hydroxyethylene compound OM99‐2 occupying binding subsites from P4 to P4′ (Hong et al., 2000), peptidomimetic ethanolamine NB‐216 (Lerchner et al., 2010), cyclic ethanolamine 11c (Rueeger et al., 2013) occupying subsites P3, P1, and P2′, and cyclic amidine NB‐360 (Neumann et al., 2015) occupying P1 and P3. Interaction sites with catalytic aspartates are marked in blue
2. PROFILE OF THE BACE INHIBITOR NB‐360 AND ITS EFFECT ON LOWERING Aβ
When we entered the field of cyclic amidine‐derived BACE‐1 inhibitors, we aimed to select a metabolically stable cyclic motif, which would provide opportunity to modulate the basicity of the amidine moiety. In our previous work, we found that moderately basic compounds are more likely to cross the blood–brain barrier, with only little recognition by the P‐glycoprotein drug transporter system (Lerchner et al., 2010). Consequently, we placed an electron‐withdrawing oxygen atom into the six‐membered ring next to the carbon β to the nitrogens forming the cyclic amidine moiety. By derivatization, we fine tuned the pKa value of this 3‐amino‐1,4‐oxazine core structure to the 6.5 to 7.5 range. We found that an additional trifluoromethyl–methyl substitution next to the oxygen at the same carbon β to the nitrogens provided a good combination of high membrane permeability and low sensitivity to P‐glycoprotein‐mediated transport (Veenstra et al., 2018). The S1 subsite was filled with a phenyl residue, connecting it to an S3‐filling picolinic acid with an amide bond (nomenclature according to Schechter & Berger, 1967). The amide makes an important interaction to the carbonyl oxygen of Gly230 in BACE‐1, contributing to the overall potency. However, the P1–P3 amide proved to be a major site of metabolism in this series. The resulting compound NB‐360 has low nanomolar potency in BACE enzyme inhibition assays and in cellular assays for Aβ release. NB‐360 has several thousand‐fold selectivity for BACE‐1 over the important off‐targets pepsin, cathepsin D, and cathepsin E but is equally potent on BACE‐2, an evolutionary‐related enzyme with high structural similarity to BACE‐1 (Neumann et al., 2015; Ostermann et al., 2006).
NB‐360 showed potent reduction of Aβ production when dosed p.o. to rats, mice, or APP‐transgenic (tg) mice; single doses of 3 μmol·kg−1 (equivalent to 1.5 mg·kg−1, p.o.) resulted in Aβ reduction of more than 50% in rat brain and CSF up to 8 hr. An acute p.o. dose of 30 μmol·kg−1 (14.5 mg·kg−1) provided 91% reduction at 4 hr after the dose and maintained >50% inhibition for more than 16 hr (Neumann et al., 2015). In wild‐type C57/BL6 mice, a daily dose of 100 μmol·kg−1 (45 mg·kg−1, p.o.) for 6 weeks reduced the brain level of endogenous mouse Aβ40 by 68%. NB‐360 showed comparable acute efficacy in an APP‐tg mouse strain, APP51/16 mice, which carries the transgene for human APP without any familial AD‐related mutations (Rabe et al., 2011). In young (preplaque) APP51/16 mice, a single p.o. dose of 30 μmol·kg−1 reduced Triton X‐100‐soluble human Aβ40 by 80% (4 hr after dose; Neumann et al., 2015), showing that NB‐360 potency is comparable in wild‐type and APP‐tg mice.
However, NB‐360 showed a strong hypopigmentation phenotype in chronic mouse studies (see below), considered to be a significant safety flag, in particular for long‐term use in early or asymptomatic AD patients. For this reason, development of NB‐360 was stopped before entering clinical trials. Apart from this phenotype, there were no findings that would limit chronic dosing of NB‐360 in animal studies. We therefore decided to make this well‐characterized and pharmacologically very active compound available to academic researchers, in order to advance knowledge about the effects of BACE inhibitor treatment in animal models of AD.
3. NB‐360 IN CHRONIC APP‐TRANSGENIC MOUSE STUDIES
Since its original publication, the BACE inhibitor NB‐360 has been used in several studies in various laboratories to test aspects of Aβ accumulation and dynamics, effects on markers of neuroinflammation and neurodegeneration, and neuronal activity and to investigate aspects of BACE‐2 inhibition and effects on the processing of physiological BACE substrates other than APP. The use of the same compound, at identical or similar doses, makes the results comparable and helps to establish robust relationships between the different models and the readouts. Furthermore, it should be mentioned that NB‐360 was well tolerated, for treatments up to 6‐month duration, across the various studies in different laboratories with different routes of administration. The completed studies as of today are summarized in Table 1.
Table 1.
Summary of studies performed with NB‐360 in mouse models
| Mouse model | Age at onset of treatment (months) | Treatment duration (months) | Daily dose | Main endpoints | Reference |
|---|---|---|---|---|---|
| APP51/16, female | 14.5 | 1.5 | 100 μmol·kg−1 (45 mg·kg−1) | Aβ deposition and neuroinflammation | Neumann et al. (2015) |
| APP23, female | 17.5 | 3 | 30 μmol·kg−1 (14.5 mg·kg−1) | Brain microhaemorrhages | Beckmann et al. (2016) |
| APP23, male | 15 | 6.5 | 0.5 g·kg−1 in chow | Mouse tau in CSF | Schelle et al. (2017) |
| APPPS1, male and female | 1.5 | 6 | 0.5 g·kg−1 chow | Mouse tau in CSF | Schelle et al. (2017) |
| APPPS1, male | 1.5 | 6 | 0.5 g·kg−1 chow | Mouse neurofilament light chain in CSF | Bacioglu et al. (2016) |
| APP23xPS45, female | 6–7 | 1.5 | 0.25 g·kg−1 chow | Neuronal hyperactivity and slow brain waves | Keskin et al. (2017) |
| APPPS1xVGLUT1 | 4 | 3 | 0.25 g·kg−1 chow | Aβ plaque kinetics | Peters et al. (2018) |
| APP tg‐SweArc | 10 | 3 | 0.5 g·kg−1 chow | PET of Aβ protofibrils | Meier et al. (2018) |
| SEZ6+/+ and SEZ6−/− × GFP, male and female | 3–4 | 0.75 | 0.25 g·kg−1 chow | Spine density and dynamics | Zhu et al. (2018) |
| C57/Bl6J | 4–5 | 2 | 100 μmol·kg−1 (45 mg·kg−1) | Hair depigmentation | Shimshek et al. (2016) |
Note: Aβ: amyloid‐β.
In a few cases, NB‐360 was dosed by daily p.o. gavage; however, for most of the chronic studies, NB‐360 was milled into chow at a concentration of 0.25 g·kg−1 food. We determined the Aβ reduction in wild‐type mouse brain after 5 days on NB‐360 food pellets and found endogenous mouse Aβ40 reduced by 84.4% (light phase of the day) and by 87.2% during the dark phase (Figure 2). The Aβ reduction is slightly lower in APP23‐tg mice (carrying the transgene for human APP with the “Swedish” double mutation; Sturchler‐Pierrat & Staufenbiel, 2000); we observed a 73% reduction both during the light and the dark phase (Figure 2a). Reduced activity of BACE‐1 inhibitors has been described already in mice carrying the “Swedish” APP mutation (Yamakawa, Yagishita, Futai, & Ishiura, 2010) and most likely results from the different subcellular localization of BACE‐1 processing “Swedish” APP (Haass et al., 1995). Dosing NB‐360 via the mouse food at 0.5 g·kg−1 food delivered a pharmacodynamic response (acute Aβ lowering of nearly 100% in the brain of wild‐type mice) that is comparable with a daily 100 μmol·kg−1 (45 mg·kg−1) p.o. gavage dose, whereas an NB‐360 dose of 0.25 g·kg−1 in food gives rise to an Aβ reduction of >80% in wild types (which corresponds to 20 μmol·kg−1 (10 mg·kg−1) p.o. gavage; Figure 2a).
Figure 2.
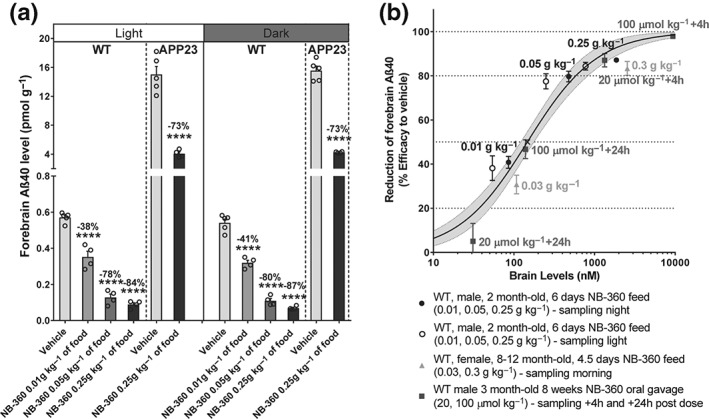
Establishment of NB‐360 treatment in food. (a) Effect of 6‐day NB‐360 dosing via food at different doses (0.01, 0.05, and 0.25 g·kg−1 NB‐360 in rodent food pellets on forebrain Aß40 levels in wild‐type (WT, n = 4–5) and APP23 (n = 4–5) male mice (2 months old) during the light and dark phase. Mean ± SEM. Statistics: one‐way ANOVA with Holm–Sidak multiple comparison test versus vehicle (***P < 0.0001) for WT and two‐tailed t test versus vehicle (****P < 0.0001) for APP23. (b) Pharmacokinetics/pharmacodynamics relationship of NB‐360 brain levels on forebrain Aß40 in WT mice (n = 4–5) at different ages, different doses, route of administration, and sampling time points as indicated. Mean ± SEM. Fitting equation: y = bottom + (top‐bottom)∕(1 + 10((LogIC50 −log X) * Hill slope)); bottom constraint = 0; top constraint = 100; and IC50 = 147 nM. The 95% confidence intervals were indicated. R 2 = 0.858. Rodent food pellets were provided by Granovit AG, Switzerland. Studies were approved by the Swiss Cantonal Veterinary Authority of Basel City, Switzerland, under Licence Number BS‐1094. C57BL/6J and transgenic, heterozygous APP23 (B6,D2‐Tg(Thy1App)23Sdz/B6.Cg‐Tg(Thy1‐APP)3Somm/J, available from Jackson Laboratory, Stock No. 030504) as well as APP23 wild‐type littermate controls were obtained from Novartis Pharma AG breeding colonies. Mice were housed in individually ventilated cage racks (maximum four mice per XJ‐type cage). The animals had access to food and water ad libitum. For brain compound and Aβ40 analysis, see Neumann et al. (2015)
4. EFFECTS OF NB‐360 TREATMENT ON Aβ PATHOLOGY
The initial investigation into the ability of NB‐360 to suppress the formation of Aβ plaques was done in the rather “mild” APP51/16 mouse model (Neumann et al., 2015). APP51/16 mice are one of the “slowest” mouse models for Aβ accumulation with a deposition rate of approximately 1.7 μg Aβ40·g−1 brain tissue·month−1, but compared with the rate of Aβ deposition in the human AD brain (0.02 μg·g−1 tissue·month−1; Roberts et al., 2017), the rate of Aβ deposition in APP51/16 mice is around 100‐fold faster. Plaque deposition in APP51/16 mice starts at around 12 months, and the phase of plaque growth lasts until the mice are very old (>20 months; Rijal Upadhaya et al., 2012). Treatment started at 14.5 months, when only a few plaques are visible in immunohistochemical analysis with an antibody staining the C‐terminus of Aβ, and mice were treated for 6 weeks. Within this time, cortical plaque area and insoluble Aβ load increased about threefold in the vehicle control group, and NB‐360 treatment completely prevented this increase and retained the Aβ load at baseline.
APP23 mice (with the “Swedish” mutation in the human APP transgene) have been treated with NB‐360 starting at 15 months of age for 6.5 months (Schelle et al., 2017) and at 17.5 months of age for 3 months (Beckmann et al., 2016). Compared with the APP51/16 mice at treatment start, APP23 mice have higher amounts of deposited, insoluble Aβ42 in the cortex: 0.14 nmol·g−1 tissue (APP51/16) versus 55 nmol·g−1 tissue in APP23 (15 months), a consequence of the higher Aβ production due to the presence of the “Swedish” mutation (Axelman, Basun, Winblad, & Lannfelt, 1994). In the vehicle groups, the amount of insoluble Aβ increased 1.5‐fold (3‐month treatment) and approximately fivefold (6‐month treatment). Treatment with NB‐360 significantly reduced the Aβ load compared with the vehicle group for both brain Aβ levels and Aβ immunostaining (Schelle et al., 2017). The doses applied with the NB‐360‐containing chow were sufficient to keep the Aβ close to the baseline.
NB‐360 was dosed to mice carrying both the APP “Arctic” and “Swedish” mutation (tg‐ArcSwe mice; Lord et al., 2006). These mice are characterized by a high tendency to form Aβ protofibrils, early intraneuronal Aβ deposition, and markedly accelerated plaque deposition, compared with tg mice with APP “Swedish” alone. Ten‐month‐old tg‐ArcSwe mice were treated with NB‐360 for 3 months. Levels of formic acid‐soluble Aβ40 and Aβ42 increased two to threefold over time in the vehicle group, whereas levels remained at baseline in the NB‐360 group. This study also addressed treatment effects on soluble protofibrils, a specific form of Aβ, by biochemical as well as PET (see below).
Studies have also been performed in mice carrying both the APP “Swedish” mutation and an additional mutation in the gene for presenilin‐1 (PS‐1). The APPPS1 mouse line carries the PS‐1 L166P mutation, and the APP23xPS45 mice contain the PS‐1 G384A mutation (Busche et al., 2008; Radde et al., 2006). The combination of the two mutations enhances the rate of plaque deposition in the double tg mice; both mouse lines show amyloid plaques as early as 1.5–2 months of age. The study in the APPPS‐1 mice was started at 1.5 months of age and continued for 6 months (Schelle et al., 2017). During this time, the brain Aβ load in the vehicle group increased dramatically (approximately 20‐fold), and almost all of this increase was inhibited in the NB‐360 treatment group, suggesting that the BACE inhibitor could cope even with very high rates of Aβ generation. Furthermore, a separate study in APPPS1 mice from 1.5 to 7.5 months of age (6‐month treatment) could confirm the attenuation of the Aβ load by stereological quantification of cortical amyloid plaques; Aβ load increased only slightly above baseline (Bacioglu et al., 2016).
Finally, in a study in APP23xPS45 mice, the treatment started when the mice were already 6–7 months old and continued for 1.5 months (Keskin et al., 2017). At this age, the mice have high insoluble Aβ load, 23 nmol·g−1 tissue for Aβ40 and 58 nmol·g−1 for Aβ42. Baseline data are not available; the comparison between vehicle group and treatment group showed 54% reduction in insoluble Aβ40 and 34% reduction in insoluble Aβ42. A similar reduction was observed on thioflavin S‐positive Aβ deposits. In the absence of baseline data, it cannot be estimated whether or not the observed difference between vehicle and treatment group accounts for the full increase in the vehicle group during the short 1.5‐month study duration. Taken together, these data show comparable efficacy of NB‐360 treatment across a wide range of APP‐tg mouse models with very different Aβ production and deposition rates.
On the basis of these data, it is hypothesized that also in humans, the primary efficacy of a BACE‐1 inhibitor (its ability to reduce Aβ deposition) will be not influenced by different Aβ generation rates, that is, in APP mutation carriers with a higher rate of Aβ production compared with those who do not carry familial AD mutations. This is not in contradiction with the finding that Aβ load was slightly increased above baseline when highly overproducing APP‐tg mice were treated with NB‐360. It needs to be taken into account that the BACE‐1 inhibition in vivo never reaches 100%. Assuming a very good efficacy of 95% inhibition, the remaining 5% account for an accumulation of 1 ng·g−1 tissue·month−1 in humans but for 85 ng·g−1 tissue·month−1 in APP51/16 mice and for 170 ng·g−1 tissue·month−1 in APP23 mice. Because NB‐360 treatment could keep the total Aβ load in APP51/16 mice at baseline, we assumed that physiological Aβ clearance mechanisms (transport, cellular uptake, and extracellular degradation) may be able to handle the even smaller amounts of Aβ being produced in humans under conditions of BACE‐1 inhibitor treatment and thereby prevent any further Aβ deposition. Previous work has shown that rather subtle changes in the human Aβ kinetics can lead to accelerated deposition and AD: The rate of Aβ production is increased by 24% in familial mutation carriers, and the rate of Aβ clearance is reduced by 30% in sporadic AD patients (Mawuenyega et al., 2010; Potter et al., 2013), whereas a 30% lower Aβ production rate in carriers of the APP A673T variant results in protection from AD (Jonsson et al., 2012; Martiskainen et al., 2017). Given the observed strong effects of NB‐360 (and other BACE inhibitors) in Aβ‐overproducing mouse models, it should be possible to compensate for these physiological imbalances by BACE‐1 inhibitor treatment in humans. A probably more interesting question related to BACE‐1 inhibitor treatment is its ability not only to stop further Aβ deposition but also to remove existing deposits. Such an effect can be only indirect and depends on the effectiveness of Aβ clearance mechanisms in the diseased brain. It is likely that differently aggregated Aβ forms are likely to have very different clearance rates, and the highly aggregated fibrillary thioflavine S‐positive core plaques may be much more resistant compared with soluble or proto‐fibrillary Aβ aggregates. Studies with NB‐360 shed some light on this: In the 6‐week study in APP23xPS45 mice, NB‐360 treatment almost completely removed the “halo” of diffuse Aβ in the surroundings of core plaques (detected with the anti amyloid fibril specific OC antibody Sigma Aldrich AB2286), but much less, if any, effect on the thioflavine S‐positive core plaques was observed within this rather short treatment period (Keskin et al., 2017). A dedicated study with a novel, antibody‐based PET tracer selective for soluble, protofibrillary forms of deposited Aβ ([124I]RmAb158‐svFv8D3) in APP tg‐SweArc mice extended these findings. Although untreated mice showed a strong increase in the PET signal, the signal in mice treated with NB‐360 remained at baseline intensity (Meier et al., 2018). However, because the study was started when little of the protofibrillar Aβ was present in the mouse brain, it can only inform us that NB‐360 treatment prevented the accumulation of this Aβ form. Until another study with significant amounts of protofibrillar Aβ present at start is conducted, we are unable to conclude that NB‐360 treatment can actually contribute to the removal of pre‐existing diffuse deposits. A solid answer to this question may be of relevance for clinical imaging with Aβ PET ligands. Since all the currently available ligands predominantly recognize fibrillary Aβ, they may be not able to inform us about treatment‐induced changes in the diffuse, nonfibrillar, or protofibrillar populations of Aβ in the AD brain. In agreement with this, a recent microamyloid–PET study performed in mice with the structurally different BACE‐1 inhibitor JNJ‐49146981 and using one of the “classical” Aβ PET ligands (18F florbetapir) did not detect any differences between the control and treatment groups (Deleye et al., 2017). However, a longitudinal in vivo two photon microscopy study using methoxy‐X04 to label Aβ in APPPS1‐tg mice, with 3‐month NB‐360 treatment during the phase of rapid Aβ deposition, gave a more detailed picture (Peters et al., 2018). The most prominent effect of NB‐360 treatment was prevention of the formation of new plaques, whereas the growth of existing plaques could only be reduced but not completely stopped. If one extrapolates back from the effect size in this highly overproducing model to the much less dramatic situation in human AD (see above), it can be predicted that BACE‐1 inhibition may completely stop growth of existing cored plaques and prevent the formation of new plaques. Whether or not a net reduction of plaque load because of BACE‐1 inhibition will be observed in human AD clinical trials remains to be seen; it will also be dependent on the duration of the clinical observation period. Very recently, it was shown that 18‐months of treatment with the potent BACE inhibitor verubecestat in mild‐to‐moderate AD patients did lead to a small but measurable reduction of Aβ load, measured with the PET ligand flutemetamol (Egan et al., 2018). This seems to indicate that physiological removal of deposited Aβ over 18 months, under conditions of little Aβ production, is slow but exists. When clinical trials with BACE‐1 inhibitors are performed in prevention settings, that is, in the early stages of Aβ deposition, a stop of further deposition is, however, likely to occur.
5. EFFECT OF NB‐360 ON AD‐RELEVANT PATHOLOGIES DOWNSTREAM OF Aβ
5.1. Neuroinflammation
Microgliosis, in addition to Aβ and tau pathology, is a hallmark of AD (Prokop, Miller, & Heppner, 2013). The change in microglia activation states in response to the ongoing disease is a subject of high current interest, and data suggest that the combination of Aβ pathology and a pro‐inflammatory response, mainly from microglia, is driving the clinical disease (Cai, Hussain, & Yan, 2014). In studies in APP‐tg mice with established plaque pathology, the number of ionized calcium‐binding adapter molecule 1(Iba1)‐positive cells, which mostly represent microglia, and the number of glial fibrillary acidic protein (GFAP)‐positive cells (astrocytes) are often found increased near plaques. The number of positive clusters of microglia and astrocytes was quantified in APP51/16 mice, treated with NB‐360 for 6 weeks (Neumann et al., 2015). We found a correlation between plaque area (as determined with NT‐12 antibody, which recognizes the C‐terminus of Aβ) and the number of microglia and astrocyte clusters. Compared with the vehicle group, NB‐360 treatment significantly reduced the number of neuroinflammatory cells to baseline values. This finding supports the concept that Aβ deposition is, directly or indirectly, the cause of glia cell activation in AD. More studies are needed to separate the relative importance of Aβ pathology and neuroinflammation on neurodegenerative processes.
5.2. Tau
There has been a long debate on the relative contribution of deposited Aβ and aggregated and hyperphosphorylated tau protein to the development of AD. Careful histopathological evaluation of the spread of the disease in humans suggests that Aβ deposition accelerates the physiologically slowly developing, age‐related tau pathology (Braak, Thal, Ghebremedhin, & Del Tredici, 2011; Thal, Capetillo‐Zarate, Del Tredici, & Braak, 2006). Data in mouse model systems have already shown that overexpression of mutant forms of tau does not induce any form of Aβ pathology, whereas APP‐tg mice show both the formation of tau aggregates, as well as elevated levels of soluble mouse tau in the CSF (Maia et al., 2013; Phinney et al., 1999). Human biomarker data show that Aβ pathology (detected as enhanced binding of Aβ‐PET ligands and/or reduction of CSF Aβ42) is detectable several years earlier than elevated levels of phospho‐tau or total tau in the CSF (Villemagne et al., 2013). Although these data seem to establish the sequence of events in the development of the human disease, it was not known whether a reduction of Aβ generation by inhibition of BACE‐1 has an effect on tau. Recent studies in old APP23 and middle‐aged APPPS‐1 mice showed elevated CSF tau, and this elevation follows the increased Aβ deposition (Maia et al., 2013; Schelle et al., 2017). The elevation of tau in these mice was markedly reduced by treatment with NB‐360 and parallels the treatment effects on deposited Aβ (Schelle et al., 2017). The results of this study further support the cause–effect relationship between Aβ and tau in AD and suggests that treatments that prevent Aβ accumulation may have a neuroprotective effect, using tau elevation in CSF as a surrogate marker. Treatment was started when CSF tau concentrations were very low, resembling a situation in human AD before neurodegeneration‐related biomarkers have started to rise. The effects of BACE‐1 inhibitor treatment at a stage of established tau pathology have not been investigated.
5.3. Neurofilament light chain
Elevated neurofilament light chain (NfL) is a more recently discovered biomarker of neurodegenerative diseases, including AD, frontotemporal dementia, amyotrophic lateral sclerosis, and multiple sclerosis (Chatterjee et al., 2018; Gille et al., 2018; Goossens et al., 2018; Siller et al., 2018). An increase in NfL in CSF and blood is interpreted as an indication of ongoing axonal damage (Lee, Taghian, & Petratos, 2014). Although both tau and NfL are both markers of neurodegeneration, they represent different degenerative processes in the brain. In ageing APPPS1 mice, NfL was shown to increase both in CSF and in plasma, and treatment with NB‐360 could significantly reduce this increase in CSF. A decrease in plasma was also observed but did not reach statistical significance (Bacioglu et al., 2016). Although more data have to be collected to support the use of NfL as a neurodegeneration biomarker in AD, the results from the APP‐tg mice indicate a possible use of NfL measurements in clinical studies.
5.4. Neuronal activity
Although numerous studies have demonstrated the effects of BACE‐1 inhibitor treatment on various details of Aβ deposition, there is clearly a need to investigate the functional consequences in a situation that has at least some similarity with the human disease. Busche and coworkers have established a method to measure neuronal activity in living APP23xPS45‐tg mice with two‐photon calcium imaging, both in single neurons around amyloid plaques and in neuronal populations in different brain regions (Busche et al., 2012; Busche et al., 2015). Neurons near amyloid plaques showed marked hyperactivity (higher firing rate), compared with “healthy” neurons. Treatment with NB‐360 for 6 weeks almost completely normalized the firing rate (Keskin et al., 2017). In a second experiment, the effect of NB‐360 treatment on long‐range circuits within the mouse brain was investigated. In the brains of wild‐type mice, there was a strong temporal correlation between the neuronal activity patterns of the various areas of the brain (occipital, somatosensory, motor, and frontal regions), reminiscent of the default network activity observed in humans (Busche et al., 2015; Chhatwal et al., 2013). In contrast, a highly asynchronous pattern was observed in the brains of 6‐ to 7‐month‐old APP23xPS45 mice, indicating a severe impairment of neuronal crosstalk because of Aβ deposition. Treatment with NB‐360 significantly restored synchronicity in the brains of these mice (Keskin et al., 2017). Both the neuronal hyperactivity and the slow wave nonsynchronicity in plaque‐bearing APP23xPS45 mice showed a correlation with the insoluble Aβ load and were, to a large part, reversible. For interpretation of this result, we assume that in these mice, despite their high plaque load, irreversible neurodegeneration has not occurred to a significant degree. Such mice may therefore be considered as mimicking the “preclinical” stage of human AD, characterized by elevated Aβ load, but showing no signs of neurodegeneration (ie no biomarker rise) or of cognitive impairment. The animal data suggest that BACE‐1 inhibitor treatment at this stage of reversible pathology may restore neuronal function.
6. EFFECTS OF NB‐360 TREATMENT ON PHYSIOLOGICAL BACE SUBSTRATES
The first characterization studies in BACE‐1 knockout mice did not detect any major impairments, in terms of viability, behaviour, and fertility, and the deletion of BACE‐1 was considered to be without functional consequences (Luo et al., 2001; Ohno et al., 2004). More recently, a more focused investigation of BACE‐1 knockout mice in combination with proteomic techniques has discovered a number of proteins (in addition to APP) that are processed by BACE‐1 in the adult mouse brain. These include, but are not limited to neuregulin‐1, APP‐like protein‐1 and APP‐like protein‐2, a close homologue of L1, and seizure‐6 (SEZ‐6; Barão et al., 2015; Dislich et al., 2015; Kuhn et al., 2012) This has raised three important questions: (a) To what extent can such findings be translated to humans? (b) To what extent will such findings also be observed as a consequence of pharmacotherapy with a BACE‐1 inhibitor? (c) To what extent such findings can limit the use of BACE‐1 inhibitors? Although we are just beginning to answer these questions, it is already clear that most of the observed mouse phenotypes are of developmental origin. Preclinical toxicological studies of the BACE‐1 inhibitor verubecestat did not produce findings that were similar to the knockout mouse phenotypes (Kennedy et al., 2016). However, this does not rule out the possiblity that the processing of specific physiological BACE‐1 substrates is affected by BACE‐1 inhibitor treatment and that this may happen in humans as well. Although no human data are currently available, a study with NB‐360 in mice has shown an effect on the processing of the SEZ‐6, and a SEZ‐6‐related effect on dendritic spine dynamics, with a treatment‐related reduction of spine density (Zhu et al., 2018). This study identified SEZ‐6 as a top candidate for a BACE‐1 inhibitor‐sensitive physiological BACE‐1 substrate. It remains to be seen whether pharmacologically active doses of BACE‐1 inhibitors in humans will have an effect on SEZ‐6 processing and whether or not this will translate into a risk in the human disease situation (Zhu, Peters, Filser, & Herms, 2018).
7. BACE‐2‐MEDIATED EFFECTS OF NB‐360
BACE‐2 is the closest homologue to BACE‐1 and shares 52% amino acid identity (Saunders, Kim, & Tanzi, 1999; Solans, Estivill, & de La Luna, 2000). BACE‐2 expression in the brain is low under physiological conditions and does not seem to have a role in APP processing. Therefore, its inhibition may not be relevant for the therapeutic treatment for AD. However, most BACE inhibitors in clinical development are not selective for BACE‐1 over BACE‐2. There is considerable peripheral BACE‐2 expression in most tissues (Bennett et al., 2000), where it is involved in different proteolytic processes (Casas et al., 2010; Rochin et al., 2013; Stutzer et al., 2013). Importantly, BACE‐2 knockout mice displayed a hypopigmented phenotype (Cai et al., 2012; Dominguez et al., 2005; Shimshek et al., 2016). This depigmentation in epidermis and hair was also observed in animals treated chronically with different BACE inhibitors (Cebers et al., 2017; Shimshek et al., 2016). At this point in time, the impact of BACE‐2 coinhibition in long‐term and preventive clinical trials in AD is unknown. Very recently, BACE‐2 expression has been shown in distinct brain regions, as well as BACE‐2 up‐regulation in pathophysiological situations (Voytyuk et al., 2018) again urging for selectivity of BACE‐1 inhibitors over BACE‐2.
8. EFFECT OF NB‐360 ON BRAIN MICROHAEMORRHAGES
Besides the aggregation of extracellular Aβ in the brain parenchyma, an accumulation of Aβ can be observed in the blood vessels of the brain in the majority of AD patients. This cerebral amyloid angiopathy may lead to vascular pathologies and is a risk factor for cerebral haemorrhages. Furthermore, major severe side effects of Aβ‐directed immunotherapies are vasogenic oedema and cerebral microhaemorrhage (Mo, Li, Yang, Liu, & Feng, 2017; Salloway et al., 2014; Sperling et al., 2012). These findings led to the implementation for regular clinical MRI monitoring for amyloid‐directed therapies (Sperling et al., 2011) In addition, the Food and Drug Administration initially requested to evaluate amyloid‐directed therapies, including BACE inhibitors, for their risk to induce microhaemorrhages in appropriate preclinical models. Many mouse models exist that display cerebral amyloid angiopathy and microhaemorrhages (Klohs, Rudin, Shimshek, & Beckmann, 2014). The APP23 line is a well‐established model for detecting these vascular alterations non‐invasively (Beckmann, Gerard, Abramowski, Cannet, & Staufenbiel, 2011). It was shown that a 3‐month daily treatment with 30 μmol·kg−1 p.o. gavage of female APP23 mice (from ages 17.5 to 20.5 months) with NB‐360 resulted in no increase in the number of microhaemorrhages (Beckmann et al., 2016). As recently reported, another BACE‐1/2 inhibitor, verubecestat, also showed no increase in the number of microhaemorrhages in 18‐ to 22‐month‐old Tg2576 mice (Villarreal et al., 2017). These results illustrate the different mechanisms of action of anti‐Aβ immunotherapy and direct inhibition of Aβ generation; the latter is not associated with a risk of cerebral microbleeds.
9. BACE‐1 FUNCTIONS INDEPENDENT OF ITS ENZYMATIC ACTIVITY
Somewhat surprisingly, BACE‐1 protein fulfils stabilizing functions for several ion channels in the mossy fibre tract of the hippocampus, including the voltage‐gated potassium channel KV3.4 (Lehnert et al., 2016). The presence of BACE‐1 protein possibly helps to target the ion channels to the presynaptic terminal and, by this, regulates neuronal excitability through direct, nonenzymatic interactions. Although studies in BACE‐1 knockout mice clearly showed a reduced expression of KV3.4 in the mouse hippocampus, compared with wild type control animals, no significant reduction was observed in young mice treated with NB‐360, at a dose that almost completely blocks Aβ generation (Hartmann et al., 2018). These early data need to be further expanded and substantiated but suggest that pharmacotherapy with a BACE‐1 inhibitor does not negatively impact on the function of these ion channels.
10. CONCLUSION
The BACE inhibitor NB‐360 has been extensively used in various different rodent models of β‐amyloidosis to investigate effects on Aβ‐related pathologies and the role of BACE‐1/2 in this function. These studies complemented the work done with BACE‐1 knockout mice, built a bridge to ongoing BACE inhibitors in clinical trials in AD, and increased our understanding of the physiological functions of BACE‐1 in adult animals. The use of the same compound at standardized doses and administration routes in different animal models allowed for insightful and robust cross‐comparisons of the results and hopefully will give rise to a deeper understanding of disease onset and progression in AD.
10.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander, Fabbro et al., 2017; Alexander, Striessnig et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Neumann U, Machauer R, Shimshek DR. The β‐secretase (BACE) inhibitor NB‐360 in preclinical models: From amyloid‐β reduction to downstream disease‐relevant effects. Br J Pharmacol. 2019;176:3435–3446. 10.1111/bph.14582
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol., 174(Suppl 1), S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174(S1), S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelman, K. , Basun, H. , Winblad, B. , & Lannfelt, L. (1994). A large Swedish family with Alzheimer's disease with a codon 670/671 amyloid precursor protein mutation. A clinical and genealogical investigation. Archives of Neurology, 51, 1193–1197. 10.1001/archneur.1994.00540240037013 [DOI] [PubMed] [Google Scholar]
- Bacioglu, M. , Maia, L. F. , Preische, O. , Schelle, J. , Apel, A. , Kaeser, S. A. , … Jucker, M. (2016). Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron, 91, 56–66. 10.1016/j.neuron.2016.05.018 [DOI] [PubMed] [Google Scholar]
- Barão, S. , Gärtner, A. , Leyva‐Díaz, E. , Demyanenko, G. , Munck, S. , Vanhoutvin, T. , … de Strooper, B. (2015). Antagonistic effects of BACE1 and APH1B‐γ‐secretase control axonal guidance by regulating growth cone collapse. Cell Reports, 12, 1367–1376. 10.1016/j.celrep.2015.07.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann, N. , Doelemeyer, A. , Zurbruegg, S. , Bigot, K. , Theil, D. , Frieauff, W. , … Shimshek, D. R. (2016). Longitudinal noninvasive magnetic resonance imaging of brain microhemorrhages in BACE inhibitor‐treated APP transgenic mice. Neurobiology of Aging, 45, 50–60. 10.1016/j.neurobiolaging.2016.05.009 [DOI] [PubMed] [Google Scholar]
- Beckmann, N. , Gerard, C. , Abramowski, D. , Cannet, C. , & Staufenbiel, M. (2011). Noninvasive magnetic resonance imaging detection of cerebral amyloid angiopathy‐related microvascular alterations using superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer's disease: Application to passive Aβ immunotherapy. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31, 1023–1031. 10.1523/JNEUROSCI.4936-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, B. D. , Babu‐Khan, S. , Loeloff, R. , Louis, J. C. , Curran, E. , Citron, M. , & Vassar, R. (2000). Expression analysis of BACE2 in brain and peripheral tissues. The Journal of Biological Chemistry, 275, 20647–20651. 10.1074/jbc.M002688200 [DOI] [PubMed] [Google Scholar]
- Braak, H. , Thal, D. R. , Ghebremedhin, E. , & Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. Journal of Neuropathology and Experimental Neurology, 70, 960–969. 10.1097/NEN.0b013e318232a379 [DOI] [PubMed] [Google Scholar]
- Busche, M. A. , Chen, X. , Henning, H. A. , Reichwald, J. , Staufenbiel, M. , Sakmann, B. , & Konnerth, A. (2012). Critical role of soluble amyloid‐β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 109, 8740–8745. 10.1073/pnas.1206171109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche, M. A. , Eichhoff, G. , Adelsberger, H. , Abramowski, D. , Wiederhold, K. H. , Haass, C. , … Garaschuk, O. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science, 321, 1686–1689. 10.1126/science.1162844 [DOI] [PubMed] [Google Scholar]
- Busche, M. A. , Kekus, M. , Adelsberger, H. , Noda, T. , Forstl, H. , Nelken, I. , & Konnerth, A. (2015). Rescue of long‐range circuit dysfunction in Alzheimer's disease models. Nature Neuroscience, 18, 1623–1630. 10.1038/nn.4137 [DOI] [PubMed] [Google Scholar]
- Cai, J. , Qi, X. , Kociok, N. , Skosyrski, S. , Emilio, A. , Ruan, Q. , … Boulton, M. E. (2012). β‐Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Molecular Medicine, 4, 980–991. 10.1002/emmm.201101084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Z. , Hussain, M. D. , & Yan, L. J. (2014). Microglia, neuroinflammation, and β‐amyloid protein in Alzheimer's disease. The International Journal of Neuroscience, 124, 307–321. 10.3109/00207454.2013.833510 [DOI] [PubMed] [Google Scholar]
- Casas, S. , Casini, P. , Piquer, S. , Altirriba, J. , Soty, M. , Cadavez, L. , … Novials, A. (2010). BACE2 plays a role in the insulin receptor trafficking in pancreatic ss‐cells. American Journal of Physiology Endocrinology and Metabolism, 299, E1087–E1095. 10.1152/ajpendo.00420.2010 [DOI] [PubMed] [Google Scholar]
- Cebers, G. , Alexander, R. C. , Haeberlein, S. B. , Han, D. , Goldwater, R. , Ereshefsky, L. , … Kugler, A. R. (2017). AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 55, 1039–1053. 10.3233/JAD-160701 [DOI] [PubMed] [Google Scholar]
- Chatterjee, P. , Goozee, K. , Sohrabi, H. R. , Shen, K. , Shah, T. , Asih, P. R. , … Martins, R. N. (2018). Association of plasma neurofilament light chain with neocortical amyloid‐β load and cognitive performance in cognitively normal elderly participants. Journal of Alzheimer's Disease: JAD, 63, 479–487. 10.3233/JAD-180025 [DOI] [PubMed] [Google Scholar]
- Chhatwal, J. P. , Schultz, A. P. , Johnson, K. , Benzinger, T. L. , Jack, C. Jr. , Ances, B. M. , … Sperling, R. A. (2013). Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology, 81, 736–744. 10.1212/WNL.0b013e3182a1aafe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleye, S. , Waldron, A. M. , Verhaeghe, J. , Bottelbergs, A. , Wyffels, L. , Van Broeck, B. , … Staelens, S. (2017). Evaluation of small‐animal PET outcome measures to detect disease modification induced by BACE inhibition in a transgenic mouse model of Alzheimer disease. Journal of Nuclear Medicine, 58, 1977–1983. 10.2967/jnumed.116.187625 [DOI] [PubMed] [Google Scholar]
- Dislich, B. , Wohlrab, F. , Bachhuber, T. , Müller, S. A. , Kuhn, P.‐H. , Hogl, S. , … Lichtenthaler, S. F. (2015). Label‐free quantitative proteomics of mouse cerebrospinal fluid detects BACE1 protease substrates in vivo. Molecular & Cellular Proteomics, 14(10), 2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, D. , Tournoy, J. , Hartmann, D. , Huth, T. , Cryns, K. , Deforce, S. , … de Strooper, B. (2005). Phenotypic and biochemical analyses of BACE1‐ and BACE2‐deficient mice. The Journal of Biological Chemistry, 280, 30797–30806. 10.1074/jbc.M505249200 [DOI] [PubMed] [Google Scholar]
- Egan, M. F. , Kost, J. , Tariot, P. N. , Aisen, P. S. , Cummings, J. L. , Vellas, B. , … Michelson, D. (2018). Randomized trial of verubecestat for mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 378, 1691–1703. 10.1056/NEJMoa1706441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A. K. , Osswald, H. L. , & Prato, G. (2016). Recent progress in the development of HIV‐1 protease inhibitors for the treatment of HIV/AIDS. Journal of Medicinal Chemistry, 59, 5172–5208. 10.1021/acs.jmedchem.5b01697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille, B. , De Schaepdryver, M. , Goossens, J. , Dedeene, L. , De Vocht, J. , Oldoni, E. , … Poesen, K. (2018). Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathology and Applied Neurobiology, 89, 307–373. [DOI] [PubMed] [Google Scholar]
- Goossens, J. , Bjerke, M. , Van Mossevelde, S. , Van den Bossche, T. , Goeman, J. , De Vil, B. , … Engelborghs, S. (2018). Diagnostic value of cerebrospinal fluid tau, neurofilament, and progranulin in definite frontotemporal lobar degeneration. Alzheimer's Research & Therapy, 10, 31–41. 10.1186/s13195-018-0364-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass, C. , Hung, A. Y. , Schlossmacher, M. G. , Oltersdorf, T. , Teplow, D. B. , & Selkoe, D. J. (1993). Normal cellular processing of the β‐amyloid precursor protein results in the secretion of the amyloid β peptide and related molecules. Annals of the New York Academy of Sciences, 695, 109–116. 10.1111/j.1749-6632.1993.tb23037.x [DOI] [PubMed] [Google Scholar]
- Haass, C. , Lemere, C. A. , Capell, A. , Citron, M. , Seubert, P. , Schenk, D. , … Selkoe, D. J. (1995). The Swedish mutation causes early‐onset Alzheimer's disease by β‐secretase cleavage within the secretory pathway. Nature Medicine, 1, 1291–1296. 10.1038/nm1295-1291 [DOI] [PubMed] [Google Scholar]
- Harding, S. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, S. , Zheng, F. , Kyncl, M. C. , Karch, S. , Voelkl, K. , Zott, B. , … Huth, T. (2018). β‐Secretase BACE1 promotes surface expression and function of Kv3.4 at hippocampal mossy fiber synapses. The Journal of Neuroscience, 38, 3480–3494. 10.1523/JNEUROSCI.2643-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, L. , Koelsch, G. , Lin, X. , Wu, S. , Terzyan, S. , Ghosh, A. K. , … Tang, J. (2000). Structure of the protease domain of memapsin 2 (β‐secretase) complexed with inhibitor. Science, 290, 150–153. 10.1126/science.290.5489.150 [DOI] [PubMed] [Google Scholar]
- Huang, H. , La, D. S. , Cheng, A. C. , Whittington, D. A. , Patel, V. F. , Chen, K. , … Fremeau, R. T. Jr. (2012). Structure‐ and property‐based design of aminooxazoline xanthenes as selective, orally efficacious, and CNS penetrable BACE inhibitors for the treatment of Alzheimer's disease. Journal of Medicinal Chemistry, 55, 9156–9169. 10.1021/jm300598e [DOI] [PubMed] [Google Scholar]
- Hussain, I. , Hawkins, J. , Harrison, D. , Hille, C. , Wayne, G. , & Cutler, L. (2007). Oral administration of a potent and selective non‐peptidic BACE‐1 inhibitor decreases β‐cleavage of amyloid precursor protein and amyloid‐β production in vivo. Journal of Neurochemistry, 100, 802–809. 10.1111/j.1471-4159.2006.04260.x [DOI] [PubMed] [Google Scholar]
- Hussain, I. , Powell, D. , Howlett, D. R. , Tew, D. G. , Meek, T. D. , & Chapman, C. (1999). Identification of a novel aspartic protease (Asp 2) as β‐secretase. Molecular and Cellular Neurosciences, 14, 419–427. 10.1006/mcne.1999.0811 [DOI] [PubMed] [Google Scholar]
- Jennings, L. D. , Cole, D. C. , Stock, J. R. , Sukhdeo, M. N. , Ellingboe, J. W. , Cowling, R. , … Bard, J. (2008). Acylguanidine inhibitors of β‐secretase: Optimization of the pyrrole ring substituents extending into the S1′ substrate binding pocket. Bioorganic & Medicinal Chemistry Letters, 18, 767–771. 10.1016/j.bmcl.2007.11.043 [DOI] [PubMed] [Google Scholar]
- Jonsson, T. , Atwal, J. K. , Steinberg, S. , Snaedal, J. , Jonsson, P. V. , Bjornsson, S. , … Stefansson, K. (2012). A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature, 488, 96–99. 10.1038/nature11283 [DOI] [PubMed] [Google Scholar]
- Kang, J. , Lemaire, H. G. , Unterbeck, A. , Salbaum, J. M. , Masters, C. L. , Grzeschik, K. H. , … Müller‐Hill, B. (1987). The precursor of Alzheimer's disease amyloid A4 protein resembles a cell‐surface receptor. Nature, 325, 733–736. 10.1038/325733a0 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. E. , Stamford, A. W. , Chen, X. , Cox, K. , Cumming, J. N. , Dockendorf, M. F. , … Forman, M. S. (2016). The BACE1 inhibitor verubecestat (MK‐8931) reduces CNS β‐amyloid in animal models and in Alzheimer's disease patients. Science Translational Medicine, 8, 363ra150–363ra150. 10.1126/scitranslmed.aad9704 [DOI] [PubMed] [Google Scholar]
- Keskin, A. D. , Kekus, M. , Adelsberger, H. , Neumann, U. , Shimshek, D. R. , Song, B. , … Busche, M. A. (2017). BACE inhibition‐dependent repair of Alzheimer's pathophysiology. Proceedings of the National Academy of Sciences of the United States of America, 114, 8631–8636. 10.1073/pnas.1708106114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klohs, J. , Rudin, M. , Shimshek, D. , & Beckmann, N. (2014). Imaging of cerebrovascular pathology in animal models of Alzheimer's disease. Frontiers in Aging Neuroscience, 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, P. H. , Koroniak, K. , Hogl, S. , Colombo, A. , Zeitschel, U. , Willem, M. , … Lichtenthaler, S. F. (2012). Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. The EMBO Journal, 31, 3157–3168. 10.1038/emboj.2012.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. Y. , Taghian, K. , & Petratos, S. (2014). Axonal degeneration in multiple sclerosis: Can we predict and prevent permanent disability? Acta Neuropathologica Communications, 2, 97 10.1186/s40478-014-0097-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert, S. , Hartmann, S. , Hessler, S. , Adelsberger, H. , Huth, T. , & Alzheimer, C. (2016). Ion channel regulation by β‐secretase BACE1—Enzymatic and non‐enzymatic effects beyond Alzheimer's disease. Channels, 10, 365–378. 10.1080/19336950.2016.1196307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchner, A. , Machauer, R. , Betschart, C. , Veenstra, S. , Rueeger, H. , McCarthy, C. , … Neumann, U. (2010). Macrocyclic BACE‐1 inhibitors acutely reduce Aβ in brain after po application. Bioorganic & Medicinal Chemistry Letters, 20, 603–607. 10.1016/j.bmcl.2009.11.092 [DOI] [PubMed] [Google Scholar]
- Lord, A. , Kalimo, H. , Eckman, C. , Zhang, X. Q. , Lannfelt, L. , & Nilsson, L. N. (2006). The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiology of Aging, 27, 67–77. 10.1016/j.neurobiolaging.2004.12.007 [DOI] [PubMed] [Google Scholar]
- Luo, Y. , Bolon, B. , Kahn, S. , Bennett, B. D. , Babu‐Khan, S. , & Denis, P. (2001). Mice deficient in BACE1, the Alzheimer's β‐secretase, have normal phenotype and abolished β‐amyloid generation. Nature Neuroscience, 4, 231232. [DOI] [PubMed] [Google Scholar]
- Maia, L. F. , Kaeser, S. A. , Reichwald, J. , Hruscha, M. , Martus, P. , Staufenbiel, M. , & Jucker, M. (2013). Changes in amyloid‐β and tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Science Translational Medicine, 5, 194re192. [DOI] [PubMed] [Google Scholar]
- Mandal, M. , Wu, Y. , Misiaszek, J. , Li, G. , Buevich, A. , Caldwell, J. P. , … Stamford, A. W. (2016). Structure‐based design of an iminoheterocyclic β‐site amyloid precursor protein cleaving enzyme (BACE) inhibitor that lowers central Aβ in nonhuman primates. Journal of Medicinal Chemistry, 59, 3231–3248. 10.1021/acs.jmedchem.5b01995 [DOI] [PubMed] [Google Scholar]
- Martiskainen, H. , Herukka, S. K. , Stancakova, A. , Paananen, J. , Soininen, H. , Kuusisto, J. , … Hiltunen, M. (2017). Decreased plasma β‐amyloid in the Alzheimer's disease APP A673T variant carriers. Annals of Neurology, 82, 128–132. 10.1002/ana.24969 [DOI] [PubMed] [Google Scholar]
- Mawuenyega, K. G. , Sigurdson, W. , Ovod, V. , Munsell, L. , Kasten, T. , Morris, J. C. , … Bateman, R. J. (2010). Decreased clearance of CNS β‐amyloid in Alzheimer's disease. Science, 330, 1774 10.1126/science.1197623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, S. R. , Syvanen, S. , Hultqvist, G. , Fang, X. T. , Roshanbin, S. , Lannfelt, L. , … Sehlin, D. (2018). Antibody‐based in vivo PET imaging detects amyloid‐β reduction in Alzheimer transgenic mice after BACE‐1 inhibition. Journal of Nuclear Medicine, 59(12), 1885–1891. 10.2967/jnumed.118.213140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo, J. J. , Li, J. Y. , Yang, Z. , Liu, Z. , & Feng, J. S. (2017). Efficacy and safety of anti‐amyloid‐β immunotherapy for Alzheimer's disease: A systematic review and network meta‐analysis. Annals of Clinical Translational Neurology, 4, 931–942. 10.1002/acn3.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, U. , Rueeger, H. , Machauer, R. , Veenstra, S. J. , Lueoend, R. M. , Tintelnot‐Blomley, M. , … Jacobson, L. H. (2015). A novel BACE inhibitor NB‐360 shows a superior pharmacological profile and robust reduction of amyloid‐β and neuroinflammation in APP transgenic mice. Molecular Neurodegeneration, 10, 44 10.1186/s13024-015-0033-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, J. T. , Hamada, Y. , Kimura, T. , & Kiso, Y. (2008). Design of potent aspartic protease inhibitors to treat various diseases. Archiv der Pharmazie, 341, 523–535. 10.1002/ardp.200700267 [DOI] [PubMed] [Google Scholar]
- Ohno, M. , Sametsky, E. A. , Younkin, L. H. , Oakley, H. , Younkin, S. G. , & Citron, M. (2004). BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron, 41, 27–33. 10.1016/S0896-6273(03)00810-9 [DOI] [PubMed] [Google Scholar]
- Ostermann, N. , Eder, J. , Eidhoff, U. , Zink, F. , Hassiepen, U. , Worpenberg, S. , … Gerhartz, B. (2006). Crystal structure of human BACE2 in complex with a hydroxyethylamine transition‐state inhibitor. Journal of Molecular Biology, 355, 249–261. 10.1016/j.jmb.2005.10.027 [DOI] [PubMed] [Google Scholar]
- Peters, F. , Salihoglu, H. , Rodrigues, E. , Herzog, E. , Blume, T. , Filser, S. , … Herms, J. (2018). BACE1 inhibition more effectively suppresses initiation than progression of β‐amyloid pathology. Acta Neuropathologica, 135, 695–710. 10.1007/s00401-017-1804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney, A. L. , Deller, T. , Stalder, M. , Calhoun, M. E. , Frotscher, M. , Sommer, B. , … Jucker, M. (1999). Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. The Journal of Neuroscience, 19, 8552–8559. 10.1523/JNEUROSCI.19-19-08552.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter, R. , Patterson, B. W. , Elbert, D. L. , Ovod, V. , Kasten, T. , Sigurdson, W. , … Bateman, R. J. (2013). Increased in vivo amyloid‐β42 production, exchange, and loss in presenilin mutation carriers. Science Translational Medicine, 5, 189ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop, S. , Miller, K. R. , & Heppner, F. L. (2013). Microglia actions in Alzheimer's disease. Acta Neuropathologica, 126, 461–477. 10.1007/s00401-013-1182-x [DOI] [PubMed] [Google Scholar]
- Rabe, S. , Reichwald, J. , Ammaturo, D. , de Strooper, B. , Saftig, P. , Neumann, U. , & Staufenbiel, M. (2011). The Swedish APP mutation alters the effect of genetically reduced BACE1 expression on the APP processing. Journal of Neurochemistry, 119, 231–239. 10.1111/j.1471-4159.2011.07412.x [DOI] [PubMed] [Google Scholar]
- Radde, R. , Bolmont, T. , Kaeser, S. A. , Coomaraswamy, J. , Lindau, D. , Stoltze, L. , … Jucker, M. (2006). Aβ42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Reports, 7, 940–946. 10.1038/sj.embor.7400784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijal Upadhaya, A. , Capetillo‐Zarate, E. , Kosterin, I. , Abramowski, D. , Kumar, S. , Yamaguchi, H. , … Thal, D. R. (2012). Dispersible amyloid β‐protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: Their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiology of Aging, 33, 2641–2660. 10.1016/j.neurobiolaging.2011.12.032 [DOI] [PubMed] [Google Scholar]
- Roberts, B. R. , Lind, M. , Wagen, A. Z. , Rembach, A. , Frugier, T. , Li, Q. X. , … Masters, C. L. (2017). Biochemically‐defined pools of amyloid‐β in sporadic Alzheimer's disease: Correlation with amyloid PET. Brain: A Journal of Neurology, 140, 1486–1498. 10.1093/brain/awx057 [DOI] [PubMed] [Google Scholar]
- Rochin, L. , Hurbain, I. , Serneels, L. , Fort, C. , Watt, B. , Leblanc, P. , … van Niel, G. (2013). BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proceedings of the National Academy of Sciences of the United States of America, 110, 10658–10663. 10.1073/pnas.1220748110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueeger, H. , Lueoend, R. , Machauer, R. , Veenstra, S. J. , Jacobson, L. H. , Staufenbiel, M. , … Neumann, U. (2013). Discovery of cyclic sulfoxide hydroxyethylamines as potent and selective β‐site APP‐cleaving enzyme 1 (BACE1) inhibitors: Structure based design and in vivo reduction of amyloid β‐peptides. Bioorganic & Medicinal Chemistry Letters, 23, 5300–5306. 10.1016/j.bmcl.2013.07.071 [DOI] [PubMed] [Google Scholar]
- Rueeger, H. , Rondeau, J.‐M. , McCarthy, C. , Möbitz, H. , Tintelnot‐Blomley, M. , Neumann, U. , & Desrayaud, S. (2011). Structure based design, synthesis and SAR of cyclic hydroxyethylamine (HEA) BACE‐1 inhibitors. Bioorganic & Medicinal Chemistry Letters, 21, 1942–1947. 10.1016/j.bmcl.2011.02.038 [DOI] [PubMed] [Google Scholar]
- Salloway, S. , Sperling, R. , Fox, N. C. , Blennow, K. , Klunk, W. , Raskind, M. , … Bapineuzumab 301 and 302 Clinical Trial Investigators . (2014). Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 370, 322–333. 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, A. J. , Kim, T.‐W. , & Tanzi, R. E. (1999). BACE maps to chromosome 11 and a BACE homolog, BACE2, reside in the obligate Down syndrome region of chromosome 21. Science, 286, 1255–1255. [Google Scholar]
- Schechter, I. , & Berger, A. (1967). On the size of the active site in proteases. I. Papain. Biochemical and Biophysical Research Communications, 27, 157–162. 10.1016/S0006-291X(67)80055-X [DOI] [PubMed] [Google Scholar]
- Schelle, J. , Hasler, L. M. , Göpfert, J. C. , Joos, T. O. , Vanderstichele, H. , Stoops, E. , … Kaeser, S. A. (2017). Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 13, 701–709. 10.1016/j.jalz.2016.09.005 [DOI] [PubMed] [Google Scholar]
- Shimshek, D. R. , Jacobson, L. H. , Kolly, C. , Zamurovic, N. , Balavenkatraman, K. K. , Morawiec, L. , … Neumann, U. (2016). Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Scientific Reports, 6, 21917 10.1038/srep21917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller, N. , Kuhle, J. , Muthuraman, M. , Barro, C. , Uphaus, T. , Groppa, S. , … Bittner, S. (2018). Serum neurofilament light chain is a biomarker of acute and chronic neuronal damage in early multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England): 10.1177/1352458518765666 [DOI] [PubMed] [Google Scholar]
- Sinha, S. , Anderson, J. P. , Barbour, R. , Basi, G. S. , Caccavello, R. , Davis, D. , … John, V. (1999). Purification and cloning of amyloid precursor protein β‐secretase from human brain. Nature, 402, 537–540. 10.1038/990114 [DOI] [PubMed] [Google Scholar]
- Solans, A. , Estivill, X. , & de La Luna, S. (2000). A new aspartyl protease on 21q22.3, BACE2, is highly similar to Alzheimer's amyloid precursor protein β‐secretase. Cytogenetics and Cell Genetics, 89, 177–184. 10.1159/000015608 [DOI] [PubMed] [Google Scholar]
- Sperling, R. , Salloway, S. , Brooks, D. J. , Tampieri, D. , Barakos, J. , Fox, N. C. , … Grundman, M. (2012). Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: A retrospective analysis. The Lancet Neurology, 11, 241–249. 10.1016/S1474-4422(12)70015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling, R. A. , Jack, C. R. Jr. , Black, S. E. , Frosch, M. P. , Greenberg, S. M. , Hyman, B. T. , … Schindler, R. J. (2011). Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: Recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 7, 367–385. 10.1016/j.jalz.2011.05.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamford, A. W. , Scott, J. D. , Li, S. W. , Babu, S. , Tadesse, D. , Hunter, R. , … Greenlee, W. J. (2012). Discovery of an orally available, brain penetrant BACE1 inhibitor that affords robust CNS Aβ reduction. ACS Medicinal Chemistry Letters, 3, 897–902. 10.1021/ml3001165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler‐Pierrat, C. , & Staufenbiel, M. (2000). Pathogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Annals of the New York Academy of Sciences, 920, 134–139. [DOI] [PubMed] [Google Scholar]
- Stutzer, I. , Selevsek, N. , Esterhazy, D. , Schmidt, A. , Aebersold, R. , & Stoffel, M. (2013). Systematic proteomic analysis identifies β‐site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic β‐cells. The Journal of Biological Chemistry, 288, 10536–10547. 10.1074/jbc.M112.444703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal, D. R. , Capetillo‐Zarate, E. , Del Tredici, K. , & Braak, H. (2006). The development of amyloid β protein deposits in the aged brain. Science of aging knowledge environment: SAGE KE, 2006, re1. [DOI] [PubMed] [Google Scholar]
- Vassar, R. , Bennett, B. D. , Babu‐Khan, S. , Kahn, S. , Mendiaz, E. A. , Denis, P. , … Citron, M. (1999). β‐Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science, 286, 735–741. 10.1126/science.286.5440.735 [DOI] [PubMed] [Google Scholar]
- Veenstra, S. J. , Rueeger, H. , Voegtle, M. , Lueoend, R. , Holzer, P. , Hurth, K. , … Machauer, R. (2018). Discovery of amino‐1,4‐oxazines as potent BACE‐1 inhibitors. Bioorganic & Medicinal Chemistry Letters, 28, 2195–2200. 10.1016/j.bmcl.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Villarreal, S. , Zhao, F. , Hyde, L. A. , Holder, D. , Forest, T. , Sondey, M. , … Kennedy, M. E. (2017). Chronic verubecestat treatment suppresses amyloid accumulation in advanced aged Tg2576‐AβPPswe mice without inducing microhemorrhage. Journal of Alzheimer's Disease: JAD, 59, 1393–1413. 10.3233/JAD-170056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne, V. L. , Burnham, S. , Bourgeat, P. , Brown, B. , Ellis, K. A. , Salvado, O. , … Masters, C. L. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: A prospective cohort study. The Lancet Neurology, 12, 357–367. 10.1016/S1474-4422(13)70044-9 [DOI] [PubMed] [Google Scholar]
- Voytyuk, I. , Mueller, S. A. , Herber, J. , Snellinx, A. , Moechars, D. , van Loo, G. , … De Strooper, B. (2018). BACE2 distribution in major brain cell types and identification of novel substrates. Life Science Alliance, 1 10.26508/lsa.201800026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. S. , Strickland, C. , Voigt, J. H. , Kennedy, M. E. , Beyer, B. M. , Senior, M. M. , … Wyss, D. F. (2010). Application of fragment‐based NMR screening, X‐ray crystallography, structure‐based design, and focused chemical library design to identify novel μM leads for the development of nM BACE‐1 (β‐site APP cleaving enzyme 1) inhibitors. Journal of Medicinal Chemistry, 53, 942–950. 10.1021/jm901472u [DOI] [PubMed] [Google Scholar]
- Yamakawa, H. , Yagishita, S. , Futai, E. , & Ishiura, S. (2010). β‐Secretase inhibitor potency is decreased by aberrant β‐cleavage location of the “Swedish mutant” amyloid precursor protein. The Journal of Biological Chemistry, 285, 1634–1642. 10.1074/jbc.M109.066753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, R. , Bienkowski, M. J. , Shuck, M. E. , Miao, H. , Tory, M. C. , & Pauley, A. M. (1999). Membrane‐anchored aspartyl protease with Alzheimer's disease β‐secretase activity. Nature, 402, 533–537. 10.1038/990107 [DOI] [PubMed] [Google Scholar]
- Zhu, K. , Peters, F. , Filser, S. , & Herms, J. (2018). Consequences of pharmacological BACE inhibition on synaptic structure and function. Biological Psychiatry. Jun 23. pii: S0006–3223(18)31515–4. 10.1016/j.biopsych.2018.04.022, 84, 478–487. [DOI] [PubMed] [Google Scholar]
- Zhu, K. , Xiang, X. , Filser, S. , Marinkovic, P. , Dorostkar, M. M. , Crux, S. , … Herms, J. (2018). β‐Site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biological Psychiatry, 83, 428–437. 10.1016/j.biopsych.2016.12.023 [DOI] [PubMed] [Google Scholar]
- Zhu, Z. , Sun, Z. Y. , Ye, Y. , Voigt, J. , Strickland, C. , Smith, E. M. , … Hunter, J. C. (2010). Discovery of cyclic acylguanidines as highly potent and selective β‐site amyloid cleaving enzyme (BACE) inhibitors: Part I—Inhibitor design and validation. Journal of Medicinal Chemistry, 53, 951–965. 10.1021/jm901408p [DOI] [PubMed] [Google Scholar]